 Galdo Bustos
Galdo Bustos Pablo Cruz
Pablo Cruz Alenka Lovy
Alenka Lovy César Cárdenas
César Cárdenas- 1Anatomy and Developmental Biology Program, Institute of Biomedical Sciences, University of Chile, Santiago, Chile
- 2Geroscience Center for Brain Health and Metabolism, Santiago, Chile
- 3Department of Neuroscience, Center for Neuroscience Research, Tufts University School of Medicine, Boston, MA, United States
- 4Buck Institute for Research on Aging, Novato, CA, United States
- 5Department of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, CA, United States
Cancer is characterized by an uncontrolled cell proliferation rate even under low nutrient availability, which is sustained by a metabolic reprograming now recognized as a hallmark of cancer. Warburg was the first to establish the relationship between cancer and mitochondria; however, he interpreted enhanced aerobic glycolysis as mitochondrial dysfunction. Today it is accepted that many cancer cell types need fully functional mitochondria to maintain their homeostasis. Calcium (Ca2+)—a key regulator of several cellular processes—has proven to be essential for mitochondrial metabolism. Inositol 1,4,5-trisphosphate receptor (IP3R)-mediated Ca2+ transfer from the endoplasmic reticulum to the mitochondria through the mitochondrial calcium uniporter (MCU) proves to be essential for the maintenance of mitochondrial function and cellular energy balance. Both IP3R and MCU are overexpressed in several cancer cell types, and the inhibition of the Ca2+ communication between these two organelles causes proliferation arrest, migration decrease, and cell death through mechanisms that are not fully understood. In this review, we summarize and analyze the current findings in this area, emphasizing the critical role of Ca2+ and mitochondrial metabolism in cancer and its potential as a novel therapeutic target.
Introduction
Mitochondria arose around two billion years ago as the result of a symbiotic interaction between an archaeon ancestor and a α-proteobacterium (1). The modern mitochondria has retained a small circular polycystronic 16 kilobase genome (2) that controls the synthesis of about 67 proteins, including 13 proteins that are core constituents of the electron transport chain (complex I–IV) (3). The terminal complex V of the electron transport chain, the ATP synthase, catalyzes the synthesis of most of the ubiquitous cellular energy currency, ATP (4). In addition, mitochondria play a vital role in metal ion homeostasis (5), programmed cell death (6–8), and the synthesis of building blocks for the generation of amino acids, lipids, and nucleotides (9). In many cancer types, glucose-derived pyruvate is transported away from mitochondria, turned to lactate and excreted (10). Detection of glutamine-derived carbon as lactate has established glutamine as an important energy source in tumor cells (11, 12). Glutamine contributes the bulk of carbon to the tricarboxylic acid (TCA) cycle through a phenomenon known as anaplerosis (13), maintaining a robust amount of citrate and malate for biosynthesis of lipids and nucleotides, respectively, in addition to providing an extra supply of reductive power (NADH) (9, 14). Also, glutamine activation of the TCA cycle helps to maintain the mitochondrial membrane potential (ΔΨm) (15), avoiding opening of the permeability transition pore and the release of pro-apoptotic factors (16). Mitochondria play a pivotal role in maintaining cellular homeostasis, and their function is essential for the viability of cancer cells.
Mitochondrial Metabolism in Cancer
A hallmark feature of cancer cells is a re-programming of their metabolism even when nutrients are available (17, 18). All major tumor suppressors and oncogenes are now recognized to have fundamental connections with metabolic pathways (10, 19, 20). Warburg suggested that cancer originates from an irreversible injury to mitochondria followed by a compensatory increase of glycolysis (21), but growing evidence indicates that most cancer cells rely on mitochondrial metabolism (20, 22–24). For example, mitochondrial function is essential for the survival of diffuse large B cell tumors (25), primary glioblastoma sphere cultures (26), pancreatic (27), and leukemic cancer (28). Moreover, a subset of cancer cells that exclusively rely on glycolysis for ATP production [with mutations either in components of the succinate dehydrogenase complex or the fumarate hydratase rendering mitochondrial oxidative phosphorylation (OXPHOS) dysfunctional] still use and need the oxidation of alpha-ketoglutarate (α-KG) in the mitochondria to generate the reducing equivalents necessary to sustain the reductive carboxylation pathway and the generation of metabolic intermediates (29). The mitochondrial function in cancer cells is sustained by a high uptake of the non-essential amino acid glutamine (15), which is converted to glutamate by glutaminases and subsequently converted to α-KG by either glutamate dehydrogenases or aminotransferases (30). α-KG then enters the TCA cycle where it is a substrate of the α-KG dehydrogenase (α-KGDH), a highly regulated enzyme that catalyzes the conversion of α-KG, coenzyme A and NAD+ to succinyl-CoA, CO2 and NADH providing electrons to the respiratory chain (14, 29). Importantly in the context of this review, α-KGDH activity is strongly Ca2+ dependent (31). Ca2+ also activates directly the isocitrate dehydrogenase (ISDH) and indirectly the pyruvate dehydrogenase (PDH) through regulation of its phosphorylation state (32, 33). In general, increases of Ca2+ in the 0.1–10 µM range up-regulates the activities of these enzymes, resulting in higher mitochondrial NADH levels (34, 35). The regulation by Ca2+ of these three key mitochondrial dehydrogenases has a strategic task in coordinating cellular workload and generation of ATP (36). Several studies demonstrate that Ca2+ signaling plays an important role in cancer progression by promoting proliferation, cell migration, metastasis and vascularization, and conferring apoptosis resistance (37). However, how Ca2+ signals may affect mitochondrial metabolism and impact the aforementioned features remains poorly understood. Although Ca2+ is the focus of this mini-review, it is not the only ion able to affect mitochondrial metabolism. For example, K+ transport modulates the coupling between mitochondrial respiration and ATP synthesis (38). In fact, inhibition of the mitochondrial potassium channel Kv1.3 or the two-pore potassium channel TASK-3 compromises mitochondrial function (39, 40). Mg2+ is also important, since the knockdown of Mrs2, a Mg2+-selective channel in the inner mitochondrial membrane (IMM), induces the loss of complex I and causes mitochondrial membrane depolarization (41).
Inositol 1,4,5-Trisphosphate Receptors (IP3Rs) and Cancer
The IP3Rs are a ubiquitous family of Ca2+ release channels composed of three isoforms (1, 2, and 3) present primarily in the endoplasmic reticulum (ER) (42) but can also be found in the nuclear envelope where they regulate gene transcription (43, 44). In addition, Ca2+ release through the IP3R regulates numerous other cellular functions including secretion, motility and autophagy (42, 45). Agonist-induced IP3R Ca2+ signals enhance mitochondrial function (46–48) primarily by stimulating the TCA cycle dehydrogenases (PDH, α-KGDH, and ISDH) (31), as well as respiratory chain components to promote OXPHOS and ATP production (49, 50). The above is possible in part by the spatial proximity that exists between these two organelles, which allows the establishment of a structural and functional coupling known as mitochondria-associated membranes (MAMs). These structures were reported in the early 1950s (51–53) and have been described as signaling platforms involved in many cellular processes including metabolism control, migration, differentiation, proliferation and cell death (54). Interestingly, many oncosupressors and oncogene proteins are located in the MAMs and interact with the IP3R (such as AKT, PML, PTEN, and mTORC2) modifying calcium release patterns and cellular fate, which has been thoroughly discussed elsewhere (55, 56).
In cancer, the expression of IP3Rs, in particular the IP3R-3 isoform, is up-regulated in glioblastoma (57), gastric (58), small and non-small lung (59), and colorectal cancer (60). Importantly, the over-expression of IP3R-3 in gastric cancer was found in cell lines established from cells that invade the peritonea, while the ones made from primary tumor cells show normal levels of IP3R expression (58). Along these lines, in colorectal cancer IP3R-3 was found in the advancing margins of the tumors, correlating with depth of invasion, lymph node metastasis and liver metastasis (60). In glioblastoma, the inhibition of IP3R with caffeine, a non-specific inhibitor of the IP3R, decreased migration in various in vitro assays and increased mean survival in a mouse xenograft model of glioblastoma (57). In addition, siRNA silencing of IP3R-3 in the colon cancer cell line CACO-2, or non-specific pharmacological inhibition of IP3R by 2APB in gastric cancer cells, induces apoptosis, while over-expression of the receptors protects the cells from apoptosis induced by staurosporine (60). Regarding breast cancer, both estradiol- and ATP-induced proliferations in the MCF7 cell line are mediated by the IP3R-3 (61, 62). Moreover, the breast cancer metastasis suppressor 1, a protein able to suppress formation of secondary tumor masses without blocking growth of neoplastic cells at orthotopic or subcutaneous sites, reduces phosphoinositide signaling, including IP3 in the MDA-MB-435 cell line (63). Clearly, IP3R plays a role in cancer progression and metastasis; however, the mechanism of its involvement has not been elucidated. A summary of the expression of IP3R in cancer and the effect of manipulating its activity is found in Table 1.
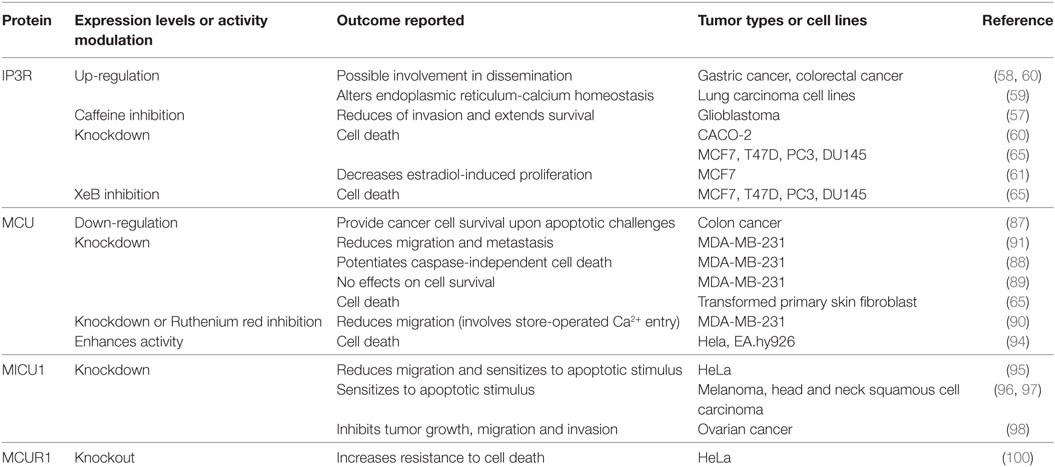
Table 1. Effects of inositol 1,4,5-trisphosphate receptor (IP3R) or MCUC component modulation on cancer cells.
Constitutive IP3R-mediated Ca2+ transfer to mitochondria is essential to maintain cell bioenergetics in normal cells, and its absence induces a bioenergetic stress that causes diminished oxidative respiration (OXPHOS) and leads to an adaptive response characterized by AMPK and autophagy activation (Figure 1A) (64). We find that breast and prostate cancer derivate cells, as well as transformed primary fibroblasts, similar to normal cells need constitutive transfer of IP3R released Ca2+ to the mitochondria to maintain the optimal activity of PDH and, therefore, sufficient amounts of NADH to support the TCA cycle (Figure 1A) (unpublished data). Genetic or pharmacological inhibition of either IP3R or MCU elicits comparable effects to those observed in normal cells, including diminished OXPHOS, AMPK activation, and induction of a pro-survival autophagy (Figures 1B,C); however, whereas normal cells were able to survive these challenges, much of the cancer cell population (60–70%) could not (Figure 1C). The cancer cell death was caused by a lack of mitochondrial function because mitochondrial substrates, which bypassed the inhibition of the TCA cycle and enabled mitochondria to work, were able to rescue the cells (65, 66). Moreover, we determined that a decrease in nucleotides, whose synthesis occurs partially in the mitochondria, added to the inability of cancer cells to arrest the cell cycle upon metabolic stress and was most likely responsible for the mitotic catastrophe that leads to cell death in cancer cells, as supplementation with nucleotides promoted survival.
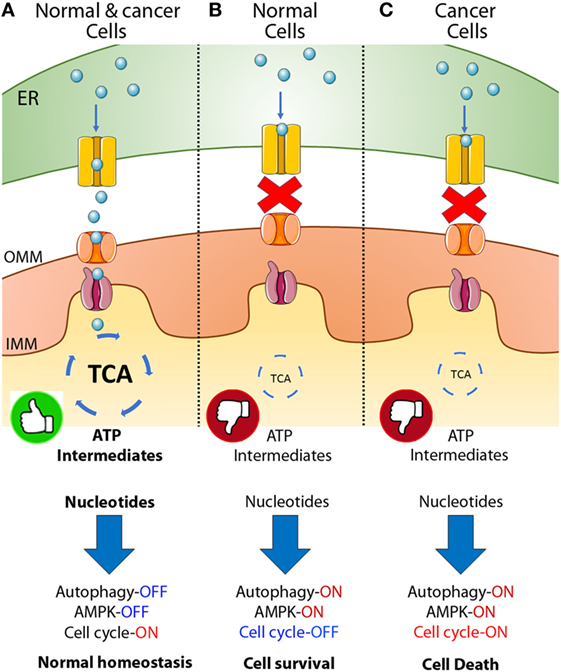
Figure 1. Inositol 1,4,5-trisphosphate receptor (IP3R)-mediated calcium transfer to mitochondria in normal and cancer cells. (A) In normal and cancer cells Ca2+ released from the endoplasmic reticulum (ER) through the IP3R enters the mitochondrial matrix through the mitochondrial calcium uniporter (MCU) and activates key dehydrogenases of the tricarboxylic acid (TCA) cycle, maintaining a robust amount of ATP and metabolic intermediates or building blocks for the generation of fatty acids, amino acids and nucleotides allowing the cells to enter the cell cycle, proliferate, and keep normal homeostasis. (B) In normal cells, the inhibition of the Ca2+ transfer to mitochondria generates a decrease in TCA cycle activity with the concomitant reduction in ATP and metabolic intermediates, inducing AMPK, autophagy and the complete shutdown of the cell cycle caused in part by a reduction in the availability of nucleotides. (C) In cancer, a similar phenomenon is observed after inhibition of Ca2+ transfer to mitochondria; decrease in TCA cycle activity, ATP, and metabolic intermediates, activation of AMPK and autophagy, and reduction in the amount of nucleotides available. However, cancer cells continue entering the cell cycle despite the metabolically unfavorable environment causing cell death (65, 66).
MCU and Cancer
In the early 1960s, it was observed that energized mitochondria were capable of taking up and accumulating Ca2+ in the matrix (67–69) and further studies found that mitochondrial Ca2+ uptake could be pharmacologically inhibited by ruthenium red and Ru360 (70–72). Nevertheless, this mitochondrial feature was ignored for many years and, only in the 1990s, did mitochondrial Ca2+ uptake begin to be considered an important mechanism for Ca2+ homeostasis (73–75). The capacity to take up and accumulate Ca2+ in the mitochondrial matrix is substantial, reaching concentrations of 100 µM (5). Nevertheless, most of the Ca2+ that enters the mitochondrial matrix is quickly chelated due to the action of phosphate; therefore, free [Ca2+] within the matrix is much lower compared to total [Ca2+], which can reach up to 1 M, as occurs in neurons. According to the chemiosmotic theory of Mitchell, the entrance of Ca2+ into the mitochondrial matrix is driven by the negative potential inside the membrane (76). However, exactly how Ca2+ enters the mitochondrial matrix was a critical question unsolved for more than 50 years.
Ca2+ crosses the outer mitochondrial membrane through the voltage-dependent anion-selective channels (VDACs), a family of channels by which different ions and molecules enter into the intermembrane space (77). VDACs play a significant role in cancer by supporting glycolytic metabolism and preventing apoptosis, which has been comprehensively reviewed recently elsewhere (78). Ca2+ then enters the mitochondrial matrix through the so-called uniporter, whose molecular identity was unknown until recently (79, 80). The discovery of the regulatory subunit mitochondrial Ca2+ uptake 1 (MICU1) was the first step in this field, discovered in 2010 by Mootha’s group, who used siRNA screening of over 13 candidates and identified an IMM protein of 54 kDa necessary for mitochondrial Ca2+ uptake (81). A year later, Mootha’s and Rizzuto’s group in parallel identified a 40 kDa protein as the pore-forming element of the uniporter, the mitochondrial calcium uniporter (MCU) (79, 80). These findings radically opened further studies, and today, we know that the mitochondrial uniporter is a multi-protein complex (MCUC) formed by MCU, MCUb, EMRE (core components of the pore) (82–84), MICU1 and 2 (81, 85), MCUR1 (86), and SLC25A23 (86) (associate regulators).
The role of the MCUC in cancer is just beginning to be explored and already with contradicting results (see Table 1) For example, the reduced expression of MCU caused by over-expression of the microRNA miR-25 in colon cancer favors cancer cell survival by preventing mitochondrial Ca2+ overload upon several apoptotic challenges (87). In contrast, several groups, using various different algorithms, find that the expression of MCU correlates with a poor prognosis, invasive behavior, and metastasis in breast cancer (88–91). Zou’s group shows that knockdown of MCU decreases migration through a mechanism that implies a reduction of the store-operated Ca2+ entry (90); however, the molecular details of this mechanism remain unclear. More recently, Tosatto et al. elegantly show that MCU silencing decreases migration and metastasis through a mechanism that includes an increase in the NAD(P)H/NADH ratio, changing the antioxidant capacity of the cell and decreasing the steady-state levels of mitochondrial reactive oxygen species (ROS). This in turn affects the stabilization of Hypoxia-inducible factor-1α (HIF-1α), with the concomitant decrease in its target genes such as the hexokinase II, the glucose-6-phosphate isomerase and the lysyl oxidase, which are critical for migration (91). The change in the NAD(P)H/NADH cannot be explained solely as the result of a decrease in the TCA cycle flux suggesting the presence of other Ca2+-sensitive mechanisms in the mitochondria that need to be explored.
The knockdown of MCU in the highly aggressive breast cancer cell line MDA-MB-231 does not affect proliferation or cell viability (88–91), unless the cells are challenged with a caspase-independent cell death inducer such as ionomycin, which shows that MCU knockdown cells are significantly more sensitive than MCU expressing cells (88). Interestingly, in transformed primary skin fibroblasts, the knockdown of MCU induces a massive cell death (64), perhaps reflecting cell type-specific differences regarding the role of MCU or the use of the mitochondria, since the MDA-MB-231 cells are highly glycolytic while the skin primary fibroblasts are more oxidative. Along these lines, primary endothelial cells derived from tissue-specific MCU or MCUR1 null mice also show a reduction in migration but, contrary to work in breast cancer, also show reduction in proliferation that correlates with the decrease in mitochondrial Ca2+ uptake and impaired mitochondrial bioenergetics (92). Interestingly, in endothelial cell lines, the oxidation of MCU Cys-97 enhances MCU channel activity increasing mitochondrial Ca2+ uptake and ROS generation. The persistent elevation of mitochondrial Ca2+ and ROS generation harms mitochondrial bioenergetic function, reducing migration and sensitizing cells to death (93). Also, a decrease in sarco/ER calcium ATPase (SERCA) activity in HeLa and EA.hy926 cancer cell lines caused by a drop in mitochondrial ATP levels (using resveratrol, piceatannol, or oligomycin) enhanced mitochondrial Ca2+ uptake causing Ca2+ overload and apoptosis, specifically in cancer cells (94).
The knockdown of MICU1, which is contrary to the MCU or MCUR1 knockdown, causes a constitutive entry of Ca2+ to the mitochondria, also reduced migration, and sensitized cells to apoptotic stimuli (95). In agreement, in melanoma, one of the most aggressive and lethal cancers, the knockdown of the ribosomal protein S3, reduced the expression of MICU1 allowing a mitochondrial Ca2+ overload that triggered apoptosis (96). Similarly, the inhibition of the enhancer of zeste homolog 2, a component of the Policomb repressive complex 2, involved in the regulation of homeotic (Hox) gene expression in head and neck squamous cell carcinoma downregulated MICU1 and caused cell death (97). Along these lines, MICU1 expression in ovarian cancer decreased PDH activity and hence the TCA cycle, shifting the cells toward glycolysis, providing chemoresistance but leading to a poor overall survival. Silencing MICU1 increases OXPHOS and inhibits tumor growth, migration, and invasion (98). Physiologically, MICU1 is a gatekeeper for MCU-mediated Ca2+ uptake, establishing a threshold that prevents Ca2+ uptake when [Ca2+] is low (<3 µM), similar to that observed during rest or during weak agonist stimulation. At higher [Ca2+] MICU1 allows the entry of Ca2+, essential to maintain cellular bioenergetics and viability (95). In fact, a recent publication of Graier group has demonstrated that cancer cells use the uncoupling proteins 2 and 3 (UCP2/3) to re-establish MICU1 function (and in turn mitochondrial Ca2+ uptake), which is desensitized by methylation mediated by PRMT1 (99), an arginine methyl transferase highly expressed in cancer (100). The presence of this compensatory mechanism confirms the importance of preserved mitochondrial function in cancer. Finally, MCUR1 knockdown inhibits cell death induced by mitochondrial Ca2+ overload and is involved in establishing the mitochondrial permeability transition (101) and, therefore, it will be interesting to determine its role in cancer. Clearly, fine-tuning of mitochondrial calcium is fundamental for mitochondrial performance and ultimately cellular fate, and it will be exciting to further decipher the role of the MCUC in cancer.
Ryanodine Receptor (RyRs) and SERCAs in Cancer
The RyRs and the SERCA are the major Ca2+ players in the ER together with the IP3R (102). RyRs are a family of Ca2+-release channels composed of three isoforms (1, 2, and 3) that have rarely been associated with cancer, despite the fact that they regulate proliferation of melanocytes and T cells (103, 104) and migration in astrocytes (105). Implications in apoptosis regulation in prostate cancer cell line LNCaP (106) and a direct correlation with tumor grades on breast cancer (107) are the strongest findings so far. It is possible that the limited participation of RyR in MAMs and Ca2+ communication with mitochondria prevents them from participating more actively in cancer.
In contrast, SERCA, consisting of three isoforms, such as SERCA1–2, which is expressed differentially in several tissues, and SERCA3, which is ubiquitously expressed (108), has been broadly associated with different types of cancer. Alteration of SERCA expression has been reported in oral squamous cancer (109), choroid plexus (110), thyroid (111), lung (59, 112), colon (113, 114), acute promyelocytic leukemia (108), cervical (115), and breast cancers (116). In colon cancer, SERCA3 expression decreases progressively during the tumorigenesis process becoming virtually null in poorly differentiated tumors (114). Similarly, ductal carcinomas show absence of SERCA3 expression compared to their normal counterparts (116) as well as acute promyelocytic leukemia cells (108) and Burkitt’s lymphoma cells (117), the details of which have been discussed thoroughly elsewhere (37, 108, 118).
Conclusion
Warburg was the first to propose a link between mitochondria and cancer by suggesting that the origin of cancer cells was an irreversible lesion in mitochondria that raises the glycolytic rate as a compensatory mechanism. Today, increasing evidence has demystified Warburg’s theory, demonstrating that mitochondria are key actors for cancer biology. To support their uncontrolled proliferation rate, cancer cells need a constant supply of building blocks to feed biomass production. This is achieved by a metabolic re-programming that favors certain metabolic pathways and in this context; mitochondrial metabolism has proven to be essential. Accumulating evidence points to the Ca2+ communication between the ER and mitochondria as key for mitochondrial function and, therefore, for cancer progression. Unfortunately, the limited access of inhibitors either for IP3R or for MCU has prevented the development of pharmacokinetics and pharmacodynamics experiments in vivo, hindering the understanding of the real potential of this pathway as a therapeutic option, as has been achieved for other ion channels and the SERCA pump (118) Thus, the design of new drugs targeting the functional Ca2+ coupling between ER and mitochondria is fundamental to further understand the role of this pathway in phenomena such as angiogenesis, metastasis, chemoresistance, and cancer relapse.
Author Contributions
GB, PC, and CC designed and outlined the structure and contents of the review. GB, PC, AL, and CC contributed to the literature review, discussion, and writing of the manuscript. All authors contributed equally to the draft revisions and final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by FONDECYT grant # 1160332 (CC), FONDAP program grant # 15150012 (CC), and NIH P30NS047243 (AL).
References
1. Margulis L. Symbiotic theory of the origin of eukaryotic organelles: criteria for proof. Symp Soc Exp Biol (1975) 29:21–38.
2. Gabaldón T, Huynen MA. Shaping the mitochondrial proteome. Biochim Biophys Acta (2004) 1659:212–20. doi:10.1016/j.bbabio.2004.07.011
3. Wallace DC. Bioenergetic origins of complexity and disease. Cold Spring Harb Symp Quant Biol (2011) 76:1–16. doi:10.1101/sqb.2011.76.010462
5. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13:566–78. doi:10.1038/nrm3412
6. Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J (1999) 18:6349–61. doi:10.1093/emboj/18.22.6349
7. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J (2001) 20:2690–701. doi:10.1093/emboj/20.11.2690
8. Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science (2003) 300:135–9. doi:10.1126/science.1081208
9. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7:11–20. doi:10.1016/j.cmet.2007.10.002
10. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
11. Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem (1979) 254:2669–76.
12. Kovačević Z. The pathway of glutamine and glutamate oxidation in isolated mitochondria from mammalian cells. Biochem J (1971) 125:757–63. doi:10.1042/bj1250757
13. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem (2002) 277:30409–12. doi:10.1074/jbc.R200006200
14. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104:19345–50. doi:10.1073/pnas.0709747104
15. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35:427–33. doi:10.1016/j.tibs.2010.05.003
16. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J (1999) 341:233–49. doi:10.1042/bj3410233
17. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev (2009) 23:537–48. doi:10.1101/gad.1756509
18. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol (2015) 17:351–9. doi:10.1038/ncb3124
19. DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev (2008) 18:54–61. doi:10.1016/j.gde.2008.02.003
20. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi:10.1038/nrc3038
21. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi:10.1136/bmj.1.4082.694-a
22. Fan Y, Dickman KG, Zong WX. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem (2010) 285:7324–33. doi:10.1074/jbc.M109.035584
23. Moreno-Sánchez R, Marín-Hernández A, Saavedra E, Pardo JP, Ralph SJ, Rodríguez-Enríquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol (2014) 50:10–23. doi:10.1016/j.biocel.2014.01.025
24. Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8:754–66. doi:10.1016/j.celrep.2014.06.043
25. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell (2012) 22:547–60. doi:10.1016/j.ccr.2012.08.014
26. Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev (2012) 26:1926–44. doi:10.1101/gad.188292.112
27. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514:628–32. doi:10.1038/nature13611
28. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell (2013) 12:329–41. doi:10.1016/j.stem.2012.12.013
29. Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep (2014) 7:1679–90. doi:10.1016/j.celrep.2014.04.037
30. Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med (2011) 89:229–36. doi:10.1007/s00109-011-0731-9
31. McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J (1979) 180:533–44. doi:10.1042/bj1800533
32. Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. J Exp Biol (1972) 128:161–3. doi:10.1242/jeb.089763
33. Denton RM, Hughes WA. Pyruvate dehydrogenase and the hormonal regulation of fat synthesis in mammalian tissues. Int J Biochem (1978) 9:545–52. doi:10.1016/0020-711X(78)90113-1
34. Duchen MR. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J (1992) 283(Pt 1):41–50. doi:10.1042/bj2830041
35. Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. J Biol Chem (1994) 269:27310–4.
36. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (2009) 1787:1309–16. doi:10.1016/j.bbabio.2009.01.005
37. Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium–cancer signalling nexus. Nat Rev Cancer (2017) 17(6):367–80. doi:10.1038/nrc.2017.18
38. Szabo I, Zoratti M. Mitochondrial channels: ion fluxes and more. Physiol Rev (2014) 94:519–608. doi:10.1152/physrev.00021.2013
39. Leanza L, Romio M, Becker KA, Azzolini M, Trentin L, Manago A, et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell (2017) 31:516–31.e10. doi:10.1016/j.ccell.2017.03.003
40. Kosztka L, Rusznak Z, Nagy D, Nagy Z, Fodor J, Szucs G, et al. Inhibition of TASK-3 (KCNK9) channel biosynthesis changes cell morphology and decreases both DNA content and mitochondrial function of melanoma cells maintained in cell culture. Melanoma Res (2011) 21:308–22. doi:10.1097/CMR.0b013e3283462713
41. Piskacek M, Zotova L, Zsurka G, Schweyen RJ. Conditional knockdown of hMRS2 results in loss of mitochondrial Mg(2+) uptake and cell death. J Cell Mol Med (2009) 13:693–700. doi:10.1111/j.1582-4934.2008.00328.x
42. Foskett JK, White C, Cheung K-H, Mak D. Inositol trisphosphate receptor Ca2+ release channels. (2007) 87:593–658. doi:10.1152/physrev.00035.2006
43. Cardenas C, Liberona JL, Molgo J, Colasante C, Mignery GA, Jaimovich E. Nuclear inositol 1,4,5-trisphosphate receptors regulate local Ca2+ transients and modulate cAMP response element binding protein phosphorylation. J Cell Sci (2005) 118:3131–40. doi:10.1242/jcs.02446
44. Cardenas C, Escobar M, Garcia A, Osorio-Reich M, Hartel S, Foskett JK, et al. Visualization of inositol 1,4,5-trisphosphate receptors on the nuclear envelope outer membrane by freeze-drying and rotary shadowing for electron microscopy. J Struct Biol (2010) 171:372–81. doi:10.1016/j.jsb.2010.05.003
45. Cardenas C, Foskett JK. Mitochondrial Ca(2+) signals in autophagy. Cell Calcium (2012) 52:44–51. doi:10.1016/j.ceca.2012.03.001
46. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca 2+ responses. Science (1998) 280:1763–6. doi:10.1126/science.280.5370.1763
47. Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J (1998) 17:4987–5000. doi:10.1093/emboj/17.17.4987
48. Spät A, Szanda G, Csordás G, Hajnóczky G. High and low calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium (2008) 44:51–63. doi:10.1016/j.ceca.2007.11.015
49. Murphy AN, Kelleher JK, Fiskum G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J Biol Chem (1990) 265:10527–34.
50. Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol (2000) 278:C423–35.
51. Bernhard W, Haguenau F, Gautier A, Oberling C. Submicroscopical structure of cytoplasmic basophils in the liver, pancreas and salivary gland; study of ultrafine slices by electron microscope. Z Zellforsch Mikrosk Anat (1952) 37:281–300. doi:10.1007/BF00343816
52. Berhard W, Rouiller C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol (1956) 2:73–8. doi:10.1083/jcb.2.4.73
53. Bernhard W, Rouiller C. Microbodies and the problem of mitochondrial regeneration in liver cells. J Biophys Biochem Cytol (1956) 2:355–60. doi:10.1083/jcb.2.4.355
54. Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta (2014) 1841:595–609. doi:10.1016/j.bbalip.2013.11.014
55. Akl H, Bultynck G. Altered Ca(2+) signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta (2013) 1835:180–93. doi:10.1016/j.bbcan.2012.12.001
56. Missiroli S, Danese A, Iannitti T, Patergnani S, Perrone M, Previati M, et al. Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of the tumor cell fate. Biochim Biophys Acta (2017) 1864:858–64. doi:10.1016/j.bbamcr.2016.12.024
57. Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res (2010) 70:1173–83. doi:10.1158/0008-5472.CAN-09-2886
58. Sakakura C, Hagiwara A, Fukuda K, Shimomura K, Takagi T, Kin S, et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res (2003) 23:3691–7.
59. Bergner A, Kellner J, Tufman A, Huber RM. Endoplasmic reticulum Ca2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J Exp Clin Cancer Res (2009) 28:25. doi:10.1186/1756-9966-28-25
60. Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium (2010) 48:315–23. doi:10.1016/j.ceca.2010.09.005
61. Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol Cancer (2010) 9:156. doi:10.1186/1476-4598-9-156
62. Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F. Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur J Cancer (2013) 49:3738–51. doi:10.1016/j.ejca.2013.07.013
63. DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, et al. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res (2005) 65:713–7.
64. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
65. Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14:2313–24. doi:10.1016/j.celrep.2016.02.030
66. Lovy A, Foskett JK, Cardenas C. InsP3R, the calcium whisperer: maintaining mitochondrial function in cancer. Mol Cell Oncol (2016) 3:e1185563. doi:10.1080/23723556.2016.1185563
67. Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A (1961) 47:1744–50. doi:10.1073/pnas.47.11.1744
68. Rossi CS, Lehninger AL. Stoichiometry of respiratory stimulation, accumulation of Ca2+ and phosphate, and oxidative phosphorylation in rat liver mitochondria. J Biol Chem (1964) 239:3971–80.
69. Vasington FD, Murphy JV. Ca++ ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem (1962) 237:2670–7.
70. Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, et al. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem (1998) 273:10223–31. doi:10.1074/jbc.273.17.10223
71. Moore CL. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun (1971) 42:298–305. doi:10.1016/0006-291X(71)90102-1
72. Reed KC, Bygrave FL. The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem J (1974) 140:143–55. doi:10.1042/bj1400143
73. Rizzuto R, Simpson AWM, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature (1992) 358:325–7. doi:10.1038/358325a0
74. Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol (1990) 425:85–115. doi:10.1113/jphysiol.1990.sp018094
75. Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci (1994) 14:348–56.
76. Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature (1967) 213:137–9. doi:10.1038/213137a0
77. Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta (2016) 1863:2498–502. doi:10.1016/j.bbamcr.2016.01.019
78. Mazure NM. VDAC in cancer. Biochim Biophys Acta (2017) 1858:665–73. doi:10.1016/j.bbabio.2017.03.002
79. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
80. De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
81. Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature (2010) 467:291–6. doi:10.1038/nature09358
82. Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol (2015) 16:545–53. doi:10.1038/nrm4039
83. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J (2013) 32:2362–76. doi:10.1038/emboj.2013.157
84. Sancak Y, Markhard ALL, Kitami T, Kovacs-Bogdan E, Kamer KJJ, Udeshi NDD, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 342:1379–82. doi:10.1126/science.1242993
85. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One (2013) 8:e55785. doi:10.1371/journal.pone.0055785
86. Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol (2012) 14:1336–43. doi:10.1038/ncb2622
87. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol (2013) 23:58–63. doi:10.1016/j.cub.2012.11.026
88. Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun (2013) 434:695–700. doi:10.1016/j.bbrc.2013.04.015
89. Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One (2014) 9:e96866. doi:10.1371/journal.pone.0096866
90. Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, et al. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun (2015) 458:186–93. doi:10.1016/j.bbrc.2015.01.092
91. Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1a. EMBO Mol Med (2016) 8:569–85. doi:10.15252/emmm
92. Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep (2016) 15:1673–85. doi:10.1016/j.celrep.2016.04.050
93. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell (2017) 65:1014–28.e7. doi:10.1016/j.molcel.2017.01.032
94. Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, Eroglu E, Klec C, Waldeck-Weiermair M, et al. Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell Physiol Biochem (2016) 39:1404–20. doi:10.1159/000447844
95. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for mcu-mediated mitochondrial Ca 2+ uptake that regulates cell survival. Cell (2012) 151:630–44. doi:10.1016/j.cell.2012.10.011
96. Tian Y, Qin L, Shi D, Sun R, Li W, Liu T, et al. RPS3 regulates melanoma cell growth and apoptosis by targeting Cyto C/Ca 2+/MICU1 dependent mitochondrial signaling. Oncotarget (2015) 6:29614–25. doi:10.18632/oncotarget.4868
97. Zhou X, Ren Y, Kong L, Cai G, Sun S, Song W, et al. Targeting EZH2 regulates tumor growth and apoptosis through modulating mitochondria dependent cell-death pathway in HNSCC. Oncotarget (2015) 6:33720–32. doi:10.18632/oncotarget.5606
98. Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S, et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun (2017) 8:14634. doi:10.1038/ncomms14634
99. Madreiter-Sokolowski CT, Klec C, Parichatikanond W, Stryeck S, Gottschalk B, Pulido S, et al. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca(2+) uptake in immortalized cells. Nat Commun (2016) 7:12897. doi:10.1038/ncomms12897
100. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer (2013) 13:37–50. doi:10.1038/nrc3409
101. Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A (2016) 113:E1872–80. doi:10.1073/pnas.1602264113
102. Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans (2012) 40:297–309. doi:10.1042/BST20110766
103. Hakamata Y, Nishimura S, Nakai J, Nakashima Y, Kita T, Imoto K. Involvement of the brain type of ryanodine receptor in T-cell proliferation. FEBS Lett (1994) 352:206–10. doi:10.1016/0014-5793(94)00955-4
104. Kang HY, Kim NS, Lee CO, Lee JY, Kang WH. Expression and function of ryanodine receptors in human melanocytes. J Cell Physiol (2000) 185:200–6. doi:10.1002/1097-4652(200011)185:2<200:AID-JCP4>3.0.CO;2-6
105. Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J (2002) 16:84–6. doi:10.1096/fj.01-0380fje
106. Mariot P, Prevarskaya N, Roudbaraki MM, Le Bourhis X, Van Coppenolle F, Vanoverberghe K, et al. Evidence of functional ryanodine receptor involved in apoptosis of prostate cancer (LNCaP) cells. Prostate (2000) 43:205–14. doi:10.1002/(SICI)1097-0045(20000515)43:3<205::AID-PROS6>3.3.CO;2-D
107. Abdul M, Ramlal S, Hoosein N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol Oncol Res (2008) 14:157–60. doi:10.1007/s12253-008-9045-9
108. Arbabian A, Brouland JP, Gélébart P, Kovàcs T, Bobe R, Enouf J, et al. Endoplasmic reticulum calcium pumps and cancer. Biofactors (2011) 37:139–49. doi:10.1002/biof.142
109. Endo Y, Uzawa K, Mochida Y, Shiiba M, Bukawa H, Yokoe H, et al. Sarcoendoplasmic reticulum Ca(2+) ATPase type 2 downregulated in human oral squamous cell carcinoma. Int J Cancer (2004) 110:225–31. doi:10.1002/ijc.20118
110. Ait-Ghezali L, Arbabian A, Jeibmann A, Hasselblatt M, Hallaert GG, Van den Broecke C, et al. Loss of endoplasmic reticulum calcium pump expression in choroid plexus tumours. Neuropathol Appl Neurobiol (2014) 40:726–35. doi:10.1111/nan.12098
111. Pacifico F, Ulianich L, De Micheli S, Treglia S, Leonardi A, Vito P, et al. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J Mol Endocrinol (2003) 30:399–409. doi:10.1677/jme.0.0300399
112. Arbabian A, Brouland J-P, Apáti Á, Pászty K, Hegedűs L, Enyedi Á, et al. Modulation of endoplasmic reticulum calcium pump expression during lung cancer cell differentiation. FEBS J (2013) 280:5408–18. doi:10.1111/febs.12064
113. Chung F-Y, Lin S-R, Lu C-Y, Yeh C-S, Chen F-M, Hsieh J-S, et al. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. Am J Surg Pathol (2006) 30(8):969–74. doi:10.1097/00000478-200608000-00006
114. Brouland J-P, Gelebart P, Kovacs T, Enouf J, Grossmann J, Papp B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am J Pathol (2005) 167:233–42. doi:10.1016/S0002-9440(10)62968-9
115. Li W, Ouyang Z, Zhang Q, Wang L, Shen Y, Wu X, et al. SBF-1 exerts strong anticervical cancer effect through inducing endoplasmic reticulum stress-associated cell death via targeting sarco/endoplasmic reticulum Ca(2+)-ATPase 2. Cell Death Dis (2014) 5:e1581. doi:10.1038/cddis.2014.538
116. Papp B, Brouland J-P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer (Auckl) (2011) 5:163–74. doi:10.4137/BCBCR.S7481
117. Dellis O, Arbabian A, Brouland J-P, Kovacs T, Rowe M, Chomienne C, et al. Modulation of B-cell endoplasmic reticulum calcium homeostasis by Epstein-Barr virus latent membrane protein-1. Mol Cancer (2009) 8:59. doi:10.1186/1476-4598-8-59
Keywords: inositol triphosphate receptors, mitochondrial Ca2+ uniporter, mitochondrial transport, TCA cycle, respiratory chain, AMPK
Citation: Bustos G, Cruz P, Lovy A and Cárdenas C (2017) Endoplasmic Reticulum–Mitochondria Calcium Communication and the Regulation of Mitochondrial Metabolism in Cancer: A Novel Potential Target. Front. Oncol. 7:199. doi: 10.3389/fonc.2017.00199
Received: 12 June 2017; Accepted: 18 August 2017;
Published: 08 September 2017
Edited by:
Jan B. Parys, KU Leuven, BelgiumReviewed by:
Ildikò Szabò, University of Padua, ItalyWolfgang F. Graier, Medical University of Graz, Austria
Copyright: © 2017 Bustos, Cruz, Lovy and Cárdenas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: César Cárdenas, amNlc2FyQHUudWNoaWxlLmNs
†These authors have contributed equally to this work.