 Rimas J. Orentas1*
Rimas J. Orentas1* Sivasish Sindiri2
Sivasish Sindiri2 Christine Duris3Xinyu Wen2Jianbin He2Jun S. Wei2
Christine Duris3Xinyu Wen2Jianbin He2Jun S. Wei2 Jason Jarzembowski3
Jason Jarzembowski3 Javed Khan2*
Javed Khan2*
- 1Lentigen Technology, Inc., a Miltenyi Biotec Company, Gaithersburg, MD, United States
- 2Genetics Branch, National Cancer Institute, Center for Cancer Research, National Institutes of Health, Bethesda, MD, United States
- 3Department of Pathology, Medical College of Wisconsin, Milwaukee, WI, United States
Adoptive immunotherapy with antibody-based therapy or with T cells transduced to express chimeric antigen receptors (CARs) is useful to the extent that the cell surface membrane protein being targeted is not expressed on normal tissues. The most successful CAR-based (anti-CD19) or antibody-based therapy (anti-CD20) in hematologic malignancies has the side effect of eliminating the normal B cell compartment. Targeting solid tumors may not provide a similar expendable marker. Beyond antibody to Her2/NEU and EGFR, very few antibody-based and no CAR-based therapies have seen broad clinical application for solid tumors. To expand the way in which the surfaceome of solid tumors can be analyzed, we created an algorithm that defines the pairwise relative overexpression of surface antigens. This enables the development of specific immunotherapies that require the expression of two discrete antigens on the surface of the tumor target. This dyad analysis was facilitated by employing the Hotelling’s T-squared test (Hotelling–Lawley multivariate analysis of variance) for two independent variables in comparison to a third constant entity (i.e., gene expression levels in normal tissues). We also present a unique consensus scoring mechanism for identifying transcripts that encode cell surface proteins. The unique application of our bioinformatics processing pipeline and statistical tools allowed us to compare the expression of two membrane protein targets as a pair, and to propose a new strategy based on implementing immunotherapies that require both antigens to be expressed on the tumor cell surface to trigger therapeutic effector mechanisms. Specifically, we found that, for MYCN amplified neuroblastoma, pairwise expression of ACVR2B or anaplastic lymphoma kinase (ALK) with GFRA3, GFRA2, Cadherin 24, or with one another provided the strongest hits. For MYCN, non-amplified stage 4 neuroblastoma, neurotrophic tyrosine kinase 1, or ALK paired with GFRA2, GFRA3, SSK1, GPR173, or with one another provided the most promising paired-hits. We propose that targeting these markers together would increase the specificity and thereby the safety of CAR-based therapy for neuroblastoma.
Introduction
The field of immunotherapy has entered a time of rapid advancement. Built upon decades of basic research, the fields of recombinant protein engineering, high-throughput screening, and gene vector biology, have allowed the implementation of engineered immunoglobulin molecules and engineered immune cells in clinically meaningful protocols. This is especially true for hematologic malignancies. Antibodies featuring engineered Fc domains or bispecific antibodies are now considered part of our current armamentarium for leukemia expressing CD19 or CD20 (1–3); as are T cells engineered to express chimeric antigen receptors (CARs) that target CD19 (4, 5). The targeting of B cell malignancies by these agents also eliminates normal mature B lymphocytes, which is a well-tolerated “on-target” side-effect. Moreover, the ability to use these approaches in conjunction with hematopoietic stem cell transplantation can take patients who are complete responders, and who may yet relapse, into the realm of true “cure” (6).
The ability to classify tumors according to their gene expression profile has resulted in an explosion of useful approaches to differentiate among tumor types, to clarify pathological anomalies, and to create meaningful sub classifications of disease that have informed further investigation. In 2000, Alizadeh et al. used DNA microarrays to classify diffuse large B-cell lymphoma into two new types, germinal center-like and activated B-cell like (7). More than a decade later, this basic principle was used to redefine how we classify medulloblastoma (8). In 2007, Wood et al. published a landmark study in which the entire transcriptome of 11 breast and 11 colorectal tumors was compared to the Reference Sequence database, thus analyzing 20,857 transcripts from 18,191 genes (9). This study identified the key conserved mutations across two specific tumor types in reference to normal transcriptomes. The need to have an even more robust definition of a “normal” transcript dataset led us to generate a database of mRNA gene expression profiles from 158 human samples (19 different organs from 30 different donors) for 18,927 unique genes (10). During the creation of this database, we demonstrated its ability to identify 19 neuroblastoma tumor-specific genes, in comparison to normal. The data for this, now expanded, set of normal and pediatric tumor gene expression analysis can be found online, hosted by the NCI, at: http://home.ccr.cancer.gov/oncology/oncogenomics/ in the “data” section. Given the ability to now rapidly analyze whole transcriptomes, our attention turned to the ability to identify new tumor-expressed targets for antibody-based or CAR-based therapy.
Using whole transcriptome-based analyses filtered for plasma membrane protein expression generates a data set referred to as the “surfaceome.” However, there is a key methodological gap remaining in this approach. There is no single database or filter that definitively identifies transcripts encoding plasma membrane proteins. Biochemical methods, wherein surface structures are chemically labeled or tagged, isolated, and then subjected to mass spectrometry are still under development, and are incomplete with respect to the catalog of surface targets identified (11, 12). Here, we present our method of consensus scoring, wherein data from multiple databases are aggregated, and a plasma membrane residence score assigned to each transcript. In our previous work, we averaged the gene expression level of each transcript across 12 types of pediatric cancers and compared each to an average collection of normal tissues (13). Here, we have updated this approach by using RNASeq data. More importantly, we have now developed a new analytical tool that allows overexpressed transcript to be scored as a dyad, that is, the two proteins that are most overexpressed in cancer versus normal tissue as a pair can now be identified.
The ability to target hematologic malignancies with immunotherapeutic agents is becoming a well-established approach. However, progress against “solid cancers” has been slower, due to at least two factors. The first is the inherent cellular complexity of the tumor lesion itself. Second, especially in reference to pediatric malignancies that have the lowest mutation rates of all cancers (14), there may be no single target that can sufficiently define a cellular target as being “cancer” or “normal.” For example, in our previous gene expression profiling work, the top 3 transcripts overexpressed in stage 4 neuroblastoma were SLC10A4, CHRNA3, and SLC29A4 (13). These encode for a Na+/bile cotransporter-like protein in the solute carrier superfamily (SLC), a subunit of the nicotinic cholinergic receptor, and an SLC family member involved in nucleoside transport, respectively. None of these hits appear safe to target, as they are likely to be broadly expressed in normal or essential tissues, even though gene expression profiling informs us that they are highly expressed in neuroblastoma as compared to normal tissue. In this report, we present a new approach, using neuroblastoma as our example, for identifying pairs of ligands that might be more safely targeted than any single target, giving another dimension of target specificity to our anticancer therapeutic approach. This approach can be similarly applied to any RNASeq data set derived from other malignancies. An important limitation to these approaches is our inability to define the “glycome” of solid tumors in a high throughput manner (15). This is an important consideration as the most promising CAR and antibody-based approaches to treat neuroblastoma to date, focus on the ganglioside GD2 (16, 17). As GD2 and other glycoform-based approaches are being refined, we propose that our current approach holds promise for developing “two-hit” gated approaches for solid tumor immunotherapy.
Materials and Methods
Neuroblastoma and Normal Tissues Sources for RNASeq
PolyA selected RNA libraries of neuroblastoma (NB) samples were prepared for RNA sequencing on an Illumina HiSeq2000 using the manufacturer’s protocol (Illumina, Inc., San Diego, CA, USA). A total of 32 stage 4 MYCN-amplified (MYCN-A) and 70 MYCN-non-amplified (MYCN-NA) neuroblastomas were analyzed. For normal tissue, 17 samples from brain and 46 from other tissues were analyzed. Raw sequencing files were converted to FASTQ format and were mapped to the human reference genome (GRch37) using Tophat2.2. Using PICARD and Samtools, a QC check was performed on the produced BAM files and PCR duplicate reads were removed.
Building a Cell Surface Annotation File
In total, 6,414 transcripts (as annotated in RefSeq) from the human genome were chosen for downstream analysis. This was accomplished by gathering information available for each transcript from the following annotation or aggregation databases:
(1) from Compendia
(a) isSurfaceomeSurfaceProtein (see Ludwig Institute for Cancer Research, Instituto de Bioinformacia e Biotecnologia, Brazil, http://www.bioinformatics-brazil.org/surfaceome/home)
(b) isCDDSurfaceProtien, Conserved Domain Database (see http://www.ncbi.nlm.nih.gov/cdd)
(c) isGOSurfaceProtein, Gene Ontology Consortium (see http://geneontology.org)
(d) isSurfaceProteinEvidence
(2) from the Pandey Lab, Johns Hopkins University (http://pandeylab.igm.jhmi.edu)
(e) AmiGo (GO) (see http://amigo.geneontology.org/amigo)
(3) Compartments, from the Jensen Lab, Novo Nordisk Foundation for Protein Research, University of Copenhagen (18)
(f) Knowledge (COMPARTMENTS, based on UniProtKB, MGI, SGD, Flybase, and WormBase, see http://compartments.jensenlab.org/Search)
(g) Experiments [derived from human protein atlas, see Li et al. (19)] (19)
(h) Prediction [aggregates WoLF PSORT (20); and YLoc (21, 22)]
(i) Text mining (text mining of Medline abstracts).
If a transcript was scored as a plasma membrane protein in at least seven of the listed databases, this transcript was considered a positive hit and entered into subsequent analysis. In total, 6,414 cell surface genes were defined for analysis.
Differential Gene Expression Using Limma/Voom and Tandem Gene Expression Analysis
Linear Models for Microarray and RNA-Seq Data (Limma) is an open-source software package that processes expression array and RNA-Seq data (using the voom function) allowing for differential gene expression analysis (23–25). The two groups analyzed were MYCN-A and stage IV MYCN-non-amplified (MYCN-NA) neuroblastoma. These were compared against normal samples. For a transcript to be considered as overexpressed, a log fold-change (FC) ≥2, expression greater than two FPKM (fragments per kilobase transcript per million mapped reads), and a highly significant p-value (pval) ≤ 0.001, was required. This analysis identified in 158 for MYCN-A neuroblastoma, and 179 for MYCN-NA neuroblastoma transcripts.
We then paired each overexpressed cell surface protein-encoding transcript with each of the other transcripts for statistical testing. Hotelling’s T-squared test (MANOVA/Hotelling–Lawley multivariate analysis of variance) was used to identify gene pairs, affected together by difference in sample conditions (tumor versus normal tissue). To assess linear dependence between the two genes in a gene pair, the Pearson correlation coefficient (only in tumor) was used. Significant gene pairs (Hotelling’s p-value ≤ 0.01) with high correlation value (r2 p-value ≤ 0.05) were chosen as final candidate gene pairs (326 gene pairs comprising 27 unique genes in MYCN-A neuroblastoma and 529 gene pairs comprising 34 unique genes in MYCN-NA neuroblastoma).
In sum, we applied the following filters to create the final set of gene pairs: (a) MANOVA pval ≤ 0.01, (b) Sort Pearson correlation and pval ≤ 0.05, (c) Median_Exp_gene1 [log2(FPKM)] ≥ 2; Median_Exp_gene2 [log2(FPKM)] ≥ 2, (d) Log_FC_Gene1 (compared to normal) ≥ 2; Log_FCGene2 (compared to normal) ≥ 2, (e) Log_FC_Gene1 (compared to only brain) ≥ 2; Log_FC_Gene2 (compared to only brain) ≥ 2, (f) Log FKPM in any vital organ was varied between ≤1.0, 1.5, and 2.0.
Pathological Analysis
Antibodies specific for cell surface antigens were obtained from the following sources and used on an automated Leica Bond staining platform as indicated at the Department of Pathology, Medical College of Wisconsin: anaplastic lymphoma kinase (ALK)-1 (Leica NCL-L-ALK, clone 5A4, 1:200 dilution, control staining on a normal tissue blocks including cerebellum, pancreas, tonsil, and lymphoma/ALL was carried with all antibodies listed unless otherwise noted, positive staining for ALK on ALL noted), Cadherin 24 (CDH24) (LC Bio LS-C168610 pAB rabbit, 1:100 dilution, positive control block and ganglion cells/nerve cells of bowel wall, endometrium surface epithelium, and hepatocellular carcinoma were also stained with positive staining for each of these three additional specimens), DLK (AbCam ab21692 pAB rabbit, 1:300 dilution, control cell block was stained and positive signal was noted additionally for placenta, with islets and ductal cells staining positive, and neutrophils staining positive), GFRA2 (Sigma HPA 024701 pAB rabbit, 1:20 dilution, control cell block and additional tissues including macrophages and sinusoids in liver, tonsil leukocytes, which were positive and ALL, weakly positive, and lung, unremarkable), GFRA3 (Sigma HPA 020731 pAB rabbit, 1:500 dilution, control cell block and additional tissue including increased staining in pancreatic ductal epithelium and islets, decreased in exocrine glandular cells, strong lymphatic and positive alveolar macrophage staining), GPR173 (Novus BioNLS51 pAB rabbit, 1:300 dilution, control block and additional stain for red cells, positive, lymphocytes, and epidermis), TrkA (Abcam ab76291, 1:100 dilution, control block and tonsil, reticular dendritic network positive, liver (negative except for arteries), positive pancreas islet cells, sinusoids of HCC, and some positivity in hepatocytes).
Tissue microarrays containing neuroblastoma were purchased from US Biomax (Derwood, MD, USA), specifically a microarray panel with neuroblastoma and peripheral nerve tissue, 32 cases (27 neuroblastoma and 5 normal peripheral nerve tissue)/64 cores (MC642), derived from retroperitoneal (38 cores), pelvic cavity (2 cores), mediastinum (6 cores), and adrenal (8 cores) disease sites were stained for each antibody. Normal and neuroblastoma cores were scored 0, 1, 2, 3 according to staining intensity. Images were processed on NDP.view 2 software (Hamamatsu Photonics, Inc.), and magnified, as indicated, for presentation. Normal controls to confirm antibody staining were from de-identified surplus tissue from the hospital core lab. All tissues were used in conformity to established ethical policies. Commercially obtained tissue (US Biomax, Derwood, MD, USA) was obtained under HIPPA approved protocols. For RNASeq, informed consent from each participant or guardian was obtained by qualified investigators at local Children’s Oncology Group institutes or at the NCI and samples were collected after approval by the local Institution Review Boards. All samples were anonymized and the study deemed exempt by the Office of Human Subject Research, NIH.
Results
Using the data analysis pipeline described in Section “Materials and Methods,” and illustrated in Figure 1, we set the final parameter of Log FKPM in any vital organ at 1.0, and received a readout of 325 unique gene pairs; comprised of 26 unique genes for MYCN-A neuroblastoma and 528 unique gene pairs, comprised of 33 unique genes, for MYCN-NA neuroblastoma. In Table 1, we present the top scoring genes that comprised the pairs we identified for both disease types as a single list. We were surprised by the brevity of the list and experimented with loosening the stringency for normal gene expression. Our analysis pipeline first filters for FPKM ≤ 2 for our composite set of normal tissues and our composite set of brain transcripts. We then require FPKM ≤ 1 for a subset of vital organs (heart, liver, lung, kidney). From our previous published work, we know that choice of the normal tissue set used for differential gene expression scoring has a profound impact on analysis outcome (26). When we loosened the vital organ stringency to FPKM ≤ 2, three more genes arose as candidates including DLK1, classified as a “Good” target (Table 1). Using this approach, investigators can set different stringency limits for normal tissues, and tumor-derived expression data sets re-filtered. One can also alternate between different sets of normal antigen expression profiles depending upon specific tissues that may be of concern.

Figure 1. Bioinformatic pipeline for identifying overexpressed gene pairs. Identification of tandem pairs of overexpressed genes (versus normal) began with building a file of transcripts known to be expressed in our cancers of interest, being advanced stage neuroblastoma (NB) with (plus) or without (minus) MYCN gene amplification (1), followed by scoring the individual genes represented in the cell surface transcriptome database, using 9 databases or filters (here, we required a positive hit in at least seven of the sources and a greater than twofold expression of the cancer-associated transcript over normal) (2), these hits were then analyzed by Hotelling–Lawley (4) to identify gene pairs expressed together, a Pearson co-efficient of tumor only to check for dependence of the transcripts in the tumor (5), and then filter sets applied (6). These variables are adjustable in the online database and allow thresholds to be set for gene expression (FPKM medians), expression of the gene pairs versus normal tissues, versus brain, and versus a subset of vital organs (liver, heart, lung, kidney), and gave a final readout of 26 unique genes for NB plus and 33 unique genes for NB minus (7). See Orentas et al. (13, 26).
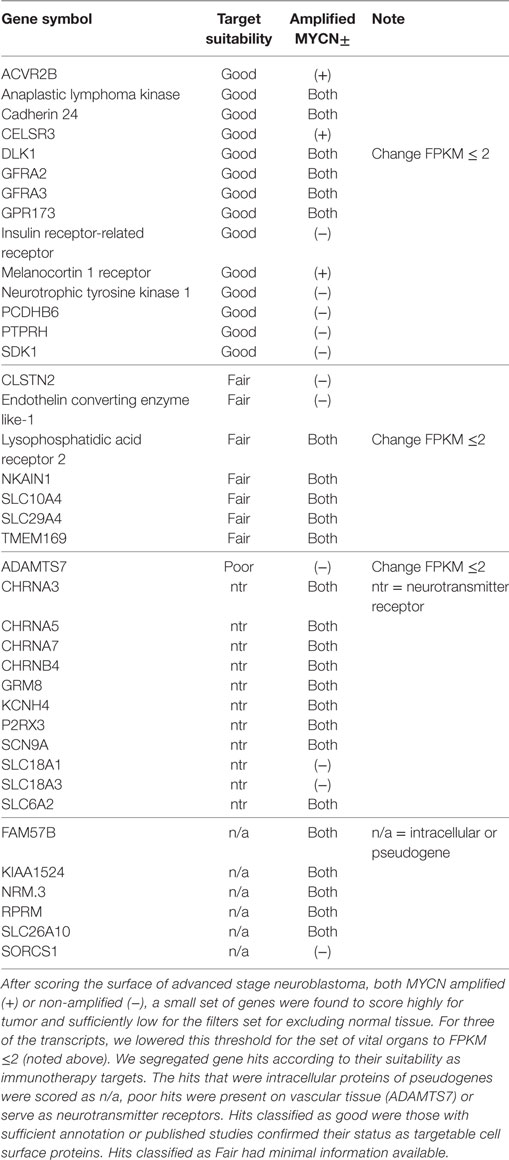
Table 1. Individual analysis of over-expressed genes scored for pairwise expression.
In examining the individual genes that comprise our gene pairs, Table 1, it is apparent that our data analysis pipeline can be further refined. Six genes were classified as “n/a” meaning that we will not consider them as targets as they are either pseudogenes or intracellular transcripts [FAM57B, KIAA1524, NRM, RPRM, SLC26A10, SORCS1 (27, 28–30, 31)]. As our pipeline still yielded these hits, we can be sure that there is more work to do in improving the annotation present in current databases and in our ability to filter them for non-plasma membrane expressed transcripts.
Targets with High Risk Profiles
Transcripts encoding genes in the “Poor” category included ADAMTS7 (32), and a series of neurotransmitter receptor proteins. Cholinergic receptors (CHRNA3, CHRNA5, CHRNA7, CHRNB4) are particularly risky. Our assumption is that they would generate severe systemic toxicity, perhaps recreating myasthenia gravis-like symptoms. Both GRM8 (glutamate receptor, metabotropic 8) and KCNH4 (potassium channel), voltage-gated subfamily H, member 4 (brain restricted) are CNS neurotransmitters. SCN9A (sodium voltage-gated channel alpha subunit 9) and P2RX3 (purogenic receptor P2X, ligand-gated ion channel 3) are involved in nociception signaling. SLC18A1 and SLC18A3 are vesicular monoamine and vesicular acetylcholine transporters, respectively. SLC6A2 is responsible for re-uptake of norepinephrine in presynaptic nerve terminals and is thus also too risky.
Targets with Moderate Risk Profiles
The group described as “Fair” targets in Table 1 each have some risk. CLSTN2 (calsyntenin 2, CDHR13, cadherin-related 13) is located in postsynaptic membrane of excitatory, primarily GABA-ergic, CNS synapses, although it appears to be developmentally regulated (33, 34). Endothelin converting enzyme like-1 is developmentally important for joint formation and innervation in humans (35). Although it is associated with the cell surface, it is also present in the endoplasmic reticulum, and its expression pattern (neuron-specific) is of concern (36). Lysophosphatidic acid receptor 2 is a G-coupled protein receptor (GPCR) that has been associated with both cancer and lung fibrosis, yet, as a class, these molecules are difficult to target and may be expressed on some normal lung endothelial cells (37, 38). NKAIN1 (Na+/K+ transporting ATPase interacting 1) has been proposed as a prostate cancer marker, but also has some restricted neuronal expression (39, 40). SLC29A4 is a plasma membrane monoamine transporter that functions a serotonin uptake transporter and, therefore, is likely associated with neuronal signaling (41). SLC10A4 is in the Na+/bile acid co-transporter family, little is known of its function, and some CNS expression by immunohisotchemical analysis has been demonstrated (42, 43). TMEM169 has little information associated with it, and thus risk cannot be evaluated.
Targets with Favorable Risk Profiles
Transcripts encoding 14 plasma membrane-associated proteins were categorized as being favorable targets for immunotherapy. We consider them here by functional groups. The first group of favorable targets are growth factor receptors known to be overexpressed on either tumors or stem cells of the neuronal and hematopoietic lineage. These are activin A receptor, type IIB (ACVR2B), glial cell-derived neurotropic factor (GDNF) family receptor alpha-2 (GFRA2, which binds neurturin), and GFRA3 (which binds artemin). ACVR2B is a transmembrane serine/threonine kinase signaling molecule in the TGF-beta signaling pathway family, which binds to activin and myostatin (44). ACVR2B is strongly expressed in renal childhood neoplasms and could be readily targeted (45). Both GFRA2 and GFRA3 are cell surface GPI-linked proteins that bind neurotrophins, forming complexes with the RET tyrosine kinase to initiate ligand-dependent signaling. Gfra1, Gfra2, and Gfra3, which signal through the RET tyrosine kinase in the presence of GDNF, neurturin, and artemin, respectively, also play a role in hematopoietic stem cell function, conferring survival signals through the Bcl2 family of proteins (46). Finally, neurotrophic tyrosine kinase 1 (NTRK1, TrkA), is a well-known neuroblastoma antigen that binds nerve growth factor (47), and whose expression has been associated with a number of human cancers, often being discovered as an oncogenic fusion protein (48).
Another favorable hit that may be considered a growth factor receptor is melanocortin 1 receptor (MC1R). However, as this receptor is also a G-coupled receptor that also crosses the membrane seven times, we classify it with the other G-coupled receptor found to be a favorable target, GPR173 (SREB3, super conserved receptor in brain or G-protein coupled receptor 173). M1CR binds alpha-melanin stimulating hormone released by sun-damaged keratinocytes, thus promoting eumelanin production in individuals with a non-mutated MC1R. MC1R also mediates anti-inflammatory properties as well and may promote anti-melanoma immunity (49). Little is known about GPCR173 other than it is expressed at the RNA level in brain and has also been called SREB1 (super conserved receptor in brain, based on low variation between species) (50). By virtue of ligand binding by MC1R and the unique extracellular domains of GPCR173, these both may serve as good targets (51).
Four of the favorable hits are adhesion receptors. CDH24 (type 2) is a cell surface protein expressing five extracellular repeat motifs and has the ability to interact with both beta-catenin, alpha-catenin, and p120 catenin (52). Frameshift mutations have been described for CHD24 in some cancers and may be associated with carcinogenesis (53). CELSR3 (cadherin, EGF LAG seven-pass G-type receptor, 3) is a non-classical cadherin in the flamingo family that does not interact with the catenins. This unique class has seven EGF-like repeats, nine cadherin domains, and two laminin repeats in their extracellular domain and seven transmembrane domains. CELSR3 interacts with molecules that govern cell motility during carcinogenesis through the WNT/planar cell polarity signaling pathway (54). CELSR3 is preferentially upregulated in pancreatic and hepatic cancer stellate cells (55) and has been described to guide axonal migration in the CNS (56). Of concern as a single target is its expression on the amacrine cells of the eye, although in depth studies are limited to zebrafish (57). Protocadherin B6 (PCDHB6) is also a neural adhesion molecule. As the name implies, this molecule family is thought to function in cell adhesion. Interestingly, other PCDH family members were demonstrated to stabilize RET signaling in neuroblastoma, indicating that PCDHs play a role in tumor cell signaling and activation (58). SDK1 (sidekick cell adhesion molecule) is a synaptic cell adhesion molecule in the Ig superfamily. SDK1 has been shown to be overexpressed in asbestos-induced lung adenocarcinoma (59, 60), Sdk1 is also expressed in retinal synaptic sites and may be dangerous for this reason. But for our dual targeting approach may prove to be a good hit.
The final category of favorable targets are transmembrane proteins known to regulate cell signaling, adhesion, or activation. These are ALK, a well-studied neuroblastoma antigen, DLK1 (Delta-like 1 homolog), a transmembrane receptor with multiple EGF repeats that regulates adipogenesis and osteogenesis, protein tyrosine phosphatase, receptor type H (PTPRH) or stomach cancer associated protein tyrosine phosphatase 1 (SAP1), and insulin receptor-related receptor (INSRR). ALK has been the focus of intensive study in neuroblastoma for many years [recently reviewed by Mossé (61)] and is an excellent immunotherapy target. DLK1, a non-canonical notch ligand, has multiple EGF domains and is proposed to govern cell growth and differentiation. Elevated expression of DLK1 was reported in neuroblastoma from patients with poor outcome (62) and induction of DLK1 expression in lung cancer activated both notch-dependent signaling and upregulated matrix metalloproteinase MMP9, which increased cellular invasive potential (63). PTPRH (SAP1) has a single intracellular catalytic domain, multiple extracellular fibronectin-type III repeats, and is known to be overexpressed in human cancer (64). In a recent study, association of SAP1 with CEACAM20 (carcinoembryonic antigen related cell adhesion molecule-20) was found to regulate the inflammatory status of gut epithelial cells by regulating the phosphorylation of CEACAM20 by c-Src, with the subsequent association with syk (spleen tyrosine kinase) signaling and induction of IL-8 production (65). INSRR functions as an alkali sensor molecule (66). Upon alterations in pH, extracellular domains rearrange and induce the autophosphorylation of internal kinase domains, thus initiating intracellular signaling. One can speculate that the kinase domain is part of the disease process or that advanced disease selects for an environment in which this gene is upregulated.
All the high-quality hits may confer upon cancer cells activation signals that may in some sense be oncogenic drivers. Many interact with the RET kinase or a pathway associated with RET signaling. The targeting of these growth-promoting proteins through immunotherapy may help prevent tumor escape by denying important growth promoting signals to cells, if escape mutants downregulate their expression.
Pairwise Associations
Having considered the quality of individual hits, we now interrogate the true power of our analysis, and that is examining pairwise associations of targets on the cancer cell surface. Table 2 lists the pairwise hits for both MYCN-A and MYCN-NA neuroblastoma (NB). What becomes immediately evident is that the MYCN-NA tumor side of the table has much higher F values and correspondingly lower p values. The highest F value for MYCN-A NB does not make the top 20 of our curated list for MYCN-NA tumors. This may indicate that without MYCN amplification more total mutations (and thus greater deviation from a normal gene expression profile) are required for MYCN-NA tumor to progress to advanced disease. For MYCN-amplified tumors, the oncogene supplies a strong internal driver already. In this case, the tumor may either arise more rapidly and have a lesser opportunity to accumulate random mutations, or may simply require less total mutations to progress to advanced disease.
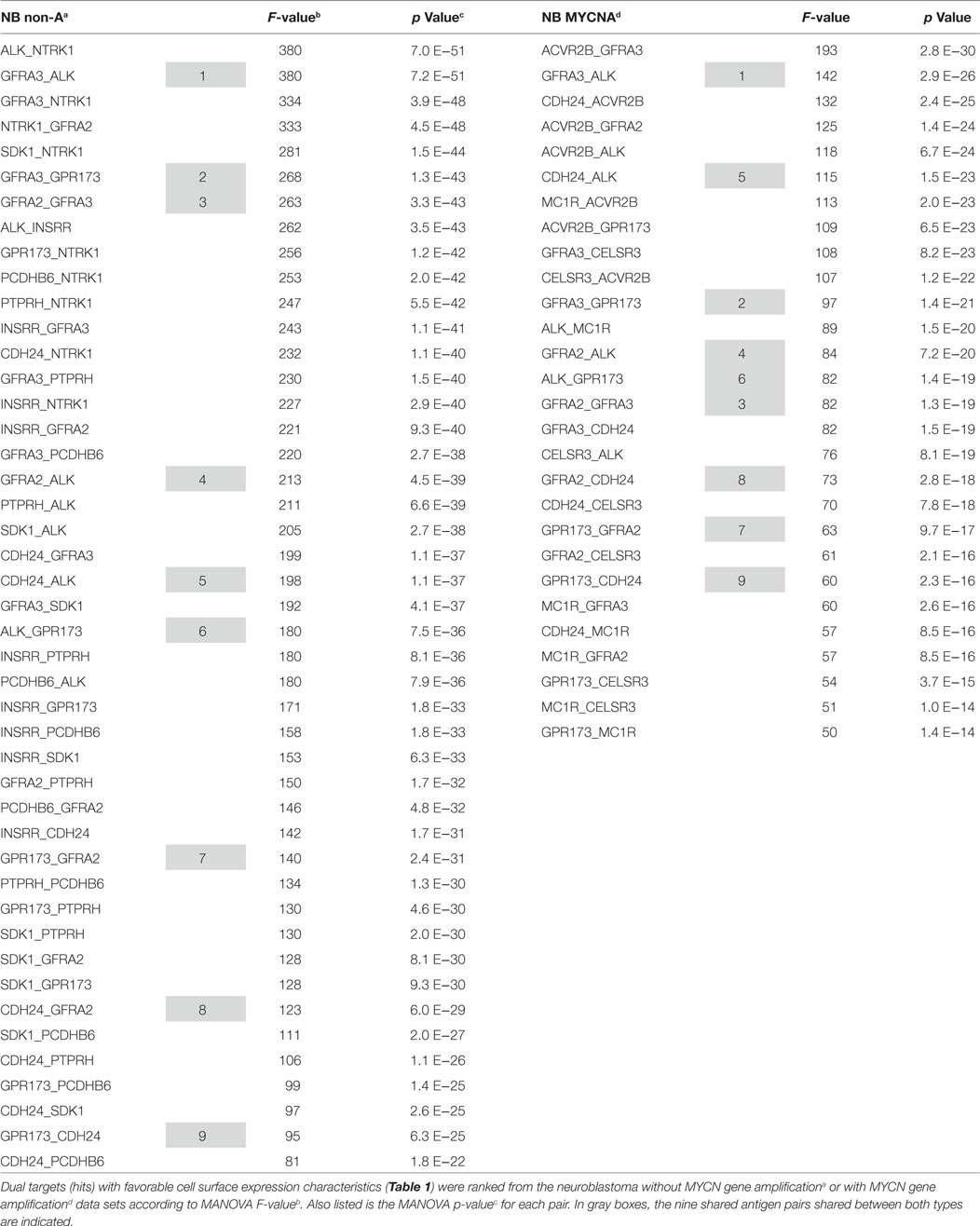
Table 2. Pairwise ranking of dual targets.
Of the 28 MYCN-A NB pairs, and the 45 MYCN-NA pairs listed in Table 2, nine are shared. As these common pairs (dyads) are upregulated together this may indicate that for both types of disease these cell surface proteins are reflective of a common cell of origin, or possibly a common disease process. ALK, GPCR173, GFRA2, GFRA3, and CDH24 are associated with each other in pairwise association 12 times in the top 25 dyads for MYCN-NA NB and 10 times for MYCN-A NB. The potential interaction of each of these molecules in RET signaling (except for ALK), indicates that this “pair” may be a linked activation complex in neuroblastoma. That these molecules are all involved in neuronal development and cellular response to migratory or chemotactic factors also is reflective of their neural crest origin. To further develop our bioinformatics analysis, we carried out pathological studies.
To examine protein expression, neuroblastoma tissue arrays were obtained and commercial antibodies that could be readily adapted to automated staining were used to probe the protein expression of ALK, DLK, NTRK1 (TrkA), GFRA2, GFRA3, GPR173, and CDH24, Figure 2. Each antibody was verified for binding activity and specificity by staining of normal tissue blocks prepared in the Department of Pathology at the Medical College of Wisconsin (see Materials and Methods). Each biopsy was then scored on a four-point scale (0, 1, 2, 3) for staining intensity. The results across all biopsies are summarized in Figure 3. A low-power image is provided to illustrate the scoring for 20 of the 54 individual cores, Figure S1 in Supplementary Material. Peripheral nerve was largely negative for ALK, DLK, GFRA2, GFRA3, GPR173, and somewhat positive for CDH24 and TrkA, Figure S2 in Supplementary Material. Staining of pathological tissues from in-house control blocks was used to confirm the staining activity of each antibody used in the neuroblastoma tissue arrays, Figure S3 in Supplementary Material. When the frequency of our bioinformatics hits for dual expression was explored in pathological sections, 6 dyads (pairwise over expressed antigens) were present in more than half of the array specimens tested. These were: (a) GPR173, GFRA3, or NTRK1 paired with CDH24, or (b) GFRA3 paired with NTRK1 or GPR173, or (c) GPR173 paired with NTRK1. These data illustrate the importance of verification of bioinformatics hits at the protein level. While we cannot conclude that these are a definitive picture of the neuroblastoma surfaceome, these data give us a place to start in designing new immunotherapeutic approaches to treat the disease.
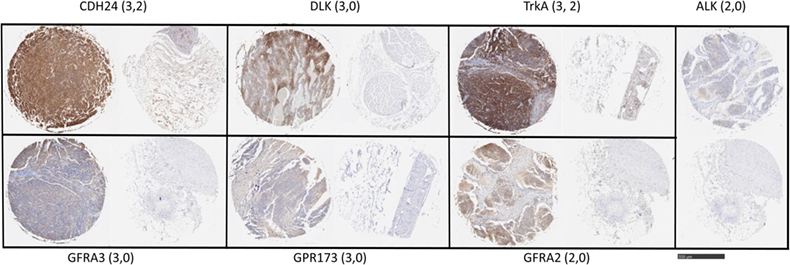
Figure 2. Target antigen expression in neuroblastoma and normal tissue arrays. Selected images from neuroblastoma tissue array were stained with the indicated antibodies. Shown are representative highest scores, in parentheses, for disease and normal (peripheral nerve) cores from the same staining array, respectively. Bar = 500 μm.
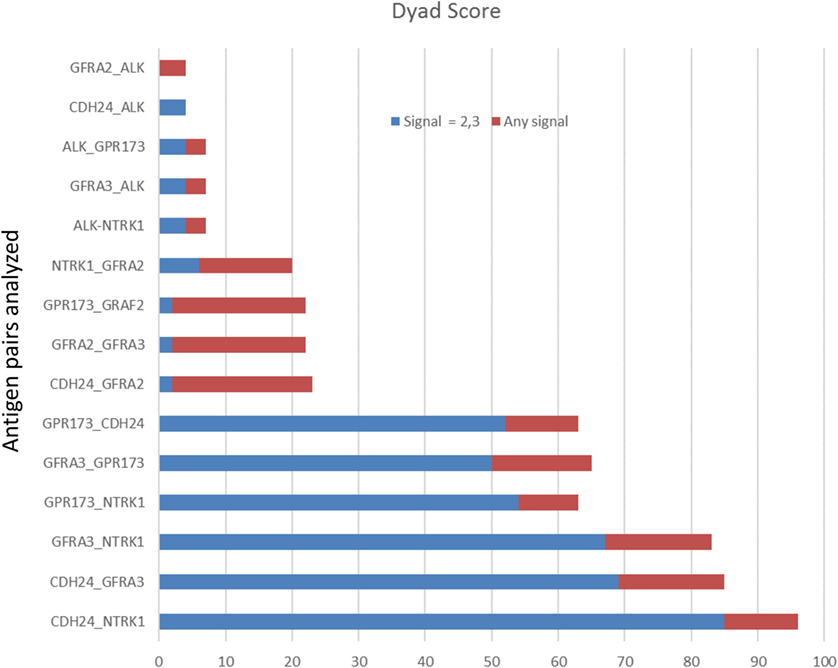
Figure 3. Target protein expression by staining intensity. Each core of the stained tissue arrays was scored as 0, 1, 2, or 3 for target protein expression. Dyad pairs identified by bioinformatics (y-axis) were placed in a scoring matrix wherein any signal above 0 (stacked red bar) or scored a 2 or 3 (blue stacked bar) was recorded and the percentage of biopsy scores expressing both antigens across each individually stained array counted. Data are presented as a percent of total specimens (n = 54) staining positive (Dyad score).
Discussion
In previous work to define transcripts overexpressed in pediatric solid tumors, we were struck by the difficulty in finding antigens that are overexpressed on tumor cells but not on normal tissue, and thus able to serve as therapeutic targets. Moreover, when creating a tumor target antigen list based on differential expression between normal and tumor tissue, the final target list can change depending on which normal tissues are included in the analysis (26). In our current bioinformatic approach, using the latest RNASeq data for neuroblastoma, we filtered for both normal tissue in aggregate, and with a greater stringency filter for a set of tissues designated as essential. We included a filter for CNS expression alone, and then for a set of vital tissues (heart, lung, kidney, liver), Figure 1. This approach yields a gene set that differs from normal to a now measurable degree, but these hits are still not entirely unique for neuroblastoma. Given the need for safety using highly active cell-based immunotherapy, the investigator is faced with a need to be able to target the tumor surfaceome in a more sophisticated manner. One approach is to create a more complex CAR activation signal, for example, one that requires two specific “hits” for activation, Figure 4. The inverse could also be attempted by creating a CAR-T cell product that is negatively regulated by an antigen that is overexpressed on a vital normal tissue. For negative signals, both intracellular phosphatases such as CD45 and the intracellular signaling motifs of the checkpoint molecules PD-1 and CTLA-4 have been proposed (67, 68). Thus, a CAR specific for tumor-associated antigen “A,” to which an active intracellular signaling domain is spliced, will react to any cell that expresses “A,” unless antigen “B” is present, which is targeted by a second CAR expressed by the same T cell, yet to which inhibitory signaling domains are linked. “Split CAR” approaches have also been proposed, wherein binding to “A” delivers only a partial signal, and full activation to the point of target cell cytolysis requires a second binding event, “C,” that is also present on the tumor cell. This process can also be approached pharmacologically, wherein a CAR specific for “A” does not contain any T cell activation signals, but does contain a small-molecule-binding domain that can induce protein–protein association. In the same lentiviral vector, a second transcript, “D,” is expressed that would contain a complementary dimerization domain linked to the intracellular activation motifs. This small molecule dimerizer thus becomes an “on switch” and is required for full CAR activity. Chemical inducers of dimerization (CID domains) have been widely developed using the rapamycin/FK506 and FK506-binding protein system and have been recently reviewed by Wu et al. (69). However, none of these options has yet been put into clinical practice.
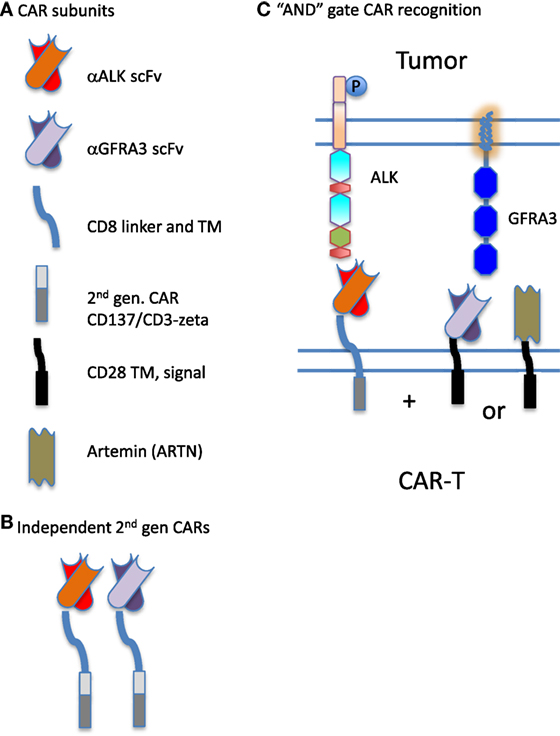
Figure 4. Synthetic biology approach to creating chimeric antigen receptor (CAR) “AND” gates. (A) CARs are composed of distinct combinatorial units, specifically: individual antigen-binding motifs [scFv specific for anaplastic lymphoma kinase (ALK) or GFRA3], linking domains (CD8 linker and TM) and intracellular signaling domains (illustrated are components of a second-generation CAR derived from CD137 and CD3-zeta, and an incomplete intracellular signaling domain composed of only CD28 transmembrane and signaling sequences). In addition to scFv, ligands for overexpressed receptors on the tumor surface such as Artemin, the ligand for GFRA3, can be used in CAR construction. (B) Two antigens on a tumor cell could be targeted by expressing two complete and independent CARs on the T cell surface, creating a logical “OR” gate for antitumor targeting. Either construct can fully activate the CAR-expressing lymphocyte (CAR-T) expressing it. (C) CAR components can also be used to split activation signals and thus require two binding interactions to fully activate the CAR-expressing lymphocyte, creating a logical “AND” gate. Here, the scFv specific for ALK expresses only one intracellular signaling domain. Complete activation of the T cell requires the binding of a second CAR. The second portion of the “AND” gate may be contributed by either an scFv specific for GFRA3 or the natural ligand of the tumor-expressed receptor, artemin (ARTN).
Although we set high filters for normal gene expression, it is apparent that the targets we have identified can be segregated into those with higher and lower risk, Table 1. Risky targets appear to reflect the neuroblastoma source tissue, which is of neural crest origin, and includes subunits of the nicotinic acetylcholine receptor (nAchR). nAchR is composed of five transmembrane proteins and serves as a primary ionotropic receptor for muscle contraction in the neuromuscular junction and is composed of different subunits in the CNS versus ganglia. Each member of the channel has four transmembrane domains with the N and C termini facing outwards into the extracellular space. The ability to target this receptor by the immune system is exemplified by myasthenia gravis, where antibodies block the ability of acetylcholine to bind to the receptor. On chromaffin cells the nAchR is formed by CHRNA3 and CHRNB4 (70), and these are the very receptors we find overexpressed in our gene expression analysis, attesting to the neural crest derivation of both chromaffin cells and neuroblastoma.
To develop a bioinformatic approach for advanced CAR-based therapy, we used gene expression data to identify targets overexpressed in tandem and scored as a pair against their expression in normal tissue. The identification of ALK was hardly surprising given its established association with neuroblastoma (61). Of interest is the discovery of GFRA2 and GFRA3 as overexpressed antigens as well. Both proteins signal in association with RET. In 2014, Cazes et al. reported that ALK triggers RET upregulation in mouse sympathetic ganglia at birth, and in human neuroblastoma (71). These authors proposed using a combination of crizotinib and vandetinib, inhibitors of ALK and RET, respectively, as a potential therapeutic approach. A phosphoproteomic analysis of neuroblastoma also found RET to be overexpressed and activated in neuroblastoma (72). Lambertz et al. recently described ALK-driven upregulation of MAPK regulators and RET in neuroblastoma, and also the RET-driven upregulation of cholinergic lineage markers (73). Once again, combined blockade of the ALK and RET pathways was proposed. Serial analysis of gene expression-analysis in a neuroblastoma cell line revealed another of our identified targets, DLK-1, to be highly upregulated. The involvement of the delta-notch signaling pathway in neuroblastoma has also been described (74). Interestingly, the combination of retinoic acid and the knockdown of DLK1 was found to induce the differentiation of neuroblastoma cells in vitro better than either intervention on its own, highlighting the tumorigenicity of DLK1 in neuroblastoma (75).
The targets identified in our tandem gene discovery process appear to be valid targets in their own right. However, using strategies that require two different antigens on the tumor cell surface to activate CAR-T adds an added layer of safety and specificity, and may open the door to new therapeutic approaches for other solid cancers. Another important finding of our study is that many of the cell surface proteins identified, comprising the neuroblastoma surfaceome, encode functional receptors. The ligands for these receptors can also be used as loci of CAR-T activation, as has been described for the IL-13-based zetakine approach (76) (Figure 4). As with any RNA-based bioinformatic approach, our findings will require extensive confirmation with protein-detection based analysis. Histochemical analysis of neuroblastoma tissue cores revealed that only a few of our identified tandem antigenic pairs were expressed on the majority of specimens. The lack of strong ALK and GFRA2 expression at the protein level highlights the need to validate analysis at the RNA level with appropriate proteomic characterization of the tumor type being studied. The proteins pairs identified in Figure 3 as having high expression by antigen staining (CDH24, NTRK1, GFRA3, and GPR173) will now serve as the starting point for the development of bispecific CAR-T approaches to neuroblastoma therapy. Our plan is to develop CARs that functions as a logical “and” gates, requiring the successful engagement of two binding moieties to initiate immune effector cell function.
Author Contributions
Studies were planned and designed by RO and JK, bioinformatic pipelines, and statistical analysis of gene expression was performed by SS, XW, JH, and JW. Pathology studies were designed and carried out by JJ and CD, JJ also responsible for normal tissue blocks, automated processing, and scoring. Final data were analyzed by RO and JK, who also wrote the manuscript.
Conflict of Interest Statement
RO is a full-time employee of Lentigen Technology, Inc., a Miltenyi Biotec Company. All other Authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Young Song for sample and data processing for RNASeq. This research was supported by the Intramural Research Program of the NIH, CCR, NIH. While at the NIH, RO was supported in part by a grant from Solving Kids’ Cancer Foundation, New York, NY, USA.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fonc.2017.00173/full#supplementary-material.
Figure S1. Low power images of neuroblastoma tissue cores. Twenty out of a total of 54 cores are shown. Each core is from retroperitoneal disease with the exception of the two most rightward cores on the second (lower) row, which were obtained from mediastinal disease. Each panel shows immunohistochemical staining for anaplastic lymphoma kinase, CDH24, DLK, GFRA2, GFRA3, GPR173, and TRKA as indicated above each panel. In the lower right of each core is the score assigned by pathological examination.
Figure S2. Low power images of peripheral nerve and high power images for TrkA staining. Six of the 10 normal peripheral nerve tissue cores used as negative control are shown. As listed on the left, each of the six cores was stained with the indicated antibody. Staining intensity score is indicated in the lower right corner of each section. In some cores, no score could be determined due to loss of tissue (ND). In the final two panels, high power images illustrate the difference between moderately strong (score of 2) and negative (score of 0) tissue. Bars indicate magnification for low power, 5 mm, and high power, 100 μm, respectively.
Figure S3. Control tissue block staining to validate staining. As described in Section “Materials and Methods,” (1) anaplastic lymphoma kinase-1-stained control tissue blocks with cerebellum, pancreas, tonsil, and lymphoma (ALCL) were utilized (CBlock1). Representative fields for ALCL (1a, strong positive) and tonsil (1b, negative) are shown. In all figures, the solid bar denotes scale. Staining from the same block shows a bar for the first field only (unless a higher power image is shown). (2) CDH24 was tested on CBlock1 with added bowel, uterus, cerebellum, and HCC. Ganglion cells/nerve cells of the bowel stained positive, endometrium surface epithelium cytoplasmic positive, and HCC was strongly positive. ALCL (2a), tonsil (2b), and uterus with strong staining of the basal layer (2c) are shown. (3) DLK1 was tested on CBlock1 with added pancreas and placenta. ALCL (3a), tonsil (3b), and placenta (3c, strong positive) are shown. Islets, ductal epithelium, and neutrophils also stained positive. (4) GFRA2 was tested on CBlock1 with added lung and liver. Staining was positive for macrophages and sinusoids in liver, tonsil leukocytes (4b), ALCL was weakly positive (4a), and lung unremarkable. (5) GFRA3 was tested on CBlock1 with added lung and liver. Increased expression in pancreatic ductal epithelium and islets, strong lymphatic vessel staining, and positive alveolar macrophages were seen. ALCL (5a) tonsil (5b), and HCC (5c) are shown. (6) GPR173 was tested on CBlock1 with added lung, liver, and skin. Positive staining was seen on red cells and lymphocytes. ALCL (6a), tonsil (6b), and HCC (6c) are shown. (7) TrkA was tested on CBlock1 and liver (7a). Positive staining was seen in tonsil reticular dendritic network (7b), pancreas islets, sinusoids of HCC, and some hepatocytes were positive, as was liver artery (7c).
References
1. Hoffman L, Gore L. Blinatumomab, a bi-specific anti-CD19/CD3 BiTE® antibody for the treatment of acute lymphoblastic leukemia: perspectives and current pediatric applications. Front Oncol (2014) 4:63. doi:10.3389/fonc.2014.00063
2. Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol (2015) 16:57–66. doi:10.1016/S1470-2045(14)71170-2
3. Robak T. GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies. Curr Opin Investig Drugs (2009) 10:588–96.
4. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi:10.1200/JCO.2014.56.2025
5. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
6. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi:10.1016/S0140-6736(14)61403-3
7. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403:503–11. doi:10.1038/35000501
8. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature (2012) 488:43–8. doi:10.1038/nature11213
9. Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science (2007) 318:1108–13. doi:10.1126/science.1145720
10. Son CG, Bilke S, Davis S, Greer BT, Wei JS, Whiteford CC, et al. Database of mRNA gene expression profiles of multiple human organs. Genome Res (2005) 15:443–50. doi:10.1101/gr.3124505
11. Hastie C, Saxton M, Akpan A, Cramer R, Masters JR, Naaby-Hansen S. Combined affinity labelling and mass spectrometry analysis of differential cell surface protein expression in normal and prostate cancer cells. Oncogene (2005) 24:5905–13. doi:10.1038/sj.onc.1208747
12. Schiess R, Mueller LN, Schmidt A, Mueller M, Wollscheid B, Aebersold R. Analysis of cell surface proteome changes via label-free, quantitative mass spectrometry. Mol Cell Proteomics (2009) 8:624–38. doi:10.1074/mcp.M800172-MCP200
13. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol (2012) 2:194. doi:10.3389/fonc.2012.00194
14. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi:10.1126/science.1235122
15. Ho W-L, Hsu W-M, Huang M-C, Kadomatsu K, Nakagawara A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblasotoma. J Hematol Oncol (2016) 9:100. doi:10.1186/s13045-016-0334-6
16. Heczey A, Louis CU, Salvado B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patietns with neuroblastoma. Mol Ther (2017). doi:10.1016/j.ymthe.2017.05.012
17. Perez Horta Z, Goldberg JL, Sondel PM. Anti-GD2 mAbs and next-generation mAB-based agents for cancer therapy. Immunotherapy (2016) 8:1097–117. doi:10.2217/imt-2016-0021
18. Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O’Donoghue SI, Schneider R, et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database (Oxford) (2014) 2014:bau012. doi:10.1093/database/bau012
19. Li J, Newberg JY, Uhlén M, Lundberg E, Murphy RF. Automated analysis and reannotation of subcellular locations in confocal images from the human protein atlas. PLoS One (2012) 7:e50514. doi:10.1371/journal.pone.0050514
20. Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res (2007) 35:W585–7. doi:10.1093/nar/gkm259
21. Briesemeister S, Rahnenfuhrer J, Kohlbacher O. Going from where to why-interpretable prediction of protein subcellular localization. Bioinformatics (2010) 26:1232–8. doi:10.1093/bioinformatics/btq115
22. Briesemeister S, Rahnenfuhrer J, Kohlbacher O. YLoc-an interpretable web server for predicting subcellular localization. Nucleic Acids Res (2010) 38:W497–502. doi:10.1093/nar/gkq477
23. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. LIMMA powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res (2015) 43:e47. doi:10.1093/nar/gkv007
24. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol (2004) 3:3. doi:10.2202/1544-6115.1027
25. Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol (2014) 15:R29. doi:10.1186/gb-2014-15-2-r29
26. Orentas RJ, Nordlund J, He J, Sindiri S, Mackall C, Fry TJ, et al. Bioinformatic description of immunotherapy targets for pediatric T-cell leukemia and the impact of normal gene sets used for comparison. Front Oncol (2014) 4:134. doi:10.3389/fonc.2014.00134
27. Yamashita-Sugahara Y, Tokuzawa Y, Nakachi Y, Kanesaki-Yatsuka Y, Matsumoto M, Mizuno Y, et al. Fam57b (family with sequence similarity 57, member B), a novel peroxisome proliferator-activated receptor γ target gene that regulates adipogenesis through ceramide synthesis. J Biol Chem (2013) 288:4522–37. doi:10.1074/jbc.M112.440792
28. Lei N, Peng B, Zhang J-Y. CIP2A regulates cell proliferation via the AKT signaling pathway in human lung cancer. Oncol Rep (2014) 32:1689–94. doi:10.3892/or.2014.3375
29. Saavedra K, Valbuena J, Olivares W, Marchant MJ, Rodríguez A, Torres-Estay V, et al. Loss of expression of reprimo, a p53-induced cell cycle arrest gene, correlates with invasive stage of tumor progression and p73 expression in gastric cancer. PLoS One (2015) 10:e0125834. doi:10.1371/journal.pone.0125834
30. Sindić A, Chang MH, Mount DB, Romero MF. Renal physiology of SLC26 anion exchangers. Curr Opin Nephrol Hypertens (2007) 16:484–90. doi:10.1097/MNH.0b013e3282e7d7d0
31. Kebede MA, Oler AT, Gregg T, Balloon AJ, Johnson A, Mitok K, et al. SORCS1 is necessary for normal insulin secretory granule biogenesis in metabolically stressed β cells. J Clin Invest (2014) 124:4240–56. doi:10.1172/JCI74072
32. Pu X, Xiao Q, Kiechl S, Chan K, Ng FL, Gor S, et al. ADAMTS7 cleavage and vascular smooth muscle cell migration is affected by a coronary-artery-disease-associated variant. Am J Hum Genet (2013) 92:366–74. doi:10.1016/j.ajhg.2013.01.012
33. Hintsch G, Zurlinden A, Meskenaite V, Steuble M, Fink-Widmer K, Kinter J, et al. The calsyntenins – a family of postsynaptic membrane proteins with distinct neuronal expression patterns. Mol Cell Neurosci (2002) 21:393–409. doi:10.1006/mcne.2002.1181
34. Ortiz-Medina H, Emond MR, Jontes JD. Zebrafish calsyntenins mediate homophilic adhesion through their amino-terminal cadherin repeats. Neuroscience (2015) 286:87–96. doi:10.1016/j.neuroscience.2014.11.030
35. Dieterich K, Quijano-Roy S, Monnier N, Zhou J, Fauré J, Smirnow DA, et al. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum Mol Genet (2013) 22:1483–92. doi:10.1093/hmg/dds514
36. Nagata K, Kiryu-Seo S, Tamada H, Okuyama-Uchimura F, Kiyama H, Saido TC. ECEL1 mutation implicates impaired axonal arborization of motor nerves in the pathogenesis of distal arthrogryposis. Acta Neuropathol (2016) 132:111–26. doi:10.1007/s00401-016-1554-0
37. Lou L, Chen YX, Jin L, Li X, Tao X, Zhu J, et al. Enhancement of invasion of hepatocellular carcinoma cells through lysophosphatidic acid receptor. J Int Med Res (2013) 41:55–63. doi:10.1177/0300060512474124
38. Ren Y, Guo L, Tang X, Apparsundaram S, Kitson C, Deguzman J, et al. Comparing the differential effects of LPA on the barrier function of human pulmonary endothelial cells. Microvasc Res (2013) 85:59–67. doi:10.1016/j.mvr.2012.10.004
39. Gorokhova S, Bibert S, Geering K, Heintz N. A novel family of transmembrane proteins interacting with beta subunits of the Na,K-ATPase. Hum Mol Genet (2007) 16:2394–410. doi:10.1093/hmg/ddm167
40. Leyten GH, Hessels D, Smit FP, Jannink SA, de Jong H, Melchers WJ, et al. Identification of a candidate gene panel for the early diagnosis of prostate cancer. Clin Cancer Res (2015) 21:3061–70. doi:10.1158/1078-0432.CCR-14-3334
41. Adamsen D, Ramaekers V, Ho HTB, Britschgi C, Rufenacht V, Meili D, et al. Autism spectrum disorder associated with low serotonin in CSF and mutations in the SLC29A4 plasma membrane monoamine transporter (PMAT) gene. Mol Autism (2014) 5:43. doi:10.1186/2040-2392-5-43
42. Abe T, Kanemitu Y, Nakasone M, Kawahata I, Yamakuni T, Nakajima A, et al. SLC10A4 is a protease-activated transporter that transports bile acids. J Biochem (2013) 154:93–101. doi:10.1093/jb/mvt031
43. Popova SN, Alafuzoff I. Distribution of SLC10A4, a synaptic vesicle protein in the human brain, and the association of this protein with Alzheimer’s disease-related neuronal degeneration. J Alzheimer Dis (2013) 37:603–10. doi:10.3233/JAD-130548
44. Han S. Crystal structure of activin receptor type IIB kinase domain. Vitam Horm (2011) 85:29–38. doi:10.1016/B978-0-12-385961-7.00002-0
45. Senanayake U, Das S, Vesely P, Alzoughbi W, Fröhlich LF, Chowdhury P, et al. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis (2012) 33:1014–21. doi:10.1093/carcin/bgs126
46. Fonseca-Pereira D, Arroz-Madeira S, Rodrigues-Campos M, Barbosa IAM, Domingues RG, Bento T, et al. The neurotrophic factor receptor RET drives haematopoietic stem cell survival and function. Nature (2014) 514:98. doi:10.1038/nature13498
47. Light JE, Koyama H, Minturn JE, Ho R, Simpson AM, Iyer R, et al. Clinical significance of NTRK family gene expression in neuroblastomas. Pediatr Blood Cancer (2012) 59:226–32. doi:10.1002/pbc.23343
48. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov (2015) 5:25–34. doi:10.1158/2159-8290.CD-14-0765
49. Nasti TH, Timares L. MC1R, eumelanin and pheomelanin: their role in determining the susceptibility to skin cancer. Photochem Photobiol (2015) 91:188–200. doi:10.1111/php.12335
50. Matsumoto M, Saito T, Takasaki J, Kamohara M, Sugimoto T, Kobayashi M, et al. An evolutionarily conserved G-protein coupled receptor family, SREB, expressed in the central nervous system. Biochem Biophys Res Commun (2000) 272:576–82. doi:10.1006/bbrc.2000.2829
51. Larco DO, Semsarzadeh NN, Cho-Clark M, Mani SK, Wu TJ. The novel actions of the metabolite GnRH-(1-5) are mediated by a G protein-coupled receptor. Front Endocrinol (2013) 4:83. doi:10.3389/fendo.2013.00083
52. Katafiasz BJ, Nieman MT, Wheelock MJ, Johnson KR. Characterization of cadherin-24, a novel alternatively spliced type II cadherin. J Biol Chem (2003) 278:27513–9. doi:10.1074/jbc.M304119200
53. An CH, Je EM, Yoo NJ, Lee SH. Frameshift mutations of cadherin genes DCHS2, CDH10 and CDH24 genes in gastric and colorectal cancers with high microsatellite instability. Pathol Oncol Res (2015) 21:181–5. doi:10.1007/s12253-014-9804-8
54. Katoh M. WNT/PCP signaling pathway and human cancer (review). Oncol Rep (2005) 14:1583–8. doi:10.3892/or.14.6.1583
55. Erkan M, Weis N, Pan Z, Schwager C, Samkharadze T, Jiang XH, et al. Organ-, inflammation- and cancer specific transcriptional fingerprints of pancreatic and hepatic stellate cells. Mol Cancer (2010) 9:88. doi:10.1186/1476-4598-9-88
56. Chai G, Goffinet AM, Tissir F. Celsr3 and Fzd3 in axon guidance. Int J Biochem Cell Biol (2015) 64:11–4. doi:10.1016/j.biocel.2015.03.013
57. Lewis AA, Mahoney JT, Wilson N, Brockerhoff SE. Identification of amacrine subtypes that express the atypical cadherin celsr3. Exp Eye Res (2015) 130:51–7. doi:10.1016/j.exer.2014.12.003
58. Schalm SS, Ballif BA, Buchanan SM, Phillips GR, Maniatis T. Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proc Natl Acad Sci U S A (2010) 107:13894–9. doi:10.1073/pnas.1007182107
59. Mäki-Nevala S, Sarhadi VK, Knuuttila A, Scheinin I, Ellonen P, Lagström S, et al. Driver gene and novel mutations in asbestos-exposed lung adenocarcinoma and malignant mesothelioma detected by exome sequencing. Lung (2016) 194:125–35. doi:10.1007/s00408-015-9814-7
60. Cadby G, Mukherjee S, Musk AW, Reid A, Garlepp M, Dick I, et al. A genome-wide association study for malignant mesothelioma risk. Lung Cancer (2013) 82:1–8. doi:10.1016/j.lungcan.2013.04.018
61. Mossé YP. Anaplastic lymphoma kinase as a cancer target in pediatric malignancies. Clin Cancer Res (2016) 22:546–52. doi:10.1158/1078-0432.CCR-14-1100
62. Wei JS, Greer BT, Westermann F, Steinberg SM, Son CG, Chen QR, et al. Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res (2004) 64:6883–91. doi:10.1158/0008-5472.CAN-04-0695
63. Li L, Tan J, Zhang Y, Han N, Di X, Xiao T, et al. DLK1 promotes lung cancer cell invasion through upregulation of MMP9 expression depending on Notch signaling. PLoS One (2014) 9:e91509. doi:10.1371/journal.pone.0091509
64. Sato T, Soejima K, Arai E, Hamamoto J, Yasuda H, Arai D, et al. Prognostic implication of PTPRH hypomethylation in non-small cell lung cancer. Oncol Rep (2015) 34:1137–45. doi:10.3892/or.2015.4082
65. Murata Y, Kotani T, Supriatna Y, Kitamura Y, Imada S, Kawahara K, et al. Protein tyrosine phosphatase SAP-1 protects against colitis through regulation of CEACAM20 in the intestinal epithelium. Proc Natl Acad Sci U S A (2015) 112:E4264–71. doi:10.1073/pnas.1510167112
66. Deyev IE, Chachina NA, Shayahmetova DM, Serova OV, Petrenko AG. Mapping of alkali-sensing sites of the insulin receptor-related receptor. The role of L2 and fibronectin domains. Biochimie (2015) 111:1–9. doi:10.1016/j.biochi.2014.12.014
67. James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature (2012) 487:64–9. doi:10.1038/nature11220
68. Fedorov VD, Themeli M, Sadelain M. PD-1-and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med (2013) 5:215ra172. doi:10.1126/scitranslmed.3006597
69. Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science (2015) 350:aab4077. doi:10.1126/science.aab4077
70. Sala F, Nistri A, Criado M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) (2008) 192:203–12. doi:10.1111/j.1748-1716.2007.01804.x
71. Cazes A, Lopez-Delisle L, Tsarovina K, Pierre-Eugene C, De Preter K, Peuchmaur M, et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget (2014) 5:2688–702. doi:10.18632/oncotarget.1883
72. DeNardo BD, Holloway MP, Ji Q, Nguyen KT, Cheng Y, Valentine MB, et al. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PLoS One (2013) 8:e82513. doi:10.1371/journal.pone.0082513
73. Lambertz I, Kumps C, Claeys S, Lindner S, Beckers A, Janssens E, et al. Upregulation of MAPK negative feedback regulators and RET in mutant ALK neuroblastoma: implications for targeted treatment. Clin Cancer Res (2015) 21:3327–39. doi:10.1158/1078-0432.CCR-14-2024
74. van Limpt V, Chan A, Caron H, Sluis PV, Boon K, Hermus MC, et al. SAGE analysis of neuroblastoma reveals a high expression of the human homologue of the Drosophila Delta gene. Med Pediatr Oncol (2000) 35:554–8. doi:10.1002/1096-911X(20001201)35:6<554::AID-MPO13>3.0.CO;2-R
75. Kim Y. Effect of retinoic acid and delta-like 1 homologue (DLK1) on differentiation in neuroblastoma. Nutr Res Prac (2010) 4:276–82. doi:10.4162/nrp.2010.4.4.276
Keywords: neuroblastoma, gene expression profiling, chimeric antigen receptor-T cells, immunotherapy, GFRA3, GFRA2, GPR173, pediatric oncology
Citation: Orentas RJ, Sindiri S, Duris C, Wen X, He J, Wei JS, Jarzembowski J and Khan J (2017) Paired Expression Analysis of Tumor Cell Surface Antigens. Front. Oncol. 7:173. doi: 10.3389/fonc.2017.00173
Received: 18 May 2017; Accepted: 31 July 2017;
Published: 21 August 2017
Edited by:
Peter Bader, University Hospital Frankfurt, GermanyReviewed by:
Claudia Rossig, Universität Münster, GermanyJaume Mora, Hospital Sant Joan de Déu Barcelona, Spain
Copyright: © 2017 Orentas, Sindiri, Duris, Wen, He, Wei, Jarzembowski and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rimas J. Orentas, cmltYXMub3JlbnRhc0BsZW50aWdlbi5jb20=;
Javed Khan, a2hhbmphdkBtYWlsLm5paC5nb3Y=