 Elzbieta Kania
Elzbieta Kania Gemma Roest
Gemma Roest Tim Vervliet
Tim Vervliet Jan B. Parys
Jan B. Parys Geert Bultynck
Geert Bultynck- Laboratory for Molecular and Cellular Signaling, Department of Cellular and Molecular Medicine, Leuven Kankerinstituut, KU Leuven, Leuven, Belgium
Calcium ions (Ca2+) play a complex role in orchestrating diverse cellular processes, including cell death and survival. To trigger signaling cascades, intracellular Ca2+ is shuffled between the cytoplasm and the major Ca2+ stores, the endoplasmic reticulum (ER), the mitochondria, and the lysosomes. A key role in the control of Ca2+ signals is attributed to the inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs), the main Ca2+-release channels in the ER. IP3Rs can transfer Ca2+ to the mitochondria, thereby not only stimulating core metabolic pathways but also increasing apoptosis sensitivity and inhibiting basal autophagy. On the other hand, IP3-induced Ca2+ release enhances autophagy flux by providing cytosolic Ca2+ required to execute autophagy upon various cellular stresses, including nutrient starvation, chemical mechanistic target of rapamycin inhibition, or drug treatment. Similarly, IP3Rs are able to amplify Ca2+ signals from the lysosomes and, therefore, impact autophagic flux in response to lysosomal channels activation. Furthermore, indirect modulation of Ca2+ release through IP3Rs may also be achieved by controlling the sarco/endoplasmic reticulum Ca2+ ATPases Ca2+ pumps of the ER. Considering the complex role of autophagy in cancer development and progression as well as in response to anticancer therapies, it becomes clear that it is important to fully understand the role of the IP3R and its cellular context in this disease. In cancer cells addicted to ER–mitochondrial Ca2+ fueling, IP3R inhibition leads to cancer cell death via mechanisms involving enhanced autophagy or mitotic catastrophe. Moreover, IP3Rs are the targets of several oncogenes and tumor suppressors and the functional loss of these genes, as occurring in many cancer types, can result in modified Ca2+ transport to the mitochondria and in modulation of the level of autophagic flux. Similarly, IP3R-mediated upregulation of autophagy can protect some cancer cells against natural killer cells-induced killing. The involvement of IP3Rs in the regulation of both autophagy and apoptosis, therefore, directly impact cancer cell biology and contribute to the molecular basis of tumor pathology.
Intracellular Ca2+ Signaling: The Endoplasmic Reticulum (ER), Mitochondria, and Lysosomes
Intracellular Ca2+ signaling controls a plethora of cellular processes, including secretion, gene transcription, metabolism, and cell death, thereby impacting cell function and cell survival (1–3). Intracellular Ca2+ signals are characterized by their spatiotemporal properties. As a function of time, Ca2+ signals can occur as transient increases in [Ca2+] (Ca2+ oscillations) (4) or as more sustained increases in [Ca2+] (global Ca2+ transients) (5). As a function of space, Ca2+ signals can occur in localized domains near the plasma membrane (PM) or organelles, such as the ER, mitochondria, lysosomes, Golgi, and nucleus (3, 6). Localized Ca2+ signaling is established in so-called microdomains due to the close apposition of different organellar compartments (7–9) or of organelles with the PM through molecular tethers (10–12).
The major intracellular Ca2+-storage organelle is the ER, where most of the intracellular Ca2+ is accumulated via active Ca2+ transport mediated by sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) followed by intraluminal Ca2+ buffering by calreticulin, calnexin, and other Ca2+-binding proteins (13, 14). These mechanisms allow for an adequate Ca2+ filling of the ER, which is required for the activity of molecular chaperones and the folding of enzymes. Hence, a depletion of the ER Ca2+ stores leads to ER stress, a condition associated with impaired protein folding capacity (13, 15, 16). To cope with this, cells engage the unfolded protein response, a concerted program triggered through the three classical ER stress sensors: inositol-requiring enzyme1α, RNA-dependent protein kinase-like ER kinase, and activating transcription factor 6 (17). Mild or transient ER stress induces activity of chaperones, folding enzymes, reactive oxygen species (ROS) scavengers, and degradative pathways, such as autophagy, while severe or persistent ER stress induces cell death (18–20).
These Ca2+-uptake mechanisms are counteracted by, on the one hand, Ca2+-leak channels and, on the other hand, Ca2+-release channels (21). Ca2+-leak channels establish a constitutive, passive Ca2+ leak from the ER, preventing ER Ca2+ overload that would result in cell death. Different ER Ca2+-leak channels have been identified, likely all contributing to this passive Ca2+ leak to some extent, although it is possible that some of these channels are restricted to certain cell types or systems (22–25). Presenilin 2, Ca2+ release-activated Ca2+ channel protein 2 (Orai2) and inositol 1,4,5-trisphosphate (IP3) receptor (IP3R) isoform 1 (IP3R1) have been proposed as major ER Ca2+-leak channels, based on a systems biology approach using HeLa cells (26). However, the ER Ca2+-leak rates in wild-type HEK293 versus HEK293 cells lacking all three IP3R isoforms, directly measured by using a genetically encoded ER Ca2+ sensor, were very similar, indicating that at least in HEK cells and these experimental conditions, IP3R does not contribute in an important way to the passive Ca2+ leak from the ER (27). These Ca2+-leak channels impact the steady-state Ca2+ content of the ER, which determines the Ca2+ available for release upon agonist stimulation (28, 29). Ca2+ release from the ER occurs through IP3Rs (30, 31) or ryanodine receptors (RyRs) (32–34). IP3Rs are ubiquitously expressed and are activated in response to IP3, which is produced from phosphatidylinositol 4,5-bisphosphate (PIP2) upon hydrolysis mediated by phospholipase C (35). This typically happens in response to cellular stimulation with hormones, growth factors, neurotransmitters, or antibodies. However, it is clear that many cells even in basal, non-stimulated conditions, display a constitutively low level of IP3-mediated signaling. The Ca2+ depletion of the ER, resulting from Ca2+ release, can trigger the activation of store-operated Ca2+ entry (SOCE) through stromal interaction molecule 1 (Stim1)-dependent activation of Ca2+ release-activated Ca2+ channel protein 1 (Orai1) channels (36–38).
Ca2+ release from the ER does not only result in a [Ca2+] rise in the cytosol but also leads to [Ca2+] increase in other organelles, including the mitochondria and the lysosomes (6, 39). This is due to contact sites between ER and mitochondria and between ER and lysosomes, decreasing the distance between these organelles and the ER (39–43). In addition to this, a highly negative potential of about −180 mV exists across the mitochondrial inner membrane, establishing a strong electrochemical driving force for Ca2+ uptake in the mitochondria (40). It is well known that IP3R-mediated Ca2+ release from the ER can “quasi-synaptically” transfer into the mitochondria (7, 44). This occurs via the so-called mitochondria-associated ER membranes (MAMs) that also harbor the IP3R and the voltage-dependent anion channel type 1 that permeates Ca2+ across the mitochondrial outer membrane (39, 41, 42). Once in the mitochondrial intermembrane space, Ca2+ is transported across the mitochondrial inner membrane via the mitochondrial Ca2+ uniporter (MCU) (45). The MCU has low inherent affinity for Ca2+ and is highly cooperative due to accessory proteins such as MICU1 (46). By comparison, Ca2+ uptake into the lysosomes is much less understood. Nevertheless, it is anticipated that a strong electrochemical gradient for H+ is present resulting from a low lysosomal pH (~4–5) which can be used for lysosomal Ca2+ accumulation via the lysosomal H+/Ca2+ exchanger (47, 48). Ca2+ can be released from these lysosomal Ca2+ stores via a variety of channels, including two-pore channels 1/2 (TPC1/2) and transient receptor potential superfamily channels such as TRPML1 (mucolipin1/MCOLN1)(47, 49–52). An important second messenger triggering lysosomal Ca2+ release through TPC1/2 is nicotinic acid adenine dinucleotide phosphate (NAADP) (53–55). TRPML1 present in endolysomal vesicles can be activated by phospholipids such as phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] (56, 57).
Ca2+ Signaling in Autophagy
ER-Derived Ca2+ Signaling in Autophagy
It is well-established that Ca2+ signaling impacts autophagy initiation and progression (Figure 1). Ca2+ signaling modulates as well basal autophagic flux as mechanistic target of rapamycin (mTOR)-controlled autophagic flux, induced by nutrient starvation or rapamycin (58–60). Moreover, compounds that directly affect Ca2+ signaling, including agonists, Ca2+ ionophores, and SERCA inhibitors that indirectly cause ER Ca2+-store depletion via the basal Ca2+ leak, can modulate the autophagic process (61). The ER, as the main intracellular Ca2+-storage organelle, has been implicated in controlling basal autophagy. This is related to Ca2+-dependent energizing of the mitochondria (62). Basal and constitutive Ca2+-release events from the ER, mediated by IP3R channels, have been involved in sustaining mitochondrial bioenergetics by driving NADH production and subsequent ATP synthesis by continuously providing Ca2+ to the mitochondria (62) (Figure 1). This is due to the presence of three mitochondrial Ca2+-dependent tricarboxylic acid (TCA) cycle enzymes, which activities are enhanced by mitochondrial Ca2+ (40). Abrogating these Ca2+ signals through pharmacological inhibition or genetic knock down of IP3Rs resulted in an increased autophagic flux due to an increase in the activity of the AMP-activated kinase (AMPK), a positive regulator of autophagy (i) inhibiting mTOR and (ii) activating the unc-51-like kinase 1 (ULK1) complex (63). However, the increased basal autophagic flux triggered upon IP3R inhibition appeared independent on mTOR (62) (Figure 1). Thus, IP3Rs exert an inhibitory role on basal autophagy, and consequently IP3R inhibition results in increased basal autophagy. An additional mechanism for this autophagy-inhibitory role of IP3Rs has been attributed to its Beclin 1-scaffolding function (64, 65). Beclin 1 regulates autophagy by forming a complex with the class III phosphatidylinositide 3-kinase Vps34, thereby stimulating phagophore nucleation, an early step in the autophagy process (66–68). It has been shown that the IP3R, independently of its Ca2+-flux properties, could serve as a sink for Beclin 1 recruitment, reducing the availability of free Beclin 1 to drive autophagy (69) (Figure 1).
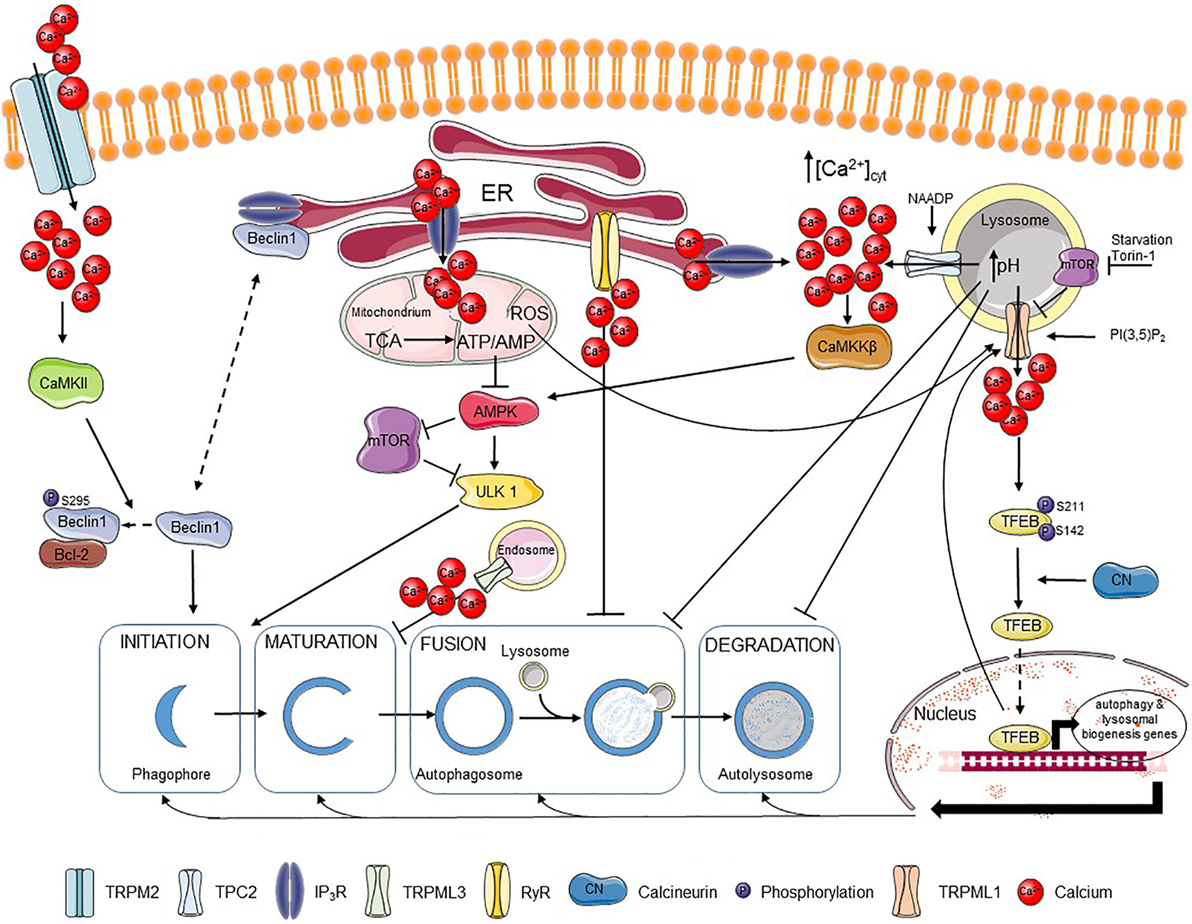
Figure 1. Ca2+ regulates different steps of autophagy. Extracellular Ca2+ can enter the cell via the TRPM2 channel, where it activates CaMKII that phosphorylates Beclin 1, promoting its binding to Bcl-2 and blocking autophagy initiation. Beclin 1 can also be scaffolded to the IP3R, which also limits autophagy promotion. IP3Rs are responsible for fueling mitochondria with Ca2+, which supports the tricarboxylic acid (TCA) cycle and the resulting ATP production. The presence of an adequate ATP/AMP ratio suppresses the AMP-activated kinase (AMPK)–mechanistic target of rapamycin (mTOR)–unc-51-like kinase 1 (ULK1) axis and thus autophagy induction. Autophagosome maturation can be inhibited by the TRPML3 channel mostly expressed in endosomes, while fusion of autophagosomes and lysosomes can be blocked by basal RyR activity. Ca2+ release through lysosomal nicotinic acid adenine dinucleotide phosphate (NAADP)-activated TPC2 channels can be further amplified by Ca2+ release from IP3Rs resulting in stimulation of the CaMKKβ–AMPK pathway and autophagosome formation. Ca2+ release from the lysosomes can, however, affect lysosomal pH and so abrogate fusion of lysosomes with autophagosomes as well as the further lysosome-dependent degradation. TRPML1, another lysosomal channel, can be regulated by different factors, including lysosomal mTOR, PI(3,5)P2 or reactive oxygen species (ROS). Ca2+ released from TRPML1 activates calcineurin, which binds and dephosphorylates TFEB and promotes its nuclear translocation, where TFEB induces transcription of various autophagy-related and lysosomal biogenesis genes. Plain black arrows indicate activatory and inhibitory pathways; dashed arrows indicate intracellular movement.
Besides IP3Rs, RyRs have also been implicated in autophagy. In hippocampal neuronal stem cells, insulin withdrawal resulted in the upregulation of the RyR3 isoform, which triggered cell death through increased autophagy. Activation of RyRs using caffeine increased autophagic cell death, while their inhibition using dantrolene suppressed this process (70). However, more recently, RyRs, both endogenously expressed in skeletal muscle cell lines and in dissociated rat hippocampal neurons or ectopically expressed in HEK cells, have been implicated in inhibition of autophagic flux, particularly at the level of autophagosome/lysosome fusion (Figure 1). RyR inhibition resulted in an increase of autophagic flux, independent of mTOR activity or of early autophagy regulators (71). This indicates that both IP3Rs and RyRs could suppress basal autophagy, but by acting at a different level: IP3Rs by suppressing autophagy at a proximal level by driving mitochondrial bioenergetics and thus decreasing AMPK activity, while RyRs block autophagy at a distal level by counteracting the fusion of autophagosomes and lysosomes.
In contrast to this, Ca2+ mobilization from the ER into the cytosol by itself can augment autophagic flux. Both physiological agonists as well as chemicals, such as Ca2+ ionophores and SERCA inhibitors, that provoke [Ca2+] rises in the cytosol originating from the ER result in the activation of Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ) that is an upstream activator of AMPK and autophagy (72). In fact, mechanisms that limited cytosolic [Ca2+] rise induced by these agents also limited autophagy induction. As such, Bcl-2, a known negative regulator of autophagy, was proposed not only to limit autophagy by scaffolding Beclin 1 but also by reducing the ER Ca2+-store content, thus suppressing cytosolic [Ca2+] rises and the extent of CaMKKβ activation (Figure 1) (73).
In addition to this, cytosolic [Ca2+] rises can promote autophagosome formation by recruiting the phosphatidylinositol 3-phosphate [PI(3)P]-binding protein, encoded by autophagy-related gene (Atg) 18 (WIPI1/WIPI2), to autophagosomal membranes. Atg18, together with Atg16L and ULK1, are recruited to early autophagosomal structures and are critical for the formation of LC3-positive autophagosomes (74). Furthermore, the autophagosomal recruitment of Atg18 was blocked by cytosolic Ca2+ chelation. This mechanism allowed for an induction of autophagy by cytosolic [Ca2+] rise independently of AMPK activation or mTOR inhibition (75). However, apart from the autophagy-inducing properties of thapsigargin and Ca2+ ionophores, these agents have also been reported to suppress autophagosome biogenesis at steps in the autophagy pathway subsequently to WIPI1 punctae formation but preceding autophagosome closure (76). This may relate to the disturbance of critical Ca2+ fluxes from the ER during the distal steps in the autophagy process, including the closure of the autophagosomal vesicles.
Beyond these roles in modulating basal autophagy, the ER and more particular ER-derived Ca2+ signals mediated via IP3Rs have been involved in driving starvation- and rapamycin-induced autophagy (58, 59). Starvation and rapamycin are two triggers that induce autophagy through inhibition of mTOR, a negative regulator of autophagy. Starvation- and rapamycin-induced autophagy resulted in enhanced Ca2+ signaling from the ER through a mechanism that involved complex formation between IP3Rs and Beclin 1 and a subsequent direct IP3R sensitization by Beclin 1 (58) (Figure 1). In turn, cytosolic Ca2+ and IP3R activity were critical for cells to be able to increase their autophagic flux in response to nutrient starvation and rapamycin.
Resveratrol (RSV) is a polyphenol found in some food products and in red wine. Several health-promoting effects have been attributed to RSV, including longevity, anti-aging, and improved cardiovascular health (77, 78). These beneficial effects of RSV have been linked to its ability to induce autophagy. RSV induces autophagy in a pleiotropic manner via both mTOR-dependent and -independent mechanisms. RSV can activate the deacetylases sirtuins, a positive regulator of AMPK (79, 80). RSV can also directly inhibit mTOR by docking onto its ATP-binding pocket and thus competing with ATP. The inhibition of mTOR and the presence of ULK1 appeared to be critical for RSV-induced autophagy (81). However, RSV can also promote autophagy in a non-canonical manner, whereby RSV induces autophagosome formation independently of Beclin 1 or its binding partner Vps34 (82, 83). Also, Ca2+ signaling has been implicated in RSV-induced autophagy (27). RSV can deplete the ER Ca2+ stores independently of the presence or absence of IP3Rs (27), which in part may be due to its inhibitory effect on mitochondrial ATP production, thereby suppressing SERCA-mediated ER Ca2+ uptake (84). Yet, although RSV triggered a Ca2+ leak from the ER independently of IP3Rs, the ability of RSV to induce autophagy was critically dependent on the presence of IP3Rs and on the availability of cytosolic Ca2+ (27). In this study, the inhibitory effect of RSV on mTOR activity was confirmed and did neither require cytosolic Ca2+ nor IP3R expression (27).
Endolysosomal-Derived Ca2+ Signaling in Autophagy
A genetic analysis of mucolipidosis type IV (MLIV), a lysosomal storage disease associated with severe neurological deficiencies, implicated that mutations in TRPML1 play a role in autophagy deregulation (85). Fibroblasts derived from MLIV patients expressing mutant TRPML1 displayed an increased autophagosome formation accompanied with a delay in the fusion of autophagosomes with lysosomes. This was proposed to contribute to an accumulation of p62 and a defective removal of ubiquitinated proteins and/or defective mitochondria (85). Also, chaperone-mediated autophagy (CMA) was defective in MLIV fibroblasts, which could be attributed to TRPML1’s ability to bind and recruit Hsc70 and Hsp40 proteins, two components critical for CMA, and a subsequent reduction in lysosomal LAMP2A-protein levels. As a consequence, oxidized proteins may accumulate in the cytosol due to their impaired degradation via CMA (86). However, in these studies, the role of TRPML1 channel activity and lysosomal Ca2+ release was not addressed.
One of the first data linking TRPML-mediated Ca2+ release and the autophagy pathway was provided from overexpression studies using TRPML3/MCOLN3 (87). This channel is mainly present in early endosomal compartments, where pH is not as low as in lysosomes. These endosomal compartments may host more functional TPRML3 channels than lysosomes, as low pH appears to inactivate TRPML3-mediated Ca2+ flux. Overexpression of TRPML3/MCOLN3 not only resulted in severe changes in the endosomal pathway, including increased endosomal pH, but also in defective autophagosome maturation (87) (Figure 1).
More recently, a direct role of lysosomal Ca2+ release through TRPML1 channels in upregulating autophagy upon mTOR inhibition has been elucidated (88). Nutrient starvation resulted in rapid peri-lysosomal [Ca2+] rises, in the close proximity of TRPML1 channels. These [Ca2+] rises were concentrated around lysosomes and could not be observed in the bulk cytosol. In turn, [Ca2+] rises resulted in the activation of the Ca2+/calmodulin-dependent phosphatase calcineurin, which can dephosphorylate the transcription factor TFEB. As a consequence, upon nutrient starvation, calcineurin dephosphorylated TFEB at two residues (Ser142 and Ser211) regulating TFEB nuclear translocation, resulting in nuclear accumulation of TFEB and activation of genes necessary for autophagy and lysosomal biogenesis (88) (Figure 1).
TRPML1 itself is also regulated during nutrient starvation. TRPML1 and the hereby associated Ca2+ flux from the lysosomes became upregulated upon nutrient deprivation, which was accompanied by transcription of autophagy-regulating genes (89). TRPML1 upregulation also occurred in response to complete inhibition of the mTOR complex 1 by Torin-1, while this was not observed upon treatment with rapamycin, a partial, allosteric inhibitor of mTORC1. A critical role for TFEB was found in the upregulation of TRPML1. Both starvation and Torin-1 treatment were able to induce TFEB dephosphorylation and its nuclear translocation, while rapamycin failed to do this. A more direct link between TFEB activation and TRPML1 upregulation was shown by overexpressing constitutively dephosphorylated and thus active TFEB in cells, which resulted in a functional upregulation of TRPML1 channels. The upregulation of TRPML1 activity by TFEB could be partially attributed to an increase in mRNA and protein expression of the channel, but likely also involved post-translational modifications or upregulation of TRPML1-interacting/modulating proteins. Moreover, a role of the lysosomal lipid PI(3,5)P2, a TRPML1-activating lipid which levels decrease upon nutrient starvation, was proposed as part of a compensatory mechanism that causes upregulation of TRPML1. At the functional level, TRPML1 activity was critical for the increase in lysosomal proteolytic activity induced by nutrient starvation (89).
Interestingly, TRPML1 activity is not only controlled by PI(3,5)P2 levels and via the TFEB transcription factor but also directly by mTOR (90). In nutrient-replete conditions, when mTOR was active and autophagy was suppressed, mTOR phosphorylated two serine residues in the C-terminal tail of TRPML1, resulting in TRPML1 channel inhibition. Upon mTOR inhibition by rapamycin, leading to autophagy induction, TRPML1 became dephosphorylated and active, resulting in lysosomal Ca2+ release. Moreover, rapamycin could induce Ca2+ release in cells expressing wild-type TRPML1 but not in cells expressing TRPML1 in which the two phosphorylable serine residues were mutated. Thus, loss of TRPML1 phosphorylation upon mTOR inhibition results in increased TRPML1 activity, driving autophagic flux (90). However, at this point, it is not clear which phosphatase is responsible for dephosphorylating the serine residues that are phosphorylated by the mTOR kinase.
Recently, it was shown that TRMPL1 can serve as a redox status sensor and can release Ca2+ upon stimulation by ROS or by mitochondrial uncouplers (91, 92). As a result of this Ca2+ release, also calcineurin-dependent TFEB activation and nuclear translocation occurred, which could be blocked by the ROS scavenger N-acetyl-cysteine (NAC), as well as by BAPTA-AM or synthetic TRPML1 inhibitors (ML-SIs). Since mitochondrial uncouplers failed to stimulate nuclear translocation of TFEB in TRPML1 knockout (KO) cells, this points out that TRPML1 is specifically required for ROS-induced activation of TFEB. By contrast, functional TRMPL1 was not required for TFEB nuclear translocation induced by mTOR inhibition through Torin-1 or nutrient starvation (91). In addition, ROS-induced autophagy and lysosome biogenesis could be impeded by NAC treatment as well as by ML-SIs or TRPML1 KO (91).
In contrast to TRPML1-mediated Ca2+ release from the lysosomes, thereby positively regulating autophagy, TPC2 has been implicated in the inhibition of autophagy (93). TPC2 channels are activated by NAADP. Overexpression of TPC2 resulted in the accumulation of autophagosomes, a phenomenon boosted by NAADP but antagonized by either the NAADP antagonist Ned19 or by knockdown (KD) of the essential autophagy gene Atg5. The effect of TPC2 on autophagosome accumulation could be attributed to an increase in lysosomal pH upon lysosomal Ca2+ release, which inhibits autophagy at the level of autophagosome–lysosome fusion (Figure 1). Therefore, lysosomal acidification could suppress TPC2-induced autophagosome accumulation (93). Similar findings of TPC2 have been observed in astrocytes, where its overexpression resulted in an increase in Beclin 1 and LC3-II levels (94). The latter may also relate to the accumulation of autophagosomes.
Ca2+ Influx from Extracellular Environment
Recently, oxidative stress has been implicated in autophagy inhibition through induction of melastatin-related transient receptor potential cation channel member 2 (TRPM2)-mediated Ca2+ influx (95). Ca2+ influx resulted in the activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which phosphorylated Beclin 1 at Ser295 and abolished its autophagy-inducing properties. The mechanism involved a decrease in Vps34 complex formation with phospho-Beclin 1 and increased Bcl-2 binding of phospho-Beclin 1 (Figure 1). Consequently to autophagy inhibition, oxidative stress triggered cell death in cells expressing TRPM2, while TRPM2 KD resulted in upregulated autophagy as a survival pathway in these cells. In addition, TRPM2/CaMKII activation further increased ROS production and contributed to mitochondrial fragmentation and loss of mitochondrial potential (95, 96).
IP3Rs and Autophagy in Cancer
IP3Rs in Cancer
IP3Rs control different hallmarks of cancer (97, 98). In particular, IP3Rs impact cell death and survival by mediating Ca2+ release from the ER and subsequently affecting mitochondria-regulated processes, including bioenergetics and apoptosis (99). Moreover, several IP3R isoforms can have distinct functions, dependent not only on their functional properties but also on their subcellular localization (100). For instance, IP3R3 has been particularly associated with pro-apoptotic Ca2+ flux from ER into mitochondria due to its localization at ER–mitochondrial contact sites (101, 102). As such, depending on the isoform and localization of the IP3R that is modulated, opposite effects can occur. Enhanced basal IP3R activity outside the MAMs may cause an increased passive Ca2+ leak from the ER. As a consequence, ER Ca2+ stores become less filled whereby less Ca2+ becomes available to be delivered to the mitochondria upon cellular exposure to a toxic, pro-apoptotic stimulus. By contrast, enhanced IP3R activity at the MAMs can increase not only mitochondrial bioenergetics but also the likelihood for pro-apoptotic Ca2+ transfers. In addition, Ca2+ transfer into the mitochondria also participate in oncogene-induced and replicative senescence, a stable proliferation arrest accompanied with distinct features like increased apoptosis resistance and altered gene expression (103–105). Here, IP3R2 and MCU were implicated in the enhanced mitochondrial Ca2+ transfer and accumulation that resulted in cellular senescence due to a decline in mitochondrial membrane potential and an increased ROS production and senescence. Therefore, loss of ITPR2, the gene encoding IP3R2, or of MCU overcame the growth arrest and escape from senescence (105, 106).
IP3Rs emerged as functional targets of an increasing number of oncogenes and tumor suppressors, which dynamically control IP3R activity and thus Ca2+ flux from ER into mitochondria (97, 107). Several oncogenes can suppress pro-apoptotic Ca2+-release events mediated by IP3Rs and this can occur via different mechanisms. First, oncogenes can directly interact with IP3Rs (97). For instance, anti-apoptotic Bcl-2 targets the central, modulatory domain of the IP3Rs, thereby suppressing excessive IP3R activity and protecting cells against pro-apoptotic Ca2+-release events (108–111). Alternatively, oncogenes can exert post-translational modifications of IP3Rs. Protein kinase B (PKB/Akt) phosphorylates IP3R3 and dampens its Ca2+ flux, suppressing pro-apoptotic Ca2+ transfer (112). Oncogenes not only prevent excessive IP3R-mediated Ca2+ release, but they can also promote basal Ca2+-signaling events that are associated with increased mitochondrial bioenergetics and, thus, increased NADH and ATP output. As such, Bcl-XL, another anti-apoptotic Bcl-2-family member sensitizes all IP3R isoforms, thereby promoting the occurrence of pro-survival Ca2+ oscillations and thus sustaining cell survival by boosting the mitochondrial metabolism (113–115).
Not only oncogenes but also tumor suppressors regulate IP3Rs (97, 107). The product of the BRCA1 gene, frequently mutated in breast cancer, binds and promotes IP3R activity. This underlies adequate apoptosis sensitivity of cells expressing wild-type BRCA1, while oncogenic mutations fail to engage IP3Rs and, thus, promote apoptosis resistance (116). Tumor suppressors can also act via post-translational modification of IP3Rs. As such, phosphatase and tensin homolog (PTEN) not only counteracts PKB/Akt activity by reducing phosphatidylinositol 3,4,5-trisphosphate (PIP3) levels but also reverses the PKB/Akt-dependent phosphorylation of IP3Rs, particularly at the MAMs where IP3R3 becomes dephosphorylated and de-repressed (117).
In addition to these events, oncogenic mutations in cancer genes can also affect IP3R expression levels. As such, the expression of mutant Ras in cells resulted in an IP3R-isoform switch, thereby reducing the pro-apoptotic IP3R3 isoform and increasing the pro-survival IP3R1 isoform (118). IP3R1 displays a higher IP3 sensitivity than IP3R3 and it is proposed to be less involved in pro-apoptotic Ca2+ transfers from ER into mitochondria than IP3R3. The elevated IP3R1-expression levels resulted in an increased Ca2+ leak from the ER and thus a slight decrease in ER Ca2+ content, which reduced Ca2+ availability for pro-apoptotic Ca2+ transfer at the ER–mitochondrial contact sites (118).
Finally, oncogenes and tumor suppressors can affect ER Ca2+ homeostasis by modulating other ER Ca2+-transport systems. For instance, the tumor suppressor p53 accumulates at ER membranes, targeting and boosting SERCA in cells exposed to toxic, chemotherapeutic, and photodynamic agents (119, 120). As a consequence, ER Ca2+ stores became overfilled and the likelihood to flood mitochondria with excess Ca2+ increased, underlying cell death induction by these agents. Cancer cells lacking p53 or having mutated p53 failed to increase SERCA activity and, thus, did not display increased mitochondrial Ca2+ overload, which contributed to the resistance of these cells to these toxic agents (119, 120). Recently, in neuroblastoma cells, acute application of cisplatin, a DNA alkylating agent, and topotecan, a topoisomerase I inhibitor, resulted in a rapid Ca2+ release from the ER stores (121). In addition to this, long-term exposure of neuroblastoma to these drugs resulted in a remodeling of Ca2+-transport systems, including an upregulation of IP3R and RyR isoforms. Blocking Ca2+ release from the ER by inhibiting these channels or by chelating cytosolic Ca2+ using a cell-permeable Ca2+ buffer, suppressed cell death in neuroblastoma treated with cisplatin and topotecan (121). For a detailed discussion on the impact of oncogenes and tumor suppressors on ER Ca2+ signaling and IP3R more specifically, we would like to refer to other recent reviews dedicated to this topic (107, 122).
In addition to this, altered IP3R expression has been implicated in a variety of cancer-associated processes. For instance, a subset of tumor tissue samples derived from breast cancer patients express higher IP3R2/IP3R3 levels compared to the adjacent non-tumorigenic tissue, which has been related to subsequent alterations in metabolic products (123, 124). IP3Rs were shown to be critical for the growth-stimulating effects of 17β-estradiol (E2) on tumorigenic MCF-7 breast cancer cells (125). E2 application exerted both acute and long-term effects on IP3Rs and Ca2+ signaling in MCF-7 cells. Acute E2 application triggered IP3R-mediated Ca2+ release, while prolonged E2 application resulted in an upregulation of IP3R3 expression. Further work indicated that IP3R3 and Ca2+-dependent K+ channels (BKCa) functioned in a concerted manner sustaining breast cancer cell proliferation by forming a macromolecular complex (126). Both IP3R3 and BKCa were critical for the proliferation of MCF-7 cells. Excitingly, in non-tumorigenic MCF-10A cells, IP3R3 and BKCa did not form such a complex and their expressions were dispensable for the proliferation of these cells (126). IP3Rs have also been implicated in the migration of cancer cells, a process associated by an epithelial–mesenchymal transition and stimulated upon loss of cell–cell contact (127). In disconnected pancreatic ductal adenocarcinoma (PDAC) cells, IP3Rs, together with Stim1-containing ER–PM junctions, redistributed to the leading edge of individual PDAC cells, supporting PDAC cell migration (127). Moreover, the selective inhibition of IP3Rs and SOCE lead to reduced cell migration underlying the importance of Ca2+ signaling in this process (127). Increased IP3R3 expression and IP3R-derived Ca2+ signals have also been shown to correlate with the invasive properties of glioblastoma cells (128, 129). Inhibition of IP3Rs with caffeine inhibited the invasion and migration of glioblastoma cells and increased the survival of mice xenografted with glioblastoma cells (128). Interestingly, not only inhibition of IP3Rs but also stimulation of IP3Rs suppresses glioblastoma cell growth and invasion. Indeed, trifluoperazine (TFP), a FDA-approved anti-psychotic drug, impeded proliferation, invasion, and motility of glioblastoma cells in vitro and in vivo by eliciting Ca2+ release from the ER through IP3R1 and IP3R2 channels, while IP3R3 channels were dispensable for TFP-induced Ca2+ mobilization. TFP-induced Ca2+ rise also depended on the presence of the calmodulin subtype 2 (CaM2) protein, which correlates with previous work revealing TFP as a calmodulin-inhibitory molecule by inducing a conformational change in Ca2+-calmodulin (130). Hence, it was proposed that TFP by targeting and antagonizing CaM2 alleviates CaM2’s inhibitory action on IP3Rs, resulting in a potent and irreversible Ca2+ release, responsible for the cell growth and invasion restraint of glioblastoma cells (129). More recently, IP3R-mediated Ca2+ signaling has been shown to be critical for normal T-cell development through repression of Sox1, an antagonist of the transcription factor Tcf1, which is important for normal T-cell development. In the absence of IP3R expression and activity, Notch signaling becomes active in T cells in post β-selection thymocytes, resulting in the development of aggressive T-cell acute lymphoblastic leukemia (131).
Autophagy in Cancer
Autophagy is a basic catabolic process, existing in all types of cells, where it functions mostly in controlling protein turnover and sustaining energetic balance (132). In cancer, however, the role of autophagy is more complex and can exert different effects depending on the stage of tumor progression, tissue origin, genetic background, etc. Therefore, autophagy in cancer can serve as both a tumor suppressive and a tumor-promoting mechanism (133–135).
The essential autophagy protein Beclin 1, encoded by the gene BECN1, has been shown to act as a haploinsufficient tumor suppressor protein (136). In fact, mice that are haplo-deficient for BECN1 develop spontaneous tumors due to an impaired basal autophagy and in humans, mutations in BECN1 occur in up to 75% of breast, ovarian, and prostate cancers (137). Also other autophagy-involved proteins, such as UV radiation resistance-associated gene (UVRAG), Atg5, and Atg7, were recently described as tumor suppressors (137). These findings strongly support an oncosuppressive role of autophagy especially at early stages of tumor development. Autophagy contributes to the maintenance of cellular homeostasis, largely by degradation of protein aggregates and dysfunctional mitochondria but also by supplying nucleotides for DNA repair processes (138, 139). This further protects cells against proteotoxicity, oxidative stress, and genomic instability—the conditions supporting tumor development. In some settings, autophagy was also shown to be necessary to execute cell death to prevent tumor transformation (140). In p53-mediated cell death, expression of the lysosomal protein damage-regulated autophagy modulator-1 (DRAM-1), responsible for autophagy induction, was critical for apoptosis to occur (141). DRAM-1 was also shown to be downregulated in a subset of epithelial cancers (141) possibly underlying a similar tumor-suppressive function as Beclin 1.
In contrast to this, cancer cells become addicted to autophagy at the later stage of tumorigenesis. Studies from Guo et al. (142, 143) revealed that in Kras-driven, genetically engineered mouse models of non-small-cell lung cancer (NSCLC), deletion of Atg7 caused accumulation of mitochondria, suppression of tumor growth and the promotion of tumor cell death. These data underscore the role of Atg7, which was required for NSCLC growth, survival, and malignancy. Furthermore, systemic genetic ablation of Atg7 in mice with established NSCLC, promoted tumor regression before damage occurred to the normal tissues (144). These findings indicate that tumors can be selectively autophagy dependent and that there exists a “therapeutic window” for autophagy modulation (144). In particular, autophagy could compensate metabolic stress by providing bioenergetics substrates for the TCA cycle and nucleotides for biosynthetic pathways, thereby supporting cancer cell survival (139). In Kras-driven NSCLC, autophagy-mediated recycling was able to sustain the levels of amino acids and several metabolites during starvation. However, further autophagy ablation caused deficiencies in mitochondrial substrates. Supplementation of glutamine, glutamate, and nucleotides was, therefore, critical to overcome autophagy deficiency caused by Atg7 deletion which indicates the role of autophagy in starvation survival (139). Autophagy can also serve as a tumor pro-survival mechanism in response to chemotherapy and other anticancer treatments. It is known that the use of anticancer agents, such as 5-fluorouracyl, bortezomib, or tamoxifen, results in elevated autophagy, which counteracts cell death induction and decreases therapy efficiency (145). Recently, it has been described that verapamil, an L-type calcium channel blocker, can induce a cytotoxic effect and autophagy in colon cancer cells. Cell death in these cells was further increased when verapamil treatment was accompanied by chloroquine, an autophagy inhibitor, or when autophagy was ablated by Atg5 and Atg7 deletion (146). Usage of chloroquine and hydroxychloroquine is a common strategy for autophagy inhibition and, therefore, the enhancement of anticancer therapy effectiveness (147). Recent studies, however, have shown that chloroquine can exert anticancer effects independently of autophagy (148). In fact, chloroquine acted by reducing intratumoral hypoxia and metastasis, specifically normalizing tumor vessels by a mechanism involving NOTCH-1. Nevertheless, the use of autophagy inhibitors, as well as genetic tools to abort autophagy, strongly support the hypothesis of tumor-promoting effects of autophagy at the later stages of tumor development.
As discussed above, autophagy is most likely playing a tumor-suppressive role at early steps of tumor development, while it tends to function as a tumor-promoting mechanism in established tumors. This division, however, is not always clear as the role of autophagy in cancer can also depend on other factors such as the genetic background of a particular tumor. Recent studies revealed that in a humanized genetically modified mouse model of PDAC, deletion of autophagy genes Atg5 and Atg7 can play different roles according to the status of p53 (149). In mice possessing an oncogenic allele of Kras, ablation of Atg5 and Atg7 prevented further tumor development. However, in mice with mutated Kras and additionally lacking p53, blockage of autophagy significantly accelerated formation of tumor lesions (149, 150). This and other examples (151) indicate that the role of autophagy strongly depends on the tumor context and this should be considered while designing autophagy modulation-based therapies. For a further discussion on the role of autophagy in cancer, we refer to more detailed reviews (152–155).
IP3Rs and Autophagy Control: A Role in Cancer?
Autophagy Contribution to Modulation of Cancer Cell Death Induced by IP3R Inhibition
Recently, it was shown that cancer cells may be addicted to basal IP3R activity and its critical role in feeding mitochondria with Ca2+, a regulator of the activity of several TCA cycle enzymes. Similarly to non-tumorigenic cells, in tumorigenic cells IP3R inhibition or KD resulted in an increased autophagic flux (156). However, while this was sufficient to sustain cell survival in non-tumorigenic cells, the increase in autophagy was not sufficient for the survival of cancer cells. Thus, pharmacological inhibition using xestospongin B or genetic KD of IP3Rs in cancer cells resulted in cancer cell death (156) (Figure 2A). Mitochondrial Ca2+ and its positive effect on the TCA cycle does not only serve to support ATP synthesis but also to support several anabolic pathways that use mitochondrial TCA cycle intermediates in their biosynthetic pathway (157). In non-tumorigenic cells, the lack of ER–mitochondrial Ca2+ transfers due to IP3R inhibition dampened cell cycle progression, thereby arresting cells at the G1/S checkpoint. Indeed, adequate ATP production is an integral part of the G1/S checkpoint and a surge in ATP production is needed for cells to progress from the G1 phase to the S phase via a mechanism that involves cyclin E upregulation (158). This surge in ATP output is achieved by mitochondrial hyperfusion at that stage of the cell cycle (159). In cells experiencing blunted ER–mitochondrial Ca2+ transfer (like upon IP3R inhibition), ATP output would be impaired and AMPK can be activated. One of the outcomes of AMPK activation besides autophagy induction is the activation of p53/p21, which downregulates cyclin E levels. As a consequence, cells cannot proceed from the G1 to S phase and are arrested (157). However, one feature of cancer cells is their uncontrolled proliferation. In fact, many cancer cells have mutations in p53 or display dysregulated cell cycle control (160). In that sense, these cancer cells become addicted to their mitochondrial metabolism to produce mitochondrial intermediates that are used for synthesis of lipids, nucleotides, and proteins, supporting the survival of the dividing cells. Cancer cells exposed to IP3R inhibitors will, thus, not slow down their cell cycling. As a consequence, these cells will continue to divide irrespective of the surge in mitochondrial ATP output and the availability of mitochondrial intermediates needed for biosynthesis upon cell division. Without sufficient lipids and nucleotides, the daughter cells will not be able to survive. Hence, IP3R inhibition caused a mitotic catastrophe in cancer cells. Consequently, cell death in these cells could be overcome by the addition of mitochondrial substrates, such as pyruvate, to the growth medium or by slowing down cell cycle progression (156). Yet, autophagy was not involved in the cell death process, as KD of essential autophagy genes did not modulate IP3R inhibition-induced cell death. This indicates that cell death did not occur via autophagic cell death and also that autophagy activation could not support survival of the cells.
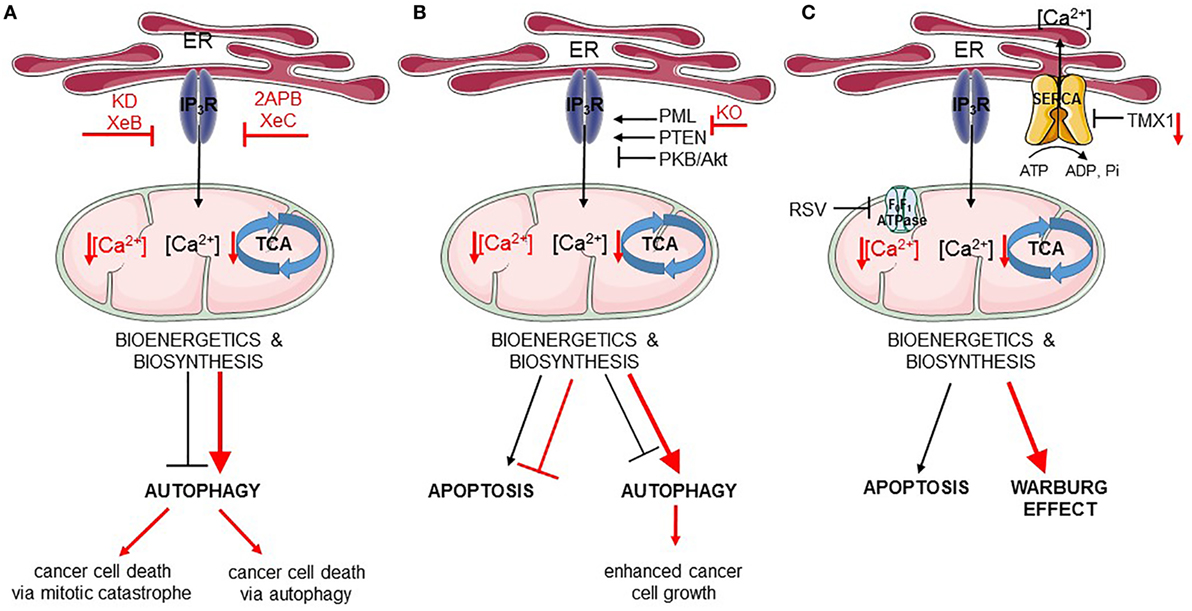
Figure 2. Endoplasmic reticulum (ER)–mitochondria Ca2+ transfer regulates apoptosis and autophagy in cancer. In all panels, the black arrows mirror the basal cellular mechanisms while the red arrows indicate modulation of the pathways by the listed chemical compounds or genetic modifications. (A) IP3Rs are engaged in autophagy and cell death regulation via the ER–mitochondrial Ca2+ flux. Ca2+ transferred to the mitochondria ensures proper tricarboxylic acid (TCA) function and, therefore, adequate bioenergetics and biosynthesis processes suppressing autophagy. Inhibition of IP3Rs by Xestospongin B (XeB) or its genetic knockdown (KD) dampens ER–mitochondrial Ca2+ transfer, which inhibits the TCA cycle and ATP production. As a consequence, autophagy is increased, but this is not sufficient for the survival of cancer cells, which undergo mitotic catastrophe. In addition to this, inhibition of IP3Rs by 2APB or Xestospongin C (XeC) also leads to impeded ER–mitochondrial Ca2+ fueling, and subsequently to further autophagy-dependent cancer cell death. (B) Fueling mitochondria with Ca2+ can be modulated by several oncogenes/tumor suppressors [promyelocytic leukemia protein (PML), PTEN, PKB/Akt]. In cancer cells lacking PML (KO, knockout), mitochondrial Ca2+ transfer is impeded resulting in downregulation of TCA cycle, stimulation of autophagy, and of cell growth. (C) Mitochondrial Ca2+ transfer can also be indirectly controlled by mitochondrial F0F1 ATPase. Its inhibition by resveratrol (RSV) impairs sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) function, thereby increasing the net Ca2+ flux from the ER and promoting mitochondrial Ca2+ overload, eventually leading to apoptosis. In addition, SERCA is negatively regulated by thioreductase TMX1. Cancer cells having low levels of TMX1, exert high SERCA activity, which correlates with decreased ER–mitochondrial Ca2+ transfer. Consequently, the TCA cycle is limited and cells switch to aerobic glycolysis (Warburg effect).
In contrast to these findings, another study revealed an important role for autophagic cell death in breast cancer cells exposed to IP3R inhibition (123) (Figure 2A). Tumorigenic breast cancer cells were sensitive to treatment with the non-specific IP3R inhibitors, 2-APB or xestospongin C, while non-tumorigenic breast cancer cells were resistant to this treatment. However, these compounds, as well as their derivatives, can indirectly cause depletion of the ER Ca2+ stores by inhibiting other Ca2+ transporters or by stimulating Ca2+ leakage out of the ER (161–164). Nevertheless, similar findings were obtained by genetically knocking down IP3R2 or IP3R3, which appear to be upregulated in tumorigenic versus non-tumorigenic breast cancer cell lines. Inhibition of IP3Rs in the tumorigenic cells resulted in excessive autophagy activation, which could be attributed to a decrease in ATP production (and thus activation of AMPK), an upregulation of Atg5 and an increase in ROS production. In these cells, excessive autophagy was responsible for cell death, as inhibition of autophagy either at the level of the Vps34 complex formation using 3-methyladenine or at the level of lysosomal degradation by Bafilomycin A1 protected cells against cell death induced by IP3R inhibition or IP3R KD (123). These in vitro findings were also translated to in vivo xenografted breast tumor models. Interestingly, the increase in IP3R2/IP3R3 expression was not only found in breast cancer cell lines but also found in patient samples consisting of breast tumor tissue compared to adjacent non-tumorous tissue. Moreover, IP3R2/IP3R3 upregulation in breast tumors correlated with an increase in lipoproteins and several metabolites (such as lactate, alanine, and lysine) in the serum of these patients compared to breast cancer patients with low IP3R2/IP3R3 levels or healthy controls (124). Unfortunately, no autophagic markers were analyzed in these samples.
Downregulation of Autophagy and Increased Apoptosis Susceptibility in Response to IP3R Modulation by Tumor Suppressors
As previously discussed, IP3Rs suppress basal autophagy by promoting mitochondrial ATP production. In addition to this, IP3Rs control the susceptibility of cells toward toxic, pro-apoptotic stimuli. By promoting Ca2+ transfer into the mitochondria, IP3Rs participate in mitochondrial Ca2+ overload, a critical factor in the opening of the mitochondrial permeability transition pore and subsequent apoptosis. In fact, mitochondrial Ca2+ overload has been shown to be a critical component of several pro-apoptotic stimuli, including chemotherapeutic drugs (165). These stimuli can trigger Ca2+ release from the ER. In addition, these compounds can trigger ER Ca2+ overload by activating SERCA in a p53-dependent manner (120). As a consequence, such cells will display an increased likelihood for mitochondrial Ca2+ accumulation and thus cell death. In fact, cancer cells lacking p53 or expressing loss-of-function p53 mutants are resistant to chemotherapeutic drugs in part due to their lack of mitochondrial Ca2+ overload, as these cells can be re-sensitized to chemotherapeutics by overexpressing MCU (120, 166). In particular, the IP3R3 isoform appears to participate in pro-apoptotic Ca2+ transfer into the mitochondria due to its presence in the MAMs (167). Moreover, IP3R3 activity in the MAMs is subjected to functional modulation by survival/oncogenes and tumor suppressors (Figure 2B). IP3R3 is phosphorylated by PKB/Akt, suppressing ER–mitochondrial Ca2+ flux and promoting apoptosis resistance (112). PKB/Akt-mediated phosphorylation of IP3R3 is counteracted by PTEN, which dephosphorylates IP3R3 particularly at the MAM fraction and, therefore, stimulates pro-apoptotic Ca2+ transfer from the ER into the mitochondria (117). Another regulator of PKB/Akt-dependent phosphorylation of IP3R3 is the tumor suppressor promyelocytic leukemia protein (PML), which also resides at the MAMs, where it recruits PP2A, which suppresses PKB/Akt activity (168). In addition to this, downregulation of IP3R3-protein levels has been implicated in cancer transformation and cell death resistance of isogenic cell pairs, in which an oncogenic mutant Ras allele was expressed (118).
More recently, further insights into the contribution of IP3R modulation in the tumor suppressive function of PML have been revealed (168, 169). PML was recruited at the MAMs in a p53-dependent manner supporting efficient ER–mitochondrial Ca2+ transfer (Figure 2B). As a consequence, cells expressing the tumor suppressor PML were susceptible to engage apoptosis upon cell stress or damage and to maintain an adequate production of ATP, preventing the growth of damaged or malignant cells. These conditions dampened AMPK activity and, thus, result in a regular basal autophagic flux. The combination of adequate apoptosis sensitivity and regular autophagy flux allows for a normal and balanced cell growth. However, in cells lacking PML, ER–mitochondrial Ca2+ transfer was suppressed, resulting in excessive apoptosis resistance and an upregulation of basal autophagy due to suppressed ATP production followed by AMPK activation and thus increased ULK1 activity. The increase in autophagy upon PML deletion in cells could be attributed to the decreased mitochondrial Ca2+ signaling, since overexpression of MCU in these cells could suppress basal autophagic flux (168). Moreover, PML-deficient cells displayed a growth advantage compared to PML-proficient cells, particularly in stress conditions such as nutrient starvation that engage the autophagy pathway. Interestingly, PML-deficient cells could be sensitized to chemotherapeutic drugs such as 5-fluorouracil by co-administration of chloroquine, an autophagy inhibitor that acts at the level of the lysosomal proteolysis. Moreover, in some promyelocytic leukemia cells, PML became fused to retinoic acid receptor α (RARα), abrogating wild-type endogenous PML function and causing neoplastic transformation. This oncogenic PML fusion was degraded by stimulating the proteasome using arsenic trioxide. Treatment of cells expressing oncogenic PML fusion protein with this drug not only resulted in PML-RARα degradation but also rescued wild-type PML levels, which was then present in the MAMs and able to promote ER–mitochondrial Ca2+ transfer (168).
Another tumor suppressor actively involved in autophagy regulation is Beclin 1. Interestingly, a target of Beclin 1 is the IP3R, which becomes sensitized upon Beclin 1 binding, a process enhanced during nutrient starvation (58). Thus, cancer cells, commonly deficient in Beclin 1, will not only be able to form less complexes with the lipid kinase Vps34 but also with the IP3Rs, what may lead to decreased autophagy. However, at this point, it is not clear how decreased IP3R/Beclin 1-complex formation contributes to basal autophagy and how this impacts tumorigenesis.
Indirect Impact of IP3R on Autophagy in Cancer Cells: Effects of ER Ca2+ Modulation
Resveratrol, a natural polyphenol, is well known to induce cancer cell death by engaging autophagy induction via different mechanisms (82, 170–172), including the modulation of Ca2+ signaling (173). More recently, further insights into RSV-induced cancer cell death via Ca2+ signaling have been obtained (84). In particular, cancer cells can display increased ER–mitochondrial contact sites, potentially to facilitate the transfer of basal Ca2+ signals to accommodate their increased need for mitochondrial TCA cycle activity, which also provides substrates for several biosynthetic pathways. Exposing cancer cells to RSV resulted in a rapid depletion of the ER Ca2+ stores. The underlying mechanisms appeared to involve the direct inhibition of the mitochondrial F0F1-type ATP synthase, resulting in a rapid drop in ATP levels, in particular at the ER–mitochondrial contact sites (Figure 2C). SERCA activity is thereby impaired, leading to a net increase of Ca2+ delivery to the mitochondria, as for a given IP3-induced Ca2+ release less Ca2+ will be pumped back in the ER. Thus, mitochondrial Ca2+ levels will increase, thereby promoting cell death. The concept of SERCA modulating ER–mitochondrial Ca2+ transfer in cell death and tumor biology has been nicely illustrated in a recent study of Raturi et al. (174). It has been shown that SERCA activity was dynamically regulated at the ER–mitochondrial contact sites by different factors, including palmitoylated calnexin, which positively regulated SERCA, and the thioreductase TMX1, which negatively regulated SERCA. In normal cells, TMX1 levels are high, thereby suppressing SERCA activity and thus promoting ER–mitochondrial Ca2+ transfer. This supports mitochondrial metabolism on the one hand and adequate apoptotic susceptibility on the other hand. Interestingly, many tumors display low TMX1 levels, which results in increased SERCA activity, particularly at the MAMs, leading to dampened ER–mitochondrial Ca2+ transfer (Figure 2C). This was proposed to contribute to the Warburg effect and increased glycolysis, as the activity of TCA cycle enzymes became suppressed, while the need for glucose metabolism remained high to sustain cell growth and proliferation (174, 175). Nevertheless, it is possible that these findings relate to concepts identified for PML at the ER–mitochondrial interface. Indeed, tumors with low TMX1 levels may display increased autophagy and decreased apoptosis susceptibility due to suppressed ER–mitochondrial Ca2+ fluxes. However, further work is needed to reconcile these concepts.
IP3R-Regulated Autophagy As a Protection against Natural Killer (NK)-Induced Cancer Cell Death
Recently, ITPR1, the gene encoding IP3R1, has been implicated as a major resistance mechanism of renal carcinoma cells against the lytic action of NK cells by activating autophagy (176–178). Many renal carcinoma cells are characterized by a dysfunctional von Hippel–Lindau gene (pVHL), which encodes a protein that has many functions, including targeting the family of hypoxia-inducible factor transcription factors for degradation by the proteasome. Thus, in cells lacking pVHL, HIF1α/HIF2α become stabilized. Currently, an emerging concept in anticancer therapies is the use of NK cells. Tumor cells contain several resistance factors against NK-induced cancer cell killing, including stabilized HIF2α. Strikingly, ITPR1 appeared to be one of the most important target genes of HIF2α in a renal carcinoma cell line with dysfunctional pVHL gene and it conferred resistance against NK-induced lysis. In particular, renal carcinoma cells upregulated pro-survival autophagy in a HIF2α/IP3R1-dependent manner in response to NK treatment, while cells in which IP3R1 was knocked down failed to stimulate autophagy and became susceptible to NK-induced lysis. The IP3R1-dependent induction of autophagy protected the carcinoma cells against the deleterious action of NK cells by degrading the lytic granzyme B. In renal carcinoma cells with functional pVHL, HIF2α levels are very low due to its targeting to the proteasome, thus failing to upregulate ITPR1 expression and abrogating autophagy induction as resistance mechanism against NK cells and the lytic action of granzyme B. Thus, antagonizing IP3R function in renal carcinoma cells lacking functional pVHL may provide a manner to sensitize these cells to lysis by NK cells by counteracting the induction of autophagy as a resistance mechanism. Furthermore, also NK cells by themselves require functional autophagy for maturation and survival (179). Upon deletion of Atg5, NK cells accumulated damaged mitochondria, which lead to their death due to excessive ROS production. Furthermore, silencing Atg7 and the resulting disruption of the interaction between Atg7 and phosphorylated forkhead box O1 (FOXO1) prevented autophagy and contributed to incomplete maturation of NK cells (179).
Conclusion
IP3Rs play a critical role in autophagy due to their localization at the ER and the ER–mitochondrial contact sites and the resulting Ca2+-signaling regulation. On the one hand, they suppress autophagy by continuously sustaining the mitochondria with Ca2+, needed for mitochondrial metabolism and energy production. On the other hand, they participate in the increased autophagic flux induced upon cellular stress, including nutrient starvation, chemical mTOR inhibition, or RSV treatment, by providing cytosolic Ca2+ that is needed to drive autophagic flux. Given autophagy’s critical role in tumor development and progression, it is not surprising that IP3R function can affect these processes through autophagy modulation. Cancer cells appear to be addicted to IP3R-mediated Ca2+ release to sustain their mitochondrial metabolism and related anabolic pathways. Cancer cells exposed to IP3R inhibition can undergo cell death, which in some cases could be due to excessive autophagy induction, while in other cases cell death could be the result of an uncontrolled cell proliferation and thus be due to mitotic catastrophe. Also, IP3Rs are modulated by tumor suppressors, such as PML, as part of a homeostatic program to support normal cell growth by balancing adequate apoptosis susceptibility and regulated autophagy flux. However, loss of these tumor suppressors results in dampened IP3R function and thus defective ER–mitochondrial Ca2+ transfer. As a consequence, cells become resistant to cell death inducers, including genotoxic stress and cell damage, by a combination of increased apoptosis resistance and an increased autophagy flux serving as a pro-survival function. This phenomenon will contribute to neoplastic transformation and tumorigenesis. The role of IP3Rs in autophagy seem also to be exploited by renal carcinoma cells with dysfunctional pVHL, which induce autophagy in a HIF2α/IP3R-dependent manner as a resistance mechanism that protects these cancers against NK-induced killing. Finally, it should be noted that IP3R function and the net IP3R-mediated Ca2+ delivery into the mitochondria is dependent on the activity of SERCA, which is also present in the MAMs. Thus, SERCA modulation at the ER–mitochondrial contact sites will affect the net Ca2+ transfer into the mitochondria and, thus, ultimately affect the Ca2+-dependent mitochondrial functions, such as bioenergetics, autophagy, and apoptosis. SERCA inhibition in the MAMs will increase mitochondrial Ca2+ accumulation, which will drive the mitochondrial metabolism and bioenergetic output and thus suppress autophagy. In cancer cells, de-inhibition of SERCA at the ER–mitochondrial contact sites can be part of the Warburg effect but also of the increase in basal autophagy that could promote neoplastic behavior by promoting cell survival and excessively protecting cancer cells against cell stress. Overall, IP3Rs impact several cancer hallmarks through autophagy modulation.
Author Contributions
All authors contributed to the conception of the work. GB and EK drafted the manuscript. EK produced the figures. GR, TV, and JP critically revised manuscript and figures and provided important intellectual content. All authors concur with the final version of the manuscript and agree to be held accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
GR and TV are recipients of a Ph.D. fellowship and a post-doctoral fellowship of the Research Foundation-Flanders (FWO), respectively. Work performed in the laboratory of the authors was supported by FWO grants G.07310.9N, G.0634.13 and G.0927.15, by the Research Council of the KU Leuven via concerted action 09/012 and grant OT/14/101 and by the Interuniversity Attraction Poles Program (Belgian Science Policy; IAP-P7/13). JP and GB are part of the Scientific Research Community “Calcium Signaling in health, disease & therapy” (CaSign) supported by the FWO (W0.019.17N).
References
1. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi:10.1038/35036035
2. Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev (2016) 96(4):1261–96. doi:10.1152/physrev.00006.2016
3. La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium (2016) 60(2):74–87. doi:10.1016/j.ceca.2016.04.005
4. Berridge MJ. Inositol trisphosphate and calcium oscillations. Biochem Soc Symp (2007) 74:1–7. doi:10.1042/BSS0740001
5. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol (2003) 4(7):517–29. doi:10.1038/nrm1155
6. Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci (2016) 41(12):1035–49. doi:10.1016/j.tibs.2016.09.001
7. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science (1993) 262(5134):744–7. doi:10.1126/science.8235595
8. Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium (1999) 26(5):193–9. doi:10.1054/ceca.1999.0076
9. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev (2006) 86(1):369–408. doi:10.1152/physrev.00004.2005
10. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456(7222):605–10. doi:10.1038/nature07534
11. de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal (2008) 10(3):621–33. doi:10.1089/ars.2007.1934
12. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta (2014) 1843(10):2253–62. doi:10.1016/j.bbamcr.2014.03.009
13. Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol (2011) 3(6):a004317. doi:10.1101/cshperspect.a004317
14. Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol (2011) 3(5):a004184. doi:10.1101/cshperspect.a004184
15. Kania E, Pajak B, Orzechowski A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed Res Int (2015) 2015:352794. doi:10.1155/2015/352794
16. Krebs J, Agellon LB, Michalak M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun (2015) 460(1):114–21. doi:10.1016/j.bbrc.2015.02.004
17. Dufey E, Urra H, Hetz C. ER proteostasis addiction in cancer biology: novel concepts. Semin Cancer Biol (2015) 33:40–7. doi:10.1016/j.semcancer.2015.04.003
18. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol (2006) 26(24):9220–31. doi:10.1128/MCB.01453-06
19. Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta (2014) 1843(10):2143–9. doi:10.1016/j.bbamcr.2014.01.012
20. Rivas A, Vidal RL, Hetz C. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets (2015) 19(9):1203–18. doi:10.1517/14728222.2015.1053869
21. Sammels E, Parys JB, Missiaen L, De Smedt H, Bultynck G. Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium (2010) 47(4):297–314. doi:10.1016/j.ceca.2010.02.001
22. Van Coppenolle F, Vanden Abeele F, Slomianny C, Flourakis M, Hesketh J, Dewailly E, et al. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci (2004) 117(Pt 18):4135–42. doi:10.1242/jcs.01274
23. Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell (2006) 126(5):981–93. doi:10.1016/j.cell.2006.06.059
24. D’Hondt C, Ponsaerts R, De Smedt H, Vinken M, De Vuyst E, De Bock M, et al. Pannexin channels in ATP release and beyond: an unexpected rendezvous at the endoplasmic reticulum. Cell Signal (2011) 23(2):305–16. doi:10.1016/j.cellsig.2010.07.018
25. Leon-Aparicio D, Pacheco J, Chavez-Reyes J, Galindo JM, Valdes J, Vaca L, et al. Orai3 channel is the 2-APB-induced endoplasmic reticulum calcium leak. Cell Calcium (2017) 65:91–101. doi:10.1016/j.ceca.2017.01.012
26. Bandara S, Malmersjo S, Meyer T. Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci Signal (2013) 6(283):ra56. doi:10.1126/scisignal.2003649
27. Luyten T, Welkenhuyzen K, Roest G, Kania E, Wang L, Bittremieux M, et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2+. Biochim Biophys Acta (2017) 1864(6):947–56. doi:10.1016/j.bbamcr.2017.02.013
28. Corbett EF, Michalak M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem Sci (2000) 25(7):307–11. doi:10.1016/S0968-0004(00)01588-7
29. Vervloessem T, Ivanova H, Luyten T, Parys JB, Bultynck G. The selective Bcl-2 inhibitor venetoclax, a BH3 mimetic, does not dysregulate intracellular Ca2+ signaling. Biochim Biophys Acta (2017) 1864(6):968–76. doi:10.1016/j.bbamcr.2016.11.024
30. Parys JB, De Smedt H. Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol (2012) 740:255–79. doi:10.1007/978-94-007-2888-2_11
31. Fedorenko OA, Popugaeva E, Enomoto M, Stathopulos PB, Ikura M, Bezprozvanny I. Intracellular calcium channels: inositol-1,4,5-trisphosphate receptors. Eur J Pharmacol (2014) 739:39–48. doi:10.1016/j.ejphar.2013.10.074
32. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol (2010) 2(11):a003996. doi:10.1101/cshperspect.a003996
33. Amador FJ, Stathopulos PB, Enomoto M, Ikura M. Ryanodine receptor calcium release channels: lessons from structure-function studies. FEBS J (2013) 280(21):5456–70. doi:10.1111/febs.12194
34. Van Petegem F. Ryanodine receptors: allosteric ion channel giants. J Mol Biol (2015) 427(1):31–53. doi:10.1016/j.jmb.2014.08.004
35. Mikoshiba K. Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul (2015) 57:217–27. doi:10.1016/j.jbior.2014.10.001
36. Prins D, Michalak M. STIM1 is cleaved by calpain. FEBS Lett (2015) 589(21):3294–301. doi:10.1016/j.febslet.2015.09.015
37. Srivats S, Balasuriya D, Pasche M, Vistal G, Edwardson JM, Taylor CW, et al. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J Cell Biol (2016) 213(1):65–79. doi:10.1083/jcb.201506022
38. van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell (2017) 65(5):885–99.e6. doi:10.1016/j.molcel.2017.01.020
39. Burgoyne T, Patel S, Eden ER. Calcium signaling at ER membrane contact sites. Biochim Biophys Acta (2015) 1853(9):2012–7. doi:10.1016/j.bbamcr.2015.01.022
40. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13(9):566–78. doi:10.1038/nrm3412
41. Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta (2013) 1833(1):213–24. doi:10.1016/j.bbamcr.2012.04.013
42. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta (2014) 1837(4):461–9. doi:10.1016/j.bbabio.2013.10.015
43. Levine TP, Patel S. Signalling at membrane contact sites: two membranes come together to handle second messengers. Curr Opin Cell Biol (2016) 39:77–83. doi:10.1016/j.ceb.2016.02.011
44. Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J (1999) 18(1):96–108. doi:10.1093/emboj/18.1.96
45. Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, et al. The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem (2013) 288(15):10750–8. doi:10.1074/jbc.R112.420752
46. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell (2014) 53(5):726–37. doi:10.1016/j.molcel.2014.01.013
47. Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J (2011) 439(3):349–74. doi:10.1042/BJ20110949
48. Lloyd-Evans E. On the move, lysosomal CAX drives Ca2+ transport and motility. J Cell Biol (2016) 212(7):755–7. doi:10.1083/jcb.201603037
49. Zhu MX, Ma J, Parrington J, Calcraft PJ, Galione A, Evans AM. Calcium signaling via two-pore channels: local or global, that is the question. Am J Physiol Cell Physiol (2010) 298(3):C430–41. doi:10.1152/ajpcell.00475.2009
50. Wang W, Zhang X, Gao Q, Xu H. TRPML1: an ion channel in the lysosome. Handb Exp Pharmacol (2014) 222:631–45. doi:10.1007/978-3-642-54215-2_24
51. Patel S. Function and dysfunction of two-pore channels. Sci Signal (2015) 8(384):re7. doi:10.1126/scisignal.aab3314
52. Waller-Evans H, Lloyd-Evans E. Regulation of TRPML1 function. Biochem Soc Trans (2015) 43(3):442–6. doi:10.1042/BST20140311
53. Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature (2009) 459(7246):596–600. doi:10.1038/nature08030
54. Morgan AJ, Davis LC, Ruas M, Galione A. TPC: the NAADP discovery channel? Biochem Soc Trans (2015) 43(3):384–9. doi:10.1042/BST20140300
55. Ruas M, Davis LC, Chen CC, Morgan AJ, Chuang KT, Walseth TF, et al. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J (2015) 34(13):1743–58. doi:10.15252/embj.201490009
56. Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun (2010) 1:38. doi:10.1038/ncomms1037
57. Feng X, Huang Y, Lu Y, Xiong J, Wong CO, Yang P, et al. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J Biol Chem (2014) 289(7):4262–72. doi:10.1074/jbc.M113.506501
58. Decuypere JP, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgo J, et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy (2011) 7(12):1472–89. doi:10.4161/auto.7.12.17909
59. Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, et al. mTOR-controlled autophagy requires intracellular Ca2+ signaling. PLoS One (2013) 8(4):e61020. doi:10.1371/journal.pone.0061020
60. Decuypere JP, Paudel RC, Parys J, Bultynck G. Intracellular Ca2+ signaling: a novel player in the canonical mTOR-controlled autophagy pathway. Commun Integr Biol (2013) 6(5):e25429. doi:10.4161/cib.25429
61. Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ (2007) 14(9):1576–82. doi:10.1038/sj.cdd.4402200
62. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142(2):270–83. doi:10.1016/j.cell.2010.06.007
63. Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy (2013) 9(2):124–37. doi:10.4161/auto.23323
64. Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ (2007) 14(5):1029–39. doi:10.1038/sj.cdd.4402099
65. Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ (2009) 16(7):1006–17. doi:10.1038/cdd.2009.34
66. Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep (2001) 2(4):330–5. doi:10.1093/embo-reports/kve061
67. Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase – Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J (2003) 376(Pt 3):577–86. doi:10.1042/BJ20030826
68. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ (2011) 18(4):571–80. doi:10.1038/cdd.2010.191
69. Decuypere JP, Bultynck G, Parys JB. A dual role for Ca2+ in autophagy regulation. Cell Calcium (2011) 50(3):242–50. doi:10.1016/j.ceca.2011.04.001
70. Chung KM, Jeong EJ, Park H, An HK, Yu SW. Mediation of autophagic cell death by type 3 ryanodine receptor (RyR3) in adult hippocampal neural stem cells. Front Cell Neurosci (2016) 10:116. doi:10.3389/fncel.2016.00116
71. Vervliet T, Pintelon I, Welkenhuyzen K, Bootman MD, Bannai H, Mikoshiba K, et al. Basal ryanodine receptor activity suppresses autophagic flux. Biochem Pharmacol (2017) 132:133–42. doi:10.1016/j.bcp.2017.03.011
72. Hoyer-Hansen M, Jaattela M. AMP-activated protein kinase: a universal regulator of autophagy? Autophagy (2007) 3(4):381–3. doi:10.4161/auto.4240
73. Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell (2007) 25(2):193–205. doi:10.1016/j.molcel.2006.12.009
74. Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy (2010) 6(4):506–22. doi:10.4161/auto.6.4.11863
75. Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal (2010) 22(6):914–25. doi:10.1016/j.cellsig.2010.01.015
76. Engedal N, Torgersen ML, Guldvik IJ, Barfeld SJ, Bakula D, Saetre F, et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy (2013) 9(10):1475–90. doi:10.4161/auto.25900
77. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell (2011) 146(5):682–95. doi:10.1016/j.cell.2011.07.030
78. Hashemzaei M, Entezari Heravi R, Rezaee R, Roohbakhsh A, Karimi G. Regulation of autophagy by some natural products as a potential therapeutic strategy for cardiovascular disorders. Eur J Pharmacol (2017) 802:44–51. doi:10.1016/j.ejphar.2017.02.038
79. Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis (2010) 1:e10. doi:10.1038/cddis.2009.8
80. Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol (2011) 192(4):615–29. doi:10.1083/jcb.201008167
81. Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, et al. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep (2016) 6:21772. doi:10.1038/srep21772
82. Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ (2008) 15(8):1318–29. doi:10.1038/cdd.2008.51
83. Scarlatti F, Maffei R, Beau I, Ghidoni R, Codogno P. Non-canonical autophagy: an exception or an underestimated form of autophagy? Autophagy (2008) 4(8):1083–5. doi:10.4161/auto.7068
84. Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, Eroglu E, Klec C, Waldeck-Weiermair M, et al. Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell Physiol Biochem (2016) 39(4):1404–20. doi:10.1159/000447844
85. Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet (2008) 17(17):2723–37. doi:10.1093/hmg/ddn174
86. Venugopal B, Mesires NT, Kennedy JC, Curcio-Morelli C, Laplante JM, Dice JF, et al. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol (2009) 219(2):344–53. doi:10.1002/jcp.21676
87. Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I, Muallem S. The Ca2+ channel TRPML3 regulates membrane trafficking and autophagy. Traffic (2009) 10(8):1157–67. doi:10.1111/j.1600-0854.2009.00924.x
88. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol (2015) 17(3):288–99. doi:10.1038/ncb3114
89. Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A (2015) 112(11):E1373–81. doi:10.1073/pnas.1419669112
90. Onyenwoke RU, Sexton JZ, Yan F, Diaz MC, Forsberg LJ, Major MB, et al. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem J (2015) 470(3):331–42. doi:10.1042/BJ20150219
91. Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun (2016) 7:12109. doi:10.1038/ncomms12109
92. Zhang X, Yu L, Xu H. Lysosome calcium in ROS regulation of autophagy. Autophagy (2016) 12(10):1954–5. doi:10.1080/15548627.2016.1212787
93. Lu Y, Hao BX, Graeff R, Wong CW, Wu WT, Yue J. Two pore channel 2 (TPC2) inhibits autophagosomal-lysosomal fusion by alkalinizing lysosomal pH. J Biol Chem (2013) 288(33):24247–63. doi:10.1074/jbc.M113.484253
94. Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem (2011) 286(32):27875–81. doi:10.1074/jbc.C110.216580
95. Wang Q, Huang L, Yue J. Oxidative stress activates the TRPM2-Ca2+-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim Biophys Acta (2017) 1864(6):957–67. doi:10.1016/j.bbamcr.2016.12.014
96. Wang Q, Guo W, Hao B, Shi X, Lu Y, Wong CW, et al. Mechanistic study of TRPM2-Ca2+-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy (2016) 12(8):1340–54. doi:10.1080/15548627.2016.1187365
97. Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta (2013) 1835(2):180–93. doi:10.1016/j.bbcan.2012.12.001
98. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B (2017) 7(1):3–17. doi:10.1016/j.apsb.2016.11.001
99. Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim Biophys Acta (2017). doi:10.1016/j.bbabio.2017.01.003
100. Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta (2014) 1843(10):2164–83. doi:10.1016/j.bbamcr.2014.03.007
101. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene (2008) 27(50):6407–18. doi:10.1038/onc.2008.308
102. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis (2012) 3:e304. doi:10.1038/cddis.2012.45
103. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi:10.1038/nrm2233
104. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol (2011) 192(4):547–56. doi:10.1083/jcb.201009094
105. Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun (2014) 5:3792. doi:10.1038/ncomms4792
106. Vervloessem T, Yule DI, Bultynck G, Parys JB. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim Biophys Acta (2015) 1853(9):1992–2005. doi:10.1016/j.bbamcr.2014.12.006
107. Bittremieux M, Parys JB, Pinton P, Bultynck G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca2+ signaling. Biochim Biophys Acta (2016) 1863(6 Pt B):1364–78. doi:10.1016/j.bbamcr.2016.01.002
108. Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell (2008) 31(2):255–65. doi:10.1016/j.molcel.2008.06.014
109. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A (2009) 106(34):14397–402. doi:10.1073/pnas.0907555106
110. Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ (2012) 19(2):295–309. doi:10.1038/cdd.2011.97
111. Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene (2016) 35(39):5079–92. doi:10.1038/onc.2016.31
112. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun (2008) 375(4):501–5. doi:10.1016/j.bbrc.2008.07.153
113. White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol (2005) 7(10):1021–8. doi:10.1038/ncb1302
114. Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci U S A (2007) 104(30):12565–70. doi:10.1073/pnas.0702489104
115. Yang J, Vais H, Gu W, Foskett JK. Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc Natl Acad Sci U S A (2016) 113(13):E1953–62. doi:10.1073/pnas.1517935113
116. Hedgepeth SC, Garcia MI, Wagner LE II, Rodriguez AM, Chintapalli SV, Snyder RR, et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem (2015) 290(11):7304–13. doi:10.1074/jbc.M114.611186
117. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ (2013) 20(12):1631–43. doi:10.1038/cdd.2013.77
118. Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci (2014) 127(Pt 7):1607–19. doi:10.1242/jcs.141408
119. Bittremieux M, Bultynck G. p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies. Oncoscience (2015) 2(3):233–8. doi:10.18632/oncoscience.139
120. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112(6):1779–84. doi:10.1073/pnas.1410723112
121. Florea AM, Varghese E, McCallum JE, Mahgoub S, Helmy I, Varghese S, et al. Calcium-regulatory proteins as modulators of chemotherapy in human neuroblastoma. Oncotarget (2017) 8(14):22876–93. doi:10.18632/oncotarget.15283
122. Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol (2014) 1(2):e956469. doi:10.4161/23723548.2014.956469
123. Singh A, Chagtoo M, Tiwari S, George N, Chakravarti B, Khan S, et al. Inhibition of inositol 1, 4, 5-trisphosphate receptor induce breast cancer cell death through deregulated autophagy and cellular bioenergetics. J Cell Biochem (2017) 118(8):2333–46. doi:10.1002/jcb.25891
124. Singh A, Sharma RK, Chagtoo M, Agarwal G, George N, Sinha N, et al. 1H NMR metabolomics reveals association of high expression of inositol 1, 4, 5 trisphosphate receptor and metabolites in breast cancer patients. PLoS One (2017) 12(1):e0169330. doi:10.1371/journal.pone.0169330
125. Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol Cancer (2010) 9:156. doi:10.1186/1476-4598-9-156
126. Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F. Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur J Cancer (2013) 49(17):3738–51. doi:10.1016/j.ejca.2013.07.013
127. Okeke E, Parker T, Dingsdale H, Concannon M, Awais M, Voronina S, et al. Epithelial-mesenchymal transition, IP3 receptors and ER-PM junctions: translocation of Ca2+ signalling complexes and regulation of migration. Biochem J (2016) 473(6):757–67. doi:10.1042/BJ20150364
128. Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res (2010) 70(3):1173–83. doi:10.1158/0008-5472.CAN-09-2886
129. Kang S, Hong J, Lee JM, Moon HE, Jeon B, Choi J, et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol Cancer Ther (2017) 16(1):217–27. doi:10.1158/1535-7163.MCT-16-0169-T
130. Vandonselaar M, Hickie RA, Quail JW, Delbaere LT. Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat Struct Biol (1994) 1(11):795–801. doi:10.1038/nsb1194-795
131. Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, et al. Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun (2014) 5:4814. doi:10.1038/ncomms5814
132. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi:10.1038/nrm3735
133. Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol (2011) 11(4):294–300. doi:10.1016/j.coph.2011.03.009
134. Liu EY, Ryan KM. Autophagy and cancer – issues we need to digest. J Cell Sci (2012) 125(Pt 10):2349–58. doi:10.1242/jcs.093708
135. Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol (2012) 226(2):255–73. doi:10.1002/path.3025
136. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A (2003) 100(25):15077–82. doi:10.1073/pnas.2436255100
137. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med (2013) 368(19):1845–6. doi:10.1056/NEJMc1303158
138. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res (2015) 21(22):5037–46. doi:10.1158/1078-0432.CCR-15-0490
139. Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev (2016) 30(15):1704–17. doi:10.1101/gad.283416.116
140. Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol (2013) 23(5):344–51. doi:10.1016/j.semcancer.2013.05.006
141. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell (2006) 126(1):121–34. doi:10.1016/j.cell.2006.05.034
142. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25(5):460–70. doi:10.1101/gad.2016311
143. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-Ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27(13):1447–61. doi:10.1101/gad.219642.113
144. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov (2014) 4(8):914–27. doi:10.1158/2159-8290.CD-14-0363
145. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med (2013) 19(7):428–46. doi:10.1016/j.molmed.2013.04.005
146. Kania E, Pajak B, O’Prey J, Sierra Gonzalez P, Litwiniuk A, Urbanska K, et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. FEBS J (2017) 284(9):1370–87. doi:10.1111/febs.14064
147. Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol (2014) 1(1):e29911. doi:10.4161/mco.29911
148. Maes H, Kuchnio A, Carmeliet P, Agostinis P. Chloroquine anticancer activity is mediated by autophagy-independent effects on the tumor vasculature. Mol Cell Oncol (2016) 3(1):e970097. doi:10.4161/23723548.2014.970097
149. Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature (2013) 504(7479):296–300. doi:10.1038/nature12865
150. McCarthy N. Autophagy: directed development. Nat Rev Cancer (2014) 14(2):74–5. doi:10.1038/nrc3673
151. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334(6062):1573–7. doi:10.1126/science.1208347
152. Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis (2011) 32(7):955–63. doi:10.1093/carcin/bgr031
153. Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell (2013) 155(6):1216–9. doi:10.1016/j.cell.2013.11.019
154. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi:10.15252/embj.201490784
155. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol (2017) 14(4):247–58. doi:10.1038/nrclinonc.2016.183
156. Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 15(1):219–20. doi:10.1016/j.celrep.2016.03.045
157. Bultynck G. Onco-IP3Rs feed cancerous cravings for mitochondrial Ca2+. Trends Biochem Sci (2016) 41(5):390–3. doi:10.1016/j.tibs.2016.03.006
158. Finkel T, Hwang PM. The Krebs cycle meets the cell cycle: mitochondria and the G1-S transition. Proc Natl Acad Sci U S A (2009) 106(29):11825–6. doi:10.1073/pnas.0906430106
159. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A (2009) 106(29):11960–5. doi:10.1073/pnas.0904875106
160. Ryan KM. p53 and autophagy in cancer: guardian of the genome meets guardian of the proteome. Eur J Cancer (2011) 47(1):44–50. doi:10.1016/j.ejca.2010.10.020
161. De Smet P, Parys JB, Callewaert G, Weidema AF, Hill E, De Smedt H, et al. Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca2+ pumps. Cell Calcium (1999) 26(1–2):9–13. doi:10.1054/ceca.1999.0047
162. Missiaen L, Callewaert G, De Smedt H, Parys JB. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium (2001) 29(2):111–6. doi:10.1054/ceca.2000.0163
163. Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol (2001) 536(Pt 1):3–19. doi:10.1111/j.1469-7793.2001.t01-1-00003.x
164. Bittremieux M, Gerasimenko JV, Schuermans M, Luyten T, Stapleton E, Alzayady KJ, et al. DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium (2017) 62:60–70. doi:10.1016/j.ceca.2017.01.015
165. Bonora M, Giorgi C, Pinton P. Novel frontiers in calcium signaling: a possible target for chemotherapy. Pharmacol Res (2015) 99:82–5. doi:10.1016/j.phrs.2015.05.008
166. Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget (2015) 6(3):1435–45. doi:10.18632/oncotarget.2935
167. Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem (2005) 280(49):40892–900. doi:10.1074/jbc.M506623200
168. Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafa R, et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep (2016) 16(9):2415–27. doi:10.1016/j.celrep.2016.07.082
169. Galluzzi L. Novel insights into PML-dependent oncosuppression. Trends Cell Biol (2016) 26(12):889–90. doi:10.1016/j.tcb.2016.09.001
170. Lang F, Qin Z, Li F, Zhang H, Fang Z, Hao E. Apoptotic cell death induced by resveratrol is partially mediated by the autophagy pathway in human ovarian cancer cells. PLoS One (2015) 10(6):e0129196. doi:10.1371/journal.pone.0129196
171. Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog (2016) 55(5):818–31. doi:10.1002/mc.22324
172. Zhong LX, Zhang Y, Wu ML, Liu YN, Zhang P, Chen XY, et al. Resveratrol and STAT inhibitor enhance autophagy in ovarian cancer cells. Cell Death Discov (2016) 2:15071. doi:10.1038/cddiscovery.2015.71
173. Zhang J, Chiu J, Zhang H, Qi T, Tang Q, Ma K, et al. Autophagic cell death induced by resveratrol depends on the Ca2+/AMPK/mTOR pathway in A549 cells. Biochem Pharmacol (2013) 86(2):317–28. doi:10.1016/j.bcp.2013.05.003
174. Raturi A, Gutierrez T, Ortiz-Sandoval C, Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J Cell Biol (2016) 214(4):433–44. doi:10.1083/jcb.201512077
175. Krols M, Bultynck G, Janssens S. ER-mitochondria contact sites: a new regulator of cellular calcium flux comes into play. J Cell Biol (2016) 214(4):367–70. doi:10.1083/jcb.201607124
176. Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res (2014) 74(23):6820–32. doi:10.1158/0008-5472.CAN-14-0303
177. Messai Y, Noman MZ, Hasmim M, Escudier B, Chouaib S. HIF-2alpha/ITPR1 axis: a new saboteur of NK-mediated lysis. Oncoimmunology (2015) 4(2):e985951. doi:10.4161/2162402X.2014.985951
178. Messai Y, Noman MZ, Janji B, Hasmim M, Escudier B, Chouaib S. The autophagy sensor ITPR1 protects renal carcinoma cells from NK-mediated killing. Autophagy (2015). doi:10.1080/15548627.2015.1017194
Keywords: Ca2+ signaling, inositol 1,4,5-trisphosphate, inositol 1,4,5-trisphosphate receptors, autophagy, apoptosis, cancer
Citation: Kania E, Roest G, Vervliet T, Parys JB and Bultynck G (2017) IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 7:140. doi: 10.3389/fonc.2017.00140
Received: 07 April 2017; Accepted: 19 June 2017;
Published: 05 July 2017
Edited by:
Jon Lane, University of Bristol, United KingdomReviewed by:
Frank Kruyt, University Medical Center Groningen, NetherlandsHamid Morjani, University of Reims Champagne-Ardenne, France
Copyright: © 2017 Kania, Roest, Vervliet, Parys and Bultynck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan B. Parys, amFuLnBhcnlzQGt1bGV1dmVuLmJl;
Geert Bultynck, Z2VlcnQuYnVsdHluY2tAa3VsZXV2ZW4uYmU=