 Alison Roos
Alison Roos Zonghui Ding
Zonghui Ding Joseph C. Loftus
Joseph C. Loftus Nhan L. Tran
Nhan L. Tran- 1Departments of Cancer Biology and Neurosurgery, Mayo Clinic Arizona, Scottsdale, AZ, United States
- 2Department of Biochemistry and Molecular Biology, Mayo Clinic Arizona, Scottsdale, AZ, United States
Glioblastoma multiforme (GBM) is the most frequent primary brain tumor in adults with a 5-year survival rate of 5% despite intensive research efforts. The poor prognosis is due, in part, to aggressive invasion into the surrounding brain parenchyma. Invasion is a complex process mediated by cell-intrinsic pathways, extrinsic microenvironmental cues, and biophysical cues from the peritumoral stromal matrix. Recent data have attributed GBM invasion to the glioma stem-like cell (GSC) subpopulation. GSCs are slowly dividing, highly invasive, therapy resistant, and are considered to give rise to tumor recurrence. GSCs are localized in a heterogeneous cellular niche, and cross talk between stromal cells and GSCs cultivates a fertile environment that promotes GSC invasion. Pro-migratory soluble factors from endothelial cells, astrocytes, macrophages, microglia, and non-stem-like tumor cells can stimulate peritumoral invasion of GSCs. Therefore, therapeutic efforts designed to target the invasive GSCs may enhance patient survival. In this review, we summarize the current understanding of extrinsic pathways and major stromal and immune players facilitating GSC maintenance and survival.
Introduction
Glioblastoma multiforme (GBM) is the most frequently occurring primary brain tumor in adults (1). GBMs are differentiated from lower grade tumors by the presence of atypical nuclei, high mitotic activity, and areas of necrosis and microvascular hyperplasia (2). The standard of care for primary GBM includes maximal safe surgical resection and a combination of radiation therapy and temozolomide (TMZ) chemotherapy (3). The prognosis and survival of patients with GBM remains dismal with a median survival time of approximately 15 months from the time of diagnosis and a 5-year survival rate of 5% (4, 5). The poor prognosis is due, in part, to GBM cells invading into the surrounding brain parenchyma and inter and intra-tumoral heterogeneity, which confer resistance to traditional and contemporary treatment modalities (6–8).
Recent analyses have been focused on the identification of genetic alterations in the patient’s tumor as a potential means for individualized therapy. Commonly, this approach utilizes biopsy samples from the tumor core, which is subsequently removed during surgical resection (8–10). Unfortunately, this approach provides little insight into the genetic alterations in the residual invasive cells that give rise to tumor recurrence or the microenvironmental factors that influence their survival. The process of invasion is complex and highly orchestrated. An invasive cell must detach from the primary tumor, adhere to extracellular matrix (ECM) components, secrete enzymes that degrade the surrounding matrix, undergo morphological changes to facilitate locomotion, and migrate through surrounding microenvironmental structures (11). To effectively improve patient survival, there is a significant need to enhance our understanding of the molecular mechanisms and microenvironmental factors that drive cell invasion and improve targeting of this population to thwart tumor recurrence.
Currently, it is assumed that recurrence results from glioma stem-like cells (GSCs), since isolated GSCs are inherently chemo- and radioresistant, express enhanced levels of neuronal stem cell markers and are highly invasive (12–17). GSCs are a small subpopulation of tumor cells that self-renew and proliferate to maintain tumor growth (18). GSCs are enriched in distinct niches in vascular and hypoxic regions and are isolated utilizing cell surface markers including CD133, CD44, and CD15 (19). Intracranial injection of CD133+ cells produces highly invasive tumors that recapitulate the heterogeneity of the parental tumor (20, 21). Additionally, CD133+ cells are enriched after radiotherapy, and CD133 expression is associated with poor patient prognosis in GBM (14, 22). Similarly, CD44 functions as a cell surface marker for several cancer stem cells and has been used to enrich for GSCs (23, 24).
Studies into genomic factors that regulate GBM invasion have compared the gene expression profiles of cells in the invasive rim to cells in the bulk tumor core both in vitro and in clinical specimens. Results have revealed differential activation of transcription factors and significant gene expression differences in antiapoptotic and survival pathways in the invasive cells relative to cells in the tumor core (25–27). However, the invasive cells do not manifest recurrence alone but recruit and exploit microenvironmental cells to sustain and promote survival and invasion. This review summarizes the paracrine, autocrine, and intrinsic molecular pathways that have been reported to facilitate GSC maintenance and survival.
GBM Perivascular Niche Supports GSC Maintenance
Glioblastoma multiforme is a vascularized tumor characterized by aggregates of proliferating endothelial cells (ECs) referred to as microvascular hyperplasia. Angiogenesis is vital for supporting and maintaining rapid tumor growth (28). GSCs migrate toward and are enriched in the abnormal tumor vascular niche and immunohistochemical staining of GBM tumors shows CD31+ ECs surrounded by CD133+ GSCs (29–31). GSCs promote tumorigenesis by secreting vascular endothelial growth factor (VEGF) that induces EC migration and subsequent angiogenesis (29). The importance of angiogenesis in the function of GSCs is demonstrated by preclinical studies with the neutralizing VEGF antibody bevacizumab, which depletes the tumor vasculature and specifically inhibits tumor growth of GSC-derived xenografts in vivo (29, 32).
GSC secretion of VEGF is induced by the CXCL12/CXCR4 ligand/receptor pair (33, 34). In response to CXCL12, CD133+ GSCs upregulate VEGF production in a PI3K/Akt-dependent manner (33). Treatment with a CXCR4 antagonist or with the PI3K inhibitor, LY294002, can reduce VEGF production and inhibit growth and angiogenesis of tumor xenografts formed by GSCs (33). Furthermore, inhibiting CXCR4 signaling suppressed the invasive phenotype of GSCs and sensitizes these cells to radiation (35).
Endothelial cells secrete several factors that confer pro-survival and invasive properties to GSCs (29). For instance, the angiopoietin (Ang1)-Tie2 receptor interaction plays a critical role in the invasive phenotype of GSCs. In response to EC-derived Ang1, the tyrosine kinase Tie2 receptor is activated on GSCs and promotes the expression of adhesion proteins, including N-cadherin and integrin β1, to facilitate GSC invasion (36). In fact, integrin β1 has been reported to be critical for diffuse infiltration in GBM (37). In addition, sonic hedgehog (Shh) secreted by CD31+ ECs within the perivascular niche can promote sustained GBM tumor growth in vivo and self-renewal of GSCs by activating Patched1 and GLI signaling (38, 39). Inhibition of Shh-GLI signaling reduces GSC self-renewal and in vivo tumorigenesis suggesting that Shh, in part, is important for GSC survival. Additionally, paracrine factors secreted from ECs in the vascular niche activate the mTOR pathway and promote expansion of GSCs (40).
GSCs can physically interact with vascular cells in the niche, and this interaction initiates, supports, and maintains tumor growth and promotes angiogenesis (29). L1CAM, a neural adhesion molecule that regulates neural growth and migration during development, is overexpressed in GBM and is required for GSC survival and proliferation (41). GSCs directly interact with ECs, induce EC migration, and promote angiogenesis via an L1CAM-integrin αvβ3 mechanism (42). Knockdown of L1CAM results in downregulation of Olig2, a critical transcription factor for proliferation and maintenance of GSCs (41, 43, 44). In addition, activated Notch signaling promotes self-renewal and the invasive GSC phenotype (45). Immunofluorescence staining of primary GBMs demonstrates that GSCs expressing high levels of the Notch1 and Notch2 receptors are localized adjacent to Notch-expressing ECs (46). ECs express the Notch ligands DLL4 and JAGGED1 that activate Notch receptors on the surface of GSCs through cell-to-cell contact and promote GSC self-renewal (46). EC-secreted nitric oxide also activates Notch signaling that results in GSC self-renewal and glioma initiation (47). Inhibition of Notch signaling with a γ-secretase inhibitor results in decreased GSC self-renewal, depletes ECs in the vascular niche, and promotes GSC sensitivity to radiation therapy (48, 49).
Hypoxia Induces GSC Survival
Hypoxia and multifocal necrosis are hallmark features of GBM tumors and arise through the unregulated proliferation of tumor cells without sufficient supporting vasculature (50). Hypoxia plays a critical role in tumor progression, metabolism, metastasis, invasion, and therapeutic resistance (51, 52). Necrotic areas are surrounded by hypercellular regions termed pseudopalisades, which are microscopic structures unique to GBM. Pseudopalisades express higher levels of stem cell markers and are hypothesized to be waves of cells migrating away from hypoxic areas (53, 54). In GBM, GSCs have been reported to be enriched in hypoxic regions that promote maintenance and induces the expansion of GSCs (55, 56). Studies have shown that the hypoxic niche promotes the reprogramming of glioma non-stem cells into a cancer stem cell-like phenotype and regulates GSC self-renewal (30, 55, 57). Cellular responses to hypoxia are largely regulated by a family of transcription factors known as hypoxia-inducible factors (HIFs) (58). The expression of HIF2α increases with hypoxia, correlates with CD133 expression, and enhances the self-renewal capacity of GSCs (56, 59). HIF2α mediates the hypoxia-induced expression of stem cell markers specifically in GSCs, including Sox2, Oct4, and c-Myc (30, 57).
Hypoxia also induces the expression of HIF1α, and ablation of HIF1α expression decreases the CD133+ population and inhibits the invasive capability of glioma cells (56, 60). HIF1α is required for hypoxia-mediated maintenance of GSCs via activation of Notch pathway. Ablation of HIF1α or inactivation of the Notch pathway inhibits the hypoxia-mediated maintenance of GSCs (61). Furthermore, HIF1α can interact with and stabilize the intracellular domain of Notch, thus activating the Notch signaling pathway (61). In fact, hypoxia-dependent activation of Notch results in a decrease in E-cadherin expression and subsequent cell invasion (62). Treatment with a Notch inhibitor blocks hypoxia-induced invasion and promotes GSC apoptosis (62, 63).
Acidosis Regulates GSC Invasion and Stemness
The pH of GBM tumors is much lower compared to normal tissue (64). Low pH derives from enhanced metabolism and low oxygenation of GBM tumors. Low pH promotes the mRNA expression of HIF2α and stem cell markers, including Oct4, Olig2, and Nanog (64). Tissue pH also regulates the expression of VEGF in a MAPK-dependent mechanism, which potentiates tumor growth (65, 66). Acidosis and hypoxia cooperate to induce HIF transcription factors and promote GSC maintenance through the stress-induced chaperone protein HSP90 pathway. Furthermore, inhibition of HSP90 by shRNA knockdown or pharmacological inhibitors impairs the self-renewal and tumorigenic capacities of GSCs induced by acidosis (67).
The sodium–hydrogen exchanger isoform 1 (NHE1) protein is one factor that mediates the cellular response to pH (68). GBM cells express high levels of NHE1 to maintain homeostatic intracellular pH levels. NHE1 potentiates GBM cell migration by altering matrix metalloproteinase (MMP) activity and through direct interaction with the ERM complex protein ezrin (69).
Tumor-Associated Macrophages/Microglia (TAMs) Support GSC Maintenance
The central nervous system has resident immune cells, or microglia, that are responsible for eliciting an immune response (70, 71). Immunohistochemical staining of GBM tumors has shown high levels of infiltrating microglia, which constitute around 40% of brain tumor mass (72, 73). Upon recruitment to the tumor microenvironment, microglia become immunosuppressive and promote angiogenesis and invasion (74, 75). Ablation of microglia with clodronate liposomes or minocycline inhibits the invasion of GBM cells, supporting an important role for microglia in GBM invasion (75–77). GSCs play a vital role in the recruitment of microglia and macrophages and also secrete factors that promote pro-tumorigenic functions of microglia (78). GSCs upregulate interleukin 6 (IL6) release from microglia via toll-like receptor 4 (TLR4) signaling, which enhances invasion and glioma growth (79). Glioma cells also produce macrophage colony-stimulating factor, which induces microglia secretion of insulin-like growth factor binding protein 1 that promotes angiogenesis (80).
The ECM surrounding brain tumors serves as an impediment to invasion. MMPs, including gelatinases and membrane-type MMPs (MT-MMPs), are necessary to digest ECM components and facilitate invasion into the surrounding brain parenchyma (81). GSCs express high levels of the gelatinase MMP2, which is synthesized as a proenzyme and must be activated by cleavage. In a paracrine mechanism, GBM cells activate TLR signaling in microglia, which results in MT1-MMP expression (76, 77). Microglia-derived MT1-MMP activates GSC-derived MMP2 and promotes GBM invasion (82). Microglia also express high levels of osteopontin (OPN), a known ECM component that regulates GBM invasion. OPN interacts with CD44 and induces cleavage of CD44 in proneural GBM. The CD44 intracellular domain promotes GLSC self-renewal and an aggressive tumor phenotype, in part, via activation of HIF2α (83).
Microglia also release several growth factors that affect glioma cell invasion and survival. Treatment with microglia-conditioned media activates the protein tyrosine kinase 2 beta (Pyk2) signaling pathway in glioma cells (84). Pyk2 promotes cell invasion by activating MAP4K4 signaling in glioma cells (85, 86). In addition, microglia produce the epidermal growth factor (EGF) and enhance EGF-signaling pathways in glioma cells. Pharmacological inhibition of EGFR prevents microglial-induced invasion in glioma cells (87). The transforming growth factor-beta (TGFβ) is another factor that is secreted by microglia. TGFβ pathway is dysregulated in glioma and plays a key role in invasion via upregulation of MMPs and αvβ3 integrin expression (88–90). Microglia secrete TGFβ1 that activates the TGFβR2 pathway in GSCs and results in increased MMP9 expression and subsequent invasion (91, 92). TGFβ inhibitors block GSC-dependent tumor initiation (23).
In addition to microglia, tumor-associated macrophages are enriched in GSC niches (93, 94). These peripheral monocyte-derived macrophages are recruited by GSCs to the microenvironment via a periostin/integrin αvβ3-dependent mechanism (95). Upon recruitment to the GSC niche, TAMs adopt a pro-tumorigenic M2 phenotype and secrete IL6 and IL10 to promote the proliferation of GSCs in hypoxic niches (96).
Astrocytes Support GSC Maintenance
GSCs synergize with resident brain cells to promote tumorigenesis. There are reports that astrocytes promote the invasive capacity of GSCs, in part, by direct cell contact and secreting proteins associated with cell invasion (97, 98). Astrocytes secrete chemokines and cytokines, including IL6 and TGFβ2, that promote GSC invasion (97). Astrocyte-derived IL6 decreases the radiosensitivity of GSCs in vitro in a Stat3-dependent mechanism (99). Treatment with WP1066, a JAK/STAT3 inhibitor, enhances radiosensitivity of GSCs in a xenograft model (99). GFAP-positive astrocytes also secrete Shh, which activates GLI signaling and GSC stemness (38). Cross talk between astrocytes and glioma cells results in the activation of astrocyte-derived MMP2 and subsequent glioma migration. In a pathway mediated by the urokinase-type plasminogen activator-plasmin cascade, glioma-derived plasmin activates astrocyte-secreted MMP2. MMP2 degrades ECM components and facilitates glioma cell dispersal (98).
ECM Factors Mediate GSC Invasion
GSCs interact with the ECM during invasion. Although the composition of the brain tumor ECM is still being elucidated, there are high levels of hyaluronic acid (HA) surrounding invasive GBM tumors compared to normal brain tissue (100). HA is a glycosaminoglycan that provides structural support and regulates cell adhesion and migration (101). HA interacts with specific cognate receptors including CD44 and receptor for hyaluronate-mediated motility (RHAMM) (102). High levels of CD44 and RHAMM correlate with poor patient prognosis in GBM (102). HA interaction with CD44 or RHAMM activates downstream signaling cascades in GSCs that result in self-renewal, multidrug resistance, and cell invasion (102–106). Blocking the glioma cell-HA interaction inhibits GBM invasion and anchorage-independent growth (107). In addition, brevican, an extracellular hyaluronan-binding protein specific to the brain that is overexpressed in GBM, is enriched in the GSC niche (108, 109). Cleavage of brevican enhances the invasion of glioma cells (110). In contrast, brevican knockdown inhibits the tumorigenicity of GBM cells suggesting that brevican may play an important role in glioma progression (108, 109).
GSCs can secrete MMPs to remodel the surrounding ECM and create space for migration. GSCs express high levels TLR2, which upon activation enhances GSC invasion via upregulation of MMP2 and MMP9 (111). GSCs also express high levels of MMP13, which specifically enhances migration and invasion (112).
ADAM proteins are characterized by their metalloproteinase activity and integrin-receptor binding and have been implicated in several important cellular processes, including cell migration (113). ADAM17 has been shown to promote self-renewal capability of GSCs and inhibit GSC differentiation via Notch signaling (114). ADAM17 also promotes the invasion of CD133+ GSCs isolated from human glioblastoma cell line U87 through the EGFR/PI3K/AKT signaling pathway (115). ADAM9 has been found to mediate tenascin-C induced invasiveness of GSCs, and inhibition of ADAM9 attenuates GSC invasion (116).
Conclusion
Until the recent survival benefits afforded by the introduction of Novocure Optune, the survival of patients with primary GBM has remained unchanged since the Stupp protocol was implemented in 2005 (3, 117). Recent clinical trials, including altering TMZ doses or targeting specific genomic alterations, have failed to improve patient overall survival (118–120). This suggests that a paradigm shift for treating this devastating disease is warranted.
Glioblastoma multiforme tumors are cellularly heterogeneous and consist of GSCs, non-GSCs, microglia/TAMs, astrocytes, and neurons (Figure 1). GBM tumors appear to have a cellular hierarchy with GSCs at the apex. The GSC niches are symbiotic environments, with GSCs and non-tumoral cells reciprocally promoting tumorigenesis.
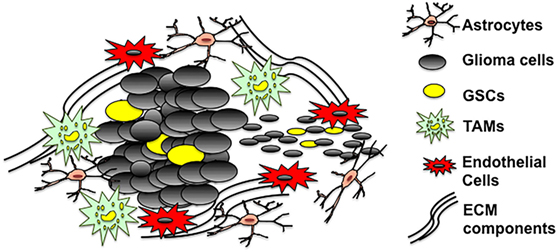
Figure 1. The GSC microenvironment. GSCs are enriched in both hypoxic and vascular niches surrounded by extracellular matrix (ECM) components and cells of different lineages including astrocytes, non-stem glioma cells, tumor-associated microglia/macrophages, and endothelial cells. The tumor microenvironment provides a fertile environment that supports and propagates GSCs.
Currently, it is thought that tumor recurrence derives from GSCs. GSCs are highly invasive and resistant to the current modes of therapy. This subpopulation is maintained and supported through direct interactions and paracrine signaling from microenvironmental cells (Figure 2). Microglia, the brain resident immune cells, cooperate with GSCs to promote invasion and tumor progression. In addition to secreting factors that potentiate GSC invasion and self-renewal, glioma cells and microglia share pro-tumorigenic pathways that may serve as nodes of intervention. Microglia express high levels of the TNFR receptor TROY, which activates microglial migration in a PYK2-dependent manner (121). Targeting microglia migration toward the GSC niche with propentofylline, a small molecule inhibitor of TROY signaling, mitigates the pro-tumorigenic functions of microglia and decreases tumor growth (122). Moreover, TROY is overexpressed in glioma cells and inhibiting TROY signaling blocks invasion (123, 124). Thus, targeting TROY could serve a dual function of inhibiting paracrine and autocrine regulators of glioma invasion.
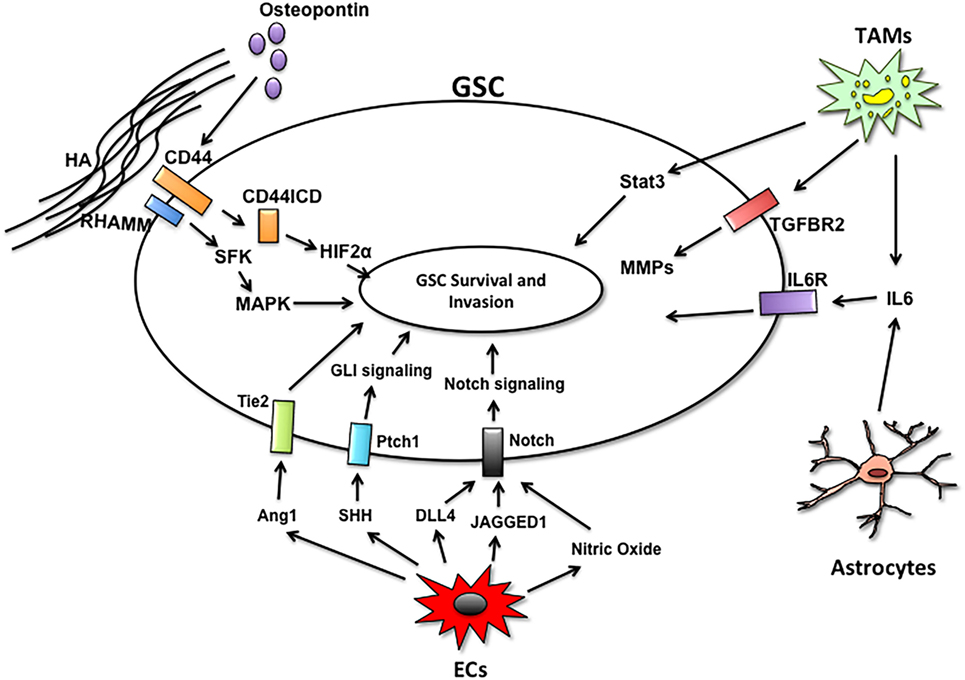
Figure 2. Microenvironmental cells and paracrine pathways facilitating GSC invasion. The GSC niches are comprised of endothelial cells (ECs), tumor-associated macrophages/microglia (TAMs), astrocytes, and extracellular matrix components. Non-tumor cells within the niche support GSC survival and invasion by secreting factors that activate key signaling pathways in GSCs to maintain self-renewal and survival.
GSCs are enriched in microvascular niches, which support GSC survival and invasion. Although preclinical models have demonstrated survival benefits of antiangiogenic therapies, clinical trials with early administration of bevacizumab failed to improve overall survival in patients with newly diagnosed GBM (125). Additionally, a phase II clinical trial with Tivozanib monotherapy demonstrated Tivozanib could alter the tumor vasculature but did not have antitumoral effect in patients with recurrent GBM (126). Therefore, it is vital that future studies consider not only resistance mechanisms to antiangiogenic therapy but also the symbiosis between GSCs and the microenvironment as a whole and assess the efficacy of antiangiogenic therapies in conjunction with other modes of therapy.
Although recent therapeutic approaches have improved GBM survival, patients still succumb to this devastating disease (117). Our understanding of the impact of the Optune system on GSCs has yet to be determined. If Optune was to be considered as part of standard of care, studies into the effect of tumor treating fields on GSCs and tumor microenvironmental cells are warranted. Additionally, deducing paracrine-regulated pathways mediating GSC survival is critical for mitigating tumor recurrence. Further investigations into the relationships between GSCs and their niches are imperative. A better understanding of the interaction of GSCs and niches will provide new insights to develop new therapeutic strategies for GBM patients. Targeting glioblastoma stem-cell niches may represent a novel strategy for treating GBM.
Author Contributions
AR wrote most of the manuscript and created the figures. ZD contributed to the text and figures. JL and NT edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the NIH grant NS86853.
References
1. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol (2014) 16(Suppl 4):iv1–63. doi:10.1093/neuonc/nou223
2. Schittenhelm J. Recent advances in subtyping tumors of the central nervous system using molecular data. Expert Rev Mol Diagn (2017) 17(1):83–94. doi:10.1080/14737159.2017.1266259
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi:10.1056/NEJMoa043330
4. Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol (2014) 71(11):1437–44. doi:10.1001/jamaneurol.2014.1701
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin (2015) 65(1):5–29. doi:10.3322/caac.21254
6. Xie Q, Mittal S, Berens ME. Targeting adaptive glioblastoma: an overview of proliferation and invasion. Neuro Oncol (2014) 16(12):1575–84. doi:10.1093/neuonc/nou147
7. Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel) (2013) 6(12):1475–506. doi:10.3390/ph6121475
8. Ene CI, Holland EC. Personalized medicine for gliomas. Surg Neurol Int (2015) 6(Suppl 1):S89–95. doi:10.4103/2152-7806.151351
9. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi:10.1016/j.cell.2013.09.034
10. Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol (2003) 21(8):1624–36. doi:10.1200/JCO.2003.05.063
11. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer (2003) 3(5):362–74. doi:10.1038/nrc1075
12. Kitai R, Horita R, Sato K, Yoshida K, Arishima H, Higashino Y, et al. Nestin expression in astrocytic tumors delineates tumor infiltration. Brain Tumor Pathol (2010) 27(1):17–21. doi:10.1007/s10014-009-0261-0
13. Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN, et al. Elevated invasive potential of glioblastoma stem cells. Biochem Biophys Res Commun (2011) 406(4):643–8. doi:10.1016/j.bbrc.2011.02.123
14. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444(7120):756–60. doi:10.1038/nature05236
15. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488(7412):522–6. doi:10.1038/nature11287
16. Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ (2006) 13(7):1238–41. doi:10.1038/sj.cdd.4401872
17. Todorova PK, Mukherjee B, Burma S. MET signaling promotes DNA repair and radiation resistance in glioblastoma stem-like cells. Ann Transl Med (2017) 5(3):61. doi:10.21037/atm.2017.01.67
18. Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene (2004) 23(43):7267–73. doi:10.1038/sj.onc.1207946
19. Bar EE. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol (2011) 21(2):119–29. doi:10.1111/j.1750-3639.2010.00460.x
20. Huang Q, Zhang QB, Dong J, Wu YY, Shen YT, Zhao YD, et al. Glioma stem cells are more aggressive in recurrent tumors with malignant progression than in the primary tumor, and both can be maintained long-term in vitro. BMC Cancer (2008) 8:304. doi:10.1186/1471-2407-8-304
21. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res (2003) 63(18):5821–8.
22. Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res (2008) 14(1):123–9. doi:10.1158/1078-0432.CCR-07-0932
23. Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell (2010) 18(6):655–68. doi:10.1016/j.ccr.2010.10.023
24. Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med (2015) 4(9):1033–43. doi:10.5966/sctm.2015-0048
25. Demuth T, Rennert JL, Hoelzinger DB, Reavie LB, Nakada M, Beaudry C, et al. Glioma cells on the run – the migratory transcriptome of 10 human glioma cell lines. BMC Genomics (2008) 9:54. doi:10.1186/1471-2164-9-54
26. Dhruv HD, McDonough Winslow WS, Armstrong B, Tuncali S, Eschbacher J, Kislin K, et al. Reciprocal activation of transcription factors underlies the dichotomy between proliferation and invasion of glioma cells. PLoS One (2013) 8(8):e72134. doi:10.1371/journal.pone.0072134
27. Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, et al. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia (2005) 7(1):7–16. doi:10.1593/neo.04535
28. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci (2007) 8(8):610–22. doi:10.1038/nrn2175
29. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell (2007) 11(1):69–82. doi:10.1016/j.ccr.2006.11.020
30. Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain (2010) 133(Pt 4):983–95. doi:10.1093/brain/awq042
31. Hira VV, Ploegmakers KJ, Grevers F, Verbovsek U, Silvestre-Roig C, Aronica E, et al. CD133+ and Nestin+ glioma stem-like cells reside around CD31+ arterioles in niches that express SDF-1alpha, CXCR4, osteopontin and cathepsin K. J Histochem Cytochem (2015) 63(7):481–93. doi:10.1369/0022155415581689
32. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res (2006) 66(16):7843–8. doi:10.1158/0008-5472.CAN-06-1010
33. Ping YF, Yao XH, Jiang JY, Zhao LT, Yu SC, Jiang T, et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J Pathol (2011) 224(3):344–54. doi:10.1002/path.2908
34. Bian XW, Yao XH, Ping YF, Chen JH, Wang JM. Glioblastoma stem cells produce angiogenic factors through activation of chemokine receptor CXCR4. J Neuropathol Exp Neurol (2007) 66(5):459–459. doi:10.1097/01.jnen.0000268979.21782.6e
35. Yadav VN, Zamler D, Baker GJ, Kadiyala P, Erdreich-Epstein A, DeCarvalho AC, et al. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: a genetic knockdown study. Oncotarget (2016) 7(50):83701–19. doi:10.18632/oncotarget.13295
36. Liu D, Martin V, Fueyo J, Lee OH, Xu J, Cortes-Santiago N, et al. Tie2/TEK modulates the interaction of glioma and brain tumor stem cells with endothelial cells and promotes an invasive phenotype. Oncotarget (2010) 1(8):700–9. doi:10.18632/oncotarget.101204
37. Paulus W, Baur I, Beutler AS, Reeves SA. Diffuse brain invasion of glioma cells requires beta 1 integrins. Lab Invest (1996) 75(6):819–26.
38. Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res (2008) 68(7):2241–9. doi:10.1158/0008-5472.CAN-07-6350
39. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi:10.1016/j.cub.2006.11.033
40. Galan-Moya EM, Le Guelte A, Lima Fernandes E, Thirant C, Dwyer J, Bidere N, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep (2011) 12(5):470–6. doi:10.1038/embor.2011.39
41. Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res (2008) 68(15):6043–8. doi:10.1158/0008-5472.CAN-08-1079
42. Burgett ME, Lathia JD, Roth P, Nowacki AS, Galileo DS, Pugacheva E, et al. Direct contact with perivascular tumor cells enhances integrin alphavbeta3 signaling and migration of endothelial cells. Oncotarget (2016) 7(28):43852–67. doi:10.18632/oncotarget.9700
43. Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron (2007) 53(4):503–17. doi:10.1016/j.neuron.2007.01.009
44. Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell (2014) 157(3):580–94. doi:10.1016/j.cell.2014.02.030
45. Yu JB, Jiang H, Zhan RY. Aberrant Notch signaling in glioblastoma stem cells contributes to tumor recurrence and invasion. Mol Med Rep (2016) 14(2):1263–8. doi:10.3892/mmr.2016.5391
46. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res (2011) 71(18):6061–72. doi:10.1158/0008-5472.CAN-10-4269
47. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell (2010) 6(2):141–52. doi:10.1016/j.stem.2010.01.001
48. Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells (2010) 28(6):1019–29. doi:10.1002/stem.429
49. Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells (2010) 28(1):17–28. doi:10.1002/stem.261
50. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26(2):225–39. doi:10.1007/s10555-007-9055-1
51. Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem (2009) 107(6):1053–62. doi:10.1002/jcb.22214
52. Talasila KM, Rosland GV, Hagland HR, Eskilsson E, Flones IH, Fritah S, et al. The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol (2017) 19(3):383–93. doi:10.1093/neuonc/now175
53. Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res (2004) 64(3):920–7. doi:10.1158/0008-5472.CAN-03-2073
54. Inukai M, Hara A, Yasui Y, Kumabe T, Matsumoto T, Saegusa M. Hypoxia-mediated cancer stem cells in pseudopalisades with activation of hypoxia-inducible factor-1alpha/Akt axis in glioblastoma. Hum Pathol (2015) 46(10):1496–505. doi:10.1016/j.humpath.2015.06.008
55. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle (2009) 8(20):3274–84. doi:10.4161/cc.8.20.9701
56. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene (2009) 28(45):3949–59. doi:10.1038/onc.2009.252
57. McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res (2009) 7(4):489–97. doi:10.1158/1541-7786.MCR-08-0360
58. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell (2007) 129(3):465–72. doi:10.1016/j.cell.2007.04.019
59. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell (2009) 15(6):501–13. doi:10.1016/j.ccr.2009.03.018
60. Mendez O, Zavadil J, Esencay M, Lukyanov Y, Santovasi D, Wang SC, et al. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol Cancer (2010) 9:133. doi:10.1186/1476-4598-9-133
61. Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ (2012) 19(2):284–94. doi:10.1038/cdd.2011.95
62. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A (2008) 105(17):6392–7. doi:10.1073/pnas.0802047105
63. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells (2010) 28(1):5–16. doi:10.1002/stem.254
64. Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ (2011) 18(5):829–40. doi:10.1038/cdd.2010.150
65. Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res (2001) 61(16):6020–4.
66. Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem (2002) 277(13):11368–74. doi:10.1074/jbc.M108347200
67. Filatova A, Seidel S, Bogurcu N, Graf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res (2016) 76(19):5845–56. doi:10.1158/0008-5472.CAN-15-2630
68. Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med (2010) 14(4):771–94. doi:10.1111/j.1582-4934.2009.00994.x
69. Cong D, Zhu W, Shi Y, Pointer KB, Clark PA, Shen H, et al. Upregulation of NHE1 protein expression enables glioblastoma cells to escape TMZ-mediated toxicity via increased H(+) extrusion, cell migration and survival. Carcinogenesis (2014) 35(9):2014–24. doi:10.1093/carcin/bgu089
70. London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci (2013) 7:34. doi:10.3389/fncel.2013.00034
71. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2016) 19(1):20–7. doi:10.1038/nn.4185
72. Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg (1979) 50(3):305–11. doi:10.3171/jns.1979.50.3.0305
73. Rossi ML, Hughes JT, Esiri MM, Coakham HB, Brownell DB. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol (1987) 74(3):269–77. doi:10.1007/BF00688191
74. Bettinger I, Thanos S, Paulus W. Microglia promote glioma migration. Acta Neuropathol (2002) 103(4):351–5. doi:10.1007/s00401-001-0472-x
75. Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol (2005) 64(9):754–62. doi:10.1097/01.jnen.0000178445.33972.a9
76. Liu WT, Lin CH, Hsiao M, Gean PW. Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy (2011) 7(2):166–75. doi:10.4161/auto.7.2.14043
77. Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, Glass R, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun (2011) 25(4):624–8. doi:10.1016/j.bbi.2011.01.015
78. Yi L, Xiao H, Xu M, Ye X, Hu J, Li F, et al. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J Neuroimmunol (2011) 232(1–2):75–82. doi:10.1016/j.jneuroim.2010.10.011
79. Dzaye ODA, Hu F, Derkow K, Haage V, Euskirchen P, Harms C, et al. Glioma stem cells but not bulk glioma cells upregulate IL-6 secretion in microglia/brain macrophages via toll-like receptor 4 signaling. J Neuropathol Exp Neurol (2016) 75(5):429–40. doi:10.1093/jnen/nlw016
80. Nijaguna MB, Patil V, Urbach S, Shwetha SD, Sravani K, Hegde AS, et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J Biol Chem (2015) 290(38):23401–15. doi:10.1074/jbc.M115.664037
81. Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci (2003) 8:e261–9. doi:10.2741/1016
82. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A (2009) 106(30):12530–5. doi:10.1073/pnas.0804273106
83. Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell (2014) 14(3):357–69. doi:10.1016/j.stem.2014.01.005
84. Rolon-Reyes K, Kucheryavykh YV, Cubano LA, Inyushin M, Skatchkov SN, Eaton MJ, et al. Microglia activate migration of glioma cells through a Pyk2 intracellular pathway. PLoS One (2015) 10(6):e0131059. doi:10.1371/journal.pone.0131059
85. Lipinski CA, Tran NL, Menashi E, Rohl C, Kloss J, Bay RC, et al. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia (2005) 7(5):435–45. doi:10.1593/neo.04712
86. Loftus JC, Yang Z, Kloss J, Dhruv H, Tran NL, Riggs DL. A novel interaction between Pyk2 and MAP4K4 is integrated with glioma cell migration. J Signal Transduct (2013) 2013:956580. doi:10.1155/2013/956580
87. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (2012) 18:519–27. doi:10.2119/molmed.2011.00217
88. Yamada N, Kato M, Yamashita H, Nister M, Miyazono K, Heldin CH, et al. Enhanced expression of transforming growth factor-beta and its type-I and type-II receptors in human glioblastoma. Int J Cancer (1995) 62(4):386–92. doi:10.1002/ijc.2910620405
89. Wick W, Platten M, Weller M. Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J Neurooncol (2001) 53(2):177–85. doi:10.1023/A:1012209518843
90. Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-beta signaling and its targeting for glioma treatment. Am J Cancer Res (2015) 5(3):945–55.
91. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol (2012) 189(1):444–53. doi:10.4049/jimmunol.1103248
92. Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion – an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene (2008) 27(7):918–30. doi:10.1038/sj.onc.1210683
93. Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell (2011) 8(5):482–5. doi:10.1016/j.stem.2011.04.013
94. Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget (2015) 6(17):15077–94. doi:10.18632/oncotarget.3730
95. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol (2015) 17(2):170–82. doi:10.1038/ncb3090
96. Samaras V, Piperi C, Korkolopoulou P, Zisakis A, Levidou G, Themistocleous MS, et al. Application of the ELISPOT method for comparative analysis of interleukin (IL)-6 and IL-10 secretion in peripheral blood of patients with astroglial tumors. Mol Cell Biochem (2007) 304(1–2):343–51. doi:10.1007/s11010-007-9517-3
97. Rath BH, Fair JM, Jamal M, Camphausen K, Tofilon PJ. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS One (2013) 8(1):e54752. doi:10.1371/journal.pone.0054752
98. Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, et al. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci (2003) 23(10):4034–43.
99. Rath BH, Wahba A, Camphausen K, Tofilon PJ. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med (2015) 4(11):1705–16. doi:10.1002/cam4.510
100. Delpech B, Maingonnat C, Girard N, Chauzy C, Maunoury R, Olivier A, et al. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur J Cancer (1993) 29A(7):1012–7. doi:10.1016/S0959-8049(05)80214-X
101. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol (2015) 6:201. doi:10.3389/fimmu.2015.00201
102. Park JB, Kwak HJ, Lee SH. Role of hyaluronan in glioma invasion. Cell Adh Migr (2008) 2(3):202–7. doi:10.4161/cam.2.3.6320
103. Solis MA, Chen YH, Wong TY, Bittencourt VZ, Lin YC, Huang LL. Hyaluronan regulates cell behavior: a potential niche matrix for stem cells. Biochem Res Int (2012) 2012:346972. doi:10.1155/2012/346972
104. Kim Y, Kumar S. CD44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Mol Cancer Res (2014) 12(10):1416–29. doi:10.1158/1541-7786.MCR-13-0629
105. Chanmee T, Ontong P, Kimata K, Itano N. Key roles of hyaluronan and its CD44 receptor in the stemness and survival of cancer stem cells. Front Oncol (2015) 5:180. doi:10.3389/fonc.2015.00180
106. Tilghman J, Wu H, Sang Y, Shi X, Guerrero-Cazares H, Quinones-Hinojosa A, et al. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res (2014) 74(11):3168–79. doi:10.1158/0008-5472.CAN-13-2103
107. Ward JA, Huang L, Guo H, Ghatak S, Toole BP. Perturbation of hyaluronan interactions inhibits malignant properties of glioma cells. Am J Pathol (2003) 162(5):1403–9. doi:10.1016/S0002-9440(10)64273-3
108. Lu R, Wu C, Guo L, Liu Y, Mo W, Wang H, et al. The role of brevican in glioma: promoting tumor cell motility in vitro and in vivo. BMC Cancer (2012) 12:607. doi:10.1186/1471-2407-12-607
109. Dwyer CA, Bi WL, Viapiano MS, Matthews RT. Brevican knockdown reduces late-stage glioma tumor aggressiveness. J Neurooncol (2014) 120(1):63–72. doi:10.1007/s11060-014-1541-z
110. Zhang H, Kelly G, Zerillo C, Jaworski DM, Hockfield S. Expression of a cleaved brain-specific extracellular matrix protein mediates glioma cell invasion in vivo. J Neurosci (1998) 18(7):2370–6.
111. Wang F, Zhang P, Yang L, Yu X, Ye X, Yang J, et al. Activation of toll-like receptor 2 promotes invasion by upregulating MMPs in glioma stem cells. Am J Transl Res (2015) 7(3):607–15.
112. Inoue A, Takahashi H, Harada H, Kohno S, Ohue S, Kobayashi K, et al. Cancer stem-like cells of glioblastoma characteristically express MMP-13 and display highly invasive activity. Int J Oncol (2010) 37(5):1121–31. doi:10.3892/ijo_00000764
113. Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev (2003) 17(1):7–30. doi:10.1101/gad.1039703
114. Chen X, Chen L, Zhang R, Yi Y, Ma Y, Yan K, et al. ADAM17 regulates self-renewal and differentiation of U87 glioblastoma stem cells. Neurosci Lett (2013) 537:44–9. doi:10.1016/j.neulet.2013.01.021
115. Chen X, Chen L, Chen J, Hu W, Gao H, Xie B, et al. ADAM17 promotes U87 glioblastoma stem cell migration and invasion. Brain Res (2013) 1538:151–8. doi:10.1016/j.brainres.2013.02.025
116. Sarkar S, Zemp FJ, Senger D, Robbins SM, Yong VW. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro Oncol (2015) 17(8):1095–105. doi:10.1093/neuonc/nou362
117. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA (2015) 314(23):2535–43. doi:10.1001/jama.2015.16669
118. Wick W, Platten M, Weller M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro Oncol (2009) 11(1):69–79. doi:10.1215/15228517-2008-078
119. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol (2013) 31(32):4085–91. doi:10.1200/JCO.2013.49.6968
120. Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol (2010) 98(1):93–9. doi:10.1007/s11060-009-0067-2
121. Jacobs VL, Liu Y, De Leo JA. Propentofylline targets TROY, a novel microglial signaling pathway. PLoS One (2012) 7(5):e37955. doi:10.1371/journal.pone.0037955
122. Jacobs VL, Landry RP, Liu Y, Romero-Sandoval EA, De Leo JA. Propentofylline decreases tumor growth in a rodent model of glioblastoma multiforme by a direct mechanism on microglia. Neuro Oncol (2012) 14(2):119–31. doi:10.1093/neuonc/nor194
123. Loftus JC, Dhruv H, Tuncali S, Kloss J, Yang Z, Schumacher CA, et al. TROY (TNFRSF19) promotes glioblastoma survival signaling and therapeutic resistance. Mol Cancer Res (2013) 11(8):865–74. doi:10.1158/1541-7786.MCR-13-0008
124. Dhruv HD, Roos A, Tomboc PJ, Tuncali S, Chavez A, Mathews I, et al. Propentofylline inhibits glioblastoma cell invasion and survival by targeting the TROY signaling pathway. J Neurooncol (2016) 126(3):397–404. doi:10.1007/s11060-015-1981-0
125. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med (2014) 370(8):699–708. doi:10.1056/NEJMoa1308573
Keywords: glioblastoma, invasion, glioma stem-like cells, survival, stem-cell niches
Citation: Roos A, Ding Z, Loftus JC and Tran NL (2017) Molecular and Microenvironmental Determinants of Glioma Stem-Like Cell Survival and Invasion. Front. Oncol. 7:120. doi: 10.3389/fonc.2017.00120
Received: 28 March 2017; Accepted: 24 May 2017;
Published: 16 June 2017
Edited by:
Cornelis Johannes Forrendinis Van Noorden, Academic Medical Center, NetherlandsReviewed by:
Vinesh Puliyappadamba, University of Alabama, United StatesSunit Das, University of Toronto, Canada
Copyright: © 2017 Roos, Ding, Loftus and Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nhan L. Tran, dHJhbi5uaGFuQG1heW8uZWR1