 Carl White
Carl White- Physiology and Biophysics, Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, IL, United States
Cell migration is one of the many processes orchestrated by calcium (Ca2+) signaling, and its dysregulation drives the increased invasive and metastatic potential of cancer cells. The ability of Ca2+ to function effectively as a regulator of migration requires the generation of temporally complex signals within spatially restricted microdomains. The generation and maintenance of these Ca2+ signals require a specific structural architecture and tightly regulated communication between the extracellular space, intracellular organelles, and cytoplasmic compartments. New insights into how Ca2+ microdomains are shaped by interorganellar Ca2+ communication have shed light on how Ca2+ coordinates cell migration by directing cellular polarization and the rearrangement of structural proteins. Importantly, we are beginning to understand how cancer subverts normal migration through the activity of oncogenes and tumor suppressors that impinge directly on the physiological function or expression levels of Ca2+ signaling proteins. In this review, we present and discuss research at the forefront of interorganellar Ca2+ signaling as it relates to cell migration, metastasis, and cancer progression, with special focus on endoplasmic reticulum-to-mitochondrial Ca2+ transfer.
Introduction
Pathology is frequently associated with the dysregulation of intracellular calcium (Ca2+) signaling (1). Cancer is no exception, with many primary tumor cells and cell lines displaying aberrant expression of Ca2+ signaling genes (2, 3). While it is unlikely that somatic mutations affecting any one individual Ca2+ signaling gene are sufficient to drive tumorigenesis (4, 5), remodeling of the Ca2+ signal in cancer appears almost universal and confers survival advantages (3, 6). And so, it may be that dysfunctional Ca2+ signaling is indeed a determinant of tumorigenesis when coincident with cancer-driving oncogene and tumor suppressor mutations.
Also, many oncoproteins and tumor suppressor proteins can themselves directly modulate Ca2+ signaling. They achieve this, in large part, by interacting with Ca2+ channels, pumps, and exchangers localized at the plasma membrane and various intracellular compartments. The Bcl-2 family of oncoproteins has been the most extensively studied in this respect and found to regulate Ca2+ signaling in ways that complement their roles as apoptotic regulators, as recently reviewed (7). Similarly, oncogenic Ras (8, 9) and the tumor suppressors promyelocytic leukemia (PML) (10), p53 (11), and BRCA1 (12) can all regulate apoptosis by impinging on the Ca2+ signal. Many of these proteins are enriched in spatially restricted domains created by the close apposition between the endoplasmic reticulum (ER) and the mitochondria, known as mitochondria-associated membranes (MAMs), where they function to modulate the flow of Ca2+ from the ER to mitochondria.
Ca2+ signaling also plays a role in cancer cell invasion and metastasis. Several different plasma membrane and ER-localized Ca2+ channels regulate the activity of effectors involved in motility and adhesion. Most of this regulation occurs by modifying the cytoplasmic Ca2+ signal and has been reviewed previously (13, 14). The significance of ER-mitochondrial Ca2+ communication in invasion and metastasis, however, has only recently emerged. This review will assess the literature relating to ER-mitochondrial Ca2+ communication. Our goal is to outline a theoretical framework that mechanistically links cancer-driven changes in ER-mitochondrial Ca2+ communication to its invasive and metastatic properties. We have made every attempt to include and reference original studies specifically related to this topic. When discussing a well-established concept, however, we direct readers to an appropriate review article.
Ca2+ Signaling Regulates Multiple Steps of the Invasion-Metastatic Cascade
Tumor metastasis directly accounts for the vast majority of cancer deaths (15). Metastasis is characterized by a sequence of events known as the invasion-metastatic cascade (16, 17). During this process, cancer cells lose their attachment to other cells and the extracellular matrix (ECM), acquire migratory capabilities and invade neighboring tissues by degrading and moving through the ECM, and ultimately transit to secondary sites by finding their way into the blood and lymphatic circulation. Importantly, Ca2+ signaling plays a key role at a number of points in the invasion-metastatic cascade.
Adhesion and Epithelial–Mesenchymal Transition (EMT)
The invasion-metastatic cascade begins with the loss of cell–ECM and cell–cell adhesion. Cells are linked to the ECM at focal adhesion points by structural complexes connecting membrane spanning integrins to the cytoskeleton. And so, the rate of focal adhesion assembly and disassembly governs the cell’s migratory ability. The process of disassembly is Ca2+ sensitive and triggered by Ca2+ oscillations that promote the association of focal adhesion kinase (FAK), a regulator of focal adhesion turnover, with the focal adhesion complex (18). The Ca2+ oscillations, which are spatially restricted at the focal adhesions (19), increase the residency of FAK at these sites through Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent regulation of its phosphorylation status (19–22).
The loss of cell–cell adhesion is also mediated through the process of EMT (23). The EMT process converts polarized epithelial cells into highly motile mesenchymal cells, defined by the induction of the mesenchymal markers N-cadherin, vimentin, and transcription factors, Snail, Slug, and Twist. Induction of EMT in the MDA-MB-231 breast cancer cell line was dependent on increased store-operated Ca2+ entry (SOCE) driven by expression of the SOCE proteins, stromal interaction molecule 1 (STIM1) and Orai1 (24). The SOCE pathway is activated in response to depletion of ER Ca2+ stores, which causes the ER-localized STIM to bind to and open the plasma membrane Ca2+ channel Orai (25). In contrast, SOCE was not important for EMT in MDA-MB-468 breast cancer cells (26). In these cells, the Ca2+ permeable transient receptor potential canonical type 1 channel was implicated as a sensitizer to EMT (26). Moreover, a subsequent study of MDA-MB-468 cells showed a requirement for Ca2+-permeable transient receptor potential melastatin-like 7 (TRPM7) channels in increasing vimentin expression through the signal transducer and activator of transcription 3 pathway (27). Collectively, these studies define a role for Ca2+ signaling in EMT and hint that EMT regulation by Ca2+ is likely to involve diverse mechanisms that are highly dependent on cell type and stimulus.
Migration and ECM Degradation
In the second step of the invasion-metastatic cascade, freely migrating cancer cells invade the surrounding stromal tissue. Migrating cells move by a cyclical process that begins with the extension of leading edge protrusions, known as lamellipodia. Lamellipodia attach to the substratum and contraction of the trailing rear edge moves the cell toward the lamellipodia (28, 29). At the leading edge, local Ca2+ signals control forward movement by regulating lamellipodia retraction and adhesion cycling through the activation of actin filament contraction (30, 31). Actin dynamics can also be influenced more indirectly by Ca2+ signaling, through activation of Ca2+-dependent kinases (30, 32) and regulation of Rac1, RhoA, and Cdc42 GTPases (33–36). In addition to forward motion, directional steering is also dependent on spatially restricted Ca2+ signals. These events, termed “Ca2+ flickers,” are triggered by Ca2+ influx through TRPM7 channels and amplified by ER Ca2+ release via inositol 1,4,5-trisphosphate receptor type 2 activation (37). While these spatially localized Ca2+ fluxes play important roles at the leading edge, there is on average, a front-to-rear increase in Ca2+ concentration (38–40). At the trailing edge, Ca2+ signaling is determined, in large part, by Ca2+ influx through L-type Ca2+ channels, which serves to maintain contractility and stabilize directional movement (40).
Invasive cancer cells migrate through their surrounding tissue. They do this by proteolytically degrading the ECM with enzymes that include matrix metalloproteinases (MMPs) and cathepsins (41). Importantly, Ca2+ influx determines how these enzymes influence metastasis. In a prostate cancer cell study, the metastatic potential was dependent on the expression of MMP2, MMP9, and cathepsin B regulated by Ca2+ influx through the transient receptor potential melastatin 2 (TRPV2) channel (42). Another study identified a role for SOCE (43). Activation of STIM/Orai was shown to influence melanoma metastasis by maintaining levels of membrane type1 MMP (MT1-MMP) at the plasma membrane (43).
Involvement of ER-Mitochondrial Ca2+ Flux in Cancer Cell Invasion and Metastasis
Cancer causes transcriptional and functional changes that often affect regulators of cytoplasmic Ca2+, including the TRP channels and components of STIM/Orai-mediated Ca2+ entry, for reviews see Ref. (2, 13, 14, 44). These changes are likely to have the greatest impact on the spatiotemporal profile of the cytoplasmic Ca2+ signal and affect the invasion-metastatic cascade by impinging on the cytoplasmically localized effectors outlined above (22, 45). As alluded to earlier, cancer is also associated with altered mitochondrial Ca2+ handling, brought about by changes in the expression profile of the mitochondrial Ca2+ uptake machinery, as well as oncoproteins and tumor suppressors at the MAM (46). Recent investigations using primary tumor models and cancer cell lines support the concept that the survival advantages of altered mitochondrial Ca2+ derive from effects on cellular metabolism (47) and apoptosis sensitivity (48–52). In the following section, we assess the evidence that altered ER-mitochondrial Ca2+ is also a determinant of increased invasive and metastatic potential (Figure 1).
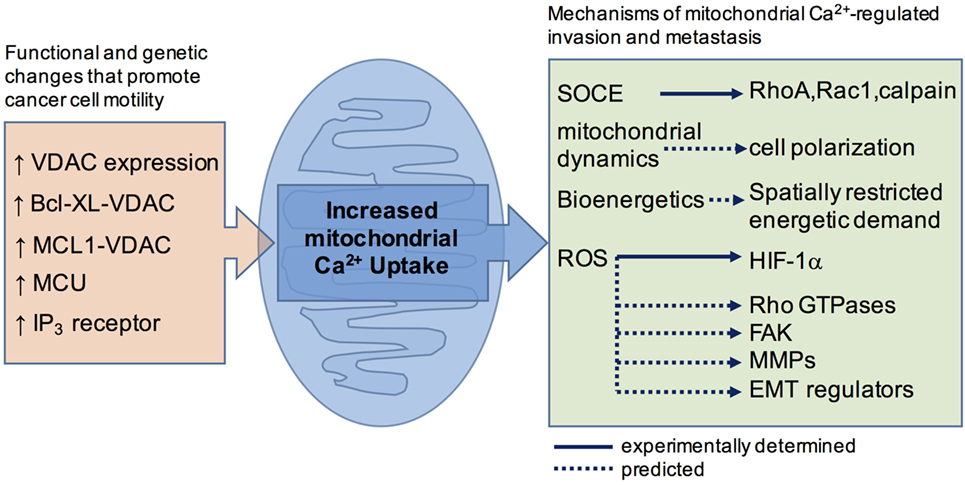
Figure 1. A schematic summary showing how cancer-associated functional and genetic changes that promote mitochondrial Ca2+ uptake are linked, or are predicted to link, to motility effector.
Mitochondria-Associated Membrane
The ER-localized inositol 1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor Ca2+ release channels deliver Ca2+ to the mitochondria (53–55). Structural elements, which include physical tethers linking both membranes (56, 57) and protein–protein interactions that bridge the ER release and mitochondrial uptake machinery (58, 59), facilitate the Ca2+ transfer. To get to the matrix, Ca2+ first moves across the outer mitochondrial membrane through the voltage-dependent anion channel (VDAC) (60–62). The VDAC is a porin channel and diffusion pathway for ions and metabolites (62). Despite its large pore size, VDAC can function as a highly regulated Ca2+ permeability (63, 64) that directly couples to IP3R-dependent Ca2+ release through interactions with the mitochondrial chaperone GRP75 (58). From the intermembrane space, Ca2+ moves across the inner membrane through the mitochondrial Ca2+ uniporter (MCU) (65, 66). Cancer remodels the MAM architecture by changing the expression levels of ER and mitochondrial Ca2+ channel proteins or their associated binding partners and regulators (46). Restructuring the MAMs and the resultant effects on mitochondrial Ca2+ homeostasis impinges on many processes including metabolism, bioenergetics, cell death, proliferation, mitochondrial dynamics, and cytoplasmic Ca2+ signaling. For the purpose of this review, we restrict our focus to those effects that most profoundly impact, or are likely to impact, invasion and metastasis.
Voltage-Dependent Anion Channel
Expression of the VDAC1 isoform is robustly increased in many cancer cell types (67) and reliably predicts survival outcomes in breast, colon, and lung cancers (68, 69). Increased VDAC likely promotes cancer cell growth primarily by influencing mitochondrial metabolism and apoptosis (70–72), processes that are also tightly regulated by VDAC interactions with hexokinase and members of the Bcl-2 family (72–74). Importantly, VDAC1 knockdown reduced cancer cell migration in vitro and suppressed tumor growth in vivo (75, 76). A decrease in VDAC expression would be expected to limit mitochondrial Ca2+ uptake (61). Indeed, VDAC influences cell migration by a mechanism that involves the regulation of mitochondrial Ca2+ uptake by interactions with Bcl-2 family proteins. The structural determinants and functional correlates of the VDAC-Bcl-2 protein interactions have been well characterized, as reviewed in Ref. 67. Antiapoptotic Bcl-XL and MCL1 both bind to VDAC1 and VDAC3 isoforms to promote mitochondrial Ca2+ uptake and drive cell migration (77–80). Importantly, disrupting the Bcl-XL-VDAC and MCL1-VDAC interactions was found to inhibit migration of triple negative breast cancer cells (80) and non-small cell lung carcinoma cells, respectively (78). These data raise the possibility of suppressing invasion and metastasis by targeting VDAC-Bcl-2 protein interactions.
Inositol 1,4,5-Trisphosphate Receptors
The type 3 IP3R isoform (IP3R-3), which is absent in normal colorectal mucosa, is expressed in colorectal carcinoma. Moreover, the expression is greatest at the invasive margin and strongly correlated with metastasis and patient survival (81). The IP3R-3 is also overexpressed in human glioblastoma tissue (82). Inhibiting IP3Rs in glioblastoma cell lines reduced invasion in vitro; it also reduced invasion in vivo and prolonged survival by suppressing tumor growth (82). These studies, however, did not define the mechanisms by which increased IP3R-3 expression directs invasion and metastasis. Interestingly, the IP3R-3 is enriched in at the MAM in some cell types (83), and it may preferentially deliver Ca2+ to the mitochondria under certain conditions (84). It is possible then that increased IP3R abundance promotes invasion and metastasis by increasing Ca2+ delivery to the mitochondria.
Mitochondrial Ca2+ Uniporter
The MCU machinery includes the MCU pore-forming subunit (65, 66) or its dominant-negative MCUb (85), together with associated regulators EMRE (86, 87) and MICU1-3 (88–91). Analysis of gene expression databases revealed that MCU levels are increased in several subtypes of breast cancer and correlated with tumor size, invasive and metastatic indices, and patient survival (92–94). While expression changes in the MCU regulators MCU1-3 and EMRE did not correlate with tumor size and invasiveness (92), poorer patient survival did correlate with increased MCU in combination with decreased MICU1 (94). The involvement of MCU in cancer progression was demonstrated in vivo by Tosatto et al., who showed that breast cancer tumor xenografts derived from MCU-deleted cells grew more slowly and were less likely to metastasize (92). In vitro experiments that knocked-down or inhibited MCU in breast cancer cell lines decreased mitochondrial Ca2+ uptake to inhibit migration and invasive potential without affecting cell survival (92, 94), proliferation, or apoptosis (95).
Mechanisms of Mitochondrial Ca2+-Regulated Invasion and Metastasis
Store-Operated Ca2+ Entry (SOCE)
Subplasmalemmal mitochondria regulate the activation and inactivation properties of SOCE by buffering incoming Ca2+ (96–98). Activation of SOCE is dependent on ER Ca2+ depletion, suggesting that ER, SOCE, and mitochondria are functionally coupled. Indeed, by limiting Ca2+ accumulation around the mouth of the IP3R, mitochondrial Ca2+ uptake prevents Ca2+-dependent inactivation of IP3Rs, which further depletes ER Ca2+ stores to promote SOCE (99). Given the Ca2+ communication between ER, mitochondrial and SOCE pathways, it is not surprising that MCU knockdown in MDA-MB-231 (93) and Hs578t (95) breast cancer lines inhibited both mitochondrial Ca2+ accumulation and SOCE. In the Hs578t cells, this caused a loss of cell polarity and migration associated with decreased RhoA, Rac1, and calpain activities (95). In these experiments, inhibiting SOCE (93, 95) or chelating intracellular Ca2+ (95) recapitulated the effects of MCU knockdown on migration. These data are consistent with studies defining STIM and Orai as key players in regulating invasion and metastasis (43, 100, 101) and suggest that altered MCU expression in cancer cells can influence downstream motility effectors by regulating SOCE.
Mitochondrial Dynamics
Mitochondria redistribute to the leading edge of cancer cells to support the increased bioenergetic demands at the invadopodia (102–105). Interestingly, the translocation of mitochondria to subplasmalemmal sites also plays a critical role during immune cell activation, where they regulate Ca2+ influx through SOCE (106). It is yet to be determined, however, if mitochondrial positioning, and its influence on Ca2+ influx, affects the polarization of cytoplasmic Ca2+ signaling in migrating cancer cells.
The translocation of mitochondria is dependent on increased mitochondrial fission, a process also promoted by mitochondrial Ca2+ accumulation (107, 108). Evidence that MCU plays a role in fission comes from the observation that fission is inhibited by the pharmacological block of the MCU (109, 110) and enhanced by loss-of-function mutations in MICU1, which promote mitochondrial Ca2+ uptake (111). Mechanistically, mitochondrial Ca2+ might influence fission by regulating the activity of dynamin-related protein 1 (Drp1). The ability of Drp1 to promote fission is dependent on phosphorylation at serine 616 (S616) and dephosphorylation of serine 637 (S637) (112, 113). Cytoplasmic Ca2+ signaling is known to regulate the phosphorylation status of Drp1 through calcineurin-dependent dephosphorylation of S637 (112, 114), and more recently it was found that blocking the MCU suppressed fission by decreasing Drp1 phosphorylation at S616 (115). These observations are relevant to this review because Drp1 is widely associated with tumor invasion and metastatic potential (104, 116–118), and increased S616 is found in breast cancer and lymph node metastases (104). Although speculative, it is possible that increased mitochondrial Ca2+ uptake in cancer cells links to invasion and metastasis through the processes of fission and mitochondrial localization.
Bioenergetics
Mitochondrial Ca2+ activates several Ca2+-dependent enzymes involved in the tricarboxylic acid (TCA) cycle (119). Work by Cárdenas et al. showed that constitutive low-level ER-mitochondrial Ca2+ transfer maintains flux through the TCA cycle to fuel oxidative phosphorylation and ATP production (120). In a follow-up study, the same group showed that blocking IP3Rs in cancer cells impaired oxidative phosphorylation, which killed cells by necrosis and reduced tumor growth in vivo (47). Although not examined in these studies, one might expect a similarly invoked bioenergetic crisis to inhibit cancer cell invasion and metastasis. Such an outcome is predicated based on the requirement for functional oxidative phosphorylation in tumor metastasis (121, 122), as well as the spatially restricted bioenergetic demands needed for cell migration (102–105).
Reactive Oxygen Species (ROS)
Mitochondrial ROS are generated as a consequence of normal respiration. As electrons supplied by the TCA cycle are passed down the electron transport chain, they escape, mostly at complex I and III, to react with O2 and produce ROS. Mitochondrial Ca2+ uptake can increase ROS production at complexes I, III and IV under a variety of conditions. Although the mechanisms are still unclear, a number of possibilities have been proposed, as reviewed previously (123, 124). Also, mitochondrial Ca2+ can promote the release of ROS accumulated in cristae and intermembrane spaces through Ca2+-dependent increases in matrix volume (125).
Increased mitochondrial ROS production is a known determinant of tumor growth and metastasis (126, 127) that likely drives invasion and metastasis by increasing cell migration (128–130). Importantly, increased migration is repeatedly correlated with increased ER-mitochondrial Ca2+ uptake (92–95). While excessive ROS production is toxic and excessive mitochondrial Ca2+ uptake inhibits rather than promotes migration (78, 131), physiological Ca2+-dependent ROS production is a major mitochondrially derived signal involved in regulating downstream effectors. In one study, increased ER-mitochondrial Ca2+ transfer in non-small cell lung carcinoma cells was caused by MCL1–VDAC interactions that promoted cell migration by increasing mitochondrial ROS production (78). In another study, increased mitochondrial Ca2+ uptake increased breast cancer cell xenograft growth and metastasis by increased ROS-dependent expression of HIF-1α (92). To our knowledge, HIF-1α is the only cell migration regulator that has been specifically linked to mitochondrial Ca2+-dependent ROS production. Nevertheless, many migration effectors are sensitive to ROS signaling (132). Perhaps more intriguingly, many of these, including the Rho GTPases, FAK, MMPs, and mediators of EMT, are sensitive to both ROS and Ca2+ signals, see Ref. (13, 132) for complete listings of ROS and Ca2+-sensitive targets, respectively. The degree of overlap between ROS and Ca2+-sensitive effectors highlights a need to carefully differentiate between ROS and Ca2+-dependent effects when probing the role of ER-mitochondrial Ca2+ transfer.
Conclusion
The molecular identification of the SOCE and MCU machinery, the introduction of powerful molecular tools, and the evolution of cancer genetics have all contributed to developing our understanding of how Ca2+ signals regulate cancer cell invasion and metastasis. As we have seen, a picture has emerged in which ER Ca2+ release, mitochondrial Ca2+ uptake, and plasmalemmal Ca2+ influx work together to exquisitely regulate cell motility. This complexity, however, should not dissuade efforts to examine the possibility of therapeutically targeting ER-mitochondrial Ca2+ transfer to affect metastasis. Encouragingly, many of the studies reviewed here have already demonstrated the feasibility of such an approach, showing reduced metastasis in vivo after targeting IP3Rs (47), STIM/Orai (22, 45), or MCU (92). In addition, therapeutics originally designed to promote cell death might also be useful for limiting metastasis. In this case, the Bcl-2 inhibitors, both BH3 and BH4 mimetics (80, 133), as well as recently developed MCL1 inhibitors (134), would be expected to suppress cell migration by limiting ER-mitochondrial Ca2+ transfer.
Author Contributions
CW wrote the manuscript and designed and prepared figures.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Support was provided by Rosalind Franklin University of Medicine and Science.
References
2. Hoth M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim Biophys Acta (2016) 1863:1408–17. doi:10.1016/j.bbamcr.2015.12.009
3. Stewart TA, Yapa KTDS, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta (2015) 1848:2502–11. doi:10.1016/j.bbamem.2014.08.016
4. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502:333–9. doi:10.1038/nature12634
5. Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep (2013) 3:2650. doi:10.1038/srep02650
6. Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol (2016) 29:1–6. doi:10.1016/j.coph.2016.03.002
7. Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene (2016) 35:5079–92. doi:10.1038/onc.2016.31
8. Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, et al. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A (2013) 110:20593–8. doi:10.1073/pnas.1306431110
9. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene (2014) 33:2329–40. doi:10.1038/onc.2013.192
10. Giorgi C, Ito K, Lin H-K, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330:1247–51. doi:10.1126/science.1189157
11. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112:1779–84. doi:10.1073/pnas.1410723112
12. Hedgepeth SC, Garcia MI, Wagner LE II, Rodriguez AM, Chintapalli SV, Snyder RR, et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem (2015) 290:7304–13. doi:10.1074/jbc.M114.611186
13. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer (2011) 11:609–18. doi:10.1038/nrc3105
14. Tsai F-C, Kuo G-H, Chang S-W, Tsai P-J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int (2015) 2015:409245. doi:10.1155/2015/409245
15. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi:10.1126/science.1203543
16. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res (2010) 70:5649–69. doi:10.1158/0008-5472.CAN-10-1040
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
18. Giannone G, Rondé P, Gaire M, Haiech J, Takeda K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J Biol Chem (2002) 277:26364–71. doi:10.1074/jbc.M203952200
19. Giannone G, Rondé P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, et al. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem (2004) 279:28715–23. doi:10.1074/jbc.M404054200
20. Easley CA IV, Brown CM, Horwitz AF, Tombes RM. CaMK-II promotes focal adhesion turnover and cell motility by inducing tyrosine dephosphorylation of FAK and paxillin. Cell Motil Cytoskeleton (2008) 65:662–74. doi:10.1002/cm.20294
21. Fan RS, Jácamo RO, Jiang X, Sinnett-Smith J, Rozengurt E. G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843. Mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J Biol Chem (2005) 280:24212–20. doi:10.1074/jbc.M500716200
22. Chen Y-F, Chiu W-T, Chen Y-T, Lin P-Y, Huang H-J, Chou C-Y, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A (2011) 108:15225–30. doi:10.1073/pnas.1103315108
23. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
24. Hu J, Qin K, Zhang Y, Gong J, Li N, Lv D, et al. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem Biophys Res Commun (2011) 411:786–91. doi:10.1016/j.bbrc.2011.07.025
25. Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev (2015) 95:1383–436. doi:10.1152/physrev.00020.2014
26. Davis FM, Peters AA, Grice DM, Cabot PJ, Parat M-O, Roberts-Thomson SJ, et al. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One (2012) 7:e36923. doi:10.1371/journal.pone.0036923
27. Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW, et al. Induction of epithelial–mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene (2013) 33:2307–16. doi:10.1038/onc.2013.187
28. Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol (2009) 10:538–49. doi:10.1038/nrm2729
29. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science (2003) 302:1704–9. doi:10.1126/science.1092053
30. Evans JH, Falke JJ. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc Natl Acad Sci U S A (2007) 104:16176–81. doi:10.1073/pnas.0707719104
31. Tsai F-C, Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr Biol (2012) 22:837–42. doi:10.1016/j.cub.2012.03.037
32. Price JT, Tiganis T, Agarwal A, Djakiew D, Thompson EW. Epidermal growth factor promotes MDA-MB-231 breast cancer cell migration through a phosphatidylinositol 3’-kinase and phospholipase C-dependent mechanism. Cancer Res (1999) 59:5475–8.
33. Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature (2006) 440:1069–72. doi:10.1038/nature04665
34. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, et al. Coordination of Rho GTPase activities during cell protrusion. Nature (2009) 461:99–103. doi:10.1038/nature08242
35. Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG. Calcium signaling regulates translocation and activation of Rac. J Biol Chem (2003) 278:39413–21. doi:10.1074/jbc.M302083200
36. Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat Neurosci (2006) 9:50–7. doi:10.1038/nn1619
37. Wei C, Wang X, Chen M, Ouyang K, Song L-S, Cheng H. Calcium flickers steer cell migration. Nature (2009) 457:901–5. doi:10.1038/nature07577
38. Brundage RA, Fogarty KE, Tuft RA, Fay FS. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science (1991) 254:703–6. doi:10.1126/science.1948048
39. Tsai F-C, Seki A, Yang HW, Hayer A, Carrasco S, Malmersjö S, et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat Cell Biol (2014) 16:133–44. doi:10.1038/ncb2906
40. Kim JM, Lee M, Kim N, Heo WD. Optogenetic toolkit reveals the role of Ca2+ sparklets in coordinated cell migration. Proc Natl Acad Sci U S A (2016) 113:5952–7. doi:10.1073/pnas.1518412113
41. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141:52–67. doi:10.1016/j.cell.2010.03.015
42. Monet M, Lehen’kyi V, Gackiere F, Firlej V, Vandenberghe M, Roudbaraki M, et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res (2010) 70:1225–35. doi:10.1158/0008-5472.CAN-09-2205
43. Sun J, Lu F, He H, Shen J, Messina J, Mathew R, et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J Cell Biol (2014) 207:535–48. doi:10.1083/jcb.201407082
44. Chen Y-F, Chen Y-T, Chiu W-T, Shen M-R. Remodeling of calcium signaling in tumor progression. J Biomed Sci (2013) 20:23. doi:10.1186/1423-0127-20-23
45. Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, et al. Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J (2013) 27:63–75. doi:10.1096/fj.12-213801
46. Missiroli S, Danese A, Iannitti T, Patergnani S, Perrone M, Previati M, et al. Endoplasmic reticulum-mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim Biophys Acta (2017) 1864(6):858–64. doi:10.1016/j.bbamcr.2016.12.024
47. Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14:2313–24. doi:10.1016/j.celrep.2016.02.030
48. Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget (2015) 6:1435–45. doi:10.18632/oncotarget.2935
49. Doghman-Bouguerra M, Granatiero V, Sbiera S, Sbiera I, Lacas-Gervais S, Brau F, et al. FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep (2016) 17:1264–80. doi:10.15252/embr.201541504
50. Pierro C, Cook SJ, Foets TCF, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci (2014) 127:1607–19. doi:10.1242/jcs.141408
51. Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis (2013) 4:e632. doi:10.1038/cddis.2013.140
52. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol (2013) 23:58–63. doi:10.1016/j.cub.2012.11.026
53. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13:566–78. doi:10.1038/nrm3412
54. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280:1763–6. doi:10.1126/science.280.5370.1763
55. Pacher P, Thomas AP, Hajnóczky G. Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc Natl Acad Sci U S A (2002) 99:2380–5. doi:10.1073/pnas.032423699
56. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol (2006) 174:915–21. doi:10.1083/jcb.200604016
57. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456:605–10. doi:10.1038/nature07534
58. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175:901–11. doi:10.1083/jcb.200608073
59. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A (2013) 110:12526–34. doi:10.1073/pnas.1302455110
60. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2012) 19(2):267–73. doi:10.1038/cdd.2011.92
61. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi:10.1083/jcb.200205091
62. Colombini M. VDAC structure, selectivity, and dynamics. Biochim Biophys Acta (2012) 1818:1457–65. doi:10.1016/j.bbamem.2011.12.026
63. Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem (2006) 281:17347–58. doi:10.1074/jbc.M600906200
64. Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta (2007) 1768:2510–5. doi:10.1016/j.bbamem.2007.06.002
65. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
66. De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
67. Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim Biophys Acta (2015) 1848:2547–75. doi:10.1016/j.bbamem.2014.10.040
68. Ko J-H, Gu W, Lim I, Zhou T, Bang H. Expression profiling of mitochondrial voltage-dependent anion channel-1 associated genes predicts recurrence-free survival in human carcinomas. PLoS One (2014) 9:e110094. doi:10.1371/journal.pone.0110094
69. Grills C, Jithesh PV, Blayney J, Zhang S-D, Fennell DA. Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS One (2011) 6:e14635. doi:10.1371/journal.pone.0014635
70. Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci U S A (2000) 97:4666–71. doi:10.1073/pnas.090082297
71. Weinberg F, Chandel NS. Mitochondrial metabolism and cancer. Ann N Y Acad Sci (2009) 1177:66–73. doi:10.1111/j.1749-6632.2009.05039.x
72. Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol (2001) 152:237–50. doi:10.1083/jcb.152.2.237
73. Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem (2008) 283:13482–90. doi:10.1074/jbc.M708216200
74. Pastorino JG, Hoek JB. Integration of energy metabolism and control of apoptosis in tumor cells. In: Costello L, Singh K, editors. Mitochondria and Cancer. New York: Springer (2009). p. 103–29.
75. Wu C-H, Lin Y-W, Wu T-F, Ko J-L, Wang P-H. Clinical implication of voltage-dependent anion channel 1 in uterine cervical cancer and its action on cervical cancer cells. Oncotarget (2016) 7:4210–25. doi:10.18632/oncotarget.6704
76. Arif T, Vasilkovsky L, Refaely Y, Konson A, Shoshan-Barmatz V. Silencing VDAC1 expression by siRNA inhibits cancer cell proliferation and tumor growth in vivo. Mol Ther Nucleic Acids (2014) 3:e159. doi:10.1038/mtna.2014.9
77. Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem (2013) 288:19870–81. doi:10.1074/jbc.M112.448290
78. Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis (2014) 5:e1482. doi:10.1038/cddis.2014.419
79. Rajan S, Choi M, Nguyen QT, Ye H, Liu W, Toh HT, et al. Structural transition in Bcl-xL and its potential association with mitochondrial calcium ion transport. Sci Rep (2015) 5:10609. doi:10.1038/srep10609
80. Fouqué A, Lepvrier E, Debure L, Gouriou Y, Malleter M, Delcroix V, et al. The apoptotic members CD95, BclxL, and Bcl-2 cooperate to promote cell migration by inducing Ca(2+) flux from the endoplasmic reticulum to mitochondria. Cell Death Differ (2016) 23:1702–16. doi:10.1038/cdd.2016.61
81. Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium (2010) 48:315–23. doi:10.1016/j.ceca.2010.09.005
82. Kang SS, Han K-S, Ku BM, Lee YK, Hong J, Shin HY, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res (2010) 70:1173–83. doi:10.1158/0008-5472.CAN-09-2886
83. Hayashi T, Su T-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell (2007) 131:596–610. doi:10.1016/j.cell.2007.08.036
84. Mendes CCP, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem (2005) 280:40892–900. doi:10.1074/jbc.M506623200
85. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J (2013) 32:2362–76. doi:10.1038/emboj.2013.157
86. Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 342:1379–82. doi:10.1126/science.1242993
87. Vais H, Mallilankaraman K, Mak D-OD, Hoff H, Payne R, Tanis JE, et al. EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep (2016) 14:403–10. doi:10.1016/j.celrep.2015.12.054
88. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell (2012) 151:630–44. doi:10.1016/j.cell.2012.10.011
89. Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature (2010) 467:291–6. doi:10.1038/nature09358
90. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell (2014) 53:726–37. doi:10.1016/j.molcel.2014.01.013
91. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One (2013) 8:e55785. doi:10.1371/journal.pone.0055785
92. Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med (2016) 8:569–85. doi:10.15252/emmm.201606255
93. Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, et al. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun (2015) 458:186–93. doi:10.1016/j.bbrc.2015.01.092
94. Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One (2014) 9:e96866. doi:10.1371/journal.pone.0096866
95. Prudent J, Popgeorgiev N, Gadet R, Deygas M, Rimokh R, Gillet G. Mitochondrial Ca(2+) uptake controls actin cytoskeleton dynamics during cell migration. Sci Rep (2016) 6:36570. doi:10.1038/srep36570
96. Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci U S A (2000) 97:10607–12. doi:10.1073/pnas.180143997
97. Gilabert JA, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J (2000) 19:6401–7. doi:10.1093/emboj/19.23.6401
98. Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol (1997) 137:633–48. doi:10.1083/jcb.137.3.633
99. Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallström S, et al. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J Cell Sci (2014) 127:2944–55. doi:10.1242/jcs.149807
100. Yang S, Zhang JJ, Huang X-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell (2009) 15:124–34. doi:10.1016/j.ccr.2008.12.019
101. Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, et al. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett (2013) 330:163–9. doi:10.1016/j.canlet.2012.11.040
102. Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J (2013) 104:2077–88. doi:10.1016/j.bpj.2013.03.025
103. Jung J-U, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, et al. NIK/MAP3K14 regulates mitochondrial dynamics and trafficking to promote cell invasion. Curr Biol (2016) 26:3288–302. doi:10.1016/j.cub.2016.10.009
104. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene (2013) 32:4814–24. doi:10.1038/onc.2012.494
105. Caino MC, Altieri DC. Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle (2015) 14:3242–7. doi:10.1080/15384101.2015.1084448
106. Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J Immunol (2010) 184:184–90. doi:10.4049/jimmunol.0902872
107. Chang KT, Niescier RF, Min K-T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A (2011) 108:15456–61. doi:10.1073/pnas.1106862108
108. Hom JR, Gewandter JS, Michael L, Sheu S-S, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol (2007) 212:498–508. doi:10.1002/jcp.21051
109. Liang N, Wang P, Wang S, Li S, Li Y, Wang J, et al. Role of mitochondrial calcium uniporter in regulating mitochondrial fission in the cerebral cortexes of living rats. J Neural Transm (2014) 121:593–600. doi:10.1007/s00702-014-1166-6
110. Zhao L, Li S, Wang S, Yu N, Liu J. The effect of mitochondrial calcium uniporter on mitochondrial fission in hippocampus cells ischemia/reperfusion injury. Biochem Biophys Res Commun (2015) 461:537–42. doi:10.1016/j.bbrc.2015.04.066
111. Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs A-M, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet (2014) 46:188–93. doi:10.1038/ng.2851
112. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep (2007) 8:939–44. doi:10.1038/sj.embor.7401062
113. Chang C-R, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem (2007) 282:21583–7. doi:10.1074/jbc.C700083200
114. Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A (2008) 105:15803–8. doi:10.1073/pnas.0808249105
115. Zheng X, Chen M, Meng X, Chu X, Cai C, Zou F. Phosphorylation of dynamin-related protein 1 at Ser616 regulates mitochondrial fission and is involved in mitochondrial calcium uniporter-mediated neutrophil polarization and chemotaxis. Mol Immunol (2017) 87:23–32. doi:10.1016/j.molimm.2017.03.019
116. Ferreira-da-Silva A, Valacca C, Rios E, Pópulo H, Soares P, Sobrinho-Simões M, et al. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One (2015) 10:e0122308. doi:10.1371/journal.pone.0122308
117. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J (2012) 26:2175–86. doi:10.1096/fj.11-196543
118. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell (2015) 57:537–51. doi:10.1016/j.molcel.2015.01.002
119. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry (2012) 51:2959–73. doi:10.1021/bi2018909
120. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
121. Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab (2015) 21:81–94. doi:10.1016/j.cmet.2014.12.003
122. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(992–1003):1–15. doi:10.1038/ncb3039
123. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu S-S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol (2004) 287:C817–33. doi:10.1152/ajpcell.00139.2004
124. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94:909–50. doi:10.1152/physrev.00026.2013
125. Booth DM, Enyedi B, Geiszt M, Várnai P, Hajnóczky G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell (2016) 63:240–8. doi:10.1016/j.molcel.2016.05.040
126. Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8:754–66. doi:10.1016/j.celrep.2014.06.043
127. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320:661–4. doi:10.1126/science.1156906
128. Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res (2009) 69:2375–83. doi:10.1158/0008-5472.CAN-08-3359
129. Hung W-Y, Huang K-H, Wu C-W, Chi C-W, Kao H-L, Li AF-Y, et al. Mitochondrial dysfunction promotes cell migration via reactive oxygen species-enhanced β5-integrin expression in human gastric cancer SC-M1 cells. Biochim Biophys Acta (2012) 1820:1102–10. doi:10.1016/j.bbagen.2012.04.016
130. Laurent A, Nicco C, Chéreau C, Goulvestre C, Alexandre J, Alves A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res (2005) 65:948–56.
131. Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep (2016) 15:1673–85. doi:10.1016/j.celrep.2016.04.050
132. Tochhawng L, Deng S, Pervaiz S, Yap CT. Redox regulation of cancer cell migration and invasion. Mitochondrion (2013) 13:246–53. doi:10.1016/j.mito.2012.08.002
133. Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood (2011) 117:2924–34. doi:10.1182/blood-2010-09-307405
Keywords: migration, Bcl-2, Bcl-XL, MCL1, reactive oxygen species, voltage-dependent anion channel, STIM, Orai
Citation: White C (2017) The Regulation of Tumor Cell Invasion and Metastasis by Endoplasmic Reticulum-to-Mitochondrial Ca2+ Transfer. Front. Oncol. 7:171. doi: 10.3389/fonc.2017.00171
Received: 02 June 2017; Accepted: 26 July 2017;
Published: 10 August 2017
Edited by:
Cesar Cardenas, Universidad de Chile, ChileReviewed by:
Cristina Mammucari, University of Padua, ItalyAlessandro Rimessi, University of Ferrara, Italy
Copyright: © 2017 White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl White, Y2FybC53aGl0ZUByb3NhbGluZGZyYW5rbGluLmVkdQ==