 Daniel F. Pease
Daniel F. Pease Robert A. Kratzke
Robert A. Kratzke- Hematology-Oncology-Transplant, University of Minnesota, Minneapolis, MN, United States
The limited effectiveness of conventional therapy for malignant pleural mesothelioma demands innovative approaches to this difficult disease. Even with aggressive multimodality treatment of surgery, radiation, and/or chemotherapy, the median survival is only 1–2 years depending on stage and histology. Oncolytic viral therapy has emerged in the last several decades as a rapidly advancing field of immunotherapy studied in a wide spectrum of malignancies. Mesothelioma makes an ideal candidate for studying oncolysis given the frequently localized pattern of growth and pleural location providing access to direct intratumoral injection of virus. Therefore, despite being a relatively uncommon disease, the multitude of viral studies for mesothelioma can provide insight for applying such therapy to other malignancies. This article will begin with a review of the general principles of oncolytic therapy focusing on antitumor efficacy, tumor selectivity, and immune system activation. The second half of this review will detail results of preclinical models and human studies for oncolytic virotherapy in mesothelioma.
Introduction: Standard Therapy for Mesothelioma
Mesothelioma is an uncommon malignancy of the parietal and visceral mesothelium, with about 3,300 new cases each year in the United States (1). Malignant pleural mesothelioma (MPM) accounts for 90% of cases, as inhalation asbestos exposure is the major risk factor. Most of the remaining cases arise from the peritoneum, with only 1–2% of cases occurring in the pericardium or tunica vaginalis testis (2). In the western world, incidence peaked in the early 21st century and has since leveled off in the US, while in Europe estimates are for a decrease in new cases (3–5). This is the result of concerted efforts over the last several decades to reduce asbestos exposure. Unfortunately, less developed countries that are slower to control asbestos exposure likely will continue to see an increase in incidence because of the prolonged latency period of at least 20 years before development of mesothelioma (6, 7).
The typical presenting symptoms of MPM are non-specific and include shortness of breath, chest pain, and weight loss. Characteristic findings on chest imaging are pleural abnormalities such as a unilateral effusion, calcified plaques, thickening, or masses (8). Diagnosis often requires a full-thickness pleural biopsy via pleuroscopy or video-assisted thoracoscopy. Pleural fluid cytology, although more easily obtained, is usually not sufficient. Even with adequate tissue, the pathologic evaluation can be challenging as mesothelioma is not frequently seen in most centers and has a number of different subtypes—epithelioid, sarcomatoid, biphasic—that must be differentiated from reactive processes in the pleura (9).
The management of mesothelioma is to the extent possible multimodality strategy incorporating chemotherapy, surgery, and/or radiation. The initial step is evaluating whether the disease is surgically resectable, with the goal of macroscopic complete resection. The two main surgical techniques are extrapleural pneumonectomy (EPP) or the less radical pleurectomy/decortication (P/D). Comparisons of EPP and P/D are limited to observational studies, with the largest cohort showing a survival advantage to P/D (10). A recent meta-analysis found lower short-term mortality for P/D, although the 2-year survival was not significantly different than EPP (11). In the absence of randomized trial data, the surgical approach is determined on a patient-specific basis.
Chemotherapy for MPM is recommended for all patients undergoing active therapy, with either cisplatin or carboplatin combined with pemetrexed as the standard of care. In patients not eligible for surgical resection, cisplatin/pemetrexed was shown to have a superior median overall survival compared to cisplatin monotherapy of 12.1 vs. 9.3 months (12). Carboplatin is equally efficacious to cisplatin in combination with pemetrexed, providing an alternative for older patients and those with borderline renal function (13). The addition of bevacizumab to cisplatin/pemetrexed may offer further benefit, pushing median overall survival to 18 months (14). For those patients having surgical resection, chemotherapy is given either preoperatively or postoperatively with no studies comparing the two approaches.
The role of radiation therapy is less clear, with most studies evaluating its use in the postoperative setting to reduce the risk of local recurrence (15). Trimodality therapy of preoperative chemotherapy, surgical resection, and postoperative radiation has been evaluated in small studies with variable success (16, 17). More detailed reviews of standard therapy for pleural and peritoneal mesothelioma are available elsewhere (8, 18, 19).
Despite the application of multimodality therapy to MPM, most patients are candidates for only palliative chemotherapy and have a median overall survival of 1–2 years (20). These limitations in current treatment highlight the importance of investigational therapies that may improve the prognosis of an otherwise highly fatal disease. This review will focus on the use of oncolytic viral therapy for mesothelioma.
The Principles of Oncolytic Viral Therapy
Background
The fact that viruses may inflict damage not only in healthy human tissue but also in tumor cells was first observed in the early 21st century (21). The first formal studies utilizing viruses as anticancer therapy were performed in the 1950s and documented transient tumor response in a small number of patients (22–24). However, these intriguing early results were tempered by technical and methodological constraints, and investigation declined for the next few decades (25).
A renewed interest began in the late 20th century as scientific advances in virology and molecular genetics allowed greater viral manipulation and the potential for increased efficacy (26). Many viruses have now been studied in this context, including adenovirus, herpes simplex virus (HSV), vaccinia, measles virus, and others, applied against a number of malignancies such as glioma, breast, head and neck, and lung (27–30). In 2015, a genetically modified HSV type 1 (HSV-1) (T-VEC) became the first FDA-approved oncolytic viral therapy, for use against melanoma (31).
The ideal oncolytic viral therapy is based on three basic principles (32, 33): (1) antitumor efficacy, the ability to directly infect and lyse tumor cells; (2) tumor selectivity, to preferentially infect tumor cells and minimize toxicity of infection to healthy tissue; and perhaps most importantly, (3) stimulation of the immune system, to provoke an antitumor response that will amplify the viral-directed cell death and provide ongoing tumor cell killing (Figure 1). Genetically engineering viruses to optimize tumor cell toxicity and selectivity has found success, while attaining a sustained immunotherapeutic response has proven a more difficult task.
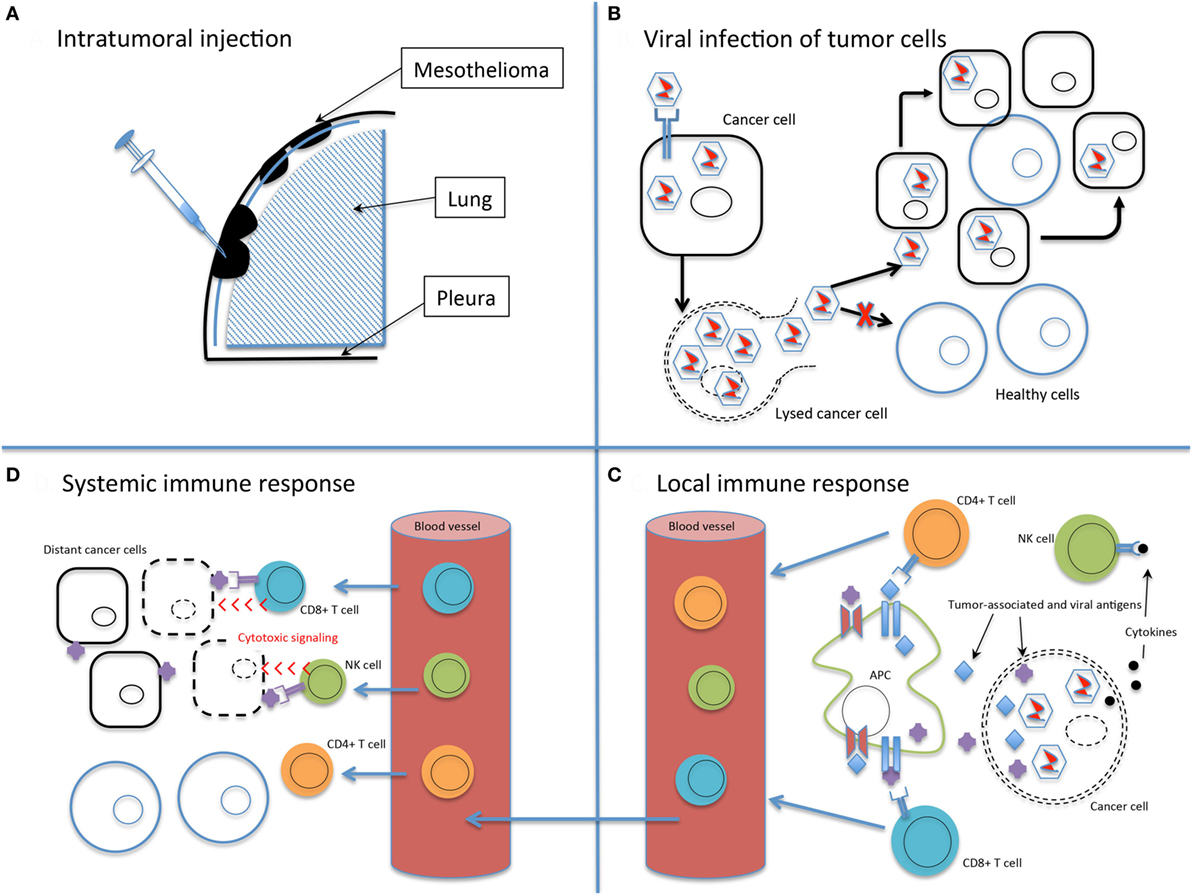
Figure 1. The basic principles of oncolytic virotherapy. (A) Administration is most commonly via direct intratumoral injection rather than systemic intravenous route to avoid viral inactivation in the bloodstream and minimize off-target infection. The pleural location of mesothelioma is particularly amenable to direct injection. (B) Viral infection of cancer cells, followed by replication, leads to cell lysis and dissemination of infection. The use of non-replicating viruses results in lysis to a lesser extent than replicating viruses. Acquired defects of the cancer cells and engineered modifications of the viral genome drive infection selectively toward cancer cells. (C) Viral infection and lysis exposes tumor-associated and viral antigens to the immune system. Antigen-presenting cells process these novel antigens via the major histocompatibility complex for presentation to CD4+ and CD8+ T cells. Cytokine release attracts NK cells. Local tumor cell death is augmented by the immune response. (D) Activated T and NK cells circulate throughout the body and recognize distant tumor cells that express the previously uncovered tumor-associated antigens. Note that the systemic immune response is not dependent on viral oncolysis.
Antitumor Efficacy
The concern for toxicity of wild-type viruses led to the first recombinant viruses being engineered as replication-incompetent strains, with the goal of delivering gene therapy but not necessarily propagation of viral infection (34). The development of techniques to enhance viral selectivity for tumor cells allowed a shift back toward using replication-competent oncolytic viruses. These virulent models allow the natural viral mechanisms to infect, replicate, and lyse tumor cells. As virions are released from lysed tumor cells, the infection spreads within the local tumor mass (33). This potentiates tumor cell killing compared to the initial input dose of viral particles and may lead to a more robust antitumor response from the immune system (32). Gene therapy with replication-incompetent viruses has a bystander effect that also may amplify cell death in the local tumor environment, although to a lesser extent than actively replicating viruses (25).
The mechanisms of tumor cell killing after viral infection are varied (25, 32). The most straightforward method is viral replication and shedding leading to eventual cell lysis. A second method of direct oncolysis is the production of cytotoxic viral proteins. Altering production of viral proteins is a target of genetic engineering to improve antitumor efficacy. For example, the adenovirus death protein (ADP) is produced during the normal adenovirus replication cycle to induce host cell death (35); a modified adenovirus designed to overexpress ADP has increased cytolytic activity in a mouse model of lung cancer (36).
A third method of antitumor efficacy is insertion of transgenes into the viral genome, so-called “armed” viruses. An early model of transgene insertion is the HSV thymidine kinase gene, which metabolizes ganciclovir into a toxic byproduct (37). Cells infected with a virus carrying this gene are rapidly lysed in the presence of ganciclovir. Both replication-deficient and replication-competent adenoviral vectors with the HSV thymidine kinase gene have been studied in humans against a number of tumors including mesothelioma, with encouraging results (38–41). Transgenes encoding cytokines such as IL-2 or TNFα to augment immune system response are also utilized (42, 43). With improved methods for oncolytic viruses to specifically target tumor cells, the use of replication-competent viruses armed with transgenes now has become common.
Tumor Selectivity
Engineering viruses to selectively target tumor cells has proven especially productive. By minimizing infection of and resulting toxicity to normal cells, larger viral doses can be administered and the therapeutic index widened. Mechanisms for engineering viruses for tumor selectivity include modification of the viral coat, exploiting abnormal signaling pathways, insertion of tumor or tissue specific promoters, and partial or entire gene deletions (Figure 2) (26, 34, 44).
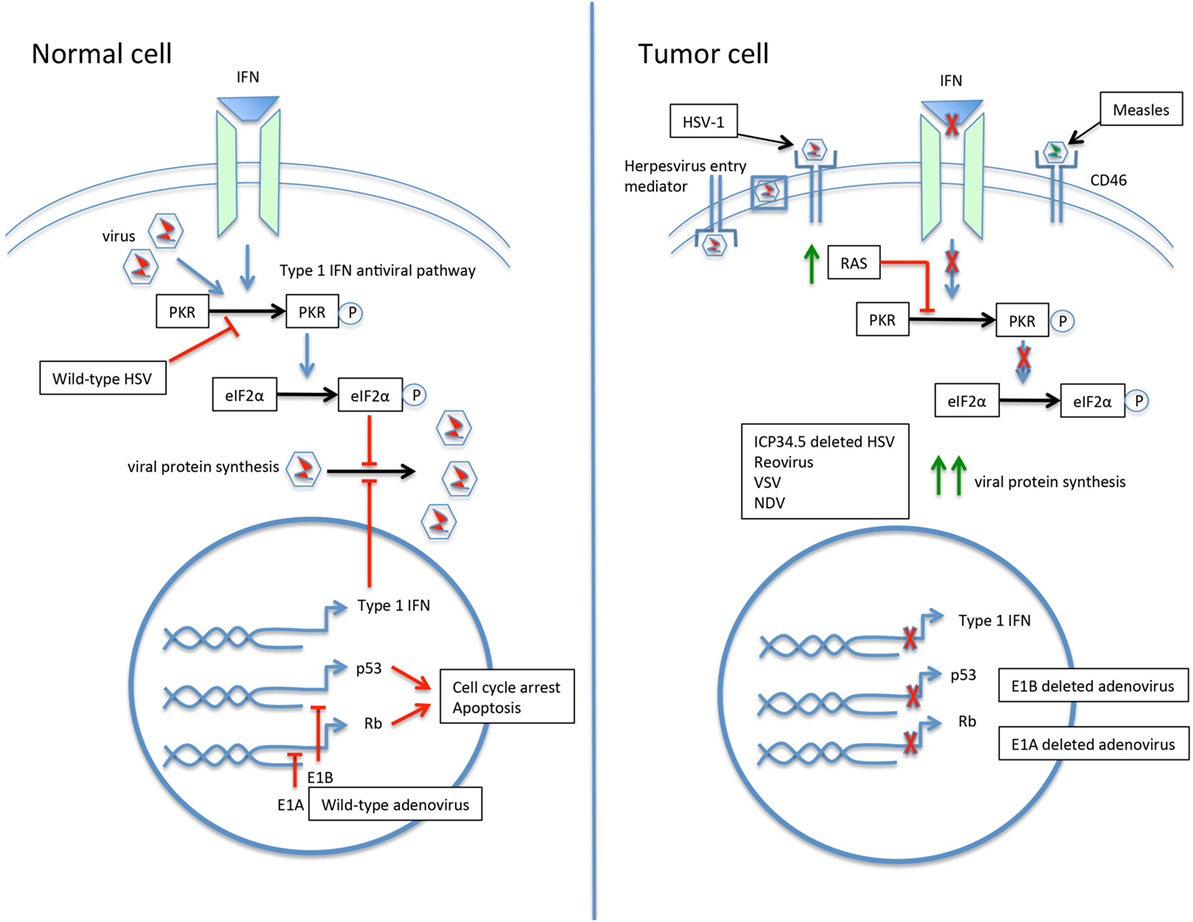
Figure 2. The selective infection of tumor cells by oncolytic viruses. In the normal cell, the response to viral infection involves activation of the type 1 interferon (IFN) and protein kinase R (PKR) pathways, resulting in upregulation of eIF2α and inhibition of viral protein synthesis. The p53 and Rb pathways are also activated. Wild-type viruses are able to inhibit various steps of the antiviral response to allow ongoing replication. For example, the herpes simplex virus (HSV) gene ICP34.5 blocks PKR signaling, and the adenovirus genes E1A and E1B inactivate Rb and p53, respectively. The tumor cell may have a number of acquired defects that allow for preferential infection by oncolytic viruses. An increased expression of cell surface proteins facilitates viral entry, such as herpesvirus entry mediator for HSV type 1 (HSV-1) and CD46 for measles virus. Defective IFN and PKR pathways lead to unimpeded viral protein synthesis. Upregulation of RAS in tumor cells results in PKR pathway inhibition. Modification of viruses can further drive tropism and minimize infection of normal cells. Deletion of the HSV gene ICP34.5 renders the virus unable to inhibit PKR in healthy cells and drives infection toward PKR-deficient tumor cells. Similarly, deletion of the adenovirus E1A or E1B genes leads to preferential infection of p53- and Rb-deficient tumor cells.
Achieving tumor selectivity does not always require a recombinant virus, as a wild-type virus may already exhibit a preference for replicating in tumor cells. This can occur through overexpression of cell surface proteins that facilitate viral entry into the tumor cell (26). Specific viruses have natural tropism for these aberrant proteins, such as HSV-1 for overexpressed herpesvirus entry mediator and nectins on carcinoma cells, measles virus for CD46, and echovirus for an integrin domain on ovarian cancer cells (45–48).
When a natural viral tropism for tumor cell surface proteins is not present, viral coat protein expression can be modified. Ligands unique to the tumor cell surface are identified and the virus engineered for uptake specifically by these ligands (34). This is used in adenoviral vectors by modification of the Ad5 fiber knob domain (49). Another example is a measles virus designed with a surface antibody targeting carcinoma embryonic antigen expressed on adenocarcinoma (50).
Wild-type viruses can also preferentially infect tumor cells by exploiting altered signaling pathways in the tumor cell (44). This illustrates how cellular changes defining malignancy, such as resistance to apoptosis and loss of p53, often overlap with virally induced cellular changes (51, 52). The environment of a tumor cell then may be advantageous by supplying cell processes necessary for viral replication. Two key antiviral pathways present in normal cells are often implicated here—protein kinase R (PKR) and interferon (IFN) signaling (44). A dysfunctional PKR pathway enhances reovirus replication, and defects in the type 1 IFN response potentiate the replication of vesicular stomatitis virus (VSV) and Newcastle disease virus (NDV) (53–55).
Altered tumor cell signaling pathways provide opportunities for viral genetic engineering. Gene deletions can remove viral genes necessary for replication in normal tissue but not required for replication in tumor cells (34). Viral gene products block the normal antiviral response through the PKR, IFN, and p53 pathways. Deletion of these viral genes restores the ability of healthy cells to prevent viral replication, while cancer cells already deficient in the antiviral pathway remain susceptible. HSV-1 modified for deletion of the ICP34.5 gene is such an example. Lacking this gene, the virus no longer blocks PKR signaling in healthy cells, leaving PKR-deficient tumor cells to be preferentially infected (26, 56). Similarly, a modified adenovirus with a gene deletion for the protein E1B no longer inactivates p53. This allows healthy cells to initiate p53-mediated apoptosis prior to viral replication; p53-deficient cancer cells are then selected for viral spread (57).
The goal of most viral gene deletions is to attenuate viral pathogenesis in normal cells. In fact, nearly all oncolytic viruses being studied for clinical use are attenuated in same manner. The first study of a virus modified specifically to improve oncolytic activity, by Martuza et al. in 1991, employed HSV-1 with deletion of the gene encoding the enzyme thymidine kinase (58). This deletion results in attenuated neurovirulence (59).
Just as genes are deleted from the viral genome to increase tumor specificity, insertion of gene promoter regions that are tumor or tissue restricted are frequent additions to achieve the same goal of specificity. This relies on the overexpression of tumor-specific proteins for activation of the promoter region of a gene that is necessary for viral replication and/or cell death. Healthy cells become relative life cycle dead ends for the virus by lacking the proteins needed to activate regulatory viral genes (44). The adenovirus E1A gene has been modified with various gene promoters including an alpha-fetoprotein gene promoter for tumor-specific replication in hepatocellular cancer cells and a prostate-specific antigen gene promoter with tissue-specific replication in prostate cancer (60, 61).
Immune System Activation
The concepts of viral antitumor efficacy and selectivity can be linked together as the first part of a two-step process necessary for successful oncolytic viral therapy. The initial viral-directed tumor cytotoxicity then must be followed by a sustained antitumor response carried out by the immune system (32). This critical second phase has been recognized for many years (62), although only recently have the mechanisms to make oncolytic viruses a more effective immunotherapy begun to be elucidated (63–65).
Tumor-induced immune tolerance is a critical part of the malignant process. This is accomplished through alteration of the tumor microenvironment by recruitment of immune-inhibitory cells and exclusion of immune-stimulating cells (66). Viral-mediated tumor cell death works to reverse this tolerance by exposing tumor-associated antigens previously restricted from presentation to the immune system, known as neoantigens, and provoking inflammatory cytokine release. Antigen-presenting cells activated by these neoantigens then direct an antitumor response by CD8+ T cells and NK cells (Figure 1) (26).
Prior to arriving at the current paradigm of immune stimulation as an essential part of virotherapy, a major concern was a robust antiviral response limiting the extent of oncolysis (67). In an effort to thwart the immune response, initial murine studies used immunocompromised models to allow adequate viral replication and cytolysis (68). The move to immune-competent models was accompanied by suppression of the immune response, such as dampening T cell response through gene deletions or administering cyclophosphamide prior to viral administration (69, 70). Current approaches aim for a balance between permitting both initial viral replication and the subsequent robust antitumor, and inevitably antiviral, immune response.
Viral genetic engineering now includes modifications to boost immune antitumor activity, often through insertion of cytokine genes. HSV expressing granulocyte macrophage colony-stimulating factor (GM-CSF) increases antigen presentation by dendritic cells and improves tumor reduction of lymphoma in a murine model (56). VSV expressing IFNβ decreased T-regulatory cells, increased CD8+ T cells, and prolonged survival in a murine lung cancer model (65). The HSV-1 protein ICP47 decreases antigen presentation on infected cells, and deletion of this gene augments antitumor effects (56, 71).
The recognition of immune stimulation by oncolytic viruses and the simultaneous development of the immune checkpoint inhibitors raise the possibility of synergy between these distinctive mechanisms of immunotherapy. A number of studies have already been completed in this new area with promising results (72–74). The remarkable success of checkpoint inhibition likely indicates the future role for oncolytic therapy as an adjunct to other more clinically advanced forms of immunotherapy.
Administration and Safety
The administration of oncolytic viral therapy must account for the setting of metastatic disease that requires a systemic immune response. Intravenous delivery of virus, while having the potential for rapid viral infection at all locations of disease, is problematic for several reasons. An immediate innate humoral immune response may lead to viral inactivation in the bloodstream, prior to infection of tumor cells. In the case of previous environmental exposure or vaccination, antiviral antibodies will provide effective at viral clearance (75–77). Even without preexisting immunity, repeated intravenous administration of virus results in production of antiviral antibody titers that quickly render vascular delivery ineffective (78).
The delivery of oncolytic viruses has predominantly been via direct intratumoral (IT) injection. IT administration has its own limitations, most apparent being the requirement of an accessible solid mass. Early viral inactivation by the innate immune system is also an issue with IT injection, although probably to a lesser extent than intravenous therapy (69, 79). Both systemic and direct viral administration must overcome a harsh local tumor environment that limits viral biodistribution (80, 81).
The main advantage of IT administration is ensuring local tumor delivery while also inducing distant tumor responses. This is true in some preclinical models and also in the phase III trial leading to approval of T-VEC (31, 65, 73). With the immune system able to provide a systemic response after local administration, any hypothetical advantage of intravenous delivery is no longer relevant.
Any effective oncolytic therapy needs to take into account effects on surrounding non-cancerous tissues. Of primary concern is the viral infection spreading to healthy cells, given the fact that cancer patients are already immunosuppressed and susceptible to infection. As previously discussed, the concept of selecting and designing a virus with tumor cell selectivity is the key to minimizing toxicity. Additional safety concerns include environmental shedding and reversion to wild-type virus. In general, studies have shown favorable toxicity profiles although perhaps at the expense of efficacy, as the field is now moving toward the use of less attenuated viruses with improved selectivity. A recent review covers safety concerns in more detail (82).
The Ideal Oncolytic Virus
In addition to optimizing antitumor efficacy, tumor selectivity, and immune system activation, a number of other viral characteristics are taken into account when choosing an oncolytic virus. These include viral genetic stability, non-integrating viruses that cannot incorporate into the host genome, a safety mechanism to inactive the virus after administration, and amenability to high titer production (44). A detailed discussion of these additional factors is beyond the scope of this review.
The ability to non-invasively image viral infectivity is of particular interest. These molecular imaging techniques allow localization of viral replication in tumor or healthy tissue, an important measure of toxicity. The viral dose and route of administration may be correlated with the level of infectivity without the need for repeat tissue biopsies (83). Two viral modifications for molecular imaging are insertion of the gene encoding green fluorescent protein (GFP) or the human sodium iodide symporter (hNIS) protein. The hNIS also offers the potential for radioiodide therapy like that used for ablation of thyroid tissue (84). Many of the mesothelioma studies detailed in the next section utilize addition of these reporter genes, allowing for monitoring of efficacy and toxicity.
With the expanding ability to genetically engineer oncolytic viruses, the use of viruses with multiple modifications is readily available. This is illustrated by T-VEC, an HSV-1 with three separate modifications—deletion of gene ICP34.5 to attenuate neurovirulence, deletion of ICP47 to increase antigen presentation on infected cells, and insertion of the gene for GM-CSF to attract antigen-presenting cells (31, 56, 85–87). This combinatorial approach to maximize efficacy through various mechanisms is now standard, as we will describe with oncolytic virotherapy for mesothelioma.
Oncolytic Viral Therapy for Mesothelioma
Malignant pleural mesothelioma provides an optimal model for the study of oncolytic virotherapy for several reasons (88). The pleural location is accessible for direct IT injection, the preferred method of administration for most viral platforms. Although distant metastases can occur, complications and death usually stem from local disease spread. Also, limited improvement in outcomes with multimodality therapy lends more urgency to experimental approaches. Given these characteristics, despite being an uncommon malignancy, an extensive amount of preclinical data with oncolytic viruses in mesothelioma models is available. This work has progressed to early phase clinical MPM studies for a number of different viruses (Table 1).
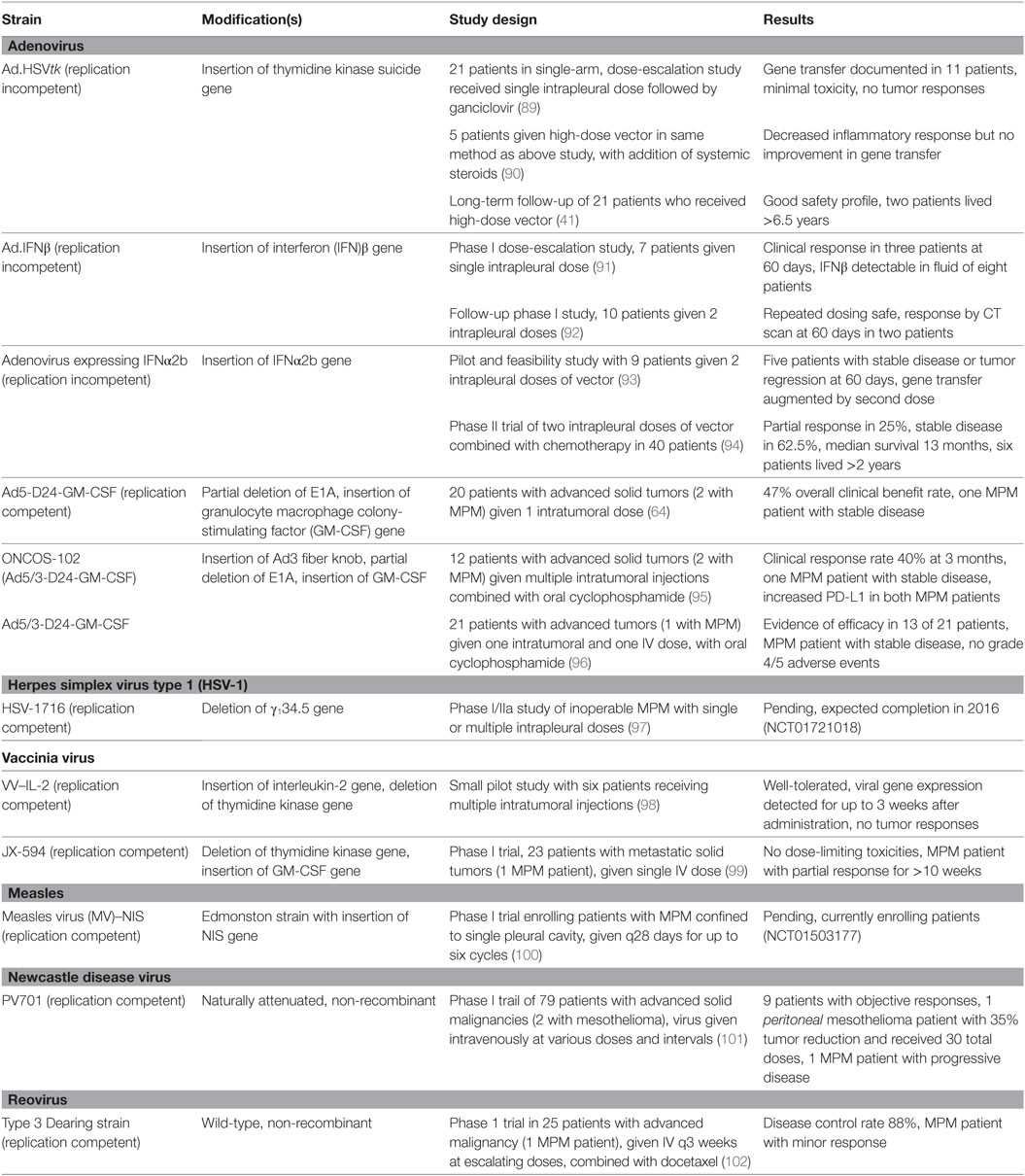
Table 1. Human clinical studies of virotherapy for malignant pleural mesothelioma (MPM).
Studies using replication-incompetent viruses are most accurately classified as gene therapy with a viral vector rather than oncolytic virotherapy, since “oncolytic” implies active viral replication. In the context of cancer, gene therapy is the transfer of genetic material to induce tumor cell death, as defined by Sterman (88). This can be accomplished in a number of ways, and oncolytic virotherapy is a subtype of gene therapy using actively replicating viruses. Given the significant overlap, studies of both replication-competent and replication-incompetent viruses are reviewed here.
Adenovirus
A non-enveloped virus with a linear, double-stranded DNA genome, adenovirus is one of the most extensively studied oncolytic viruses, rivaled only by HSV. A moderately sized genome of ~38 kilo base pairs (kb) allows for multiple modifications (26, 32). Other favorable characteristics are a stable genome, non-integration, and high-titer production. Most humans are exposed and asymptomatic upon infection, although susceptible individuals can develop upper respiratory symptoms or conjunctivitis (33).
Studies with oncolytic adenovirus have advanced to many early phase human trials including prostate, pancreatic, and colorectal carcinomas (60, 103, 104). Notably, a phase III trial for head and neck squamous cell carcinoma conducted in China combined an E1B-deleted adenovirus (H101) with chemotherapy (29). This led to approval in China of H101 for treatment of nasopharyngeal carcinoma in combination with chemotherapy.
Adenovirus for mesothelioma includes both preclinical and human studies. The first in vitro studies out of the University of Pennsylvania focused on gene therapy with the HSV-thymidine kinase suicide gene inserted into a replication-deficient adenoviral vector (Ad.HSVtk) (105, 106). This same adenovirus then proved successful in animal models (107, 108). For example, Elshami and colleagues in 1996 used a rat model of MPM to intratumorally administer Ad.HSVtk followed by systemic ganciclovir, which is metabolized into toxic byproducts by the HSVtk gene product (109). The experimental rats showed tumor regression at 20 days (average tumor weight 0.6 vs. 5.4 g) and improved mean survival (34 vs. 26 days) compared to controls.
Replication-deficient adenovirus has also been studied as a vector for cytokine gene therapy to counter the immune tolerance characteristic of mesothelioma (88). After passive immunotherapy with intrapleural or systemic delivery of IL-2, IFNα, and IFNγ for mesothelioma met with some success in phase I/II trials, administration of cytokines via gene therapy was proposed to improve efficacy (110–112). In several murine experiments, IT injection of an adenovirus with insertion of the IFNγ gene resulted in tumor regression and a CD8+ T cell-mediated response (113, 114). By using a different mechanism to induce antitumor immunity, a replication-deficient adenovirus engineered to express the costimulatory molecule CD40L showed regression of both directly injected and distant tumors, indicative of a systemic immune response (115).
More recently, a series of preclinical studies using conditionally replicating adenoviruses (CRAds), replication-competent oncolytic viruses with modifications to improve tumor selectivity, have shown antimesothelioma activity. Instead of the E1 gene being deleted to produce replication-incompetent viruses, the gene is placed under control of tumor-specific promoters. An in vitro study inserted a midkine promoter overexpressed in tumor cells and demonstrated effective oncolysis in human MPM cell lines (116). In vivo studies with murine models have used a number of CRAd modifications—promoters linked to E1 gene expression, viral capsid alterations, and insertion of GFP for viral imaging (117, 118). For example, Watanabe and colleagues in 2010 used an adenovirus engineered to express a telomerase-driven promoter linked to the E1 gene (119). This was co-administered with a replication-incompetent adenovirus with insertion of the heparanase gene to improve viral penetration through the dense extracellular matrix. The study showed significant tumor regression compared to controls as well as improved survival.
Human studies using adenoviral vectors began relatively quickly following preclinical experiments. Consecutive phase I dose-escalation studies used the replication-incompetent Ad.HSVtk gene therapy followed by ganciclovir (89, 90, 120). The response rate was low, with 1 of 26 patients having evidence of tumor regression. Seventeen patients had evidence of IT gene transfer on repeat pleural biopsy, although this was limited to the outermost tumor layers. A follow-up study of 21 patients who received “high-dose” therapy reported a good safety profile and 2 patients surviving >6.5 years (41). Although these studies proved to be safe, the low response rate indicated a need for improved gene transfer within the tumor and a more robust antitumor immune response.
Focusing on stimulating an immune response rather than delivering a suicide gene, several human trials have been completed using an adenoviral vector for gene transfer of IFNβ (Ad.IFNβ). These studies, like those for Ad.HSVtk, were with replication-incompetent virus. The initial phase I dose-escalation trial enrolled seven patients with MPM and three patients with metastatic pleural effusions due to other malignancies, administering a single intrapleural dose of Ad.IFNβ (91). At 60 days, three patients with MPM had a clinical response and four patients had progressive disease. IFNβ was detectable in the pleural fluid of eight patients, indicating successful gene transfer.
A follow-up phase I trial with Ad.IFNβ evaluated giving a second intrapleural dose (92). Repeated administration was safe, and 2 of 10 MPM patients had a clinical response by CT scan at 2 months. This lack of improvement in response with repeat dosing was likely from rapid development of neutralizing antibodies, as humoral immune responses were consistently detected but not the cellular responses more essential to antitumor immunity. Although these two IFNβ gene therapy trials were encouraging for stimulating an immune response, further dose modifications or combination therapy are needed to have a more significant impact on outcome.
When Ad.IFNβ was no longer commercially available, a subsequent phase I gene therapy study by Sterman and colleagues was completed using replication-incompetent adenovirus expressing IFNα2b (Ad.IFNα2b) (93). Clinical responses were encouraging, with five of nine patients having stable disease or tumor regression at 60 days. This led to a phase II trial combining Ad.IFNα2b with chemotherapy in 40 patients (94). Patients received two intrapleural doses of Ad.IFNα2b on days 1 and 4, followed by chemotherapy on day 15 with standard of care pemetrexed/platinum doublet for chemotherapy naïve patients (first-line treatment, 18 patients). If this was second-line chemotherapy, gemcitabine or pemetrexed was given (22 patients). Partial responses were observed in 25% of patients and stable disease in 62.5%. Although the median overall survival of 13 months was not significantly improved from standard treatments, six patients lived more than 24 months. Based on these results, a randomized phase III trial is planned.
Studies using replication-competent, oncolytic adenoviruses for MPM are scarce. A study by Cerullo and colleagues evaluated an oncolytic adenovirus (Ad5-D24-GM-CSF) modified for tumor selectivity and with insertion of a transgene for GM-CSF to augment immune response (64, 121). The virus was administered once intratumorally to 20 patients with advanced solid cancers, including 2 patients with MPM. Both patients had received prior chemotherapy. Reflecting the clinical benefit rate of 47% among all patients, one case of MPM had stable disease and lived over 1 year; the other case had progressive disease and lived about 100 days. No serious adverse events occurred.
A phase I study published in 2016 used Ad5/3-D24-GM-CSF in patients with advanced solid tumors (95). Twelve patients were enrolled including two with MPM. Multiple IT injections were given along with oral cyclophosphamide. Clinical response rate at 3 months was 40%; one MPM patient had stable disease, and the other had progressive disease. Tumor-infiltrating leukocytes increased following treatment in 11 of 12 patients. Interestingly, both patients with MPM showed increased PD-L1 expression posttreatment, relevant for potential future combination studies with immune checkpoint inhibitors.
HSV Type 1
An enveloped, double-stranded DNA virus, HSV-1 has a large 152 kb genome. About 30 kb of the genome is non-essential, which allows space for insertion of transgenes (33). The human pathogenesis of HSV-1 causes oral and genital lesions, latent infection in peripheral nerves, and less frequently CNS complications. This natural tropism for neuronal tissue led to early studies on brain tumors and also necessitates viral gene deletions to attenuate neurotoxicity in all oncolytic experiments using HSV-1 (58, 59, 122). Recombinant HSV-1 has been studied in a number of malignancies including colorectal, pancreatic carcinoma, and melanoma (31, 123, 124). In fact the only FDA-approved oncolytic virus, T-VEC for melanoma, is a modified HSV-1.
A preclinical study by Kucharczuk and colleagues in 1997 evaluated the replication-competent, neuroattenuated HSV-1716 as oncolytic virotherapy for mesothelioma (125). Neuroattenuation was achieved by deletion of both γ134.5 genes encoding the protein ICP34.5. The virus efficiently replicated in and lysed human mesothelioma cells in vitro. The same human mesothelioma cell line was then used to establish intraperitoneal tumors in immunodeficient mice. Fourteen days later, the mice were given HSV-1716 by intraperitoneal injection, resulting in decreased tumor burden and increased survival compared to controls. No viral dissemination was detected in non-tumor tissue.
Another preclinical study evaluated three different replication-competent, oncolytic herpesviruses: G207, NV1020, and NV1066 (126). G207 has both γ134.5 genes deleted along with inactivation of the ICP6 gene for additional attenuation in non-replicating tissues. NV1020, originally designed as an HSV vaccine, has deletions encoding the genes ICP0, ICP4, latency-associated transcripts, one copy of γ134.5, and UL24, all resulting in loss of virulence. NV1066 has single copy deletions of ICP0, ICP4, and γ134.5, plus the addition of GFP for viral imaging. Each virus was tested against 11 different MPM cell lines in vitro, including each histologic subtype of MPM—epithelioid, sarcomatoid, biphasic, and mixed. All three viruses were cytotoxic to each cell line, even at low multiplicities of infection (the ratio of viral particles to tumor cells). A murine model of MPM treated with NV1066 decreased tumor burden and increased survival.
Adusumilli and colleagues at Memorial Sloan-Kettering Cancer Center did several additional elegant in vitro studies combining NV1066 with other therapies. The first study evaluated viral replication and cytotoxicity in 10 human MPM cell lines infected with NV1066 with or without cisplatin (127). The combination proved synergistic, at least partly attributable to cisplatin-induced DNA damage upregulating the protein GADD34 that in turn potentiates replication and cytotoxicity of the mutant HSV-1.
The second study combined NV1066 and radiation therapy in multiple human MPM cell lines and found synergistic or additive effects depending on the cell line, based on the same mechanism of GADD34 upregulation (128). A murine flank tumor model demonstrated reduced tumor growth with the combination compared to controls or either therapy alone. Importantly, in both of these studies, cytotoxicity was maintained with dose reductions, which may allow for decreased toxicity if such combination therapy advances to further trials.
Human studies of oncolytic herpesviruses for mesothelioma have not been completed. An ongoing phase I/IIa study of HSV-1716 for inoperable MPM began recruiting in 2012. The virus is delivered into the pleural cavity in single or multiple doses, with safety as primary outcome and efficacy as secondary outcome. Study completion is expected in 2016 (97).
Vaccinia Virus
An enveloped, double-stranded DNA virus in the poxvirus family, vaccinia virus has a large ~190 kb genome that facilitates insertion and deletion modifications to improve oncolytic efficacy. Vaccinia replicates in the cytoplasm, posing no risk for host genome integration. An attenuated vaccinia virus was used to eradicate smallpox. Pathogenesis of the wild-type virus in immunocompetent humans is limited to a mild viral syndrome of fever, rash, and myalgias (26, 33).
Vaccinia viruses have been studied in a number of solid tumors in humans including breast, melanoma, and prostate (129–131). Some of these recombinant viruses are described as vaccines, since the objective is stimulation of an antitumor immune response rather than active viral replication causing oncolysis. JX-594, the most clinically advanced oncolytic vaccinia virus, has deletion of the thymidine kinase gene and an inserted transgene to express GM-CSF (132). A phase III trial combining JX-594 with sorafenib for treatment of hepatocellular carcinoma is now recruiting patients (133, 134).
Preclinical and human studies have evaluated treatment of mesothelioma with vaccinia viruses. The replication-competent vaccinia virus GLV-1h68 has deletions of the hemagglutinin and thymidine kinase genes for attenuation and insertion of three transgenes including GFP for viral imaging (135). GLV-1h68 successfully replicated in and lysed multiple MPM cell lines in vitro (136). The same study then established a murine model of MPM followed by intrapleural delivery of the virus that resulted in decreased tumor burden and increased survival. The GLV-1h153 virus, a modification of the parent virus GLV-1h68 by insertion of the hNIS gene, proved similarly effective for in vitro and murine models with the addition of radioiodine-based imaging for viral infection (137).
A study by Acuna and colleagues in 2014 evaluated vaccinia virus as adjuvant therapy following surgery in a murine model of malignant peritoneal mesothelioma (138). An oncolytic strain with deletions in thymidine kinase and vaccinia growth factor genes for tumor selectivity was used, the double-deleted vaccinia virus (vvDD). A single intraperitoneal dose of vvDD prolonged survival compared to controls. When combined with incomplete cytoreductive surgery, survival was not significantly prolonged compared to either vvDD alone or surgery alone. This led the investigators to propose that the effectiveness of vvDD as an adjuvant therapy following surgery may be limited to microscopic disease after complete surgical resection, although further studies have not yet been completed to confirm this hypothesis.
Human studies with vaccinia virus from MPM are limited. A small pilot study published in 2000 used a vaccinia virus with insertion of the IL-2 gene into the thymidine kinase gene region, a replication-competent virus with tumor cell selectivity (98). Six patients received multiple IT injections. Treatment was well tolerated, and viral gene expression was detected for up to 3 weeks after injection despite the development of antivaccinia antibodies; however, no tumor responses were seen.
An early phase I trial with the oncolytic JX-594 vaccinia virus enrolled 23 patients with metastatic solid tumors, including 1 patient with MPM (99). In contrast to IT or intrapleural administration in nearly every other study, this virus was given intravenously as vaccinia has natural mechanisms to prevent inactivation in blood (139, 140). Following a single intravenous administration, the patient with MPM had a partial response for greater than 10 weeks.
Measles Virus
An enveloped RNA virus with a small ~15 kb genome, measles virus (MV) is a well-known human pathogen that occasionally causes serious illness in non-vaccinated individuals. The attenuated Edmonston strain is used for oncolytic virotherapy given its proven safety profile and also natural tumor specificity due to the upregulation of CD46 on tumor cell surface that the virus uses for cellular uptake (26, 47, 48). Other favorable characteristics of MV are a stable genome and cytoplasmic replication.
Oncolytic measles viruses have been studied in both solid and hematologic malignancies (141, 142). The most visible success thus far is a preliminary report from the Mayo Clinic of two relapsed, refractory myeloma patients given attenuated MV intravenously, with one patient achieving a complete remission lasting 9 months (143). This phase I/II study for myeloma patients is continuing to enroll patients (144).
Several preclinical studies with MV in MPM have been completed. The first in vitro experiment used the live attenuated Schwartz strain to evaluate oncolytic activity and immune response against human mesothelioma cells and normal mesothelial cells (145). The mesothelioma cells were more susceptible to infection and viral-induced cell death than the mesothelial cells, attributed to increased CD46 expression on the cancerous cells. Dendritic cells phagocytized the apoptotic MV-infected mesothelioma cells, resulting in dendritic cell maturation and priming of CD8+ T cells. Although in vitro, these results were encouraging for MV-stimulating antitumor immunogenicity.
Li and colleagues used a murine model of mesothelioma to study Edmonston strain MV with insertion of the IFNβ and NIS genes (MV–mIFNβ–NIS) (146). After confirming infectivity and replication of the virus in vitro, mice were injected subcutaneously in the flank with mesothelioma cells. After tumors grew to 5 mm, different MVs were injected intratumorally. Tumors injected with MV–mIFNβ had increased immune cell infiltration and decreased angiogenesis compared to tumors injected with the parent MV without mIFNβ expression. These pathological findings correlated with median survival, which increased from 20 days for control mice, to 45 days for mice receiving MV without mIFNβ, to 65 days with MV-mIFNβ. A peritoneal mesothelioma mouse model showed similar improvements in survival for each virus. In addition, the NIS gene facilitated non-invasive radioiodine imaging.
A more recent in vitro study published in 2015 evaluated the mechanism of MV for tumor cell selectivity. Twenty-two MPM cell lines were tested for infectivity and replication of MV, along with four healthy cell lines. Interestingly, the amount of CD46 expression did not predict for MV infectivity, contrary to previous assumptions. A better correlate for sensitivity to MV was the ability to mount a complete type 1 IFN response. Cell lines unable to generate or respond to IFNα or IFNβ, the case for 70% of the MPM lines, were more susceptible to MV infection (147). These data have implications for predicting response in future studies of MPM to oncolytic MV.
No human studies of MV for mesothelioma have yet to be completed. A phase I trial using the attenuated Edmonston strain with insertion of the NIS gene (MV–NIS) is currently enrolling patients with MPM confined to a single pleural cavity (100). The virus is administered intrapleurally once every 28 days for up to six cycles. Primary and secondary objectives are maximum tolerated dose, safety, and toxicity; tertiary objectives are measurements of viral activity, immune response, and efficacy.
Other Oncolytic Viruses for Mesothelioma
Adenovirus, HSV-1, vaccinia virus, and MV are the most extensively studied virotherapy vectors for mesothelioma. A more limited number of studies have evaluated additional viruses including VSV, NDV, reovirus, and Sendai virus.
An RNA virus in the Rhabdoviridae family, VSV has no known pathogenesis in humans. Exposure is possible in those working with livestock or mice; otherwise the general population is immune naïve. This lack of pre-formed immune response is an advantage when introducing VSV as an oncolytic virus (65). VSV displays natural tumor selectivity via induction of the antiviral type 1 IFN pathway. In healthy cells with intact IFN signaling, this prevents viral replication, whereas tumor cells with a defective IFN response allow viral infection to proceed unimpeded (148).
Recombinant VSV engineered to express IFNβ (VSV–IFNβ) augments both the antiviral defense in healthy tissue and the immune response against tumor cells. Several preclinical studies have evaluated VSV–IFNβ against mesothelioma. A murine model of subcutaneous and intraperitoneal tumors injected with VSV–IFNβ showed reduced tumor growth and increased survival compared to controls (149). Safety was also enhanced, with less neurotoxicity with mouse IFNβ.
A second study looked at mesothelioma cell lines in vitro and correlated cytotoxicity from VSV–IFNβ with the extent of IFN responsiveness (150). Partial responsiveness, measured by upregulation of PKR and other elements after viral infection, led to resistance to cytolysis. Conversely, downregulation of p48 and PKR caused sensitivity to the virus. The authors proposed that testing tumor cells for IFN responsiveness might provide a predictive marker for this virotherapy.
NDV is an RNA avian paramyxovirus that causes serious disease in fowl but only mild disease in humans. Similar to VSV, the tumor specificity of NDV is dictated through a defective type I IFN pathway in tumor cells (151). A preclinical study in mesothelioma with NDV engineered to express GFP showed effective oncolysis against multiple mesothelioma cell lines in vitro (152). An orthotopic model of MPM in mice was then treated with either single or multiple intrapleural doses of NDV. Animals receiving multiple treatments had decreased tumor burden, measured by bioluminescence imaging of GFP. Survival was longest in those receiving multiple treatments and shortest in the control group.
A phase I trial using a replication-competent NDV enrolled 79 patients with advanced solid malignancies, including 2 cases of mesothelioma (101). The virus was administered intravenously at various dose levels and intervals. Of the 9 patients with objective tumor responses, 1 patient with peritoneal mesothelioma received over 30 doses of virus with a 35% tumor reduction, improved performance status, and no cumulative toxicity. A posttreatment tumor biopsy showed active NDV replication. Despite this encouraging result, no further human studies with NDV for mesothelioma have been completed.
Reovirus and Sendai virus are two additional RNA viruses that have been studied in combination with chemotherapy for mesothelioma. In a murine model of MPM, Sendai virus with cisplatin showed synergistic effects (153). A phase I trial evaluated intravenous reovirus plus docetaxel in 25 patients with advanced cancer (102). The one patient with mesothelioma had a minor response.
The Future of Oncolytic Virotherapy for Mesothelioma
The current paradigm for treatment of MPM emphasizes a multimodality approaching with surgery, radiation, or chemotherapy. Most studies of oncolytic therapy for MPM have been as monotherapy, necessary to confirm viral activity, dosing, and safety in preclinical and early-phase human trials. However, a number of studies have successfully combined virotherapy for mesothelioma with chemotherapy (94, 127, 154), radiation (128), and surgery (138, 155). Given the documented safety but overall limited efficacy thus far when administered as monotherapy, future studies will likely use oncolytic viruses as an adjuvant to more established therapy (156).
Combining the immune checkpoint inhibitors with oncolytic viruses is of exceptional interest given the synergistic mechanisms of immune activation. In fact since the recent approval of the oncolytic virus T-VEC for melanoma, a study has already shown improved efficacy with T-VEC when given with the CTLA-4 inhibitor ipilimumab (72). A study by Patel and colleagues finding increased tumor expression of PD-L1 after treatment with VSV–IFNβ in a murine model of non-small-cell lung cancer indicates the potential of PD-1/PD-L1 agents to increase efficacy (65). Early phase trials with immune checkpoint inhibitors for mesothelioma are in process, with promising preliminary results (157).
The ability to molecularly engineer recombinant viruses to improve safety and efficacy has led to rapid advances in oncolytic virotherapy over the last several decades. Preclinical work for mesothelioma is now starting to move into more significant human studies with several clinical trials currently recruiting patients (97, 100). As the field continues to develop, more studies are needed to determine how oncolytic virotherapy is best utilized alongside current treatment for mesothelioma.
Author Contributions
DP wrote the first draft of this article in consultation with RK. RK conceived and edited the article, as well as drafting the format.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Teta MJ, Mink PJ, Lau E, Sceurman BK, Foster ED. US mesothelioma patterns 1973-2002: indicators of change and insights into background rates. Eur J Cancer Prev (2008) 17:525–34. doi:10.1097/CEJ.0b013e3282f0c0a2
2. Mezei G, Chang ET, Mowat FS, Moolgavkar SH. Epidemiology of mesothelioma of the pericardium and tunica vaginalis testis. Ann Epidemiol (2017) 27:348–59.e11. doi:10.1016/j.annepidem.2017.04.001
3. Henley SJ, Larson TC, Wu M, Antao VC, Lewis M, Pinheiro GA, et al. Mesothelioma incidence in 50 states and the District of Columbia, United States, 2003-2008. Int J Occup Environ Health (2013) 19:1–10. doi:10.1179/2049396712Y.0000000016
4. Lehnert M, Kraywinkel K, Heinze E, Wiethege T, Johnen G, Fiebig J, et al. Incidence of malignant mesothelioma in Germany 2009-2013. Cancer Causes Control (2017) 28:97–105. doi:10.1007/s10552-016-0838-y
5. Hodgson JT, McElvenny DM, Darnton AJ, Price MJ, Peto J. The expected burden of mesothelioma mortality in Great Britain from 2002 to 2050. Br J Cancer (2005) 92:587–93. doi:10.1038/sj.bjc.6602307
6. Robinson BM. Malignant pleural mesothelioma: an epidemiological perspective. Ann Cardiothorac Surg (2012) 1:491–6. doi:10.3978/j.issn.2225-319X.2012.11.04
7. Lanphear BP, Buncher CR. Latent period for malignant mesothelioma of occupational origin. J Occup Med (1992) 34:718–21.
8. Baas P, Fennell D, Kerr KM, Van Schil PE, Haas RL, Peters S, et al. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2015) 26(Suppl 5):v31–9. doi:10.1093/annonc/mdv199
9. Churg A, Roggli V, Galateau-Salle F, Cagle PHT, Gibbs AR, Hasleton PHS, et al. Tumors of the pleura: mesothelial tumours. In: Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors. Pathology and Genetics of Tumors of the Lung, Pleura, Thymus, and Heart. World Health Organization Classification of Tumours. Lyon, France: IARC (2004). p. 125–36.
10. Flores RM, Pass HI, Seshan VE, Dycoco J, Zakowski M, Carbone M, et al. Extrapleural pneumonectomy versus pleurectomy/decortication in the surgical management of malignant pleural mesothelioma: results in 663 patients. J Thorac Cardiovasc Surg (2008) 135:620–6, 626.e1–3. doi:10.1016/j.jtcvs.2007.10.054
11. Taioli E, Wolf AS, Flores RM. Meta-analysis of survival after pleurectomy decortication versus extrapleural pneumonectomy in mesothelioma. Ann Thorac Surg (2015) 99:472–80. doi:10.1016/j.athoracsur.2014.09.056
12. Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol (2003) 21:2636–44. doi:10.1200/JCO.2003.11.136
13. Santoro A, O’Brien ME, Stahel RA, Nackaerts K, Baas P, Karthaus M, et al. Pemetrexed plus cisplatin or pemetrexed plus carboplatin for chemonaïve patients with malignant pleural mesothelioma: results of the International Expanded Access Program. J Thorac Oncol (2008) 3:756–63. doi:10.1097/JTO.0b013e31817c73d6
14. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet (2016) 387:1405–14. doi:10.1016/S0140-6736(15)01238-6
15. Rusch VW, Rosenzweig K, Venkatraman E, Leon L, Raben A, Harrison L, et al. A phase II trial of surgical resection and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J Thorac Cardiovasc Surg (2001) 122:788–95. doi:10.1067/mtc.2001.116560
16. Cao C, Tian D, Manganas C, Matthews P, Yan TD. Systematic review of trimodality therapy for patients with malignant pleural mesothelioma. Ann Cardiothorac Surg (2012) 1:428–37. doi:10.3978/j.issn.2225-319X.2012.11.07
17. Rimner A, Zauderer MG, Gomez DR, Adusumilli PS, Parhar PK, Wu AJ, et al. Phase II study of hemithoracic intensity-modulated pleural radiation therapy (IMPRINT) as part of lung-sparing multimodality therapy in patients with malignant pleural mesothelioma. J Clin Oncol (2016) 34:2761–8. doi:10.1200/JCO.2016.67.2675
18. Bibby AC, Tsim S, Kanellakis N, Ball H, Talbot DC, Blyth KG, et al. Malignant pleural mesothelioma: an update on investigation, diagnosis and treatment. Eur Respir Rev (2016) 25:472–86. doi:10.1183/16000617.0063-2016
19. Sugarbaker PH, Turaga KK, Alexander HR Jr, Deraco M, Hesdorffer M. Management of malignant peritoneal mesothelioma using cytoreductive surgery and perioperative chemotherapy. J Oncol Pract (2016) 12:928–35. doi:10.1200/JOP.2016.011908
20. Rusch VW, Giroux D, Kennedy C, Ruffini E, Cangir AK, Rice D, et al. Initial analysis of the international association for the study of lung cancer mesothelioma database. J Thorac Oncol (2012) 7:1631–9. doi:10.1097/JTO.0b013e31826915f1
21. DePace N. Sulla scomparsa di un enorme cancro vegetante del collo dell’utero senza cura chirurgia. Ginecologia (1912) 9:82–8.
22. Pack GT. Note on the experimental use of rabies vaccine for melanomatosis. AMA Arch Derm Syphilol (1950) 62:694–5. doi:10.1001/archderm.1950.01530180083015
23. Huebner RJ, Rowe WP, Schatten WE, Smith RR, Thomas LB. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer (1956) 9:1211–8. doi:10.1002/1097-0142(195611/12)9:6<1211::AID-CNCR2820090624>3.0.CO;2-7
24. Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer (1952) 5:1025–34. doi:10.1002/1097-0142(195209)5:5<1025::AID-CNCR2820050518>3.0.CO;2-Q
25. Kirn DH, McCormick F. Replicating viruses as selective cancer therapeutics. Mol Med Today (1996) 2:519–27. doi:10.1016/S1357-4310(97)81456-6
26. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov (2015) 14:642–62. doi:10.1038/nrd4663
27. Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol (2014) 16:334–51. doi:10.1093/neuonc/not310
28. Hartkopf AD, Fehm T, Wallwiener D, Lauer UM. Oncolytic virotherapy of breast cancer. Gynecol Oncol (2011) 123:164–71. doi:10.1016/j.ygyno.2011.06.021
29. Xia ZJ, Chang JH, Zhang L, Jiang WQ, Guan ZZ, Liu JW, et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng (2004) 23:1666–70.
30. Patel MR, Jacobson BA, Belgum H, Raza A, Sadiq A, Drees J, et al. Measles vaccine strains for virotherapy of non-small-cell lung carcinoma. J Thorac Oncol (2014) 9:1101–10. doi:10.1097/JTO.0000000000000214
31. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33:2780–8. doi:10.1200/JCO.2014.58.3377
32. Mullen JT, Tanabe KK. Viral oncolysis. Oncologist (2002) 7:106–19. doi:10.1634/theoncologist.7-2-106
33. Smyth Templeton N. Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. 4th ed. Baton Rouge, USA: CRC Press (2015).
34. Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med (2001) 7:781–7. doi:10.1038/89901
35. Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology (1996) 220:152–62. doi:10.1006/viro.1996.0295
36. Doronin K, Toth K, Kuppuswamy M, Ward P, Tollefson AE, Wold WS. Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein. J Virol (2000) 74:6147–55. doi:10.1128/JVI.74.13.6147-6155.2000
38. Sung MW, Yeh HC, Thung SN, Schwartz ME, Mandeli JP, Chen SH, et al. Intratumoral adenovirus-mediated suicide gene transfer for hepatic metastases from colorectal adenocarcinoma: results of a phase I clinical trial. Mol Ther (2001) 4:182–91. doi:10.1006/mthe.2001.0444
39. Kubo H, Gardner TA, Wada Y, Koeneman KS, Gotoh A, Yang L, et al. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther (2003) 14:227–41. doi:10.1089/10430340360535788
40. Germano IM, Fable J, Gultekin SH, Silvers A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncol (2003) 65:279–89. doi:10.1023/B:NEON.0000003657.95085.56
41. Sterman DH, Recio A, Vachani A, Sun J, Cheung L, DeLong P, et al. Long-term follow-up of patients with malignant pleural mesothelioma receiving high-dose adenovirus herpes simplex thymidine kinase/ganciclovir suicide gene therapy. Clin Cancer Res (2005) 11:7444–53. doi:10.1158/1078-0432.CCR-05-0405
42. Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A (2000) 97:2208–13. doi:10.1073/pnas.040557897
43. Hirvinen M, Rajecki M, Kapanen M, Parviainen S, Rouvinen-Lagerström N, Diaconu I, et al. Immunological effects of a tumor necrosis factor alpha-armed oncolytic adenovirus. Hum Gene Ther (2015) 26:134–44. doi:10.1089/hum.2014.069
44. Thorne SH, Hermiston T, Kirn D. Oncolytic virotherapy: approaches to tumor targeting and enhancing antitumor effects. Semin Oncol (2005) 32:537–48. doi:10.1053/j.seminoncol.2005.09.007
45. Shafren DR, Sylvester D, Johansson ES, Campbell IG, Barry RD. Oncolysis of human ovarian cancers by echovirus type 1. Int J Cancer (2005) 115:320–8. doi:10.1002/ijc.20866
46. Yu Z, Chan MK, O-charoenrat P, Eisenberg DP, Shah JP, Singh B, et al. Enhanced nectin-1 expression and herpes oncolytic sensitivity in highly migratory and invasive carcinoma. Clin Cancer Res (2005) 11:4889–97. doi:10.1158/1078-0432.CCR-05-0309
47. Dörig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell (1993) 75:295–305. doi:10.1016/0092-8674(93)80071-L
48. Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res (2004) 64:4919–26. doi:10.1158/0008-5472.CAN-04-0884
49. Jakubczak JL, Rollence ML, Stewart DA, Jafari JD, Von Seggern DJ, Nemerow GR, et al. Adenovirus type 5 viral particles pseudotyped with mutagenized fiber proteins show diminished infectivity of coxsackie B-adenovirus receptor-bearing cells. J Virol (2001) 75:2972–81. doi:10.1128/JVI.75.6.2972-2981.2001
50. Hammond AL, Plemper RK, Zhang J, Schneider U, Russell SJ, Cattaneo R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol (2001) 75:2087–96. doi:10.1128/JVI.75.5.2087-2096.2001
51. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
52. Kirn D. Replication-selective oncolytic adenoviruses: virotherapy aimed at genetic targets in cancer. Oncogene (2000) 19:6660–9. doi:10.1038/sj.onc.1204094
53. Wilden H, Fournier P, Zawatzky R, Schirrmacher V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int J Oncol (2009) 34:971–82. doi:10.3892/ijo_00000223
54. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J (1998) 17:3351–62. doi:10.1093/emboj/17.12.3351
55. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med (2000) 6:821–5. doi:10.1038/77558
56. Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther (2003) 10:292–303. doi:10.1038/sj.gt.3301885
57. Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science (1996) 274:373–6. doi:10.1126/science.274.5286.373
58. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (1991) 252:854–6. doi:10.1126/science.1851332
59. MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ’a’ sequence. J Gen Virol (1991) 72(Pt 3):631–9. doi:10.1099/0022-1317-72-3-631
60. DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res (2001) 61:7464–72.
61. Hallenbeck PL, Chang YN, Hay C, Golightly D, Stewart D, Lin J, et al. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum Gene Ther (1999) 10:1721–33. doi:10.1089/10430349950017725
62. Asada T. Treatment of human cancer with mumps virus. Cancer (1974) 34:1907–28. doi:10.1002/1097-0142(197412)34:6<1907::AID-CNCR2820340609>3.0.CO;2-4
63. Tuve S, Liu Y, Tragoolpua K, Jacobs JD, Yumul RC, Li ZY, et al. In situ adenovirus vaccination engages T effector cells against cancer. Vaccine (2009) 27:4225–39. doi:10.1016/j.vaccine.2009.03.074
64. Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res (2010) 70:4297–309. doi:10.1158/0008-5472.CAN-09-3567
65. Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K, et al. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget (2015) 6:33165–77. doi:10.18632/oncotarget.5320
66. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi:10.1038/ni.2703
67. Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther (2009) 20:1119–32. doi:10.1089/hum.2009.135
68. Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood (2001) 97:3746–54. doi:10.1182/blood.V97.12.3746
69. Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A (2006) 103:12873–8. doi:10.1073/pnas.0605496103
70. Berger C, Xuereb S, Johnson DC, Watanabe KS, Kiem HP, Greenberg PD, et al. Expression of herpes simplex virus ICP47 and human cytomegalovirus US11 prevents recognition of transgene products by CD8(+) cytotoxic T lymphocytes. J Virol (2000) 74:4465–73. doi:10.1128/JVI.74.10.4465-4473.2000
71. Tomazin R, van Schoot NE, Goldsmith K, Jugovic P, Sempé P, Früh K, et al. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol (1998) 72:2560–3.
72. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol (2016) 34(22):2619–26. doi:10.1200/JCO.2016.67.1529
73. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med (2014) 6:226ra32. doi:10.1126/scitranslmed.3008095
74. Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res (2015) 21:5543–51. doi:10.1158/1078-0432.CCR-14-2009
75. Dörner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity (2007) 27:384–92. doi:10.1016/j.immuni.2007.09.002
76. Peng KW, Myers R, Greenslade A, Mader E, Greiner S, Federspiel MJ, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther (2013) 20:255–61. doi:10.1038/gt.2012.31
77. Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther (2003) 10:983–90. doi:10.1038/sj.gt.3302038
78. Raki M, Sarkioja M, Escutenaire S, Kangasniemi L, Haavisto E, Kanerva A, et al. Switching the fiber knob of oncolytic adenoviruses to avoid neutralizing antibodies in human cancer patients. J Gene Med (2011) 13:253–61. doi:10.1002/jgm.1565
79. Friedman A, Tian JP, Fulci G, Chiocca EA, Wang J. Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res (2006) 66:2314–9. doi:10.1158/0008-5472.CAN-05-2661
80. Shen BH, Hermiston TW. Effect of hypoxia on Ad5 infection, transgene expression and replication. Gene Ther (2005) 12:902–10. doi:10.1038/sj.gt.3302448
81. Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res (2007) 67:10664–8. doi:10.1158/0008-5472.CAN-07-3107
82. Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG. Oncolytic viruses: from bench to bedside with a focus on safety. Hum Vaccin Immunother (2015) 11:1573–84. doi:10.1080/21645515.2015.1037058
83. Haddad D, Fong Y. Molecular imaging of oncolytic viral therapy. Mol Ther Oncolytics (2015) 1:14007. doi:10.1038/mto.2014.7
84. Miller A, Russell SJ. The use of the NIS reporter gene for optimizing oncolytic virotherapy. Expert Opin Biol Ther (2016) 16:15–32. doi:10.1517/14712598.2016.1100162
85. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science (1990) 250:1262–6. doi:10.1126/science.2173860
86. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med (1998) 187:341–8. doi:10.1084/jem.187.3.341
87. Kaufman HL, Ruby CE, Hughes T, Slingluff CL Jr. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J Immunother Cancer (2014) 2:11. doi:10.1186/2051-1426-2-11
88. Sterman DH. Gene therapy for malignant pleural mesothelioma. Hematol Oncol Clin North Am (2005) 19:1147–73,viii. doi:10.1016/j.hoc.2005.09.004
89. Sterman DH, Treat J, Litzky LA, Amin KM, Coonrod L, Molnar-Kimber K, et al. Adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy in patients with localized malignancy: results of a phase I clinical trial in malignant mesothelioma. Hum Gene Ther (1998) 9:1083–92. doi:10.1089/hum.1998.9.7-1083
90. Sterman DH, Molnar-Kimber K, Iyengar T, Chang M, Lanuti M, Amin KM, et al. A pilot study of systemic corticosteroid administration in conjunction with intrapleural adenoviral vector administration in patients with malignant pleural mesothelioma. Cancer Gene Ther (2000) 7:1511–8. doi:10.1038/sj.cgt.7700269
91. Sterman DH, Recio A, Carroll RG, Gillespie CT, Haas A, Vachani A, et al. A phase I clinical trial of single-dose intrapleural IFN-beta gene transfer for malignant pleural mesothelioma and metastatic pleural effusions: high rate of antitumor immune responses. Clin Cancer Res (2007) 13:4456–66. doi:10.1158/1078-0432.CCR-07-0403
92. Sterman DH, Recio A, Haas AR, Vachani A, Katz SI, Gillespie CT, et al. A phase I trial of repeated intrapleural adenoviral-mediated interferon-beta gene transfer for mesothelioma and metastatic pleural effusions. Mol Ther (2010) 18:852–60. doi:10.1038/mt.2009.309
93. Sterman DH, Haas A, Moon E, Recio A, Schwed D, Vachani A, et al. A trial of intrapleural adenoviral-mediated Interferon-α2b gene transfer for malignant pleural mesothelioma. Am J Respir Crit Care Med (2011) 184:1395–9. doi:10.1164/rccm.201103-0554CR
94. Sterman DH, Alley E, Stevenson JP, Friedberg J, Metzger S, Recio A, et al. Pilot and feasibility trial evaluating immuno-gene therapy of malignant mesothelioma using intrapleural delivery of adenovirus-IFNα combined with chemotherapy. Clin Cancer Res (2016) 22(15):3791–800. doi:10.1158/1078-0432.CCR-15-2133
95. Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T, et al. Phase I study with ONCOS-102 for the treatment of solid tumors – an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer (2016) 4:17. doi:10.1186/s40425-016-0121-5
96. Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther (2010) 18:1874–84. doi:10.1038/mt.2010.161
97. ClinicalTrials.gov [Internet]. Identifier NCT01721018, Intrapleural Administration of HSV1716 to Treat Patients with Malignant Pleural Mesothelioma; 2012 [cited 2017 Apr 4]; [about 4 screens]. Bethesda, MD: National Library of Medicine (US); 2000. Available from: https://clinicaltrials.gov/ct2/show/NCT01721018
98. Mukherjee S, Haenel T, Himbeck R, Scott B, Ramshaw I, Lake RA, et al. Replication-restricted vaccinia as a cytokine gene therapy vector in cancer: persistent transgene expression despite antibody generation. Cancer Gene Ther (2000) 7:663–70. doi:10.1038/sj.cgt.7700133
99. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature (2011) 477:99–102. doi:10.1038/nature10358
100. ClinicalTrials.gov [Internet]. Identifier NCT01503177, Intrapleural Measles Virus Therapy in Patients With Malignant Pleural Mesothelioma; 2011 [cited 2017 Apr 4]; [about 5 screens]. Bethesda, MD: National Library of Medicine (US); 2000. Available from: https://clinicaltrials.gov/ct2/show/NCT01503177
101. Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL, et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol (2002) 20:2251–66. doi:10.1200/JCO.2002.08.042
102. Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res (2010) 16:5564–72. doi:10.1158/1078-0432.CCR-10-1233
103. Hecht JR, Bedford R, Abbruzzese JL, Lahoti S, Reid TR, Soetikno RM, et al. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res (2003) 9:555–61.
104. Reid T, Galanis E, Abbruzzese J, Sze D, Andrews J, Romel L, et al. Intra-arterial administration of a replication-selective adenovirus (dl1520) in patients with colorectal carcinoma metastatic to the liver: a phase I trial. Gene Ther (2001) 8:1618–26. doi:10.1038/sj.gt.3301512
105. Smythe WR, Hwang HC, Amin KM, Eck SL, Davidson BL, Wilson JM, et al. Use of recombinant adenovirus to transfer the herpes simplex virus thymidine kinase (HSVtk) gene to thoracic neoplasms: an effective in vitro drug sensitization system. Cancer Res (1994) 54:2055–9.
106. Smythe WR, Kaiser LR, Hwang HC, Amin KM, Pilewski JM, Eck SJ, et al. Successful adenovirus-mediated gene transfer in an in vivo model of human malignant mesothelioma. Ann Thorac Surg (1994) 57:1395–401. doi:10.1016/0003-4975(94)90090-6
107. Smythe WR, Hwang HC, Elshami AA, Amin KM, Eck SL, Davidson BL, et al. Treatment of experimental human mesothelioma using adenovirus transfer of the herpes simplex thymidine kinase gene. Ann Surg (1995) 222:78–86. doi:10.1097/00000658-199507000-00013
108. Hwang HC, Smythe WR, Elshami AA, Kucharczuk JC, Amin KM, Williams JP, et al. Gene therapy using adenovirus carrying the herpes simplex-thymidine kinase gene to treat in vivo models of human malignant mesothelioma and lung cancer. Am J Respir Cell Mol Biol (1995) 13:7–16. doi:10.1165/ajrcmb.13.1.7598939
109. Elshami AA, Kucharczuk JC, Zhang HB, Smythe WR, Hwang HC, Litzky LA, et al. Treatment of pleural mesothelioma in an immunocompetent rat model utilizing adenoviral transfer of the herpes simplex virus thymidine kinase gene. Hum Gene Ther (1996) 7:141–8. doi:10.1089/hum.1996.7.2-141
110. Christmas TI, Manning LS, Garlepp MJ, Musk AW, Robinson BW. Effect of interferon-alpha 2a on malignant mesothelioma. J Interferon Res (1993) 13:9–12. doi:10.1089/jir.1993.13.9
111. Astoul P, Picat-Joossen D, Viallat JR, Boutin C. Intrapleural administration of interleukin-2 for the treatment of patients with malignant pleural mesothelioma: a Phase II study. Cancer (1998) 83:2099–104. doi:10.1002/(SICI)1097-0142(19981115)83:10<2099::AID-CNCR8>3.0.CO;2-3
112. Boutin C, Nussbaum E, Monnet I, Bignon J, Vanderschueren R, Guerin JC, et al. Intrapleural treatment with recombinant gamma-interferon in early stage malignant pleural mesothelioma. Cancer (1994) 74:2460–7. doi:10.1002/1097-0142(19941101)74:9<2460::AID-CNCR2820740912>3.0.CO;2-N
113. Cordier Kellerman L, Valeyrie L, Fernandez N, Opolon P, Sabourin JC, Maubec E, et al. Regression of AK7 malignant mesothelioma established in immunocompetent mice following intratumoral gene transfer of interferon gamma. Cancer Gene Ther (2003) 10:481–90. doi:10.1038/sj.cgt.7700594
114. Odaka M, Sterman DH, Wiewrodt R, Zhang Y, Kiefer M, Amin KM, et al. Eradication of intraperitoneal and distant tumor by adenovirus-mediated interferon-beta gene therapy is attributable to induction of systemic immunity. Cancer Res (2001) 61:6201–12.
115. Friedlander PL, Delaune CL, Abadie JM, Toups M, LaCour J, Marrero L, et al. Efficacy of CD40 ligand gene therapy in malignant mesothelioma. Am J Respir Cell Mol Biol (2003) 29:321–30. doi:10.1165/rcmb.2002-0226OC
116. Gotoh A, Kanno T, Nagaya H, Nakano T, Tabata C, Fukuoka K, et al. Gene therapy using adenovirus against malignant mesothelioma. Anticancer Res (2012) 32:3743–7.
117. Kubo S, Kawasaki Y, Yamaoka N, Tagawa M, Kasahara N, Terada N, et al. Complete regression of human malignant mesothelioma xenografts following local injection of midkine promoter-driven oncolytic adenovirus. J Gene Med (2010) 12:681–92. doi:10.1002/jgm.1486
118. Takagi-Kimura M, Yamano T, Tamamoto A, Okamura N, Okamura H, Hashimoto-Tamaoki T, et al. Enhanced antitumor efficacy of fiber-modified, midkine promoter-regulated oncolytic adenovirus in human malignant mesothelioma. Cancer Sci (2013) 104:1433–9. doi:10.1111/cas.12267
119. Watanabe Y, Kojima T, Kagawa S, Uno F, Hashimoto Y, Kyo S, et al. A novel translational approach for human malignant pleural mesothelioma: heparanase-assisted dual virotherapy. Oncogene (2010) 29:1145–54. doi:10.1038/onc.2009.415
120. Treat J, Kaiser LR, Sterman DH, Litzky L, Davis A, Wilson JM, et al. Treatment of advanced mesothelioma with the recombinant adenovirus H5.010RSVTK: a phase 1 trial (BB-IND 6274). Hum Gene Ther (1996) 7:2047–57. doi:10.1089/hum.1996.7.16-2047
121. Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med (2000) 6:1134–9. doi:10.1038/80474
122. Kesari S, Randazzo BP, Valyi-Nagy T, Huang QS, Brown SM, MacLean AR, et al. Therapy of experimental human brain tumors using a neuroattenuated herpes simplex virus mutant. Lab Invest (1995) 73:636–48.
123. Kemeny N, Brown K, Covey A, Kim T, Bhargava A, Brody L, et al. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther (2006) 17:1214–24. doi:10.1089/hum.2006.17.1214
124. Gayral M, Lulka H, Hanoun N, Biollay C, Sèlves J, Vignolle-Vidoni A, et al. Targeted oncolytic herpes simplex virus type 1 eradicates experimental pancreatic tumors. Hum Gene Ther (2015) 26:104–13. doi:10.1089/hum.2014.072
125. Kucharczuk JC, Randazzo B, Chang MY, Amin KM, Elshami AA, Sterman DH, et al. Use of a "replication-restricted" herpes virus to treat experimental human malignant mesothelioma. Cancer Res (1997) 57:466–71.
126. Adusumilli PS, Stiles BM, Chan MK, Mullerad M, Eisenberg DP, Ben-Porat L, et al. Imaging and therapy of malignant pleural mesothelioma using replication-competent herpes simplex viruses. J Gene Med (2006) 8:603–15. doi:10.1002/jgm.877
127. Adusumilli PS, Chan MK, Chun YS, Hezel M, Chou TC, Rusch VW, et al. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol Ther (2006) 5:48–53. doi:10.4161/cbt.5.1.2237
128. Adusumilli PS, Chan MK, Hezel M, Yu Z, Stiles BM, Chou TC, et al. Radiation-induced cellular DNA damage repair response enhances viral gene therapy efficacy in the treatment of malignant pleural mesothelioma. Ann Surg Oncol (2007) 14:258–69. doi:10.1245/s10434-006-9127-4
129. Scholl SM, Balloul JM, Le Goc G, Bizouarne N, Schatz C, Kieny MP, et al. Recombinant vaccinia virus encoding human MUC1 and IL2 as immunotherapy in patients with breast cancer. J Immunother (2000) 23:570–80. doi:10.1097/00002371-200009000-00007
130. Kaufman HL, Deraffele G, Mitcham J, Moroziewicz D, Cohen SM, Hurst-Wicker KS, et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J Clin Invest (2005) 115:1903–12. doi:10.1172/JCI24624
131. Eder JP, Kantoff PW, Roper K, Xu GX, Bubley GJ, Boyden J, et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res (2000) 6:1632–8.
132. Chan WM, McFadden G. Oncolytic poxviruses. Annu Rev Virol (2014) 1:119–41. doi:10.1146/annurev-virology-031413-085442
133. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19:329–36. doi:10.1038/nm.3089
134. ClinicalTrials.gov [Internet]. Identifier NCT02562755, Hepatocellular Carcinoma Study Comparing Vaccinia Virus Based Immunotherapy Plus Sorafenib vs Sorafenib Alone (PHOCUS); 2015 [cited 2017 Apr 4]; [about 4 screens]. Bethesda, MD: National Library of Medicine (US); 2000. Available from: https://clinicaltrials.gov/ct2/show/NCT02562755
135. Zhang Q, Yu YA, Wang E, Chen N, Danner RL, Munson PJ, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res (2007) 67:10038–46. doi:10.1158/0008-5472.CAN-07-0146
136. Kelly KJ, Woo Y, Brader P, Yu Z, Riedl C, Lin SF, et al. Novel oncolytic agent GLV-1h68 is effective against malignant pleural mesothelioma. Hum Gene Ther (2008) 19:774–82. doi:10.1089/hum.2008.036
137. Belin LJ, Ady JW, Lewis C, Marano D, Gholami S, Mojica K, et al. An oncolytic vaccinia virus expressing the human sodium iodine symporter prolongs survival and facilitates SPECT/CT imaging in an orthotopic model of malignant pleural mesothelioma. Surgery (2013) 154:486–95. doi:10.1016/j.surg.2013.06.004
138. Acuna SA, Ottolino-Perry K, Çako B, Tang N, Angarita FA, McCart JA. Oncolytic vaccinia virus as an adjuvant treatment to cytoreductive surgery for malignant peritoneal mesothelioma. Ann Surg Oncol (2014) 21:2259–66. doi:10.1245/s10434-014-3651-4
139. Vanderplasschen A, Hollinshead M, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J Gen Virol (1997) 78(Pt 8):2041–8. doi:10.1099/0022-1317-78-8-2041
140. Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci U S A (1998) 95:7544–9. doi:10.1073/pnas.95.13.7544
141. Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res (2002) 62:4656–62.
142. Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther (2010) 18:1155–64. doi:10.1038/mt.2010.43
143. Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc (2014) 89:926–33. doi:10.1016/j.mayocp.2014.04.003
144. ClinicalTrials.gov [Internet]. Identifier NCT00450814, Vaccine Therapy With or Without Cyclophosphamide in Treating Patients With Recurrent or Refractory Multiple Myeloma; 2007 Mar 20 [cited 2017 Apr 4]; [about 7 screens]. Bethesda, MD: National Library of Medicine (US); 2000. Available from: https://clinicaltrials.gov/ct2/show/NCT00450814
145. Gauvrit A, Brandler S, Sapede-Peroz C, Boisgerault N, Tangy F, Gregoire M. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res (2008) 68:4882–92. doi:10.1158/0008-5472.CAN-07-6265
146. Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther (2010) 17:550–8. doi:10.1038/cgt.2010.10
147. Achard C, Boisgerault N, Delaunay T, Roulois D, Nedellec S, Royer PJ, et al. Sensitivity of human pleural mesothelioma to oncolytic measles virus depends on defects of the type I interferon response. Oncotarget (2015) 6:44892–904. doi:10.18632/oncotarget.6285
148. Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol (2004) 17:516–27. doi:10.1089/vim.2004.17.516
149. Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, et al. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res (2009) 69:7713–20. doi:10.1158/0008-5472.CAN-09-1013
150. Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther (2010) 21:51–64. doi:10.1089/hum.2009.088
151. Elankumaran S, Chavan V, Qiao D, Shobana R, Moorkanat G, Biswas M, et al. Type I interferon-sensitive recombinant newcastle disease virus for oncolytic virotherapy. J Virol (2010) 84:3835–44. doi:10.1128/JVI.01553-09
152. Silberhumer GR, Brader P, Wong J, Serganova IS, Gönen M, Gonzalez SJ, et al. Genetically engineered oncolytic Newcastle disease virus effectively induces sustained remission of malignant pleural mesothelioma. Mol Cancer Ther (2010) 9:2761–9. doi:10.1158/1535-7163.MCT-10-0090
153. Kitamura A, Matsushita K, Takiguchi Y, Shimada H, Tada Y, Yamanaka M, et al. Synergistic effect of non-transmissible Sendai virus vector encoding the c-myc suppressor FUSE-binding protein-interacting repressor plus cisplatin in the treatment of malignant pleural mesothelioma. Cancer Sci (2011) 102:1366–73. doi:10.1111/j.1349-7006.2011.01931.x
154. Yamanaka M, Tada Y, Kawamura K, Li Q, Okamoto S, Chai K, et al. E1B-55 kDa-defective adenoviruses activate p53 in mesothelioma and enhance cytotoxicity of anticancer agents. J Thorac Oncol (2012) 7:1850–7. doi:10.1097/JTO.0b013e3182725fa4
155. Kruklitis RJ, Singhal S, Delong P, Kapoor V, Sterman DH, Kaiser LR, et al. Immuno-gene therapy with interferon-beta before surgical debulking delays recurrence and improves survival in a murine model of malignant mesothelioma. J Thorac Cardiovasc Surg (2004) 127:123–30. doi:10.1016/j.jtcvs.2003.08.034
156. Boisgerault N, Achard C, Delaunay T, Cellerin L, Tangy F, Grégoire M, et al. Oncolytic virotherapy for human malignant mesothelioma: recent advances. Oncolytic Virother (2015) 4:133–40. doi:10.2147/OV.S66091
Keywords: mesothelioma, oncolytic, virotherapy, novel, measles virus, adenovirus, herpes simplex virus type 1, vaccinia virus
Citation: Pease DF and Kratzke RA (2017) Oncolytic Viral Therapy for Mesothelioma. Front. Oncol. 7:179. doi: 10.3389/fonc.2017.00179
Received: 13 June 2017; Accepted: 04 August 2017;
Published: 24 August 2017
Edited by:
Benjamin Gesundheit, Cell-El Ltd., IsraelReviewed by:
Joel S. Greenberger, University of Pittsburgh Medical Center, United StatesTullio Florio, Università di Genova, Italy
Copyright: © 2017 Pease and Kratzke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert A. Kratzke, a3JhdHowMDNAdW1uLmVkdQ==