 Shi Hao Tan1
Shi Hao Tan1 Takaomi Sanda
Takaomi Sanda- 1Cancer Science Institute of Singapore, National University of Singapore, Singapore, Singapore
- 2Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
T-cell acute lymphoblastic leukemia (T-ALL) is a hematological malignancy characterized by the clonal proliferation of immature T-cell precursors. T-ALL has many similar pathophysiological features to acute myeloid leukemia, which has been extensively studied in the establishment of the cancer stem cell (CSC) theory, but the CSC concept in T-ALL is still debatable. Although leukemia-initiating cells (LICs), which can generate leukemia in a xenograft setting, have been found in both human T-ALL patients and animal models, the nature and origin of LICs are largely unknown. In this review, we discuss recent studies on LICs in T-ALL and the potential mechanisms of LIC emergence in this disease. We focus on the oncogenic transcription factors TAL1, LMO2, and NOTCH1 and highlight the significance of the transcriptional regulatory programs in normal hematopoietic stem cells and T-ALL.
Introduction
Since the establishment of functional repopulation assays in the late 1990s, accumulating studies have demonstrated the existence of cancer stem cells (CSCs) that possess self-renewal capability and the potential to generate differentiated daughter cells (1–4). Purification of a unique cell population based on the expression of specific cell surface markers enabled the prospective isolation of CSCs in various types of cancers. A prime example is acute myeloid leukemia (AML), which has been extensively studied as a model disease for the establishment of the CSC theory. Although T-cell acute lymphoblastic leukemia (T-ALL) has many similarities in pathophysiological features to AML, the CSC concept in T-ALL has not been firmly established. Leukemia-initiating cells (LICs), which can generate leukemia in a xenograft setting, have been confirmed in both human T-ALL patients and mouse models (5–12), but common stem cell markers have not been identified in this disease.
Unlike AML, which arises from the bone marrow, T-ALL clones originally emerge in the thymus, which does not provide a niche for hematopoietic stem cells (HSCs) (13–15). In many T-ALL cases, oncogenes are driven by a chromosomal translocation involving the T-cell receptor (TCR) gene locus, which is associated with somatic recombination in immature thymocytes (16). This suggests that T-ALL arises from committed T-cell precursors, but not from multi-potent HSCs. It is likely that developing thymocytes acquire stemness capability as a consequence of genetic and epigenetic abnormalities. On the other hand, recent studies have shown that early thymocytes can self-renew under certain condition (17, 18). Therefore, it is also possible that T-ALL arises from thymocytes that already possess self-renewal potential.
Genetic Abnormalities in T-ALL: Overview
Acute lymphoblastic leukemia (ALL) is the most common type of childhood malignancy (19). Approximately 20% of ALL cases are classified as T-ALL. T-ALL is an aggressive malignancy characterized by the clonal proliferation of immature T-cell precursors that arise from the thymus and infiltrate into the bone marrow and peripheral blood (13–16). Enormous progress has been made in the treatment of T-ALL in the past few decades, with long-term remission observed in approximately 80% of children and 60% of adult patients (20, 21). However, a substantial fraction of T-ALL patients fail to respond to induction therapy or relapse within 2 years of diagnosis. The prognosis for this group of patients is very poor, with a 5-year survival rate of less than 25% (22).
T-ALL development requires multi-step genetic alterations of crucial oncogenes and tumor suppressors via different recurrent mechanisms, such as chromosomal translocations, intrachromosomal rearrangements, and mutations in protein-coding genes or enhancer elements, as well as epigenetic abnormalities (13–16). These alterations commonly affect genes that are required for cell growth, survival, and differentiation during normal T-cell development (14, 16). Results from recent genome-wide sequencing studies across different types of cancers indicate that ALL exhibits the fewest genomic abnormalities compared with other hematological malignancies and solid tumors (23, 24). This suggests that relatively few molecular alterations are crucial and significant enough to hijack the normal developmental program and promote malignant transformation.
Molecular Abnormalities That Delineate the T-ALL Subgroups
Chromosomal translocation is a hallmark of T-ALL (16, 25). The most commonly observed translocations involve the TCR loci on chromosome 14q11.2 (TCR alpha/delta), 7q34 (TCR beta), and 7p14 (TCR gamma). They are often fused to a range of oncogenic transcription factors that are important during different stages of normal hematopoiesis and lymphocyte development (13–16), resulting in constitutive and ectopic expression of these factors. The affected genes include transcription factors from the basic helix-loop-helix family, including TAL1, TAL2, and LYL1; the homeobox family, including TLX1, TLX3; the HOXA genes; NKX2-1; MYB; and the LIM domain-only (LMO) genes LMO1 and LMO2.
Cytogenetic analysis coupled with gene expression profiling has been used to classify T-ALL into several subgroups: TAL1/LMO1/2-, TLX1/3-, HOXA/MEISI-, LMO2/LYL1, and NKX2-1-positive T-ALL cases (25–27). Briefly, TAL1, LMO2, and LYL1 are essential regulators of hematopoiesis (28–33). Those factors can be oncogenic when abnormally or ectopically overexpressed in immature T-cells (8, 34, 35), as we discuss later. Besides translocation, TAL1 is aberrantly induced by intrachromosomal rearrangement or mutations in the enhancer (36–38). TLX genes are expressed during embryogenesis and required for normal development of the spleen (39). Overexpression of TLX1 leads to T-ALL and exhibits aneuploidy in a mouse model (40). The HOX genes are a family of homeodomain containing transcription factors, which are expressed in HSCs and immature progenitors compartments (41). HOX cofactors such as MEIS1 which is important to improve binding selectivity and specificity of HOX proteins are also found to be overexpressed in T-ALL (42). Notably, these subgroups are mutually exclusive to each other and reflect the arrest of T-cell differentiation at different stages, including (a) early blockage at the CD4−CD8− double-negative (DN) stage of thymocyte development for the LMO2/LYL1 group, (b) early cortical T-ALL (CD1a+, CD4+, and CD8+) with expression of TLX1/3 or NKX2-1, and (c) late cortical T-ALL (CD3+, CD4+, and CD8+) with expression of TAL1 (26, 43). More recently, the early T-cell precursor (ETP) subtype has been defined based on cell surface markers and gene expression profiles (43). ETP is enriched in the LMO2/LYL1 group but can be also found in other subgroups (27).
Activation of the NOTCH1 Pathway
Another major molecular abnormality in T-ALL is the mutations that affect the NOTCH1 pathway (13–16). NOTCH1 signaling is essential for normal T-cell precursor development and is strictly regulated in a ligand-dependent manner. Remarkably, activating mutations affecting NOTCH1 are observed in more than 50% of T-ALL cases (44). Aberrant activation of NOTCH1 was originally identified in T-ALL cases harboring the t(7;9)(q34;q34.3) chromosomal translocation, through which the intracellular form of NOTCH1 (ICN1) gene fuses to the TCR beta regulatory element, leading to expression of a constitutively active, truncated form of NOTCH1 (45). However, the majority of aberrant NOTCH1 activation observed in T-ALL occurs due to mutations in its heterodimerization (HD) domain and/or the PEST domain (44). Mutations in the HD domain cause the NOTCH1 receptor to be susceptible to proteolytic cleavage and release of the ICN1 protein, while the PEST domain mutations inhibit the proteasomal degradation of ICN1 by the FBXW7 ubiquitin ligase, thus lengthening its half-life in T-ALL cells. Additionally, deletions or inactivating mutations of FBXW7 are frequently observed in T-ALL (46, 47).
The oncogenic roles of NOTCH1 signaling in T-ALL have been extensively studied both in humans and in animal models. Overexpression of ICN1 protein in mouse hematopoietic progenitor cells leads to very rapid onset of T-ALL (48). Subsequent studies have identified the direct transcriptional targets of NOTCH1 in T-ALL, which are enriched in genes responsible for cell proliferation, metabolism, and protein synthesis, including MYC and HES1 (49–53). These studies implicated NOTCH1 as a driver oncogene in T-ALL.
Epigenetic Regulators and Other Molecular Abnormalities
Alterations in genes that encode for epigenetic regulators such as EZH2, SUZ12, and EED have been also identified in T-ALL (54–57). These genes make up the core components of the polycomb repressor complex 2 that mediates the repressive histone mark H3 lysine 27 trimethylation (H3K27me3). Loss-of function mutations in these genes can lead to accelerated leukemia onset in mice (54, 55), suggesting that they act as tumor suppressors in T-ALL. Recent studies have shown that the KDM6A/UTX, which is responsible for demethylating H3K27me3, have cases of inactivating lesions and downregulation of this gene accelerates NOTCH1-driven leukemia in mice (55, 56). In contrast, another study showed that KDM6A/UTX acts as a pro-oncogenic cofactor when it is recruited by TAL1 in T-ALL to activate target gene expressions (57).
Other recurrent molecular abnormalities include genes that encode for proteins involved in the JAK-STAT signaling pathway, such as IL7R, JAK1, JAK3, and STAT5B; genes that are involved in PI3K-AKT signaling pathways, such as PI3K and PTEN; and genes involved in RAS-MAPK signaling pathways, such as HRAS, KRAS, and PTPN11 (13–16). Additionally, recent sequencing studies discovered several new alternations including mutations in CCND3, CTCF, and MYB genes (27), and SPI1/PU.1 fusions (58).
CSC and LIC Concepts
The concept of CSCs originates from the observation that tumors consist of a hierarchically organized, heterogeneous population of cells with a minority of biologically distinct subsets capable of self-renewing and giving rise to clonal daughter cells (1–4). A number of studies have shown the existence of CSCs in various types of cancers. The CSC model also indicates that this rare cell population is able to tolerate therapeutic agents such as chemotherapy and radiation that eradicate the bulk of the rapidly proliferating tumor cells, thus resulting in inevitable cancer relapse in the long term (1–4, 59).
The most definitive property of stem cells lies in their self-renewal ability (1–4). Self-renewal in normal cells or CSCs gives rise either to one stem and one differentiated daughter cell via asymmetric division or to two stem cells via symmetric division. The general consensus in stem cell research is that CSCs are able to initiate and maintain clonal growth in long-term repopulation assays where the cancer cells are serially transplanted into immunodeficient recipient mice. The purification of a unique cell population based on the expression of specific cell surface markers has allowed researchers to isolate CSCs in various cancers, including AML and breast cancer (60–62). However, such populations have not been well characterized in many other cancers, including T-ALL. Hence, other terms, such as “tumor-initiating cells (TICs)” or “LICs,” have been coined to refer to the ability of transplanted cells to initiate tumor formation or leukemia in animals and are more preferentially used in experimental settings (1). Notably, the TIC/LIC concept is distinct from the “cell-of-origin” idea, as TIC/LIC strictly refers to cells in which tumorigenesis can be initiated (63), whereas the cell of origin that received the first oncogenic “hit” would progressively accumulate mutations during clonal evolution of the tumor. The acquisition of stem cell-like properties may occur at a much later stage of tumorigenesis in the evolved cells than the original cell that received only the first hit. In this regard, John Dick has proposed that TICs/LICs should be defined by their ability to (a) generate tumors in xenograft models that are representative of the parent tumors, (b) generate tumors upon serial passages in xenograft models, and, lastly, (c) give rise to daughter cells that can proliferate but might not be able to establish tumors after serial passages (1).
LICs in Human AML and ALL: Discovery and Challenges
The presence of LICs was first reported by Dick and his colleagues in the late 1990s in studies of AML (60, 61). In a series of seminal studies, they showed that a rare subset of CD34+CD38− cells isolated from AML patients was able to initiate the disease when transplanted into severe combined immunodeficient (SCID) mice (60). Crucially, the more differentiated CD34+CD38+ cells were unable to generate leukemia. In the initial study, secondary transplant of leukemic cells from SCID mice failed to generate leukemia. However, using a more immunocompromised non-obese diabetic (NOD)/SCID mouse model, the authors demonstrated that CD34+CD38− cells have self-renewal properties (61). Furthermore, this group has shown that the engrafted CD34+CD38− cells were able to give rise to more differentiated leukemic cells (61). Thus, this study demonstrated the presence of a leukemic hierarchy, with the CD34+CD38− LICs at the top of the pyramid.
These results have also been challenged by studies utilizing more immunocompromised mouse models. For example, in the NOD/LtSz-scid IL-2Rγchainnull (NSG) mouse model, AML LICs are not only present exclusively in CD34+CD38− cells (64). Results from this model showed that LICs can also be found in more differentiated CD34− and CD38+ cells. The concept of LICs was also challenged by a study in which leukemic cells from Ras-induced T-cell lymphoma or an Eμ-Myc model of pre-B/B-cell lymphoma were shown to engraft in non-congenic animals regardless of the number of cells injected (65). The authors stressed the need to interpret data from serial transplantations more carefully, since failure to show engraftment could simply be due to the inability of the human cells to adapt to the microenvironment in the mouse.
The identification of LICs in ALL is even more challenging. To date, the identity and presence of LICs in human ALL has not been firmly established and is still debatable. Early studies in B-cell ALL (B-ALL) reported that the relatively immature CD34+CD19− cells could contain LICs (66, 67). However, recent studies have found that more mature CD34+CD19+ leukemic blasts could initiate leukemia in ETV6-RUNX1- or TEL-AML1-positive B-ALL cases (68). In addition, a more recent study on MLL-AF4-positive infant ALL indicated that the LICs capable of reconstituting transplanted mice are exclusively CD19+ but exhibit variable CD34 expression (69). These studies highlight the heterogeneity of LICs in B-ALL cases and suggest that different cytogenetic abnormalities might play a role in determining the type of LICs present.
Similarly, the nature of LICs in human T-ALL has not been well characterized. An early study suggested that CD34+CD4− and CD34+CD7− cells, which make up a fraction of the leukemic cells from pediatric T-ALL patients, had leukemia-initiating properties when engrafted into NOD/SCID mice (5). A follow-up study investigating LIC activity in cortical/mature T-ALL patients reported that the CD34+CD7− population from these patients contained normal hematopoietic cells that were able to differentiate into different lineages, while the CD34+CD7+ cells possessed LIC capability (6). Dick and Chiu et al. have also reported that the CD7+CD1a− subset is enriched for LIC activity and exhibits glucocorticoid resistance (7).
LICs in Animal Models of T-ALL
Although the findings on LICs in primary human T-ALL are limited, several studies have been performed on transgenic animal models of T-ALL.
LICs in the Tal1-Induced Mouse Models of T-ALL
One of the most commonly used T-ALL mouse models in the study of LICs is the Tal1 transgenic mouse model; approximately 30% of these mice develop leukemia after a long latency period (8, 34, 35). Notably, tumor onset and progression can be accelerated by co-expressing the oncogene Lmo1 or Lmo2. Tremblay and Hoang et al. have found that overexpression of Tal1 and Lmo1 resulted in a marked expansion of T-cells making up the CD4−CD8− DN1, DN3, and DN4 populations and blocked differentiation into the CD4+CD8+ double-positive (DP) stage (8). The leukemia cells contain LICs that can generate leukemia in transplanted mice. Interestingly, they demonstrated that LICs are enriched in the DN population, especially DN3 and DN4, compared with the DP population and that these LICs could give rise to more differentiated leukemic cells (8). This study suggested that committed DN-stage T-cells with ectopic expression of Tal1 and Lmo1 exhibit self-renewal properties while retaining the potential to differentiate. A subsequent study by Kelliher and her colleagues utilizing the Tal1/Lmo2 mouse model of T-ALL also showed that the DN3 and DN4 populations of leukemia cells possess LIC properties and drive T-ALL leukemogenesis (9, 10). In support of these data in double transgenic mice, McCormack and Curtis et al. demonstrated that Lmo2 single transgenic mice show an increase in thymic progenitors in the DN3 subset while also displaying properties of LICs in serial transplantation experiments (11). Interestingly, several genes, such as Hhex and Lyl1, that are normally expressed in HSCs were expressed in the self-renewing cells. This suggests that an HSC-like transcriptional program might be induced in T-ALL cells. Taken together, these studies indicated that DN3 thymocytes gained self-renewal potential.
Significance of NOTCH1 Activation in Mouse Models of T-ALL
Notably, gain-of-function mutations of the Notch1 gene are frequently found in the Tal1/Lmo1 mouse model of T-ALL (8, 70), similar to observations in human T-ALL (44). Tremblay and Hoang et al. reported that Notch1 mutations occurred mostly at the DN4 preleukemic stage and that the mutations could also be observed during overt leukemia in the same mice (8). Interestingly, leukemia development and Notch1 mutations were abolished in the absence of CD3e. Similarly, Cui and Mackall have reported that forced expression of TCR during early stages of T-cell development caused T-ALL in 100% and all cases harbored Notch1 mutations (71). These results suggested that pre-TCR and TCR signaling have a permissive role in the acquisition of Notch1 mutations and that active NOTCH1 signaling confers clonal dominance upon leukemia development.
Importantly, Tremblay and Hoang et al. showed that Notch1/Tal1/Lmo1 triple transgenic mice developed leukemia significantly faster than single or double transgenic animals (8). The DN1–DN2 and DN3–DN4 subsets from Notch1/Tal1/Lmo1 triple transgenic mice were able to induce T-ALL in secondary hosts with high efficiency compared with Tal1/Lmo1 double transgenic mice (8). A subsequent study from the same group further suggested that Notch1 drives self-renewal of thymocytes from the Tal1/Lmo1 mouse model via its target genes Hes1 and Myc (12). Treatment of the leukemic cells before and throughout the transplantation period with γ-secretase inhibitor, which inhibits the catalytic cleavage of NOTCH1, completely abolished the LIC function of the leukemic T-ALL cells. Given the importance of active NOTCH1 signaling in primary human T-ALL patient samples, these studies support the hypothesis that Notch1-activating mutations are important for the cells to gain clonal dominance during disease development.
Notably, a recent study by Pear and his colleagues showed that LICs in T-ALL induced by the overexpression of a mutant form of NOTCH1 in adult mouse bone marrow progenitor cells are enriched in a single-positive (SP) T-cell population consisting of the CD8+CD4−HSAhi fraction of cells (72). Thus, the types of LICs generated could be different from those found in the Tal1/Lmo transgenic mouse model.
LICs in Other Animal Models of T-ALL
Additionally, several other animal models of T-ALL have been used to analyze LICs. In studies of Pten-null mice, which develop T-ALL with 100% penetrance, LICs are identified as cKitmidCD3+ cells and often overexpress Myc due to a recurrent chromosomal translocation at t(14;15). The self-renewal properties of these LICs could also be abolished via targeting both the deregulated PI3K signaling pathway and Myc expression concurrently (73, 74).
Apart from studies in T-ALL mouse models, a T-ALL zebrafish model has also been employed to investigate the presence of LICs in T-ALL. Langenau and Look et al. reported that the Myc-induced T-ALL zebrafish model demonstrates very similar molecular characteristics to human T-ALL patients that overexpress TAL1 and LMO2 (75). More recently, Langenau and his colleagues used syngeneic clonal zebrafish that can be transplanted into hosts without prior irradiation to show that the proportion of LICs in the Myc-induced T-ALL zebrafish model is much higher than previously reported (76). Further studies by the same group demonstrated that abnormal activation of the AKT-mTORC1 signaling pathway is the main underlying cause of the acquisition of LIC potential (77). These results support the mouse studies on LICs in T-ALL.
The Role of Microenvironment in T-ALL Pathogenesis
Another important consideration in the study of LICs is the interaction between leukemia cells and non-leukemia cells in the microenvironment. Bone marrow niche is essential for the maintenance and regulation of normal HSCs (78, 79). AML and ALL cells also home and expand in the bone marrow. Several studies have shown that signals from the bone marrow niche can dictate the survival of LICs and their responses to various types of treatment administered (80, 81).
Notably, two recent studies have elucidated the roles of bone marrow niche in T-ALL pathogenesis and implicated the CXCL12-CXCR4 signaling axis in the maintenance and progression of T-ALL (82, 83). CXCL12 is a chemokine secreted from endothelial and mesenchymal cells in the bone marrow and binds to its G protein-coupled receptor CXCR4 (79). Pitt et al. showed that in the bone marrow, T-ALL cells reside in close contact with stroma cells that secrete Cxcl12 (82). Deletion of the Cxcr4 receptor resulted in a reduction of leukemia burden and their infiltration into the bone marrow, thymus, and spleen in mouse model of T-ALL (82). Treatment of patient-derived human T-ALL cells in xenografts with a CXCR4 antagonist also produced the same result. Importantly, the authors observed a reduction in LIC activity in the absence of Cxcr4 in mice (82). Passaro et al. independently showed that depletion of CXCR4 affected T-ALL cell migration and expansion (83). Furthermore, the authors reported that calcineurin regulates CXCR4 expression in a cortactin-dependent manner (83). Those studies demonstrated the roles of the bone marrow niche in the maintenance of T-ALL.
Self-Renewal Capability of T-ALL Cells: Does it Already Exist in the Thymus or is it Acquired?
One of the fundamental questions in LIC research is whether the LICs are derived from cells that already have self-renewal potential, such as HSCs, or whether they emerge from differentiated cells by newly acquiring stemness capability. T-ALL is derived from committed T-cell precursors in the thymus, which does not provide a niche for HSCs. The chromosomal translocation involving the TCR gene locus found in many T-ALL cases is associated with somatic recombination in immature thymocytes (16). These findings suggest that developing thymocytes likely acquire stemness capability as a consequence of genetic and epigenetic abnormalities. Tal1 and Lmo1/2 transgenic mice show an increased number of thymic progenitors that can generate leukemia, indicating that these oncogenic transcription factors are capable of inducing LIC ability in immature thymocytes.
On the other hand, recent studies have shown that normal thymocytes can self-renew in the absence of competitive precursor replacement (17, 18, 84). In general, HSCs differentiate into common lymphoid progenitor (CLP) cells in the bone marrow. CLPs migrate into the thymus and are committed to T-cell precursors that can differentiate into the DN to DP stage of thymocytes. In this well-accepted model, a continuous supply of lymphoid progenitor cells from the bone marrow is necessary to support T-cell development. Interestingly, Martins and Rodewald et al. recently reported that in Rag2−/−γc−/−KitW/Wv mice, which do not produce lymphoid progenitors from the bone marrow, a transplanted wild-type thymus sustained T-cell development for a long period of time (17). Similarly, Peaudecerf and Rocha et al. reported that in Rag2−/−γc−/−IL7 receptor−/− mice engrafted with a wild-type thymus, persistent development of donor T-cells was observed (18). In this setting, host lymphoid progenitors can still migrate into the thymus and replace donor thymocytes but cannot differentiate after the DN2 stage, because IL7R signaling is required for the proliferation of early T-cell progenitors. Thus, competitive replacement by the host lymphoid progenitors is restricted to the DN1 and DN2 stages in this mouse model. This indicates that the donor thymus, which contains DN3 thymocytes, sustained T-cell development. Although this mechanism may be activated only when the competitive DN3 thymocytes are absent, these studies indicate that the thymus harbors cell populations with self-renewal potential that are capable of reconstituting the full diversity of T-cells.
Importantly, a large fraction of mice develop T-ALL in these settings (85). Tal1 and Lmo2 expression is strongly upregulated in these mouse T-ALL cells, and Notch1 mutations are also frequently found. This is consistent with observations in Tal1 and Lmo2 transgenic mice, which exhibit LICs in the DN3 subset and acquire Notch1 mutations (8). One possible mechanism is that differentiation arrest and expansion of DN3 thymocytes caused by overexpression of oncogenic transcription factors result in a loss of competitive replacement by bone marrow-derived progenitor cells, leading to activation of self-renewal machinery and malignant transformation. Alternatively, a loss of competitive replacement may result in the failure to silence the transcription factors that are normally expressed in stem and progenitor cells. Although the intrinsic mechanism of self-renewal in thymocytes is still unclear, these studies suggest that in T-ALL, LICs may arise from thymocytes that already have self-renewal potential via cellular competition.
Transcriptional Regulatory Programs in HSCs and T-Cell Differentiation
Mouse studies have suggested that cellular competition potentially triggers the self-renewal capability of immature thymocytes, which may eventually lead to malignant transformation via the acquisition of genetic abnormalities such as Notch1 mutations. In human T-ALL, a loss of competition may be caused by overexpression of oncogenic transcription factors such as TAL1 and LMO2. Notably, these transcription factors themselves are also involved in the stem cell regulatory program during normal hematopoiesis.
In general, cellular differentiation of hematopoietic cells is associated with developmental restrictions that can be illustrated by the analogy of a “ball rolling down a hill” (86). During the differentiation process, HSCs lose their self-renewal and lineage potential. This process is regulated by an epigenetic and transcriptional network (87–89). A number of hematopoietic transcription factors are involved in this process. For example, TAL1 has been implicated as an essential regulator of hematopoiesis (33). TAL1 is expressed in normal HSCs, progenitor cells, and erythro-megakaryocytic lineages. Studies in knockout mouse models have revealed that this factor is required for hematopoietic specification and the genesis of hematopoietic cells (28, 29). In normal hematopoietic cells, TAL1 forms a large transcriptional complex with E-protein, LMO2, LDB1, and GATA (90). Several other transcription factors, including RUNX1 and the ETS family proteins, also frequently co-regulate downstream target genes (91).
Interestingly, these transcription factors co-occupy their own regulatory elements and positively regulate each other, thus forming an interconnected auto-regulatory loop (87, 88, 92). This structure is also termed a “core regulatory circuit” (CRC) and has been reported in other stem cells (93–95). For example, in embryonic stem cells, three key transcription factors that establish stem cell identity, OCT4, SOX2, and NANOG regulate each other (93, 94). This mechanism is thought to reinforce and stabilize downstream gene expression by “interlocking” the regulatory loop and is likely required for stem cell properties (92). Importantly, ectopic expression of these transcription factors can reprogram somatic cells back into stem cells, as has been established for the production of induced pluripotent stem cells (96). Similarly, recent studies have demonstrated that adult somatic fibroblasts can be reprogrammed into multi-potent hematopoietic stem progenitor cells by ectopic overexpression of TAL1, LMO2, RUNX1, GATA2, and ERG (“iHSPCs”) (97). This clearly indicates that a relatively small number of transcription factors are sufficient to control cell fate and identity.
In contrast to the regulatory circuit in HSCs, a very different type of transcriptional program is formed in developing thymocytes to regulate genes that are essential for T-cell differentiation (98). This process requires a number of transcription factors working in a cascade as well as the interactions in the microenvironment (Figure 1). Briefly, the NOTCH ligand expressed on thymic stromal cells induces expression of the transcription factors TCF7 and GATA3, which regulate other key transcription factors such as BCL11B and LEF1 (98). During this process, stem cell transcription factors such as TAL1 and LMO2 are gradually silenced, resulting in the loss of stem and progenitor cell potential. Meanwhile, E-proteins (E2A and HEB) are functionally and transcriptionally upregulated to induce RAG1, RAG2, and PTCRA, for example, which are required for somatic TCR recombination (99, 100). Such orchestrated stage-specific regulation of transcription factors mediates the T-cell differentiation process like a “ball rolling down a hill.” TAL1 and LMO2 silencing and E-protein upregulation are crucial to controlling the reciprocal switch from self-renewal to lineage-specific genetic programs. In other words, ectopic expression of TAL1 and LMO2 in developing thymocytes may rewrite the internal regulatory program.
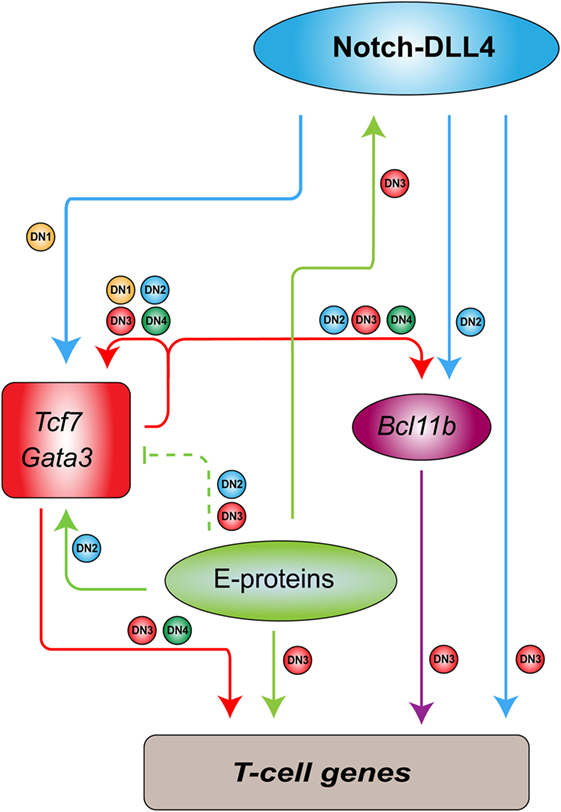
Figure 1. Transcriptional regulatory program in developing thymocytes [adapted and modified from figures by Yui and Rothenberg (98)]. In mouse models, Notch-DLL4 ligand expressed on thymic stroma cells induces the expressions of Tcf7 and Gata3, which regulate additional transcription factors such as Bcl11b. These factors, together with E-proteins and Notch1, stimulate the expressions of T-cell genes in a differentiation stage-specific manner. Arrows show activation or positive regulation. Dashed lines indicate “soft repression” of the maximal activity of the target. Small circles beside the lines correspond to the differentiation stages [double-negative (DN1–4)] at which the regulation occurs.
Aberrant Transcriptional Regulatory Program in TAL1/LMO-Positive T-ALL
Interestingly, TAL1 and LMO2 function as oncogenes in T-ALL cells, similar to their behavior in normal HSCs (33). TAL1 is expressed in 40–60% of T-ALL cases due to chromosomal translocation, intrachromosomal rearrangement, or mutations in non-coding elements (16, 36–38). These alterations replace an endogenous regulatory element controlling TAL1 expression with a new, potent enhancer that drives ectopic expression of this oncogene. Similarly, LMO2 or its related gene LMO1 is ectopically expressed in T-ALL cells due to chromosomal translocation or mutations in the regulatory elements (16, 101, 102). LMO1 or LMO2 is often expressed together with TAL1. In T-ALL cells, TAL1 and LMO proteins form a transcriptional complex with E-proteins and GATA3 (103, 104). Their regulatory partners in normal HSCs, RUNX1, ETS1, and MYB are also endogenously expressed in T-cells (98). We previously reported that TAL1, GATA3, RUNX1, and MYB co-occupy their own regulatory elements and positively regulate each other, forming the interconnected auto-regulatory structure (Figure 2) (105). These factors coordinately regulate downstream target genes. All these mechanisms are essentially the same as the machinery observed in normal HSCs.
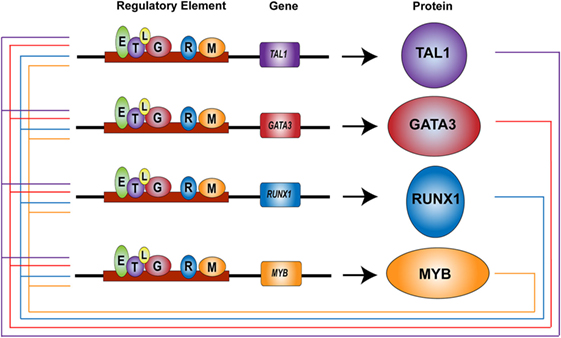
Figure 2. Core regulatory circuit in T-cell acute lymphoblastic leukemia (T-ALL) (38, 105). TAL1, GATA3, RUNX1, and MYB proteins (circles) bind at their own regulatory elements (boxes) and positively regulate each other, thus forming an interconnected auto-regulatory loop structure in T-ALL cells. TAL1 (T), GATA3 (G), RUNX1 (R), MYB (M), E-protein (E), LMO1/2 (L).
At the same time, TAL1 counteracts the function of E-proteins by sequestering them, thus preventing them from transcriptionally inducing genes required for T-cell differentiation (99, 100). In this context, E-proteins act as tumor suppressors, as several groups have shown that E2a-deficient mice develop T-cell lymphoma and that this deficiency accelerates leukemia onset and progression in Tal1-transgenic mice (10, 106). Our recent study also revealed that in human T-ALL cells, TAL1 opposes the expression of E-protein target genes (105). Thus, the imbalance between the oncogenic TAL1 complex and E-protein is a primary determinant underlying the molecular pathogenesis of T-ALL (Figure 3) (107). Together, ectopic expression of TAL1 and LMO1/2 leads to the induction of HSC-like machinery and disruption of the T-cell differentiation program.
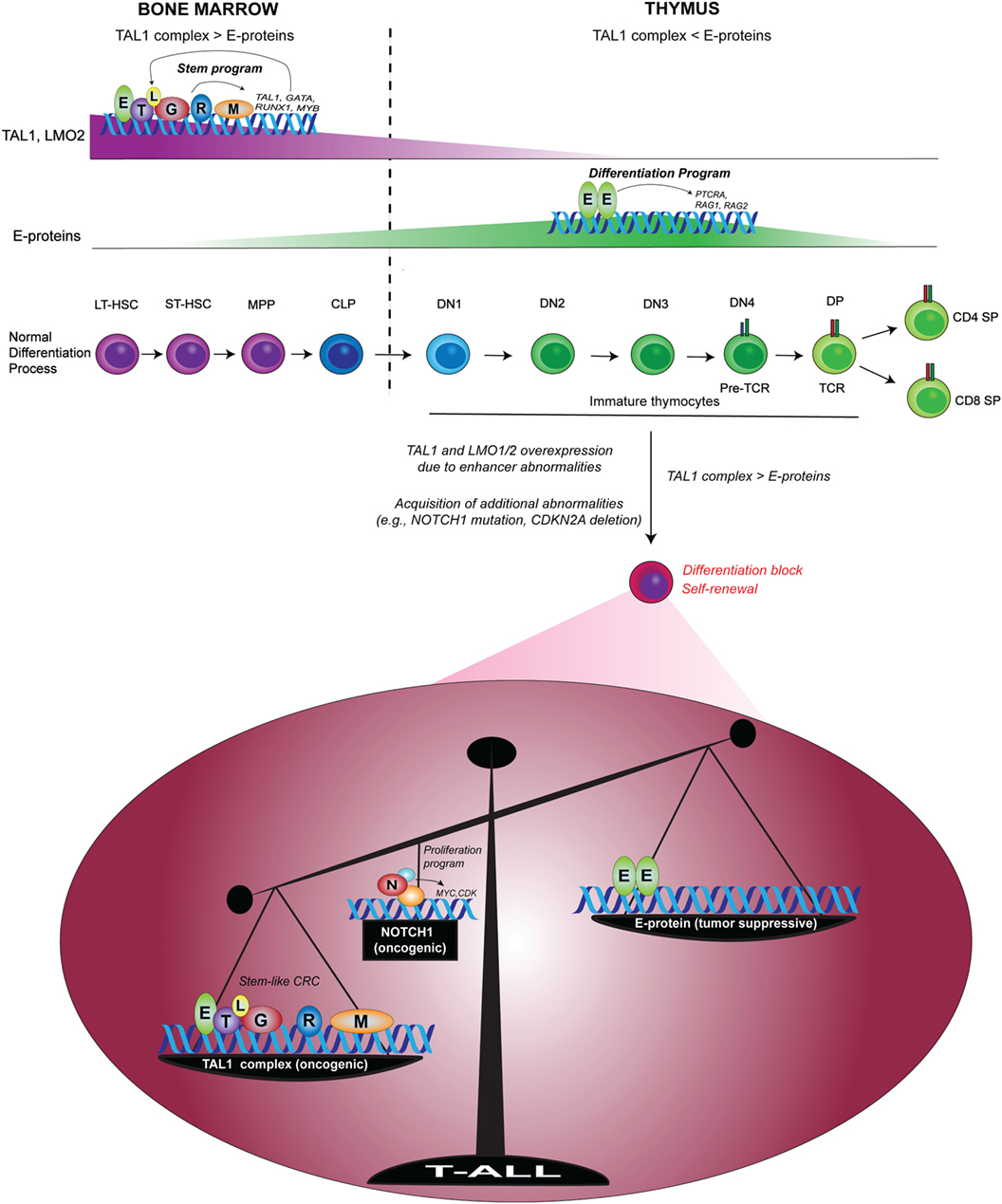
Figure 3. Imbalance between the oncogenic TAL1 complex and E-protein tumor suppressor in T-cell acute lymphoblastic leukemia (T-ALL) [modified from a figure by Sanda and Leong (107)]. In normal hematopoiesis, TAL1 forms a transcriptional complex with E-protein, GATA2, RUNX1, MYB, and LMO2 to drive a regulatory program in HSCs and progenitor cells via the auto-regulatory loop. Upon the progression of T-cell commitment in the thymus, TAL1 and LMO2 expressions are silenced, while E-proteins are functionally and transcriptionally upregulated. E-protein dimers induce the expressions of RAG1, RAG2, and PTRCA to prompt the differentiation program of T-cells. In T-ALL, enhancer abnormalities (chromosomal translocation, intrachromosomal rearrangement or mutations in the enhancer) cause ectopic expressions of TAL1 and/or LMO1/2, leading to the formation of TAL1 complex and the inhibition of E-protein dimers. T-ALL cells also acquire additional abnormalities such as genetic mutations of NOTCH1 and deletion of CDKN2A. TAL1 and its regulatory partners form a stem cell-like core regulatory circuit (CRC) and NOTCH1 activates a different set of genes such as MYC. The functional imbalance between the oncogenic TAL1 complex and E-protein tumor suppressors possibly contributes to the induction of self-renewal program and the blockade of T-cell differentiation machinery. Mutated NOTCH1 boosts this oncogenic mechanism. LT-HSC, long-term HSC; ST-HSC, short-term HSC; MPP, multipotent progenitor; CLP, common lymphoid progenitor; DN, CD4−CD8− double-negative; DP, CD4+CD8+ double-positive; SP, CD4+ or CD8+ single-positive; T, TAL1; E, E-protein; L, LMO1/2; G, GATA; R, RUNX1; M, MYB; N, NOTCH1.
Potential Stem Cell Signature Induced by TAL1 in T-ALL
In this regard, it would be interesting to identify genes that are abnormally induced by the TAL1 complex in T-ALL cells. Recently, our group used a targeted approach to identify regulatory elements that are differentially controlled by TAL1 and E-proteins (108). From this analysis, we discovered an enhancer situated within a cluster of seven genes belonging to the GTPase of Immunity Associated Protein (GIMAP) family. This region is associated with active histone marks in T-ALL cells but not in the normal human thymus, suggesting that the GIMAP enhancer is aberrantly activated in T-ALL cells. Importantly, GIMAP genes are expressed in mouse HSCs and CD4 or CD8 SP mature T-cells, while they are downregulated in DN3-4 stage thymocytes where TAL1 is also silenced. Using an in vivo reporter system in zebrafish, we showed that the GIMAP enhancer can be activated in normal hematopoietic stem and progenitor cells but not in the thymus. In addition, a reporter assay in human T-ALL cell lines indicated that the GIMAP enhancer is activated by TAL1 and its regulatory partners (GATA3 and RUNX1) and is repressed by E-proteins (E2A and HEB). Although ectopic expression of human GIMAP genes in immature zebrafish thymocytes did not induce tumor formation, their overexpression accelerated leukemia development in the presence of the MYC oncogene. Thus, our results revealed that aberrant activation of the GIMAP enhancer contributes to T-cell leukemogenesis.
While GIMAP genes have been known to be involved in the development of mature T- and B-lymphocytes (109–112), another group has also implicated their importance in HSC survival and maintenance (113). The work of Chen et al. on Gimap5−/− mice demonstrated that Gimap5 regulates the survival of HSCs and other early hematopoietic progenitors by stabilizing the Mcl-1 protein, which is an anti-apoptotic Bcl-2 family member (113). The HSCs in Gimap5-deficient mice exhibited defective long-term repopulation capacity, as demonstrated by their impaired engrafting ability. This study provided insights into the critical roles of GIMAP genes in the survival of HSCs and early progenitor cells. Notably, NOTCH1 was also identified as a positive regulator of the GIMAP genes in T-ALL cells (114, 115). A functional study by Chadwick et al. showed that Gimap5 mediates apoptosis protection in T-ALL cells upon its upregulation by NOTCH1 (114). Together with our findings, these studies suggest that as a consequence of TAL1/LMO overexpression and activation of the NOTCH1 pathway, the GIMAP genes could be reactivated in immature thymocytes in which they are normally repressed, possibly by E-proteins, thereby contributing to leukemogenesis.
Another gene that has been implicated in stem cells and is also aberrantly activated by the TAL1 complex in T-ALL is the ALDH1A2 gene (105, 116). Based on our ChIP-seq and gene expression data, this gene was one of the top candidate genes directly regulated by TAL1 in human T-ALL cells (105). ALDH activity has been proposed to be a universal CSC marker, as demonstrated by the tumorigenic and self-renewal properties of ALDH+ cells isolated from leukemia and many solid tumors (117–119). Among the 19 isoforms in the ALDH family, only a few of them, including ALDH1A2 are involved in retinoic acid signaling, which has been known to be associated with the stemness characteristics of CSCs. Another group and our recent study indicated that ALDH1A2 is induced by TAL1 via an internal enhancer in T-ALL cells (116) (and Zhang and Tan et al., unpublished data). Although the role of GIMAPs and ALDH1A2 in the self-renewal potential of malignant T-cells is yet to be elucidated, their ability to mark stem cells and T-ALL cells may be used as a signature of the aberrant transcriptional program induced by T-ALL oncogenes.
Conclusion and Future Prospective
The transformation mechanism in T-ALL is very efficient. T-ALL oncogenes alter the intrinsic transcriptional regulatory program by disrupting the differentiation machinery and by introducing the stem cell-like properties into developing thymocytes. This may initiate or reactivate the self-renewal ability that potentially exists in thymocytes. This process is mediated by a relatively small number of oncogenic transcription factors and seems not require the accumulation of a large number of genetic and chromosomal abnormalities until it obtains the hallmarks of cancer.
In other words, this mechanism poses a potential severe risk hidden in the thymus. Thymocytes may always be “primed” to initiate leukemogenesis. As recently reported (17, 18, 84), the competitive replacement of thymocytes via a continuous supply of lymphoid progenitor cells from the bone marrow plays an important tumor suppressive role in homeostasis. Further investigation is necessary to elucidate the loss-of-competition mechanism in human T-ALL. In particular, it is of great interest to analyze whether T-ALL develops from a self-renewal pool prior to the TCR rearrangement or pre-leukemic clones, which harbor the TCR translocation newly acquire the self-renewal capability. Single cell sequencing analysis is ideal to detect the emergence of those clones. Another important consideration is the mechanism of self-renewal in the ETP subtype of T-ALL. ETP cases show a very different genomic landscape and gene expression signature as compared to non-ETP cases. For example, mutations of NOTCH1 are less frequently found in ETP (27), thus suggesting that different oncogenic mechanisms are involved. Establishment of proper model systems is needed to analyze LICs in this particular subtype.
The mechanisms described above can be also therapeutic targets to eliminate LICs in T-ALL. Disruption of the transcriptional complex involving TAL1 would efficiently block the formation of the CRC and revert the functional imbalance between oncogenic TAL1 complex and E-protein tumor suppressors. Rabbit and his colleagues have developed a peptide and intracellular antibody targeting LMO2 protein to disassociate the TAL1–LMO2 complex (32, 120, 121). Inhibition of transcriptional machinery by small-molecule inhibitors of CDK7 or BRD4 concurrently reduces expressions of multiple oncogenic transcription factors in T-ALL, thereby leading to cell death (122, 123). Moreover, targeting CXCR4/CXCL12 signaling is an ideal strategy to disrupt the interaction between T-ALL cells and stroma cells in the bone marrow niche, as recently reported (82, 83). Additionally, identification of specific cell surface markers associated with LIC capability in T-ALL is critical for developing better therapeutic strategy.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Nature Publishing Group Language Editing for editing the manuscript. We thank members of the Sanda Laboratory for discussions and critical reviews.
Funding
This research is supported by the National Research Foundation, Prime Minister’s Office, Singapore, under its Competitive Research Programme (NRF-NRFF2013-02), the National Medical Research Council, Ministry of Health’s CS-IRG grant (NMRC/CIRG/1443/2016), and the RNA Biology Center at CSI Singapore, NUS, from funding by the Singapore Ministry of Education’s Tier 3 grant (MOE2014-T3-1-006).
References
1. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell (2014) 14(3):275–91. doi:10.1016/j.stem.2014.02.006
2. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med (2011) 17(3):313–9. doi:10.1038/nm.2304
3. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov (2014) 13(7):497–512. doi:10.1038/nrd4253
4. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer (2012) 12(2):133–43. doi:10.1038/nrc3184
5. Cox CV, Martin HM, Kearns PR, Virgo P, Evely RS, Blair A. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood (2007) 109(2):674–82. doi:10.1182/blood-2006-06-030445
6. Gerby B, Clappier E, Armstrong F, Deswarte C, Calvo J, Poglio S, et al. Expression of CD34 and CD7 on human T-cell acute lymphoblastic leukemia discriminates functionally heterogeneous cell populations. Leukemia (2011) 25(8):1249–58. doi:10.1038/leu.2011.93
7. Chiu PP, Jiang H, Dick JE. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood (2010) 116(24):5268–79. doi:10.1182/blood-2010-06-292300
8. Tremblay M, Tremblay CS, Herblot S, Aplan PD, Hébert J, Perreault C, et al. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev (2010) 24(11):1093–105. doi:10.1101/gad.1897910
9. Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC, Kelliher MA. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood (2011) 118(6):1579–90. doi:10.1182/blood-2010-08-300343
10. O’Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell (2004) 5(6):587–96. doi:10.1016/j.ccr.2004.05.023
11. McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science (2010) 327(5967):879–83. doi:10.1126/science.1182378
12. Gerby B, Tremblay CS, Tremblay M, Rojas-Sutterlin S, Herblot S, Hébert J, et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet (2014) 10(12):e1004768. doi:10.1371/journal.pgen.1004768
13. Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol (2008) 8(5):380–90. doi:10.1038/nri2304
14. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer (2016) 16(8):494–507. doi:10.1038/nrc.2016.63
15. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol (2017) 35(9):975–83. doi:10.1200/JCO.2016.70.7836
16. Look AT. Oncogenic transcription factors in the human acute leukemias. Science (1997) 278(5340):1059–64. doi:10.1126/science.278.5340.1059
17. Martins VC, Ruggiero E, Schlenner SM, Madan V, Schmidt M, Fink PJ, et al. Thymus-autonomous T cell development in the absence of progenitor import. J Exp Med (2012) 209(8):1409–17. doi:10.1084/jem.20120846
18. Peaudecerf L, Lemos S, Galgano A, Krenn G, Vasseur F, Di Santo JP, et al. Thymocytes may persist and differentiate without any input from bone marrow progenitors. J Exp Med (2012) 209(8):1401–8. doi:10.1084/jem.20120845
19. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet (2008) 371(9617):1030–43. doi:10.1016/S0140-6736(08)60457-2
20. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol (2011) 29(5):551–65. doi:10.1200/JCO.2010.30.7405
21. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol (2011) 29(5):532–43. doi:10.1200/JCO.2010.30.1382
22. Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia (2008) 22(12):2142–50. doi:10.1038/leu.2008.251
23. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature (2014) 505(7484):495–501. doi:10.1038/nature12912
24. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi:10.1038/nature12477
25. Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol (2005) 23(26):6306–15. doi:10.1200/JCO.2005.05.047
26. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell (2002) 1(1):75–87. doi:10.1016/S1535-6108(02)00018-1
27. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet (2017) 49(8):1211–8. doi:10.1038/ng.3909
28. Porcher C, Liao EC, Fujiwara Y, Zon LI, Orkin SH. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development (1999) 126(20):4603–15.
29. Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature (1995) 373(6513):432–4. doi:10.1038/373432a0
30. Warren AJ, Colledge WH, Carlton MB, Evans MJ, Smith AJ, Rabbitts TH. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell (1994) 78(1):45–57. doi:10.1016/0092-8674(94)90571-1
31. Capron C, Lecluse Y, Kaushik AL, Foudi A, Lacout C, Sekkai D, et al. The SCL relative LYL-1 is required for fetal and adult hematopoietic stem cell function and B-cell differentiation. Blood (2006) 107(12):4678–86. doi:10.1182/blood-2005-08-3145
32. Chambers J, Rabbitts TH. LMO2 at 25 years: a paradigm of chromosomal translocation proteins. Open Biol (2015) 5(6):150062. doi:10.1098/rsob.150062
33. Porcher C, Chagraoui H, Kristiansen MS. SCL/TAL1: a multifaceted regulator from blood development to disease. Blood (2017) 129(15):2051–60. doi:10.1182/blood-2016-12-754051
34. Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J (1996) 15(19):5160–6.
35. Aplan PD, Jones CA, Chervinsky DS, Zhao X, Ellsworth M, Wu C, et al. An scl gene product lacking the transactivation domain induces bony abnormalities and cooperates with LMO1 to generate T-cell malignancies in transgenic mice. EMBO J (1997) 16(9):2408–19. doi:10.1093/emboj/16.9.2408
36. Begley CG, Aplan PD, Davey MP, Nakahara K, Tchorz K, Kurtzberg J, et al. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor delta-chain diversity region and results in a previously unreported fusion transcript. Proc Natl Acad Sci U S A (1989) 86(6):2031–5. doi:10.1073/pnas.86.6.2031
37. Breit TM, Mol EJ, Wolvers-Tettero IL, Ludwig WD, van Wering ER, van Dongen JJ. Site-specific deletions involving the tal-1 and sil genes are restricted to cells of the T cell receptor alpha/beta lineage: T cell receptor delta gene deletion mechanism affects multiple genes. J Exp Med (1993) 177(4):965–77. doi:10.1084/jem.177.4.965
38. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science (2014) 346(6215):1373–7. doi:10.1126/science.1259037
39. Roberts CW, Shutter JR, Korsmeyer SJ. Hox11 controls the genesis of the spleen. Nature (1994) 368(6473):747–9. doi:10.1038/368747a0
40. De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med (2010) 16(11):1321–7. doi:10.1038/nm.2246
41. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia (2013) 27(5):1000–8. doi:10.1038/leu.2012.356
42. Dik WA, Brahim W, Braun C, Asnafi V, Dastugue N, Bernard OA, et al. CALM-AF10+ T-ALL expression profiles are characterized by overexpression of HOXA and BMI1 oncogenes. Leukemia (2005) 19(11):1948–57. doi:10.1038/sj.leu.2403891
43. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol (2009) 10(2):147–56. doi:10.1016/S1470-2045(08)70314-0
44. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306(5694):269–71. doi:10.1126/science.1102160
45. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell (1991) 66(4):649–61. doi:10.1016/0092-8674(91)90111-B
46. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med (2007) 204(8):1813–24. doi:10.1084/jem.20070876
47. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med (2007) 204(8):1825–35. doi:10.1084/jem.20070872
48. Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med (1996) 183(5):2283–91. doi:10.1084/jem.183.5.2283
49. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A (2006) 103(48):18261–6. doi:10.1073/pnas.0606108103
50. Wendorff AA, Koch U, Wunderlich FT, Wirth S, Dubey C, Brüning JC, et al. Hes1 is a critical but context-dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity (2010) 33(5):671–84. doi:10.1016/j.immuni.2010.11.014
51. Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol (2006) 26(21):8022–31. doi:10.1128/MCB.01091-06
52. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev (2006) 20(15):2096–109. doi:10.1101/gad.1450406
53. Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O’Neil J, et al. Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood (2010) 115(9):1735–45. doi:10.1182/blood-2009-07-235143
54. Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med (2012) 18(2):298–301. doi:10.1038/nm.2651
55. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature (2014) 514(7523):513–7. doi:10.1038/nature13605
56. Van der Meulen J, Sanghvi V, Mavrakis K, Durinck K, Fang F, Matthijssens F, et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood (2015) 125(1):13–21. doi:10.1182/blood-2014-05-577270
57. Benyoucef A, Palii CG, Wang C, Porter CJ, Chu A, Dai F, et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev (2016) 30(5):508–21. doi:10.1101/gad.276790.115
58. Seki M, Kimura S, Isobe T, Yoshida K, Ueno H, Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet (2017) 49(8):1274–81. doi:10.1038/ng.3900
59. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res (2006) 66(19):9339–44. doi:10.1158/0008-5472.CAN-06-3126
60. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367(6464):645–8. doi:10.1038/367645a0
61. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med (1997) 3(7):730–7. doi:10.1038/nm0797-730
62. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100(7):3983–8. doi:10.1073/pnas.0530291100
64. Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest (2011) 121(1):384–95. doi:10.1172/JCI41495
65. Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science (2007) 317(5836):337. doi:10.1126/science.1142596
66. Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood (2004) 104(9):2919–25. doi:10.1182/blood-2004-03-0901
67. Cox CV, Diamanti P, Evely RS, Kearns PR, Blair A. Expression of CD133 on leukemia-initiating cells in childhood ALL. Blood (2009) 113(14):3287–96. doi:10.1182/blood-2008-04-154187
68. Castor A, Nilsson L, Astrand-Grundström I, Buitenhuis M, Ramirez C, Anderson K, et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med (2005) 11(6):630–7. doi:10.1038/nm1253
69. Bardini M, Woll PS, Corral L, Luc S, Wittmann L, Ma Z, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia (2015) 29(1):38–50. doi:10.1038/leu.2014.154
70. O’Neil J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, et al. Activating Notch1 mutations in mouse models of T-ALL. Blood (2006) 107(2):781–5. doi:10.1182/blood-2005-06-2553
71. Cui Y, Onozawa M, Garber HR, Samsel L, Wang Z, McCoy JP, et al. Thymic expression of a T-cell receptor targeting a tumor-associated antigen coexpressed in the thymus induces T-ALL. Blood (2015) 125(19):2958–67. doi:10.1182/blood-2014-10-609271
72. Chiang MY, Shestova O, Xu L, Aster JC, Pear WS. Divergent effects of supraphysiologic Notch signals on leukemia stem cells and hematopoietic stem cells. Blood (2013) 121(6):905–17. doi:10.1182/blood-2012-03-416503
73. Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature (2008) 453(7194):529–33. doi:10.1038/nature06933
74. Schubbert S, Cardenas A, Chen H, Garcia C, Guo W, Bradner J, et al. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res (2014) 74(23):7048–59. doi:10.1158/0008-5472.CAN-14-1470
75. Langenau DM, Feng H, Berghmans S, Kanki JP, Kutok JL, Look AT. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A (2005) 102(17):6068–73. doi:10.1073/pnas.0408708102
76. Smith AC, Raimondi AR, Salthouse CD, Ignatius MS, Blackburn JS, Mizgirev IV, et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood (2010) 115(16):3296–303. doi:10.1182/blood-2009-10-246488
77. Blackburn JS, Liu S, Wilder JL, Dobrinski KP, Lobbardi R, Moore FE, et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell (2014) 25(3):366–78. doi:10.1016/j.ccr.2014.01.032
78. Scadden DT. Nice neighborhood: emerging concepts of the stem cell niche. Cell (2014) 157(1):41–50. doi:10.1016/j.cell.2014.02.013
79. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505(7483):327–34. doi:10.1038/nature12984
80. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest (2011) 121(1):396–409. doi:10.1172/JCI35721
81. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia (2009) 23(12):2233–41. doi:10.1038/leu.2009.175
82. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell (2015) 27(6):755–68. doi:10.1016/j.ccell.2015.05.002
83. Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C, et al. CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell (2015) 27(6):769–79. doi:10.1016/j.ccell.2015.05.003
84. Boehm T. Self-renewal of thymocytes in the absence of competitive precursor replenishment. J Exp Med (2012) 209(8):1397–400. doi:10.1084/jem.20121412
85. Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature (2014) 509(7501):465–70. doi:10.1038/nature13317
86. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell (2012) 10(2):120–36. doi:10.1016/j.stem.2012.01.006
87. Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell (2011) 144(2):296–309. doi:10.1016/j.cell.2011.01.004
88. Wilkinson AC, Gottgens B. Transcriptional regulation of haematopoietic stem cells. Adv Exp Med Biol (2013) 786:187–212. doi:10.1007/978-94-007-6621-1_11
89. Rieger MA, Schroeder T. Hematopoiesis. Cold Spring Harb Perspect Biol (2012) 4(12):a008250. doi:10.1101/cshperspect.a008250
90. Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, et al. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J (1997) 16(11):3145–57. doi:10.1093/emboj/16.11.3145
91. Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell (2010) 7(4):532–44. doi:10.1016/j.stem.2010.07.016
92. Moignard V, Woodhouse S, Fisher J, Gottgens B. Transcriptional hierarchies regulating early blood cell development. Blood Cells Mol Dis (2013) 51(4):239–47. doi:10.1016/j.bcmd.2013.07.007
93. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell (2005) 122(6):947–56. doi:10.1016/j.cell.2005.08.020
94. Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, Danford TW, et al. Core transcriptional regulatory circuitry in human hepatocytes. Mol Syst Biol (2006) 2(2006):0017. doi:10.1038/msb4100059
95. Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res (2016) 26(3):385–96. doi:10.1101/gr.197590.115
96. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
97. Batta K, Florkowska M, Kouskoff V, Lacaud G. Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep (2014) 9(5):1871–84. doi:10.1016/j.celrep.2014.11.002
98. Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol (2014) 14(8):529–45. doi:10.1038/nri3702
100. Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol (2005) 6(11):1079–86. doi:10.1038/ni1260
101. Abraham BJ, Hnisz D, Weintraub AS, Kwiatkowski N, Li CH, Li Z, et al. Small genomic insertions form enhancers that misregulate oncogenes. Nat Commun (2017) 8:14385. doi:10.1038/ncomms14385
102. Li Z, Abraham BJ, Berezovskaya A, Farah N, Liu Y, Leon T, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T-ALL. Leukemia (2017). doi:10.1038/leu.2017.75
103. Grutz GG, Bucher K, Lavenir I, Larson T, Larson R, Rabbitts TH. The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. EMBO J (1998) 17(16):4594–605. doi:10.1093/emboj/17.16.4594
104. Hsu HL, Wadman I, Baer R. Formation of in vivo complexes between the TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad Sci U S A (1994) 91(8):3181–5. doi:10.1073/pnas.91.8.3181
105. Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell (2012) 22(2):209–21. doi:10.1016/j.ccr.2012.06.007
106. Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, et al. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol (1997) 17(8):4782–91. doi:10.1128/MCB.17.8.4782
107. Sanda T, Leong WZ. TAL1 as a master oncogenic transcription factor in T-cell acute lymphoblastic leukemia. Exp Hematol (2017) 53:7–15. doi:10.1016/j.exphem.2017.06.001
108. Liau WS, Tan SH, Ngoc PC, Wang CQ, Tergaonkar V, Feng H, et al. Aberrant activation of the GIMAP enhancer by oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Leukemia (2017) 31(8):1798–807. doi:10.1038/leu.2016.392
109. Nitta T, Takahama Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol (2007) 28(2):58–65. doi:10.1016/j.it.2006.12.002
110. Cambot M, Aresta S, Kahn-Perles B, de Gunzburg J, Romeo PH. Human immune associated nucleotide 1: a member of a new guanosine triphosphatase family expressed in resting T and B cells. Blood (2002) 99(9):3293–301. doi:10.1182/blood.V99.9.3293
111. Webb LM, Datta P, Bell SE, Kitamura D, Turner M, Butcher GW. GIMAP1 is essential for the survival of naive and activated B cells in vivo. J Immunol (2016) 196(1):207–16. doi:10.4049/jimmunol.1501582
112. Yano K, Carter C, Yoshida N, Abe T, Yamada A, Nitta T, et al. Gimap3 and Gimap5 cooperate to maintain T-cell numbers in the mouse. Eur J Immunol (2014) 44(2):561–72. doi:10.1002/eji.201343750
113. Chen Y, Yu M, Dai X, Zogg M, Wen R, Weiler H, et al. Critical role for Gimap5 in the survival of mouse hematopoietic stem and progenitor cells. J Exp Med (2011) 208(5):923–35. doi:10.1084/jem.20101192
114. Chadwick N, Zeef L, Portillo V, Boros J, Hoyle S, van Doesburg JC, et al. Notch protection against apoptosis in T-ALL cells mediated by GIMAP5. Blood Cells Mol Dis (2010) 45(3):201–9. doi:10.1016/j.bcmd.2010.07.006
115. Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A (2011) 108(36):14908–13. doi:10.1073/pnas.1109023108
116. Ono Y, Fukuhara N, Yoshie O. TAL1 and LIM-only proteins synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol (1998) 18(12):6939–50. doi:10.1128/MCB.18.12.6939
117. Cheung AM, Wan TS, Leung JC, Chan LY, Huang H, Kwong YL, et al. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia (2007) 21(7):1423–30. doi:10.1038/sj.leu.2404721
118. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1(5):555–67. doi:10.1016/j.stem.2007.08.014
119. Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle (2011) 10(9):1378–84. doi:10.4161/cc.10.9.15486
120. Appert A, Nam CH, Lobato N, Priego E, Miguel RN, Blundell T, et al. Targeting LMO2 with a peptide aptamer establishes a necessary function in overt T-cell neoplasia. Cancer Res (2009) 69(11):4784–90. doi:10.1158/0008-5472.CAN-08-4774
121. Tanaka T, Sewell H, Waters S, Phillips SE, Rabbitts TH. Single domain intracellular antibodies from diverse libraries: emphasizing dual functions of LMO2 protein interactions using a single VH domain. J Biol Chem (2011) 286(5):3707–16. doi:10.1074/jbc.M110.188193
122. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature (2014) 511(7511):616–20. doi:10.1038/nature13393
Keywords: T-cell acute lymphoblastic leukemia, leukemia initiating cells, TAL1, NOTCH1, core regulatory circuit
Citation: Tan SH, Bertulfo FC and Sanda T (2017) Leukemia-Initiating Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 7:218. doi: 10.3389/fonc.2017.00218
Received: 28 June 2017; Accepted: 01 September 2017;
Published: 25 September 2017
Edited by:
Keisuke Ito, Albert Einstein College of Medicine, United StatesReviewed by:
Antonella Papa, Monash University, AustraliaRimas J. Orentas, Lentigen Technology Inc., United States
Copyright: © 2017 Tan, Bertulfo and Sanda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takaomi Sanda, dGFrYW9taV9zYW5kYUBudXMuZWR1LnNn