Abstract
Recent evidence highlights that the cancer cell energy requirements vary greatly from normal cells and that cancer cells exhibit different metabolic phenotypes with variable participation of both glycolysis and oxidative phosphorylation. NADH–ubiquinone oxidoreductase (Complex I) is the largest complex of the mitochondrial electron transport chain and contributes about 40% of the proton motive force required for mitochondrial ATP synthesis. In addition, Complex I plays an essential role in biosynthesis and redox control during proliferation, resistance to cell death, and metastasis of cancer cells. Although knowledge about the structure and assembly of Complex I is increasing, information about the role of Complex I subunits in tumorigenesis is scarce and contradictory. Several small molecule inhibitors of Complex I have been described as selective anticancer agents; however, pharmacologic and genetic interventions on Complex I have also shown pro-tumorigenic actions, involving different cellular signaling. Here, we discuss the role of Complex I in tumorigenesis, focusing on the specific participation of Complex I subunits in proliferation and metastasis of cancer cells.
Introduction: The Anatomy of Complex I
Mammalian Complex I (NADH–quinone oxidoreductase) is the largest respiratory complex of the electron transport chain (ETC) (1). It oxidizes NADH produced in the tricarboxylic acid (TCA) cycle and β-oxidation of fatty acids, regenerating the NAD+ levels in the mitochondrial matrix (2). Complex I couples electron transfer from NADH to ubiquinone to the translocation of four protons from the mitochondrial matrix to the intermembrane space (3) generating, together with the proton-pumping Complexes III and IV, the electrochemical proton gradient required for ATP synthesis (4, 5).
Complex I is a L-shaped assembly (Figure 1A) composed of a hydrophilic peripheral arm, which contains the redox centers involved in electron transfer, and a membrane arm containing the proton-translocating machinery (6, 7). Forty five subunits make up the mitochondrial Complex I (Figure 1A), including 14 conserved “core” subunits that are sufficient to catalyze energy transduction and which are shared equally between peripheral and membrane arms, and 31 “accessory” or “supernumerary” subunits distributed around the core (2, 8–10).
Figure 1
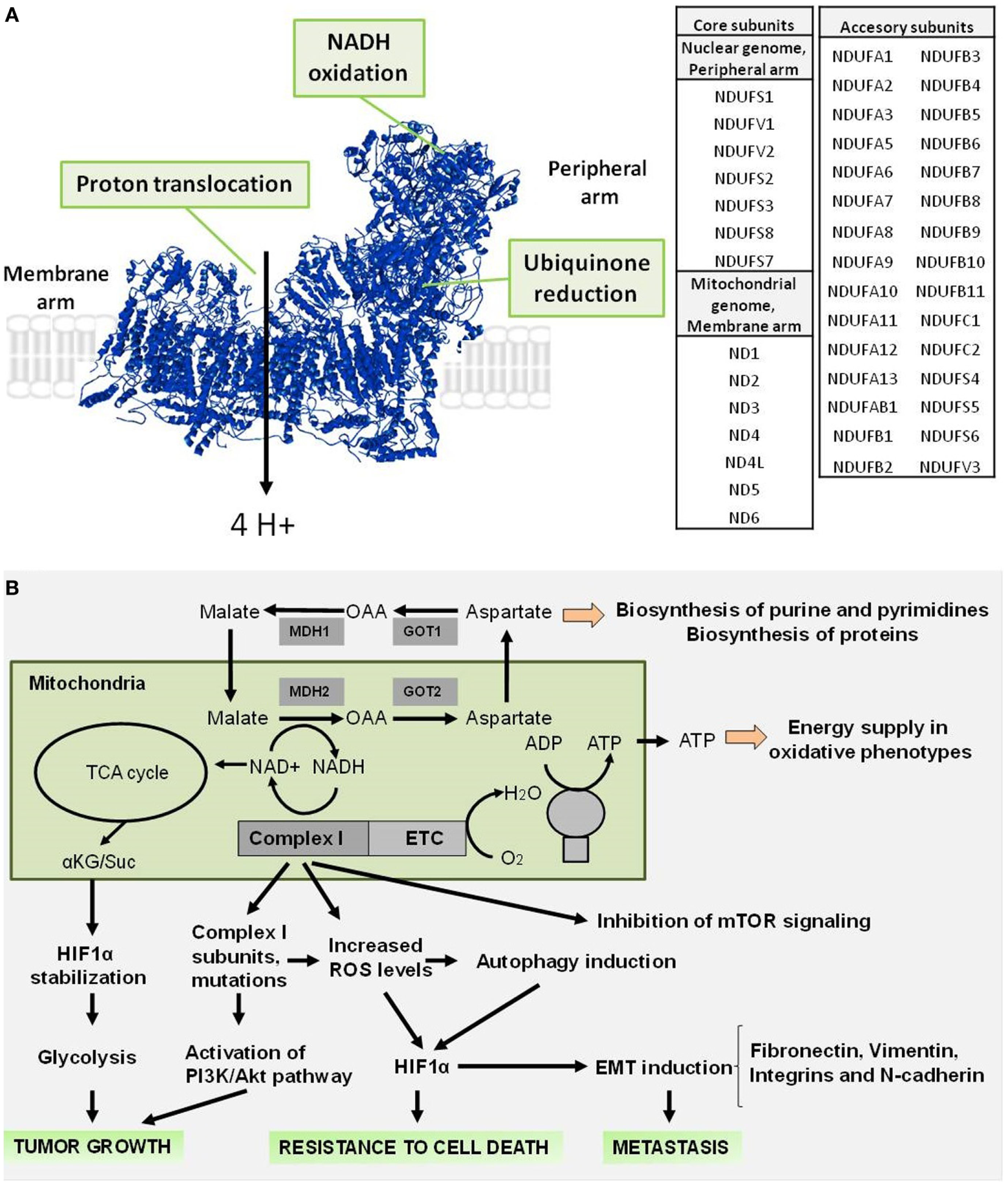
Structure of mammalian respiratory Complex I and role during tumorigenesis. (A) Structure of mammalian Complex I (PDB: 4UQ8), indicating the sites involved in the NADH oxidoreductase activity (NADH oxidation and ubiquinone reduction) in the peripheral arm and in the proton translocation in the membrane arm. A list of the core and accessory subunits that compose the mitochondrial Complex I is shown. (B) Complex I signaling involved in the supporting of tumor growth, resistance to cell death, and promoting of metastasis.
Mutations in mitochondrial and nuclear genes that encode Complex I subunits are a contributing factor in several pathological conditions such as neurodegenerative diseases (11–13), diabetes (14, 15), and cancer (12, 16). Regarding cancer, reports are controversial. On the one hand, several studies suggest that Complex I subunits are tumor suppressors (17–19). On the other hand, mutations in Complex I genes promote progression of prostate (20), thyroid (21, 22), breast (23), lung (24), renal (25, 26), colorectal (27), and head and neck tumors (28). Here, we will focus on recent advances in understanding the role of Complex I in tumorigenesis and highlight the specific participation of Complex I subunits in supporting cancer cell proliferation and metastasis.
Role of Complex I in the Proliferation of Cancer Cells
Classically, the role of NADH oxidation by Complex I activity as an entry point of electrons in the ETC has been considered essential to generate the membrane potential across the mitochondrial inner membrane that supports ATP synthesis (5). However, recent evidence suggests that the non-energetic roles of the mitochondrial respiration, and in particular Complex I activity, support proliferation by providing electron acceptors and regenerating oxidized cofactors (29, 30). Complex I activity maintains the cellular NAD+ pool and the NAD+/NADH ratio (Figure 1B) necessary to sustain the activity of the mitochondrial malate dehydrogenase (MDH2), a NAD+/NADH ratio-dependent oxidoreductase and the generation of aspartate (29, 30). Consistently, the inhibition of ETC activity using Complex I inhibitors (metformin, rotenone, and piericidin) affects the NAD+/NADH balance, producing a decrease in electron acceptors. This event limits the aspartate synthesis, which is a precursor of purine and pyrimidine synthesis (31) required for biosynthesis of nucleic acids and macromolecules during cell proliferation (29, 30).
In addition, maintenance of the NAD+/NADH ratio by Complex I is essential for the induction of adaptive mechanisms to hypoxia through hypoxia-inducible factor 1-alpha (HIF1α) stabilization (18) and to promote a metabolic remodeling toward aerobic glycolysis (Figure 1B), a phenomenon known as the Warburg effect (32, 33). Upon Complex I inhibition, NADH accumulation allosterically inhibits the TCA cycle enzyme α-ketoglutarate (αKG) dehydrogenase, thereby increasing the α-ketoglutarate/succinate (αKG/Suc) ratio, which favors the activity of the prolyl-hydroxylases in charge of the degradation of HIF1α, and causing tumor growth arrest (18, 34, 35). Similarly, this correlation between Complex I inhibition and HIF1α destabilization has been described with ETC inhibitors (36, 37). Conversely, certain mutations in mitochondrial DNA (mtDNA)-encoded core subunits that produce oxidative phosphorylation (OXPHOS) defects have pro-tumorigenic effects (38). For example, a heteroplasmic ND5 mutation produces increased resistance to apoptosis and activation of the PI3K/Akt pathway, leading to a higher tumorigenic potential (39). Similarly, ND6 mutations produce deficient Complex I activity and high reactive oxygen species (ROS) generation that makes these cells highly metastatic, a characteristic that is suppressed by ROS scavengers (40). In addition, cancer cells with mutations in ND4 and ND6 that causes a mild decrease in OXPHOS function promote tumor growth when injected in nude mice (41). This contradictory behavior of Complex I in cancer can be explained based on the type and severity of the OXPHOS dysfunction, which has been elegantly described by the Porcelli’s group (12, 17). Lack of OXPHOS caused by absence of functional Complex I due to homoplasmic mtDNA mutations (m.3571insC/MT-ND1 and m.3243A>G/MT-TL1) in osteosarcoma cells induces an imbalance of the αKG/Suc ratio, destabilizing HIF1α and reducing the ability of these cells to grow in an anchorage-independent fashion and form tumors in vivo. On the other hand, osteosarcoma cells carrying a homoplasmic mtDNA mutation (m.3460G>A/MT-ND1) that only mildly affects Complex I function and hence OXPHOS are able to form tumors at the same rate as osteosarcoma cells carrying normal mtDNA (17). Thus, a complete inhibition of Complex I that avoids generation of ROS and hinders hypoxic adaptation by rewiring of mitochondrial metabolism is apparently necessary to have an antitumorigenic effect.
Along these lines, the receptor of cyclophilin A, CD147, a transmembrane glycoprotein expressed mainly at the cell surface (42) often translocates to the cytoplasm and mitochondria in melanoma cells where it promotes Complex I activity by interacting with the NDUFS6 subunit, protecting mitochondria from damage that may trigger mitochondrial-dependent apoptosis (43). Thus, the interaction between CD147 and NDUFS6 subunit in the mitochondria may be a potential key mechanism of the multidrug resistance of cancer cells associated with CD147 (44). Additional studies are necessary to understand this interaction more thoroughly and unveil the role of Complex I and mitochondrial function in multidrug resistance.
Similarly, the signal transducer and activator of transcription 3 (STAT3), a nuclear transcription factor known for mediating tumor growth (45, 46), also translocates to the mitochondria where it is necessary for the activity of Complexes I and II and its knockdown impairs OXPHOS (47). It has been proposed that STAT3 may interact with iron sulfur clusters in the distal region of Complex I to increase its activity and reduce ROS accumulation (48), which in a murine breast cancer cell model favored cell survival and tumor formation (49). Interaction between STAT3 and Complex I subunit NDUFA13, also known as GRIM-19 (50–52), has been described (53); however, the contribution of this interaction in tumorigenesis requires further studies.
Role of Complex I in Metastasis of Cancer Cells
Metastatic cells begin dissemination with migration and invasion into surrounding tissues and lymphatic vessels to finally seed in distant organs (54). Emergent evidence indicates that the mitochondrion, especially ETC activity, contributes to several steps of metastasis in vitro and in vivo (55–57). In fact, the down-modulation of certain Complex I subunits by genetic or pharmacologic means produces enhanced migratory behavior of cancer cells and metastasis (19, 40, 58). For example, knockdown of Complex I subunit NDUFV1 increases the metastatic behavior of the already aggressive breast cancer cell line MDA-MB-231. This phenomenon (Figure 1B) was mediated by a decreased NAD+/NADH ratio, increased Akt and mTORC1 activities, and reduced levels of autophagy (59). Conversely, an increase in the NAD+/NADH ratio enhancing Complex I activity through the expression of NADH dehydrogenase Ndi1 from Saccharomyces cerevisiae in human breast cancer cells reduces the metastatic potential of these cells (59). In addition, it has been observed that a down-expression of nuclear-encoded NDUFA13 and NDUFS3 subunits in HeLa cells promotes the loss of epithelial morphology and acquisition of mesenchymal properties, a key event for the development of metastasis known as epithelial–mesenchymal transition (EMT) (60, 61). EMT is characterized by an increase of lamellipodial formation and high cell–matrix adhesion capacity due to an increased secretion of fibronectin and increased expression of its receptor integrin α5, N-cadherin, and vimentin promoting migration and invasion. These events are accompanied with an increase in ROS generation and can be reversed with the presence of ROS scavenger N-acetyl cysteine (NAC) (62). Comparably, high invasive capacities in breast cancer cell lines have been correlated with reduced levels of Complex I subunits such as NDUFA13, NDUFS3, and accessory subunit NDUFB9 (62, 63). In addition, it had been shown that highly metastatic breast cancer cells have reduced expression of nuclear-encoded NDUFB9 subunit and the knockdown of this subunit generates high levels of mitochondrial ROS, a slight decrease of NAD+/NADH ratio and a metabolic disturbance dependent on Akt/mTOR/p70S6K signaling accompanied with increased expression of mesenchymal markers (vimentin and fibronectin) and SMAD3, an upstream regulator of EMT (19). Interestingly, cybrid cancer cells harboring the pathogenic A3243T mutation in the leucine transfer RNA gene (tRNAleu), which render mitochondria OXPHOS deficient, display high motility and migration, which are associated with high levels of membrane-bound integrin β1 and increased binding to fibronectin, a non-collagenous extracellular matrix glycoprotein (64). As mutations in mtDNA represent an early event during breast tumorigenesis, producing defective OXPHOS with a metabolic shift toward glycolysis could be used as a potential biomarker for early detection and prognosis (65). In further support, several reports indicate that the inhibition of Complex I activity by pharmacologic interventions using small molecules can increase ROS generation, promoting the migration and invasion of cancer cells (62, 66, 67). For example, Ma et al. (67) described that clones of breast cancer cells generated by treatment with rotenone, exhibited mitochondrial respiratory defects, increased ROS levels, and high migration and invasion properties, which were inhibited by treatment with antioxidants such as NAC and mito-TEMPO, a mitochondria-targeted antioxidant (67). Similar effects have also been observed in hepatoma cells (66). Altogether, these data suggest that the inhibition of Complex I activity accompanied by ROS generation promotes migration, invasion, and metastasis. However, recently it has been reported that partial inhibition of Complex I with nanomolar concentrations of rotenone, which inhibited between 11 and 33% of its activity, limited instead of promoted, migration and invasion of non-small-cell lung cancer cells (68). Moreover, lung adenocarcinoma patient data have shown that elevated expression of OXPHOS-encoding genes, in particular genes of core, accessory and assembly subunits of Complex I are associated with a poor prognosis (68). In fact, cisplatin-resistant lung cancer cells exhibited high Complex I activity, elevated mitochondrial transmembrane potential, high ATP content, and increased migration and invasion compared with parental cells (68). The conflicting observations regarding the activity of Complex I in migration, invasion, and metastasis can be explained as cancer-type specific differences, but most likely they occurred as a result of the level of inhibition of Complex I, which finally determines the pro- and antitumorigenic effects (69–71). In this regard, Porporato et al. (72) elegantly demonstrate that either ETC overload with excess electrons from the TCA cycle, without uncoupling the ETC from ATP synthase, or partial ETC inhibition using low doses of Complex I inhibitor rotenone promotes a similar mitochondrial superoxide-dependent pro-metastatic phenotype in vitro and in vivo (72). In contrast, the full ECT inhibition with high doses of rotenone generates inhibition of mitochondrial respiration without superoxide production, inhibiting the migration of cancer cells (72).
Complex I as a Target for Anticancer Small Molecules
Recently reported Complex I inhibitors (Table 1) exhibit different structural characteristics (e.g., rotenoids, vanilloids, alkaloids, biguanides, annonaceous acetogenins, and polyphenols), with no obvious establishment of structural factors involved in the interaction with this respiratory complex (73). Classic Complex I inhibitors and some new small molecules such as AG311 (74) are uncharged, aromatic and highly hydrophobic small molecules (75) that can putatively interact with the binding site of ubiquinone, producing a competitive inhibition. Generally, they have a hydroquinone/quinone motif that interacts with Complex I, and this interaction is highly sensitive to small structural changes of the inhibitors (76–78). On the other hand, metformin and other biguanides represent a new class of relatively hydrophilic positively charged Complex I inhibitors that produce non-competitive inhibition by binding in an amphipathic region close to the matrix loop of ND3 subunit (75).
Table 1
| Compound | Mechanism of action | Cancer cells | Reference |
|---|---|---|---|
| JCI-20679 | Complex I inhibition mediated antitumor activity | A panel of 39 cancer cell lines | (79) |
| Celastrol | Complex I inhibition associated with reactive oxygen species (ROS) accumulation, causing cytotoxicity | Lung and liver cancer cells | (80) |
| AG311 | Complex I inhibition and hypoxia-inducible factor 1-alpha stabilization inhibition, loss of mitochondrial transmembrane potential, decrease in ATP content, antiproliferative effect, and cell death | Triple-negative breast cancer cells | (74) |
| Kalkitoxin | Disruption of cellular hypoxic signaling and angiogenesis inhibition | T47D breast cancer cell | (81) |
| BAY 87-2243 | Reduces oxygen consumption rate, partial mitochondrial depolarization, associated with increased ROS levels, AMP-activated protein kinase (AMPK) activation, and reduction in cell viability | BRAF mutant melanoma cells | (82) |
| Xanthohumol | Causes increased ROS levels due to Complex I inhibition, resulting in apoptotic cell death | Lung and cervical cancer cells | (83) |
| Verrucosidin | Induces cell death in the absence of glucose | Breast cancer cells | (84) |
| Canagliflozin | Limits cancer cell proliferation by inhibiting Complex I-dependent respiration, causing a decrease in ATP, and activation of AMPK | Lung and prostate cancer cells | (85) |
| Metformin | Inhibits cell proliferation when grown in high glucose media, induces cell death when grown in glucose deprivation | Colon rectal and lung cancer cells | (86) |
| Fenofibrate | Induces metabolic catastrophe and cell death, decreases tumor growth in intracranial glioblastoma model | Glioblastoma cells | (87) |
New small molecules and Food and Drug Administration-approved drugs reported as Complex I inhibitors with anticancer actions.
Complex I inhibition by small molecules has been suggested as a strategy to target the Warburg effect and metabolic plasticity of cancer cells (88–90). BAY 87-2243, fenofibrate, metformin, canagliflozin, and AG311 compounds produce mitochondrial depolarization, ATP depletion, and increase ROS production, which triggers the activation of AMP-activated protein kinase (AMPK) signaling (80, 82, 83, 87). In addition, AG311, kalkitoxin, and metformin trigger the inhibition of HIF1α signaling, producing selective anticancer effects (Table 1). Some Complex I inhibitors induce cell death in cancer cells by a mechanism that involves increased ROS production such as celastrol, BAY 87-2243, and xanthohumol (80, 82, 83). Interestingly, fenofibrate-induced AMPK activation produces inhibition of mTOR substrates and a decrease in autophagy markers in glioblastoma cancer cells (87). The inhibition of autophagy in these malignant cells produces an increase in fenofibrate-induced cell death, suggesting a protective role of autophagy when fenofibrate inhibits Complex I (87).
Selective delivery systems for cancer cells have been extensively explored in recent years, decreasing toxic side effects and enhancing activity of antitumor agents (91). The elevated mitochondrial membrane potential of cancer cells compared with non-tumor cells has allowed the development of small molecules that incorporate the lipophilic cation triphenylphosphonium (TTP+), which is selectively accumulated within mitochondria in a mitochondrial membrane potential-dependent manner (92). Interestingly, a metformin-TTP+ derivative (Mito-Met10) has recently shown over 1,000-fold greater potency than metformin to inhibit Complex I, correlating with greater than 1,000-fold enhanced antiproliferative effect of Mito-Met10 compared with metformin in pancreatic cancer cells (93). The mechanism of action of Mito-Met10 includes induction of ROS production and AMPK activation (93, 94). This compound lacks toxicity in vivo and is accumulated in liver, kidney, spleen, and tumor tissues (93). Similarly, norMitoMet a metformin-TTP+ derivative that lacks a methyl group on the nitrogen adjacent to the 10-carbon spacer is more efficient than its parental drug inhibiting the proliferation in pancreatic cancer cells. This compound has a putative binding site for Complex I inhibition at the ubiquinone-binding pocket (94). Given the role of Complex I in supporting proliferation and survival of cancer cells, the inhibition of its activity appears to be a promising target for anticancer action. Evidence of anticancer effects by Complex I inhibition on several cancer cell lines of Food and Drug Administration-approved drugs with known safety profile and pharmacokinetics such as canagliflozin (85), fenofibrate (87), and metformin (86) provides strong incentive for further preclinical and clinical studies.
Conclusion and Future Directions
Complex I, the main point of entry of electrons in the ETC, controls the synthesis of precursors such as aspartate by maintaining the NAD+/NADH ratio and mitochondrial ATP synthesis by proton pumping toward the intermembrane space. In cancer cells, these Complex I-dependent events contribute to tumor formation, acquisition of resistance to cell death stimuli, and promotion of metastasis by increasing ROS levels, inducing HIF1α signaling, and inhibiting mTORC1 signaling and EMT induction. Further studies are required to understand the role of Complex I in other metabolic aspects of cancer cells and the molecular mechanisms involved. In particular, recent evidence indicates that as normal cells (95), cancer cells require calcium transfer from endoplasmic reticulum (ER) to mitochondria, to maintain a continuous supply of biosynthetic precursors for proliferation, and inhibition of the ER–mitochondrial communication generates a massive selective cell death of cancer cells (96, 97). The mitochondrial calcium uptake across the mitochondrial calcium uniporter complex (MCUC) (98) is mediated by the mitochondrial transmembrane potential, a bioenergetic parameter maintained mainly by Complex I activity (1, 99). Thus, a relation between Complex I and MCUC is expected; however, no information is available in this regard. Thus, we are wondering; could Complex I activity be essential to promote calcium uptake and support TCA cycle activity in cancer cells? Could Complex I subunits interact with MCUC or other molecular components of mitochondrial calcium machinery? Is there an adaptive mechanism that maintains the mitochondrial calcium uptake under a Complex I inhibition?
One of the main features of cancer cells is their ability to avoid cell death stimuli, which they achieve by suppressing the activity of the mitochondrial permeability transition pore (mPTP), a fundamental player in the initiation of apoptosis and necrosis (100). The mPTP is a putative pore responsible for the mitochondrial permeability transition (101) that corresponds to an alteration in the permeability of the inner mitochondrial membrane and causes the release of proapoptotic factors that lead to apoptosis (100, 102). Interestingly, it has been described that the Complex I inhibitor rotenone also inhibits the mPTP, depending on the inorganic phosphate levels (103). This suggests that Complex I may act as a negative regulator of mPTP by a direct interaction (103, 104). Whether cancer cells exhibit a fine cross talk between Complex I and mPTP to acquire cell death resistance or regulate the Complex I activity is an unexplored issue that may represent a novel level of mitochondrial bioenergetics regulation.
In addition, cancer cells exhibit metabolic flexibility able to adapt their metabolism under changes of energetic substrate availability (i.e., glucose and glutamine) to promote survival and metastasis (55, 105), modifying the participation of OXPHOS and determining the sensitivity of cancer cells to Complex I inhibitors (105, 106). In this context, what molecular signaling is involved in the modulation of Complex I activity in cancer cells? Are there changes in the expression of subunit-encoding genes under Complex I activity inhibition? Finally, the specific roles of Complex I subunits during tumorigenesis are poorly understood. Increasing evidence suggests that a plethora of changes in expression levels or mild mutations in nuclear and mitochondrial genes that encode Complex I subunits give adaptive advantage to promote metastasis. In contrast, severe mutations in Complex I subunit-encoding genes or pharmacologic inhibition of Complex I activity with small molecules produces antitumorigenic effects. Taking this into consideration, Complex I inhibitors offer a promising strategy to obtain anticancer activity and overall, this is an exciting time to rationally design molecules that target mitochondrial metabolism.
Statements
Author contributions
FU and CC designed and outlined the structure and contents of the review. CC, FM, AL, and FU contributed to the literature review, discussion, and writing of the manuscript. All the authors contributed equally to the draft revisions and final approval of the version to be published.
Funding
This work was supported by FONDECYT grant #1160332 (CC), CONICYT/FONDAP #15150012 (CC), NIH P30NS047243 (AL) and a FONDECYT postdoctoral fellowship #3170813 (FU).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMPK, AMP-activated protein kinase; EMT, epithelial–mesenchymal transition; ERK, extracellular signal-regulated kinase; ETC, electron transport chain; FDA, Food and Drug Administration; HIF1α, hypoxia-inducible factor 1-alpha; mtDNA, mitochondrial DNA; NAC, N-acetyl cysteine; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; STAT3, signal transducer and activator of transcription 3; Suc, succinate; TCA cycle, tricarboxylic acid cycle; αKG, α-ketoglutarate.
References
1
Zickermann V Wirth C Nasiri H Siegmund K Schwalbe H Hunte C et al Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science (2015) 347(6217):44–9.10.1126/science.1259859
2
Hirst J . Mitochondrial complex I. Annu Rev Biochem (2013) 82:551–75.10.1146/annurev-biochem-070511-103700
3
Sazanov L . The mechanism of coupling between electron transfer and proton translocation in respiratory complex I. J Bioenerg Biomembr (2014) 46(4):247–53.10.1007/s10863-014-9554-z
4
Mitchell P . Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature (1961) 191:144–8.10.1038/191144a0
5
Brand M Nicholls D . Assessing mitochondrial dysfunction in cells. Biochem J (2011) 435(2):297–312.10.1042/bj20110162
6
Hunte C Zickermann V Brandt U . Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science (2010) 329(5990):448–51.10.1126/science.1191046
7
Sazanov L . A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol (2015) 16(6):375–88.10.1038/nrm3997
8
Letts J Sazanov L . Gaining mass: the structure of respiratory complex I-from bacterial towards mitochondrial versions. Curr Opin Struct Biol (2015) 33:135–45.10.1016/j.sbi.2015.08.008
9
Fiedorczuk K Letts J Degliesposti G Kaszuba K Skehel M Sazanov L . Atomic structure of the entire mammalian mitochondrial complex I. Nature (2016) 538(7625):406–10.10.1038/nature19794
10
Zhu J Vinothkumar K Hirst J . Structure of mammalian respiratory complex I. Nature (2016) 536(7616):354–8.10.1038/nature19095
11
Rodenburg R . Mitochondrial complex I-linked disease. Biochim Biophys Acta (2016) 1857(7):938–45.10.1016/j.bbabio.2016.02.012
12
Iommarini L Calvaruso M Kurelac I Gasparre G Porcelli A . Complex I impairment in mitochondrial diseases and cancer: parallel roads leading to different outcomes. Int J Biochem Cell Biol (2013) 45(1):47–63.10.1016/j.biocel.2012.05.016
13
Marin S Mesterman R Robinson B Rodenburg R Smeitink J Tarnopolsky M . Leigh syndrome associated with mitochondrial complex I deficiency due to novel mutations in NDUFV1 and NDUFS2. Gene (2013) 516(1):162–7.10.1016/j.gene.2012.12.024
14
Sethumadhavan S Vasquez-Vivar J Migrino R Harmann L Jacob H Lazar J . Mitochondrial DNA variant for complex I reveals a role in diabetic cardiac remodeling. J Biol Chem (2012) 287(26):22174–82.10.1074/jbc.M111.327866
15
Elango S Venugopal S Thangaraj K Viswanadha V . Novel mutations in ATPase 8, ND1 and ND5 genes associated with peripheral neuropathy of diabetes. Diabetes Res Clin Pract (2014) 103(3):e49–52.10.1016/j.diabres.2013.12.015
16
Kurelac I MacKay A Lambros M Di Cesare E Cenacchi G Ceccarelli C et al Somatic complex I disruptive mitochondrial DNA mutations are modifiers of tumorigenesis that correlate with low genomic instability in pituitary adenomas. Hum Mol Genet (2013) 22(2):226–38.10.1093/hmg/dds422
17
Iommarini L Kurelac I Capristo M Calvaruso M Giorgio V Bergamini C et al Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet (2014) 23(6):1453–66.10.1093/hmg/ddt533
18
Calabrese C Iommarini L Kurelac I Calvaruso M Capristo M Lollini P et al Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab (2013) 1(1):11.10.1186/2049-3002-1-11
19
Li L Sun H Liu X Gao S Jiang H Hu X et al Down-regulation of NDUFB9 promotes breast cancer cell proliferation, metastasis by mediating mitochondrial metabolism. PLoS One (2015) 10(12):e0144441.10.1371/journal.pone.0144441
20
Philley J Kannan A Qin W Sauter E Ikebe M Hertweck K et al Complex-I alteration and enhanced mitochondrial fusion are associated with prostate cancer progression. J Cell Physiol (2016) 231(6):1364–74.10.1002/jcp.25240
21
Gasparre G Porcelli A Bonora E Pennisi L Toller M Iommarini L et al Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A (2007) 104(21):9001–6.10.1073/pnas.0703056104
22
Evangelisti C de Biase D Kurelac I Ceccarelli C Prokisch H Meitinger T et al A mutation screening of oncogenes, tumor suppressor gene TP53 and nuclear encoded mitochondrial complex I genes in oncocytic thyroid tumors. BMC Cancer (2015) 15:157.10.1186/s12885-015-1122-3
23
Yu Y Lv F Lin H Qian G Jiang Y Pang L et al Mitochondrial ND3 G10398A mutation: a biomarker for breast cancer. Genet Mol Res (2015) 14(4):17426–31.10.4238/2015.December.21.12
24
Su C Chang Y Yang C Huang M Hsiao M . The opposite prognostic effect of NDUFS1 and NDUFS8 in lung cancer reflects the oncojanus role of mitochondrial complex I. Sci Rep (2016) 6:31357.10.1038/srep31357
25
Kim H Komiyama T Inomoto C Kamiguchi H Kajiwara H Kobayashi H et al Mutations in the mitochondrial ND1 gene are associated with postoperative prognosis of localized renal cell carcinoma. Int J Mol Sci (2016) 17(12):E2049.10.3390/ijms17122049
26
Horton T Petros J Heddi A Shoffner J Kaufman A Graham S et al Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosomes Cancer (1996) 15(2):95–101.10.1002/(SICI)1098-2264(199602)15:2<95:AID-GCC3>3.0.CO;2-Z
27
Akouchekian M Houshmand M Akbari M Kamalidehghan B Dehghan M . Analysis of mitochondrial ND1 gene in human colorectal cancer. J Res Med Sci (2011) 16(1):50–5.
28
Allegra E Garozzo A Lombardo N De Clemente M Carey T . Mutations and polymorphisms in mitochondrial DNA in head and neck cancer cell lines. Acta Otorhinolaryngol Ital (2006) 26(4):185–90.
29
Birsoy K Wang T Chen W Freinkman E Abu-Remaileh M Sabatini D . An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162(3):540–51.10.1016/j.cell.2015.07.016
30
Sullivan L Gui D Hosios A Bush L Freinkman E Vander Heiden M . Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162(3):552–63.10.1016/j.cell.2015.07.017
31
Ahn C Metallo C . Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab (2015) 3(1):1.10.1186/s40170-015-0128-2
32
Wu M Neilson A Swift A Moran R Tamagnine J Parslow D et al Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol (2007) 292(1):C125–36.10.1152/ajpcell.00247.2006
33
Mullen A Wheaton W Jin E Chen P Sullivan L Cheng T et al Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481(7381):385–8.10.1038/nature10642
34
Selak MA Armour SM MacKenzie ED Boulahbel H Watson DG Mansfield KD et al Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7(1):77–85.10.1016/j.ccr.2004.11.022
35
Gasparre G Porcelli A Lenaz G Romeo G . Relevance of mitochondrial genetics and metabolism in cancer development. Cold Spring Harb Perspect Biol (2013) 5(3):a011411.10.1101/cshperspect.a011411
36
Agani FH Pichiule P Carlos Chavez J LaManna JC . Inhibitors of mitochondrial complex I attenuate the accumulation of hypoxia-inducible factor-1 during hypoxia in Hep3B cells. Comp Biochem Physiol A Mol Integr Physiol (2002) 132(1):107–9.10.1016/S1095-6433(01)00535-9
37
Liu Y Morgan JB Coothankandaswamy V Liu R Jekabsons MB Mahdi F et al The Caulerpa pigment caulerpin inhibits HIF-1 activation and mitochondrial respiration. J Nat Prod (2009) 72(12):2104–9.10.1021/np9005794
38
Garcia-Heredia J Carnero A . Decoding Warburg’s hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget (2015) 6(39):41582–99.10.18632/oncotarget.6057
39
Sharma L Fang H Liu J Vartak R Deng J Bai Y . Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet (2011) 20(23):4605–16.10.1093/hmg/ddr395
40
Ishikawa K Takenaga K Akimoto M Koshikawa N Yamaguchi A Imanishi H et al ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320(5876):661–4.10.1126/science.1156906
41
Cruz-Bermúdez A Vallejo C Vicente-Blanco R Gallardo M Fernández-Moreno M Quintanilla M et al Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget (2015) 6(15):13628–43.10.18632/oncotarget.3698
42
Li J Peng L Wu L Kuang Y Su J Yi M et al Depletion of CD147 sensitizes human malignant melanoma cells to hydrogen peroxide-induced oxidative stress. J Dermatol Sci (2010) 58(3):204–10.10.1016/j.jdermsci.2010.03.022
43
Luo Z Zeng W Tang W Long T Zhang J Xie X et al CD147 interacts with NDUFS6 in regulating mitochondrial complex I activity and the mitochondrial apoptotic pathway in human malignant melanoma cells. Curr Mol Med (2014) 14(10):1252–64.10.2174/1566524014666141202144601
44
Kuang Y Chen X Su J Wu L Li J Chang J et al Proteome analysis of multidrug resistance of human oral squamous carcinoma cells using CD147 silencing. J Proteome Res (2008) 7(11):4784–91.10.1021/pr800355b
45
Yuan J Zhang F Niu R . Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep (2015) 5:17663.10.1038/srep17663
46
Yu H Lee H Herrmann A Buettner R Jove R . Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14(11):736–46.10.1038/nrc3818
47
Wegrzyn J Potla R Chwae Y Sepuri N Zhang Q Koeck T et al Function of mitochondrial Stat3 in cellular respiration. Science (2009) 323(5915):793–7.10.1126/science.1164551
48
Szczepanek K Chen Q Derecka M Salloum F Zhang Q Szelag M et al Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem (2011) 286(34):29610–20.10.1074/jbc.M111.226209
49
Zhang Q Raje V Yakovlev V Yacoub A Szczepanek K Meier J et al Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem (2013) 288(43):31280–8.10.1074/jbc.M113.505057
50
Fearnley I Carroll J Shannon R Runswick M Walker J Hirst J . GRIM-19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem (2001) 276(42):38345–8.10.1074/jbc.C100444200
51
Huang G Lu H Hao A Ng D Ponniah S Guo K et al GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Mol Cell Biol (2004) 24(19):8447–56.10.1128/MCB.24.19.8447-8456.2004
52
Lu H Cao X . GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol Biol Cell (2008) 19(5):1893–902.10.1091/mbc.E07-07-0683
53
Lufei C Ma J Huang G Zhang T Novotny-Diermayr V Ong C et al GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J (2003) 22(6):1325–35.10.1093/emboj/cdg135
54
Celià-Terrassa T Kang Y . Distinctive properties of metastasis-initiating cells. Genes Dev (2016) 30(8):892–908.10.1101/gad.277681.116
55
Lehuédé C Dupuy F Rabinovitch R Jones R Siegel P . Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res (2016) 76(18):5201–8.10.1158/0008-5472.CAN-16-0266
56
Viale A Corti D Draetta G . Tumors and mitochondrial respiration: a neglected connection. Cancer Res (2015) 75(18):3685–6.10.1158/0008-5472.CAN-15-0491
57
LeBleu V O’Connell J Gonzalez Herrera K Wikman H Pantel K Haigis M et al PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–1003,1–15.10.1038/ncb3039
58
Yuan Y Wang W Li H Yu Y Tao J Huang S et al Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer (2015) 15:346.10.1186/s12885-015-1349-z
59
Santidrian A Matsuno-Yagi A Ritland M Seo B LeBoeuf S Gay L et al Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest (2013) 123(3):1068–81.10.1172/JCI64264
60
Gonzalez D Medici D . Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal (2014) 7(344):re8.10.1126/scisignal.2005189
61
Li L Li W . Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther (2015) 150:33–46.10.1016/j.pharmthera.2015.01.004
62
He X Zhou A Lu H Chen Y Huang G Yue X et al Suppression of mitochondrial complex I influences cell metastatic properties. PLoS One (2013) 8(4):e61677.10.1371/journal.pone.0061677
63
Putignani L Raffa S Pescosolido R Rizza T Del Chierico F Leone L et al Preliminary evidences on mitochondrial injury and impaired oxidative metabolism in breast cancer. Mitochondrion (2012) 12(3):363–9.10.1016/j.mito.2012.02.003
64
Nunes J Peixoto J Soares P Maximo V Carvalho S Pinho S et al OXPHOS dysfunction regulates integrin-β1 modifications and enhances cell motility and migration. Hum Mol Genet (2015) 24(7):1977–90.10.1093/hmg/ddu612
65
Yadav N Chandra D . Mitochondrial DNA mutations and breast tumorigenesis. Biochim Biophys Acta (2013) 1836(2):336–44.10.1016/j.bbcan.2013.10.002
66
Lee J Lee Y Lim J Byun H Park I Kim G et al Mitochondrial respiratory dysfunction induces claudin-1 expression via reactive oxygen species-mediated heat shock factor 1 activation, leading to hepatoma cell invasiveness. J Biol Chem (2015) 290(35):21421–31.10.1074/jbc.M115.654913
67
Ma J Zhang Q Chen S Fang B Yang Q Chen C et al Mitochondrial dysfunction promotes breast cancer cell migration and invasion through HIF1α accumulation via increased production of reactive oxygen species. PLoS One (2013) 8(7):e69485.10.1371/journal.pone.0069485
68
Jeon J Kim D Shin Y Kim H Song B Lee E et al Migration and invasion of drug-resistant lung adenocarcinoma cells are dependent on mitochondrial activity. Exp Mol Med (2016) 48(12):e277.10.1038/emm.2016.129
69
Jose C Rossignol R . Rationale for mitochondria-targeting strategies in cancer bioenergetic therapies. Int J Biochem Cell Biol (2013) 45(1):123–9.10.1016/j.biocel.2012.07.005
70
Benard G Bellance N James D Parrone P Fernandez H Letellier T et al Mitochondrial bioenergetics and structural network organization. J Cell Sci (2007) 120(Pt 5):838–48.10.1242/jcs.03381
71
Tan AS Baty J Berridge M . The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim Biophys Acta (2014) 1840(4):1454–63.10.1016/j.bbagen.2013.10.016
72
Porporato P Payen V Pérez-Escuredo J De Saedeleer C Danhier P Copetti T et al A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8(3):754–66.10.1016/j.celrep.2014.06.043
73
Miyoshi H . Probing the ubiquinone reduction site in bovine mitochondrial complex I using a series of synthetic ubiquinones and inhibitors. J Bioenerg Biomembr (2001) 33(3):223–31.10.1023/A:1010735019982
74
Bastian A Matsuzaki S Humphries K Pharaoh G Doshi A Zaware N et al AG311, a small molecule inhibitor of complex I and hypoxia-induced HIF-1α stabilization. Cancer Lett (2017) 388:149–57.10.1016/j.canlet.2016.11.040
75
Bridges H Jones A Pollak M Hirst J . Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J (2014) 462(3):475–87.10.1042/BJ20140620
76
Urra F Córdova-Delgado M Lapier M Orellana-Manzano A Acevedo-Arévalo L Pessoa-Mahana H et al Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol Appl Pharmacol (2016) 291:46–57.10.1016/j.taap.2015.12.005
77
Satoh T Miyoshi H Sakamoto K Iwamura H . Comparison of the inhibitory action of synthetic capsaicin analogues with various NADH-ubiquinone oxidoreductases. Biochim Biophys Acta (1996) 1273(1):21–30.10.1016/0005-2728(95)00131-X
78
Ueno H Miyoshi H Ebisui K Iwamura H . Comparison of the inhibitory action of natural rotenone and its stereoisomers with various NADH-ubiquinone reductases. Eur J Biochem (1994) 225(1):411–7.10.1111/j.1432-1033.1994.00411.x
79
Akatsuka A Kojima N Okamura M Dan S Yamori T . A novel thiophene-3-carboxamide analog of annonaceous acetogenin exhibits antitumor activity via inhibition of mitochondrial complex I. Pharmacol Res Perspect (2016) 4(4):e00246.10.1002/prp2.246
80
Chen G Zhang X Zhao M Wang Y Cheng X Wang D et al Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer (2011) 11:170.10.1186/1471-2407-11-170
81
Morgan J Liu Y Coothankandaswamy V Mahdi F Jekabsons M Gerwick W et al Kalkitoxin inhibits angiogenesis, disrupts cellular hypoxic signaling, and blocks mitochondrial electron transport in tumor cells. Mar Drugs (2015) 13(3):1552–68.10.3390/md13031552
82
Schöckel L Glasauer A Basit F Bitschar K Truong H Erdmann G et al Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab (2015) 3:11.10.1186/s40170-015-0138-0
83
Zhang B Chu W Wei P Liu Y Wei T . Xanthohumol induces generation of reactive oxygen species and triggers apoptosis through inhibition of mitochondrial electron transfer chain complex I. Free Radic Biol Med (2015) 89:486–97.10.1016/j.freeradbiomed.2015.09.021
84
Thomas S Sharma N Gonzalez R Pao P Hofman F Chen T et al Repositioning of verrucosidin, a purported inhibitor of chaperone protein GRP78, as an inhibitor of mitochondrial electron transport chain complex I. PLoS One (2013) 8(6):e65695.10.1371/journal.pone.0065695
85
Villani L Smith B Marcinko K Ford R Broadfield L Green A et al The diabetes medication canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol Metab (2016) 5(10):1048–56.10.1016/j.molmet.2016.08.014
86
Wheaton W Weinberg S Hamanaka R Soberanes S Sullivan L Anso E et al Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife (2014) 3:e02242.10.7554/eLife.02242
87
Wilk A Wyczechowska D Zapata A Dean M Mullinax J Marrero L et al Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol (2015) 35(1):182–98.10.1128/MCB.00562-14
88
Chaube B Malvi P Singh S Mohammad N Meena A Bhat M . Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget (2015) 6(35):37281–99.10.18632/oncotarget.6134
89
Vatrinet R Iommarini L Kurelac I De Luise M Gasparre G Porcelli A . Targeting respiratory complex I to prevent the Warburg effect. Int J Biochem Cell Biol (2015) 63:41–5.10.1016/j.biocel.2015.01.017
90
Xu Q Biener-Ramanujan E Yang W Ramanujan V . Targeting metabolic plasticity in breast cancer cells via mitochondrial complex I modulation. Breast Cancer Res Treat (2015) 150(1):43–56.10.1007/s10549-015-3304-8
91
Yamada Y Harashima H . Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv Drug Deliv Rev (2008) 60(13–14):1439–62.10.1016/j.addr.2008.04.016
92
Porteous CM Logan A Evans C Ledgerwood EC Menon DK Aigbirhio F et al Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim Biophys Acta (2010) 1800(9):1009–17.10.1016/j.bbagen.2010.06.001
93
Cheng G Zielonka J Ouari O Lopez M McAllister D Boyle K et al Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res (2016) 76(13):3904–15.10.1158/0008-5472.CAN-15-2534
94
Kalyanaraman B Cheng G Hardy M Ouari O Sikora A Zielonka J et al Mitochondria-targeted metformins: anti-tumour and redox signalling mechanisms. Interface Focus (2017) 7(2):20160109.10.1098/rsfs.2016.0109
95
Cárdenas C Miller R Smith I Bui T Molgó J Müller M et al Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142(2):270–83.10.1016/j.cell.2010.06.007
96
Lovy A Foskett J Cárdenas C . InsP3R, the calcium whisperer: maintaining mitochondrial function in cancer. Mol Cell Oncol (2016) 3(4):e1185563.10.1080/23723556.2016.1185563
97
Cárdenas C Müller M McNeal A Lovy A Jaňa F Bustos G et al Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14(10):2313–24.10.1016/j.celrep.2016.02.030
98
De Stefani D Patron M Rizzuto R . Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta (2015) 1853(9):2006–11.10.1016/j.bbamcr.2015.04.008
99
Galkin A Dröse S Brandt U . The proton pumping stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim Biophys Acta (2006) 1757(12):1575–81.10.1016/j.bbabio.2006.10.001
100
Bonora M Pinton P . The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol (2014) 4:302.10.3389/fonc.2014.00302
101
Vianello A Casolo V Petrussa E Peresson C Patui S Bertolini A et al The mitochondrial permeability transition pore (PTP) – an example of multiple molecular exaptation? Biochim Biophys Acta (2012) 1817(11):2072–86.10.1016/j.bbabio.2012.06.620
102
Suh D Kim M Kim H Chung H Song Y . Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front Oncol (2013) 3:41.10.3389/fonc.2013.00041
103
Li B Chauvin C De Paulis D De Oliveira F Gharib A Vial G et al Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim Biophys Acta (2012) 1817(9):1628–34.10.1016/j.bbabio.2012.05.011
104
Batandier C Leverve X Fontaine E . Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem (2004) 279(17):17197–204.10.1074/jbc.M310329200
105
Urra F Weiss-López B Araya-Maturana R . Determinants of anti-cancer effect of mitochondrial electron transport chain inhibitors: bioenergetic profile and metabolic flexibility of cancer cells. Curr Pharm Des (2016) 22(39):5998–6008.10.2174/1381612822666160719122626
106
Gui D Sullivan L Luengo A Hosios A Bush L Gitego N et al Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab (2016) 24(5):716–27.10.1016/j.cmet.2016.09.006
Summary
Keywords
electron transport chain, mitochondrial respiration, cancer cells, metastasis, anticancer agents
Citation
Urra FA, Muñoz F, Lovy A and Cárdenas C (2017) The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 7:118. doi: 10.3389/fonc.2017.00118
Received
22 February 2017
Accepted
19 May 2017
Published
08 June 2017
Volume
7 - 2017
Edited by
Cristina Mammucari, University of Padua, Italy
Reviewed by
Amadou K. S. Camara, Medical College of Wisconsin, United States; Eirini Lionaki, Foundation for Research and Technology Hellas, Greece
Updates
Copyright
© 2017 Urra, Muñoz, Lovy and Cárdenas.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: César Cárdenas, jcesar@u.uchile.cl
Specialty section: This article was submitted to Molecular and Cellular Oncology, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.