Abstract
Metastasis is the dissemination of cells from the primary tumor to other locations within the body, and continues to be the predominant cause of death among cancer patients. Metastatic progression within the adult central nervous system is 10 times more frequent than primary brain tumors. Metastases affecting the brain parenchyma and leptomeninges are associated with grave prognosis, and even after successful control of the primary tumor the median survival is a dismal 2–3 months with treatment options typically limited to palliative care. Current treatment options for brain metastases (BM) and disseminated brain tumors are scarce, and the improvement of novel targeted therapies requires a broader understanding of the biological complexity that characterizes metastatic progression. In this review, we provide insight into patterns of BM progression and leptomeningeal spread, outlining the development of clinically relevant in vivo models and their contribution to the discovery of innovative cancer therapies. In vivo models paired with manipulation of in vitro methods have expanded the tools available for investigators to develop agents that can be used to prevent or treat metastatic disease. The knowledge gained from the use of such models can ultimately lead to the prevention of metastatic dissemination and can extend patient survival by transforming a uniformly fatal systemic disease into a locally controlled and eminently more treatable one.
Introduction
A metastatic tumor is a secondary tumor formed from cells that have escaped from a primary tumor elsewhere in the body. Metastases are the most frequent neoplasm to affect the adult central nervous system (CNS), occurring 10 times more than primary brain tumors (1). Metastases commonly arise within the brain parenchyma, where tumor cells travel via the arterial circulation and are deposited at terminal “watershed areas” (2). Metastatic spread to the leptomeninges is a rare presentation of CNS metastasis, developing from the infiltration of metastatic cells into cerebrospinal fluid (CSF) encasing the brain and spine. Approximately 40% of cancer patients will develop brain metastases (BM) (3), and 5–8% will develop leptomeningeal metastases (LM) throughout the progression of the disease (4). The primary cancers that have the highest propensity to develop BM are lung (40–60%), breast (15–30%), and melanoma (5–15%) (5). Up to 24% of hematological malignancies result in LM, 10–32% from primary CNS tumors, and of solid tumors the most common origins are lung (14–29%), breast (11–64%), melanoma (6–18%), and the gastrointestinal tract (4–14%) (6–10). Both BM and LM are associated with poor clinical outcome. The median survival rate of untreated BM and LM patients is 4–6 weeks, yet even when receiving standard interventions (palliative radiotherapy and intrathecal chemotherapy) survival is merely increased to 8–16 weeks (6, 11). The incidence of BM and LM has increased in recent years due to both the improved efficacy of primary tumor interventions, which in turn increases survival and time for metastatic development, as well as the lack of available treatments that are capable of penetrating the blood–brain barrier (BBB) to target the metastatic cells (7, 12). Moreover, administration of current genotoxic treatments can select for increasingly chemoresistant metastatic populations, contributing to their growing resistance over time (7, 12, 13).
The Metastatic Process
Migration
A tumor cell can obtain a metastatic phenotype through several mechanisms. Briefly, this process requires a cell to undergo a (1) loss of cell–cell adhesion, (2) acquisition of motility, and (3) ability to digest through surrounding tissues to enter/exit the circulation. Different strategies can be employed by tumor cells to achieve invasive phenotypes. When intercellular junctions have been lost, cells can migrate as single entities (14). These single cells can adopt two main morphological types to promote their motility, amoeboid, and mesenchymal. Amoeboid cells are rounded or abnormally shaped and produce “bleb”-like protrusions to aid migration (13). A typically accepted mechanism to achieve a mesenchymal phenotype is through the epithelial–mesenchymal transition (EMT) (13). Initiated by external and internal factors (environmental cues, transcription factors, etc.), this transition induces a shift from an epithelial state to a more motile mesenchymal phenotype, characterized by an elongated, spindle-like form (15). Conversely, recent studies have addressed the necessity of EMT in metastasis where, due to the transient, non-linear of the process, tumor cells may not require full completion of EMT to become metastatic. Studies have shown that forced induction of EMT through overexpression of EMT-regulating transcription factors causes a loss of tumor-initiating properties in the mesenchymal tumor cell (16–18). As such, EMT has been proposed to include a spectrum of phenotypes, where a tumor cell will undergo partial phase shifts to promote migration as well as maintain their tumor-initiation capacity (19). Cells can migrate collectively with intact cell–cell contacts, where either a single cell or multiple cells will serve as a leader to a line or sheet of follower cells. Collectively migrating cells can be of either epithelial or mesenchymal phenotypes, which may differ between the leader and follower cells (15). In some cases, EMT may not be required at all to achieve migration. Utilizing a lineage-tracing Cre system, Fischer et al. determined that the majority of cells forming lung metastases were of an epithelial phenotype (20). Similar results were found by Zheng et al., using a genetically engineered model of pancreatic cancer and EMT inhibited by SNAIL1 or TWIST deletion (21).
Invasion into the Circulation
These metastasizing cells can either secrete various matrix metalloproteinases and enzymes to remodel surrounding tissue or, in the case of amoeboid cells activate contractile actin:myosin core networks to squeeze between intercellular spaces, allowing them to intravasate into the surrounding tissue, and invade adjacent blood or lymphatic vessels (22). Once in the circulation, the majority of metastasizing cells will succumb to a myriad of lethal barriers, ranging from host’s immune response to shearing forces within the vessel (23). These metastasizing cells, otherwise known as circulating tumor cells (CTCs), have adopted successful defensive strategies. One of the methods employed by single CTCs involves platelet aggregation, where the CTC will express thrombin to collect platelets and form protective layer from immune surveillance and hemodynamic shearing forces (24). Metastasizing cells that have invaded the circulation as collective groups can form clusters of CTCs or microemboli, arising from oligoclonal tumor cells and are rarer but appear to have a higher metastatic potential than single CTCs (25). These clusters provide protection similar to platelet shields but also an added benefit of avoiding anchorage-dependent apoptosis (26).
Colonization
As the cell arrests at the new site, both through homing mechanisms as well as physical restraints (27), the cell will extravasate into the tissue. Depending on the environmental cues the cell will either remain in a dormant state or colonize the tissue where initial seeding of the brain will form micrometastases and subsequent development of tumor-associated vasculature (neoangiogenesis) will give rise to macrometastases (28). The clonality of the resulting metastasis is dependant on the nature of the seeding cell. A single cell can give rise to a clonal metastasis, whereas a polyclonal metastasis can develop from a CTC cluster or seeding of the same region by multiple single cells (29). Recent studies have identified metastases to be primarily polyclonal, including prostate (30), breast (31), and pancreatic (32), which is consistent with the concept of enhanced survival and metastatic seeding potential by CTC clusters over single metastatic cells (29).
To enter the CNS, metastasizing cells must overcome additional barriers during extravasation and colonization: the BBB and brain–CSF barrier (BCSFB). It is thought that the properties required to exit the circulation are rate limiting; though millions of cells can be shed into the circulation, only a very small percentage are able to colonize the secondary environment (33). However, once these obstacles are overcome, the CNS becomes a sanctuary site for these metastasizing cells, allowing their escape and protection from typical cytotoxic agents and immune surveillance that are unable to cross an intact BBB and BCSFB (7).
The flow of arterial blood largely determines the spread of metastatic cells throughout the brain parenchyma: 85% of BM arise within the cerebrum, 5–10% within the cerebellum, and 3–5% within the brainstem (2). The type of primary tumor can also dictate the distribution and number of metastases within the brain. For instance, lung cancers typically result in multiple lesions within the occipital lobe and cerebellum, whereas breast cancer results in metastases within the brain parenchyma, leptomeninges, cerebellum, and brain stem (5, 34). Within the meninges, LM are generated either by diffuse, non-adherent single cells or clusters or nodules (35).
Brain metastases typically develop from solid and hematological tumors, whereas LM can arise from primary CNS tumors (e.g., medulloblastoma, glioma, and PNET), systemic cancers (lymphoma and leukemia), or solid tumors. In LM, metastatic cells gain access to the CSF (and subsequently the leptomeninges) through several methods: the most common route is hematogenous or lymphatic systems; however, cells are also able to enter the CSF by escaping from adjacent bone tumors (i.e., the skull or spine) into the dural sinus or epidural plexus (36). Once in the CSF these cells can travel throughout the CNS, either remaining within the leptomeninges or invading the brain parenchyma, spinal cord, or nerve roots (Figure 1).
Figure 1
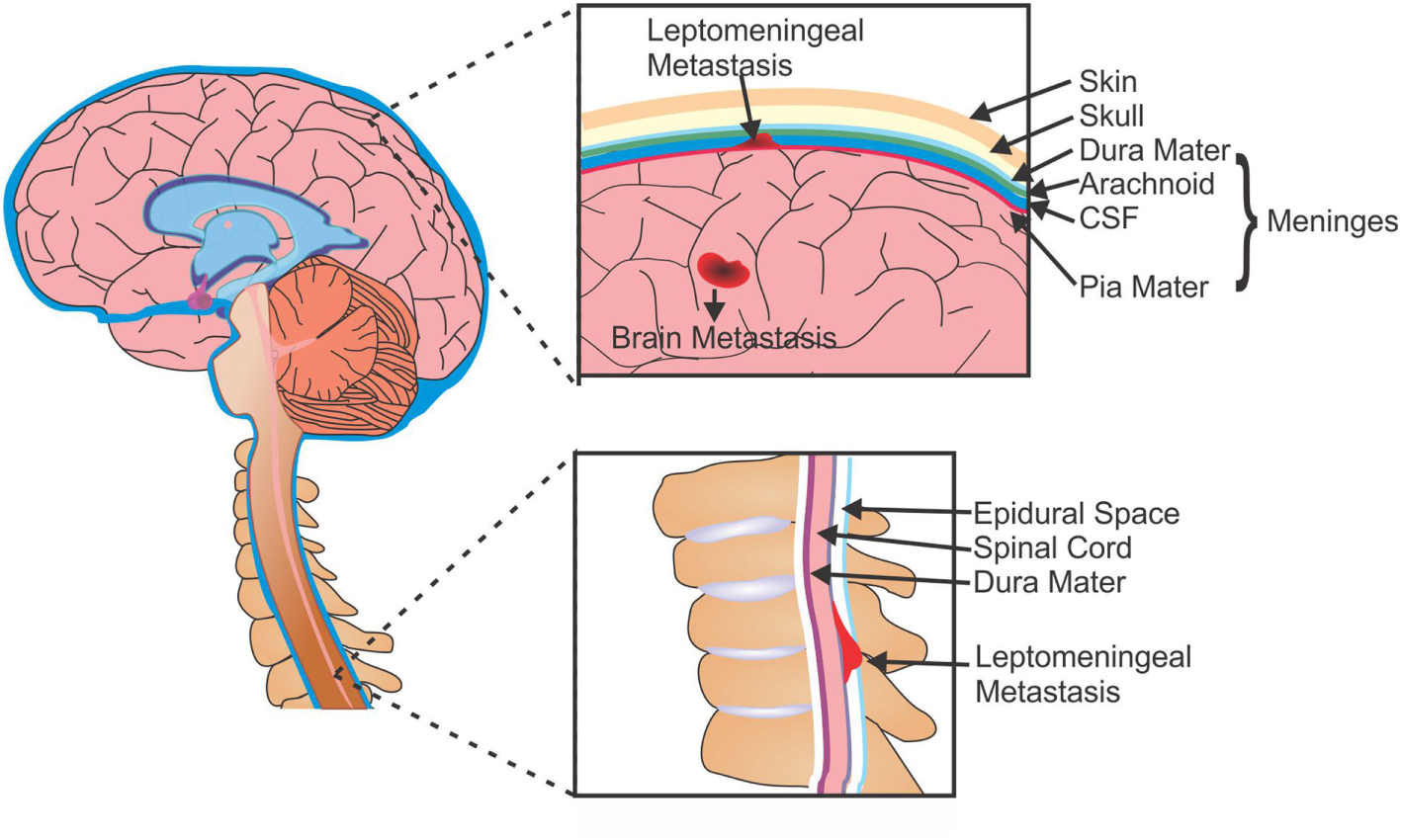
Metastases of the brain and leptomeninges. Cells from primary tumors are able to metastasize to the brain and can form secondary tumors within the parenchyma (BM), or can enter the CSF and spread to the membranes surrounding the brain and spine (LM). CSF, cerebrospinal fluid; BM, brain metastasis; LM, leptomeningeal metastasis.
A significant controversy in cancer research surrounds the origin of metastatic spread; do metastases develop in a linear fashion after primary tumor formation, or in parallel to the primary tumor? The linear model of dissemination is intimated in cancers where there are close genetic similarities between the primary and secondary tumors, whereas the parallel model is suggested in cases of genetic diversity. A third theory suggests metastasis-to-metastasis seeding (37). Although from several studies the general thought appears to lean toward parallel progression of metastatic dissemination, recent phylogenetic studies have shown multiple modes of dissemination (38–42).
Considerations for the Development of BM and LM Models
The complex nature of the metastatic process has led to the generation of several 2- and 3-dimensional in vitro assays that strive to recapitulate the various stages of metastasis under more stable conditions. For instance angiogenesis or neovascularization, the process of forming new blood vessels branching from existing vasculature, can be recognized through a tube formation assay, where cells plated on an extracellular matrix layer that mimics the in vivo environment and will form tubule-like structures that resemble vessels (43). Cancer cell migration, an integral component of metastatic cells, can be modeled in multiple assays including transwell or Boyden chambers, scratch wound, zone exclusion, or microfluidics. These assays have been invaluable as tools to not only delineate the intricacies of the mechanistic regulation of metastasis but also serve as screening platforms for therapeutic targets, unfortunately they also face several limitations. The involvement of complex host/cell interactions throughout BM development are more accurately examined in vivo, such as anatomical barriers (BBB and BCSFB), stromal/environmental determinants, immune signaling and response, and cytokines/growth factors (44). As such, animal models represent a vital tool in a scientist’s repertoire for translational research. A clinically relevant in vivo model can enable researchers to identify the genetic events that contribute to metastatic development within the CNS and provide a platform to identify and screen novel therapeutics (44).
Although the genetic mouse models have become an important tool in studying the functional significance of defined mutations in the development of BM and LM, such models lack the ability to recapitulate the genetic heterogeneity of primary human tumors. Furthermore, the genetically engineered mouse models (GEMMs) are limited by complex breeding schemes, incomplete tumor penetrance, and variable tumor onset (45). In contrast, patient-derived xenograft (PDX) models for many cancer subtypes (46–51) have been generated through injection of patient tumor cells into an appropriate microenvironment. Tumors generated through PDX models have been shown to retain the molecular identity and recapitulate the complex heterogeneity of the original patient tumor. In addition, PDX models allow for a more accurate evaluation of tumor growth patterns, metastatic properties, and their changes in response to therapeutic intervention (52, 53). Currently, various xenograft models have been developed that are capable of reproducing specific individual stages of metastasis, providing a more detailed understanding of the intricacies involved in the process. For instance, the avian embryo provides a unique model support system for many metastatic features, including growth, invasion, and angiogenesis. The chorioallantoic membrane, a vascularized embryonic tissue, shows easy engraftment of human cells, and the embryo itself provides an immunodeficient environment (54–56). The use of zebrafish xenograft models has also risen over the last few years, providing a novel high throughput and inexpensive platform for drug discovery and in vivo imaging (57, 58). Despite the novelty of these unique models, the use of mice and rats (murine) has remained a standby host species in modeling metastasis, providing highly reproducible disease development and easy to manipulate/inject due to size (59). The advent of transgenic and immunodeficient strains significantly increased the success rate of tumor transplantation and human–mouse xenograft model development (60).
When developing an appropriate in vivo model for LM and BM, several biological and technical factors must be considered.
-
Material injected: commercial cells are a commonly utilized cell source, providing an unlimited number of cells for cost-effective use. Unfortunately, these lines have undergone significant selective pressures from years of culturing that they rarely represent the genotypic or phenotypic profile of the original patient sample and are sometimes even misidentified or cross-contaminated (61). Conversely, patient-derived cell lines provide a much more accurate representation of clonal heterogeneity existing within the original tumor, as the length of culture remains minimal, though these lines also face difficulties due to poor growth and engraftment, and a limited life span (61). Moreover, a significant concern with patient-derived cell lines is that the majority of patients have received some form of therapy, thus there are very few samples that have not faced a selection pressure from exposure to a chemotherapeutic (62).
-
The number of cells delivered, a property easily controlled by the researcher, can also play a large role in the time it takes for engraftment. A larger the cell number injected may permit for a shorter incubation period; however, this may not accurately represent the slower growth observed with the clinical presentation of metastatic progression. Conversely, a low cell number may not be engrafted easily, reducing the success rate of engraftment or cell collection (62).
-
Host selection: the choice of host when establishing a metastasis model can be key to successful engraftment rates. Murine models can be divided into two broad categories: (1) syngeneic and (2) xenogeneic. Syngeneic models utilize cancer cell lines of the same genetic background as the host and are typically generated through chemical or spontaneous induction. These models offer researchers the ability to study oncogenesis and metastatic progression in the presence of a functioning immune response and potential to identify therapeutics that can target the immune system. Unfortunately, this model is solely mouse related, which can have difficulties with correlations to human disease. On the contrary, xenograft models are developed from the administration of human cancer cells into an immunocompromised host. The lack of an immune response, which would otherwise attack the foreign cells injected and limit engraftment, permits a high rate of human tumor transplantation and study of human cancer cell behavior in a live host but lacks information on the interaction between the immune system and tumor cells.
-
The route of injection (Figure 2): the location of cell delivery and subsequent tumor engraftment and metastatic progression is another decision vital to model development. Due to circulation patterns, some locations for metastatic spread are more likely to over others, such as tail vein injections resulting primary in lung metastases (62). Certain hosts do not possess the proper/compatible physiology to represent clinical disease progression, whereas injections in some areas may not even be feasible for a particular host due to anatomical differences. Another criteria is host size, where a larger animal may allow for easy and safe repeated access to the injection route (59).
Figure 2
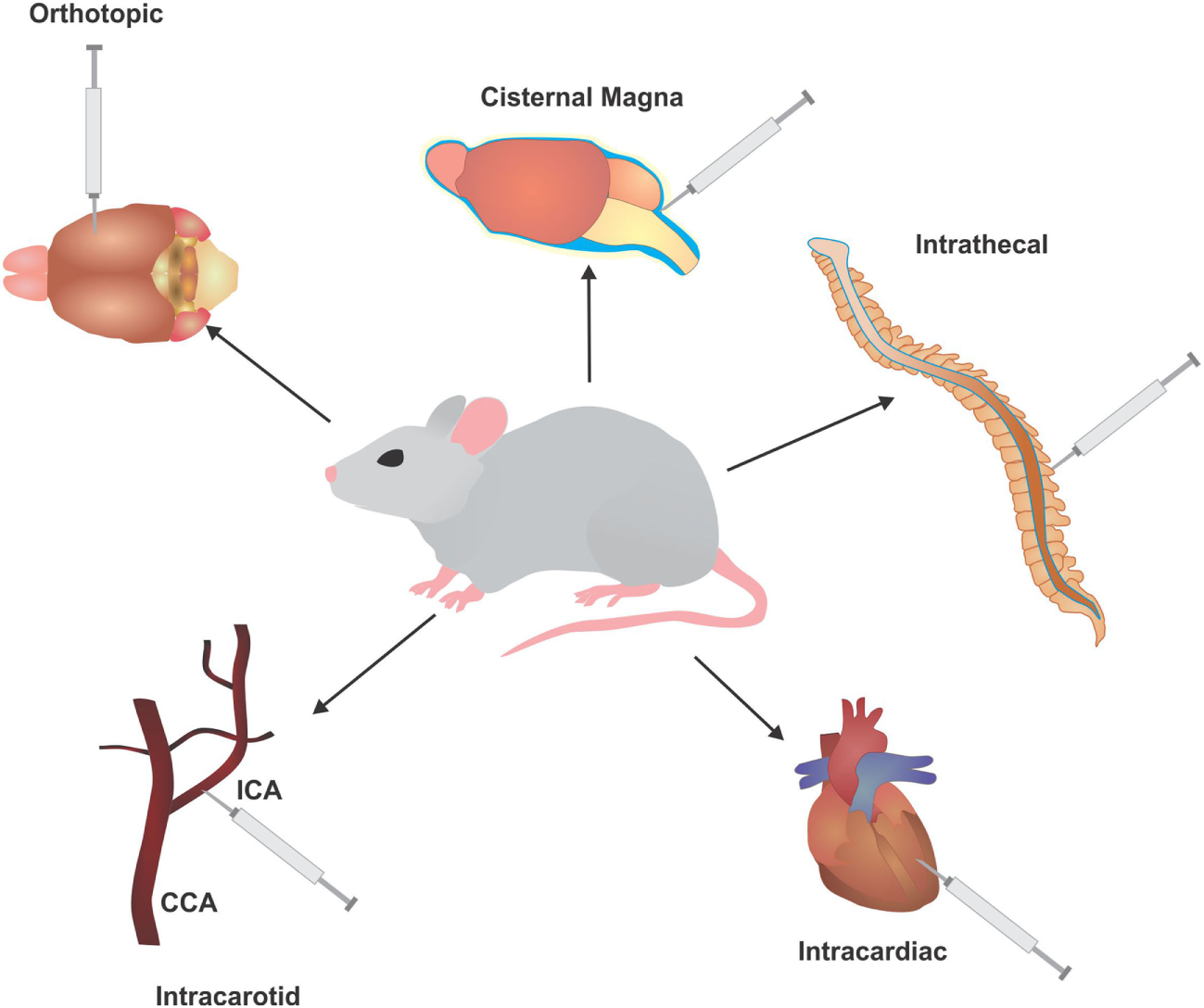
Injection routes utilized to develop in vivo models of leptomeningeal metastases (LM) and brain metastases (BM). Common injection routes used in murine models to develop BM and LM, typically involving injection of cells of directly into the circulation or cerebrospinal fluid to bypass the initial stages of metastasis. ICA, intracarotid artery; CCA, common carotid artery.
When modeling metastasis, the best route of injection would replicate tumor formation at the primary site first and subsequent metastatic development. Several such models have been established with commercial mouse and human cell lines, unfortunately this method can be laden with difficulties in capturing the metastatic cells at the desired secondary site. To overcome this, successive rounds of in vivo selection are performed with cells harvested from the secondary site and reinjected, selecting for cells that are aggressively metastatic with each round (63, 64).
-
(a)
Intracardiac/intracarotid: a common method for BM development is direct injection of tumor cells into the circulation. This method is more of an assessment of brain colonization and not full metastasis, as it selectively ignores the ability of cells to undergo EMT and intravasate into the circulation. In addition, the number of cells injected into the circulation is several folds higher than the number of cells that would typically escape from the primary tumor. Nonetheless, this method allows for selection of highly metastatic populations that are able to cross the BBB and BCSF to engraft into the brain. Intracardiac injections (via the left ventricle) allow cells to freely enter the circulation and have indiscriminate access to all organs of the body, allowing cells to seed metastases in different areas (65). Injection of cells into the intracarotid artery allows cells to travel directly to the brain, and primarily produces BM and LM (66–69).
-
(b)
Orthotopic: orthotopic injections place cells directly into the originating environment of the primary tumor. For BM development, the most common route of inoculation of tumor cells derived from a BM or primary brain tumor is directly into the brain parenchyma (intracranial). This surpasses all barriers encountered in the initial and mid stages of metastasis, allowing the cells to begin colonization, but creating a significant selection bias by giving cells that may not be capable of surviving the metastatic cascade an opportunity to engraft. When utilizing tumor cells from other primary cancers, the injection site will follow accordingly for better representation of the metastatic cascade. For instance, melanoma cells can be injected subcutaneously, lung cancer cells injected intrathoracically, and breast cancer cells injected into the fatpad (70–72).
-
(c)
Intrathecal: a common method for LM modeling is intrathecal, allowing direct entry of cells into the CSF via the cisterna magna or spinal canal (73, 74). The larger size of a rat provides safe, easy, and repeated access to the CSF through these methods. For instance, an arachnoid catheter can be implanted into the great cistern and passed along the spinal cord (75).
Keeping these factors in mind, several groups have proven successful in developing clinically relevant models of LM and BM. Massague et al. performed several rounds of selection on human and mouse cancer lines through injection into the cisterna magna, allowing the cells to propagate within the leptomeningeal space before collecting the cells from the basilar meninges and thecal sac. After the third round of selection, cells were then injected intracardially, where hematogenously disseminated cells were found to consistently form LM as opposed to BM, faithfully replicating many clinical and histopathological aspects of LM (76). A more recent study preformed by this group utilized LM models to dissect the molecular processed involved in leptomeningeal dissemination of breast and lung cancers. They determined that cancer cells within the CSF express complement component 3 to promote disruption of the BCSF and is predictive of leptomeningeal relapse (77).
Sandén et al. were able to propagate a cell line from a primary patient sample of Group 3 MB with overexpression of c-Myc, a gene that the worst clinical outcome for MB. Intracerebellar injection of this cell line resulted in primary brain tumors that recapitulated both epigenetic and phenotypic characteristics of the original patient sample. In addition, similar to the clinical progression of the disease, researchers were able to observe metastatic spread to the meninges and down the spinal axis even before it was observed in the surviving patient, thus providing an opportunity to study early stages of spinal dissemination and allow for preclinical evaluation of targeted therapeutic interventions (45).
Recently, Singh et al. successfully established cell lines from primary patient samples of lung BMs, where specific in vitro culture conditions were utilized to enrich for a metastatic subpopulation of cancer stem cells within these BM, termed brain metastasis-initiating cells (BMICs). Using these BMICs, they developed a novel PDX model of lung-to-brain metastasis, where through intracardiac injections the researchers were able to obtain macrometastases, whereas intrathoracic injection of BMICs not only reformed tumors within the lung but also developed micrometastatic growths within the brain (78).
Modeling Therapy Response
Originally described in 1800s (79, 80), few substantial advancements have been made since then in understanding the progression of LM, and this progress has added very little to the improvement upon the dismal survival rates for patients. A similar lack of therapeutic progress is seen with BMs. The typical treatment strategy for BM and LM is palliative care for poor risk patients, as treatment options offering any significant extension of survival are presently not available. Poor risk patients may receive radiation therapy, analgesics and corticosteroids for persistent pain and headaches, antidepressants, antinauseants, and anticonvulsants (36). Good risk patients will receive treatments tailored to control the tumor, including stereotactic radiosurgery, whole-brain radiotherapy, surgical resection, and systemic treatments (81). Systemic therapies include chemotherapies, small molecules (Table 1), and immunotherapies and can be administered depending on various patient factors, such as tumor histology and the patient’s prior treatment history to theoretically target both the active systemic disease as well as the LM and BM (7). Intrathecal administration (direct injection into the spinal canal) of anticancer agents guarantees the treatment will enter the CSF; however, this route can have limited efficacy. An alternative intraventricular administration shows improved CSF drug levels, especially in bulky tumors, and less variability between patients (7).
-
Chemotherapy: various chemotherapeutic agents have been employed to treat LM and BM, often used in a combination of 2–3 along with whole-brain radiotherapy. For BM, standard chemotherapies are administered based on the primary cancer. For instance cisplatin, cyclophosphamide, etopside, prednisone, and irinotecan have all been administered for BM and LM of lung, breast, and melanoma cancers (81). Capecitabine, belonging to the class of fluoropyrimidines of chemotherapies, is administered for several cancer types (106), and a phase II trial has shown the combination of lapatinib and capecitabine as a first line treatment for HER2-positive breast cancer (89). Temozolomide has shown a modest therapeutic effect when administered alone, however shows much more promise when used in conjunction with whole-brain radiotherapy and/or other anticancer agents, as discussed in a thorough review by Zhu et al. (107). For LM, standard chemotherapies administered are methotrexate, cytarabine (Ara-C), thiotepa, all safe for intrathecal administration (108).
-
Small molecules: the mutational status of the primary tumor can determine the type of small molecule administered to BM and LM. Tyrosine kinase inhibitors (TKI) such as gefitinib, osimertinib, and erlotinib target EGFR mutations found in lung cancers, lapatinib is a dual TKI that targets HER2/neu and EGFR mutations and is applied to advanced or metastatic breast cancer, vemurafenib and dabrafenib target BRAF mutations in melanoma, and crizotinib, ceritinib, and alectinib target anaplastic lymphoma kinase fusions in lung cancers (7, 81).
-
Biologics: recent studies have shown promising results with the administration of monoclonal antibodies and immune-modulating therapies, activating T-cell responses to target BM and LM in a non-cytotoxic manner. Bevacizumab is a monoclonal antibody that targets high levels of VEGF in several cancers to inhibit angiogenesis (109). Rituximab and trastuzumab are non-cytotoxic monoclonal antibodies that are administered intrathecally, targeting CD20 and HER2/neu, respectively (7); however, trastuzumab has been linked with increased incidence of brain metastasis. Checkpoint inhibitors such as ipilimumab and nivolumab are a new class of immunotherapies showing great promise in phase trials, targeting CTLA-4 and PD-L1 in metastasis of kidney, melanoma, and non-small cell lung cancer (6, 110). A more extensive coverage of the ongoing research on immunotherapies directed at BM is comprehensively discussed in the review by Farber et al. (111).
Table 1
| Drug | Mechanism of action | Mode of delivery | Clinical uses | Reference |
|---|---|---|---|---|
| Chemotherapy | ||||
|
|
||||
| Cisplatin | Inhibitor of DNA replication | Intravenous infusion | Treatment of BM in patients with breast cancer, NSCLC, and melanoma | Franciosi et al. (82) |
|
|
||||
| Cyclophosphamide | Inhibitor of DNA replication | Intravenous infusion; oral administration | Treatment of BM in patients with breast cancer | Rosner et al. (83) |
|
|
||||
| Irinotecan | Inhibitor of DNA replication and transcription | Intravenous infusion | Treatment of BM in patients with small cell lung cancer | Sevinc et al. (84) |
|
|
||||
| Methotrexate | S-phase specific cytotoxic chemotherapy | Intraventricular or intrathecal infusion | Treatment of patients with neoplastic meningitis | Grossman et al. (8) |
|
|
||||
| Cytarabine (Ara-C) | Antimetabolite, blocks activity of DNA polymerase | Intraventricular or intrathecal infusion | Targeting leptomeningeal dissemination of patients with glioma, breast cancers, and NSCLC | Zhao et al. (85) |
| Niwińska et al. (86) | ||||
| Disruption of PI3K/Akt/mTOR pathway | Nagpal et al. (87) | |||
|
|
||||
| Thiotepa | Non-specific cell cycle inhibitor | Intraventricular or intrathecal infusion | Treatment of patients with neoplastic meningitis | Grossman et al. (8) |
|
|
||||
| Capecitabine | Antimetabolite, blocks activity of DNA polymerase | Oral administration | Treatment of BM in patients with HER2-positive breast cancer | Petrelli et al. (88) Bachelot et al. (89) |
|
|
||||
| Small molecules | ||||
|
|
||||
| Gefitinib | Inhibitor of EGFR-associated tyrosine kinase | Oral administration | Targeting brain metastasis in patients with NSCLC | Ceresoli et al. (90) |
|
|
||||
| Osimertinib | Inhibitor of EGFR-activating mutations and EGFR with T790M mutation | Oral administration | Targeting brain metastasis in NSCLC patients with EGFR T790M mutation | Koba et al. (91) |
|
|
||||
| Erlotinib | Inhibitor of EGFR-associated tyrosine kinase | Oral administration | Targeting brain metastasis in patients with NSCLC | Sperduto et al. (92) |
|
|
||||
| Lapatinib | Inhibitor of EGFR and HER2/neu | Oral administration | Targeting brain metastasis in patients with HER2+ breast cancer | Lin et al. (93) |
| Lin et al. (94) | ||||
|
|
||||
| Vemurafenib | Inhibitor of BRAF, resulting in disruption of BRAF/MEK/ERK pathway | Oral administration | Targeting brain metastasis in patients with V600E BRAF mutation-positive melanoma | Dummer et al. (95) |
|
|
||||
| Dabrafenib | Inhibitor of BRAF, resulting in disruption of BRAF/MEK/ERK pathway | Oral administration | Targeting brain metastasis in patients with V600E BRAF mutation-positive melanoma | Long et al. (96) |
|
|
||||
| Crizotinib | Inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) | Oral administration | Targeting brain metastasis in patients with EML4-ALK fusion NSCLC | Yoshida et al. (97) |
|
|
||||
| Ceritinib | Inhibitor of ALK | Oral administration | Targeting brain metastasis in patients with ALK-positive NSCLC | Melosky et al. (98) |
|
|
||||
| Alectinib | Inhibitor of ALK | Oral administration | Targeting brain metastasis in patients with ALK-positive NSCLC that are resistant to crizotinib | Gadgeel et al. (99) |
|
|
||||
| Biologics | ||||
|
|
||||
| Bevacizumab | Monoclonal antibody, blocks angiogenesis though inhibition of VEGF-A | Intravenous infusion | Targeting brain metastasis in patients with NSCLC | Besse et al. (100) |
|
|
||||
| Rituximab | Monoclonal antibody targeting CD20 | Intraventricular or intrathecal infusion | Targeting leptomeningeal dissemination of patients with lymphoma | Schulz et al. (101) |
|
|
||||
| Trastuzumab | Monoclonal antibody targeting HER2/neu | Intravenous infusion or subcutaneous injection | Preventing brain metastasis development in HER2-overexpressing metastatic breast cancer | Park et al. (102) |
|
|
||||
| Ipilimumab | Monoclonal antibody targeting CTLA-4 | Intravenous infusion | Targeting brain metastasis in patients with melanoma | Margolin et al. (103) |
|
|
||||
| Nivolumab | Monoclonal antibody targeting PD-1 | Intravenous infusion | Targeting brain metastasis in patients with NSCLC | Dudnik et al. (104) |
|
|
||||
| Pembrolizumab | Monoclonal antibody targeting PD-1 | Intravenous infusion | Targeting brain metastasis in patients with melanoma or NSCLC | Goldberg et al. (105) |
List of selected chemotherapies, small molecules, and immunotherapies administered in the treatment of brain metastases (BM) and leptomeningeal metastases.
One of the major hurdles in developing novel therapies for brain tumors has been the paucity of overlapping actionable targets between treatment-naïve and recurrent tumors. Using a sleeping beauty transposon system in Ptch+/− mice with mutated Trp53, Wu et al. were able to generate GEMM of sonic hedgehog MB with increased tumor penetrance and reduced latency period (112). The introduction of humanized therapy protocols combining surgical resection and fractionated craniospinal irradiation led to generation of a mouse model that recapitulated both tumor initiation as well as disease progression, including rise of local and distal metastasis (113). The development of novel therapeutics is further complicated by the hurdles encountered throughout delivery of anticancer drugs to the brain. The BBB and BCSFB are substantial obstacles that need to be overcome to identify feasible cancer therapies. Both barriers differ in composition, permeability, and function. The BBB is a barrier formed by the endothelial cells of the brain capillaries, closely associated with pericytes, perivascular astrocytes, and microglia, and separates the circulating blood from the brain interstitial fluid (114). The BCSFB is composed of modified cuboidal epithelium of the choroid plexus, serving to secrete and separate CSF from circulating blood (114). Both barriers express transporters, multi-specific carriers, receptors, and enzymes that help to regulate diffusion and transport of polar molecules, essential nutrients, and wastes, and restrain the passage of anticancer compounds into the brain (114). Several factors within the composition of a drug also impede its ability to penetrate the BBB and BCSFB, including lipid solubility, molecular weight, polarity, and protein binding (7).
Various methods have been employed when designing drugs and delivery systems to increase drug efficacy in crossing the BBB and BCSF and targeting BM and LM. One method utilized to avoid these barriers is the use of transporters, where a drug that is not able to cross the BBB is coupled to a substance that can. This process can improve the peripheral pharmacokinetics, yet results in a hybrid compound that may not be recognized by the transporter or is destroyed as a foreign body (115). Another method is the formulation of a compound to be highly lipid soluble and with low molecular weight, increasing the likelihood of drug transport by transmembrane diffusion. Typical clinical therapeutic drugs are small, lipid soluble molecules. Unfortunately, once across the BBB the drug must then enter the surrounding aqueous interstitial fluid of the brain to be effective, resulting in drugs that are too lipid soluble being sequestered to the capillary bed and unable to reach areas beyond the BBB (116). The evidence of exosome-based communication in neural cells (117) opened up a possibility of potentially developing therapies that deliver short interfering RNA (siRNA) against specific targets to the brain. Despite a lack of clinical trial testing the efficacy of exosome-based therapies for cancer has been initiated, a study published by Alvarez-Erviti et al. (118) demonstrated a prominent reduction of both mRNA and protein levels of BACE1 within multiple brain cell lineages post siRNA delivery to the brain (118). Other delivery strategies include targeting peptides, regulatory proteins, and oligonucleotides (115).
Conclusion
Brain metastases and LM are a common complication of cancer progression, associated with poor survival and limited treatment options. Elucidation of the molecular mechanisms underlying brain metastasis development, which includes the shifting microenvironment and interactions with the immune system the disseminated cell encounters, is supremely difficult to capture with an in vitro system. Consequently, experimental in vivo models are heavily relied on to serve as platforms to explore the nature of metastatic dissemination in a more comprehensive manner. Unique models have been generated with fish, mice, chicks, and companion animals (cats and dogs), all providing much needed knowledge to the field. However, there are several shortcomings associated with in vivo models, including the lack of feasible models that recapitulate the clinical progression of BM development in its entirety, and the obvious dissimilarities between the biological makeup of an animal and human. As such one must be aware of the benefits and caveats associated with available models to properly interpret results. Nonetheless, these animal models have provided significant knowledge of the characterization of metastatic disease progression in a live host and are a fundamental component to the identification, study, and testing of new cancer regimens.
Statements
Author contributions
Conceptualization of review, drafting of work, revision of manuscript, figure design, final approval of manuscript, and agreement for accountability for content of work: MS. Conceptualization of review, revision of manuscript, final approval of manuscript, and agreement for accountability for content of work: DB, CV, and SS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Sjøbakk TE Vettukattil R Gulati M Gulati S Lundgren S Gribbestad IS et al Metabolic profiles of brain metastases. Int J Mol Sci (2013) 14:2104–18.10.3390/ijms14012104
2
Patchell RA . The management of brain metastases. Cancer Treat Rev (2003) 29:533–40.10.1016/S0305-7372(03)00105-1
3
Gavrilovic IT Posner JB . Brain metastases: epidemiology and pathophysiology. J Neurooncol (2005) 75:5–14.10.1007/s11060-004-8093-6
4
Glantz MJ Hall WA Cole BF Chozick BS Shannon CM Wahlberg L et al Diagnosis, management, and survival of patients with leptomeningeal cancer based on cerebrospinal fluid-flow status. Cancer (1995) 75:2919–31.10.1002/1097-0142(19950615)75:12<2919::AID-CNCR2820751220>3.0.CO;2-9
5
Palmieri D , editor. Central Nervous System Metastasis, the Biological Basis and Clinical Considerations Vol. 18 Cancer Metastasis – Biology and Treatment. (Chap. 1). Netherlands: Springer (2012). p. 1–13.
6
Leal T Chang JE Mehta M Robins HI . Leptomeningeal metastasis: challenges in diagnosis and treatment. Curr Cancer Ther Rev (2011) 7:319–27.10.2174/157339411797642597
7
Groves MD . New strategies in the management of leptomeningeal metastases. Arch Neurol (2010) 67:305–12.10.1001/archneurol.2010.18
8
Grossman SA Finkelstein DM Ruckdeschel JC Trump DL Moynihan T Ettinger DS . Randomized prospective comparison of intraventricular methotrexate and thiotepa in patients with previously untreated neoplastic meningitis. Eastern Cooperative Oncology Group. J Clin Oncol (1993) 11:561–9.10.1200/JCO.1993.11.3.561
9
Nugent JL Bunn PA Jr Matthews MJ Ihde DC Cohen MH Gazdar A et al CNS metastases in small cell bronchogenic carcinoma: increasing frequency and changing pattern with lengthening survival. Cancer (1979) 44:1885–93.10.1002/1097-0142(197911)44:5<1885::AID-CNCR2820440550>3.0.CO;2-F
10
Kaplan JG DeSouza TG Farkash A Shafran B Pack D Rehman F et al Leptomeningeal metastases: comparison of clinical features and laboratory data of solid tumors, lymphomas and leukemias. J Neurooncol (1990) 9:225–9.10.1007/BF02341153
11
Soffietti R Ruda R Mutani R . Management of brain metastases. J Neurol (2002) 249:1357–69.10.1007/s00415-002-0870-6
12
Langley RR Fidler IJ . The biology of brain metastasis. Clin Chem (2013) 59:180–9.10.1373/clinchem.2012.193342
13
Heerboth S Housman G Leary M Longacre M Byler S Lapinska K et al EMT and tumor metastasis. Clin Transl Med (2015) 4:6.10.1186/s40169-015-0048-3
14
Friedl P Alexander S . Cancer invasion and the microenvironment: plasticity and reciprocity. Cell (2011) 147:992–1009.10.1016/j.cell.2011.11.016
15
Clark AG Vignjevic DM . Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol (2015) 36:13–22.10.1016/j.ceb.2015.06.004
16
Celià-Terrassa T Meca-Cortés O Mateo F Martínez de Paz A Rubio N Arnal-Estapé A et al Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest (2012) 122:1849–68.10.1172/JCI59218
17
Ocaña OH Córcoles R Fabra A Moreno-Bueno G Acloque H Vega S et al Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell (2012) 22:709–24.10.1016/j.ccr.2012.10.012
18
Tsai JH Donaher JL Murphy DA Chau S Yang J . Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell (2012) 22:725–36.10.1016/j.ccr.2012.09.022
19
Lambert AW Pattabiraman DR Weinberg RA . Emerging biological principles of metastasis. Cell (2017) 168:670–91.10.1016/j.cell.2016.11.037
20
Fischer KR Durrans A Lee S Sheng J Li F Wong ST et al Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature (2015) 527:472–6.10.1038/nature15748
21
Zheng X Carstens JL Kim J Scheible M Kaye J Sugimoto H et al Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature (2015) 527:525–30.10.1038/nature16064
22
Friedl P Wolf K . Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer (2003) 3:362–74.10.1038/nrc1075
23
Luzzi KJ MacDonald IC Schmidt EE Kerkvliet N Morris VL Chambers AF et al Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol (1998) 153:865–73.10.1016/S0002-9440(10)65628-3
24
Palumbo JS Talmage KE Massari JV La Jeunesse CM Flick MJ Kombrinck KW et al Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood (2007) 110:133–41.10.1182/blood-2007-01-065995
25
Aceto N Bardia A Miyamoto DT Donaldson MC Wittner BS Spencer JA et al Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158:1110–22.10.1016/j.cell.2014.07.013
26
Liotta LA Saidel MG Kleinerman J . The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res (1976) 36:889–94.
27
Ramakrishna R Rostomily R . Seed, soil, and beyond: the basic biology of brain metastasis. Surg Neurol Int (2013) 4:S256–64.10.4103/2152-7806.111303
28
Singh M Manoranjan B Mahendram S McFarlane N Venugopal C Singh SK . Brain metastasis-initiating cells: survival of the fittest. Int J Mol Sci (2014) 15:9117–33.10.3390/ijms15059117
29
Cheung KJ Ewald AJ . A collective route to metastasis: seeding by tumor cell clusters. Science (2016) 352:167–9.10.1126/science.aaf6546
30
Gundem G Van Loo P Kremeyer B Alexandrov LB Tubio JMC Papaemmanuil E et al The evolutionary history of lethal metastatic prostate cancer. Nature (2015) 520:353–7.10.1038/nature14347
31
Cheung KJ Padmanaban V Silvestri V Schipper K Cohen JD Fairchild AN et al Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A (2016) 113:E854–63.10.1073/pnas.1508541113
32
Maddipati R Stanger BZ . Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov (2015) 5:1086–97.10.1158/2159-8290.CD-15-0120
33
Vanharanta S Massague J . Origins of metastatic traits. Cancer Cell (2013) 24:410–21.10.1016/j.ccr.2013.09.007
34
Barnholtz-Sloan JS Sloan AE Davis FG Vigneau FD Lai P Sawaya RE . Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol (2004) 22:2865–72.10.1200/JCO.2004.12.149
35
Mack F Baumert BG Schäfer N Hattingen E Scheffler B Herrlinger U et al Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat Rev (2016) 43:83–91.10.1016/j.ctrv.2015.12.004
36
DeAngelis LM Boutros D . Leptomeningeal metastasis. Cancer Invest (2005) 23:145–54.10.1081/CNV-50458
37
Naxerova K Jain RK . Using tumour phylogenetics to identify the roots of metastasis in humans. Nat Rev Clin Oncol (2015) 12:258–72.10.1038/nrclinonc.2014.238
38
Sanborn JZ Chung J Purdom E Wang NJ Kakavand H Wilmott JS et al Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A (2015) 112:10995–1000.10.1073/pnas.1508074112
39
Krøigård AB Larsen MJ Brasch-Andersen C Lænkholm AV Knoop AS Jensen JD et al Genomic analyses of breast cancer progression reveal distinct routes of metastasis emergence. Sci Rep (2017) 7:43813.10.1038/srep43813
40
Brown D Smeets D Székely B Larsimont D Szász AM Adnet PY et al Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat Commun (2017) 8:14944.10.1038/ncomms14944
41
Budczies J von Winterfeld M Klauschen F Bockmayr M Lennerz JK Denkert C et al The landscape of metastatic progression patterns across major human cancers. Oncotarget (2015) 6:570–83.10.18632/oncotarget.2677
42
Klein CA . Parallel progression of primary tumours and metastases. Nat Rev Cancer (2009) 9:302–12.10.1038/nrc2627
43
DeCicco-Skinner KL Henry GH Cataisson C Tabib T Gwilliam JC Watson NJ et al Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp (2014) 91:e51312.10.3791/51312
44
Huszthy PC Daphu I Niclou SP Stieber D Nigro JM Sakariassen PØ et al In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol (2012) 14:979–93.10.1093/neuonc/nos135
45
Sandén E Dyberg C Krona C Gallo-Oller G Olsen TK Enríquez Pérez J et al Establishment and characterization of an orthotopic patient-derived group 3 medulloblastoma model for preclinical drug evaluation. Sci Rep (2017) 7:46366.10.1038/srep46366
46
Joo KM Kim J Jin J Kim M Seol HJ Muradov J et al Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep (2013) 3:260–73.10.1016/j.celrep.2012.12.013
47
Wakimoto H Mohapatra G Kanai R Curry WT Jr Yip S Nitta M et al Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol (2012) 14:132–44.10.1093/neuonc/nor195
48
Zhao X Liu Z Yu L Zhang Y Baxter P Voicu H et al Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol (2012) 14:574–83.10.1093/neuonc/nos061
49
Yu L Baxter PA Voicu H Gurusiddappa S Zhao Y Adesina A et al A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol (2010) 12:580–94.10.1093/neuonc/nop056
50
Shu Q Wong KK Su JM Adesina AM Yu LT Tsang YT et al Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells (2008) 26:1414–24.10.1634/stemcells.2007-1009
51
Singh SK Hawkins C Clarke ID Squire JA Bayani J Hide T et al Identification of human brain tumour initiating cells. Nature (2004) 432:396–401.10.1038/nature03128
52
Hoffman RM . Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer (2015) 15:451–2.10.1038/nrc3972
53
Talmadge JE Singh RK Fidler IJ Raz A . Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol (2007) 170:793–804.10.2353/ajpath.2007.060929
54
Lokman NA Elder AS Ricciardelli C Oehler MK . Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int J Mol Sci (2012) 13:9959–70.10.3390/ijms13089959
55
Palmer TD Lewis J Zijlstra A . Quantitative analysis of cancer metastasis using an avian embryo model. J Vis Exp (2011) (51):e2815.10.3791/2815
56
Wilson SM Chambers AF . Experimental metastasis assays in the chick embryo. Curr Protoc Cell Biol (2004) 19:19.16.10.1002/0471143030.cb1906s21
57
Brown HK Schiavone K Tazzyman S Heymann D Chico TJ . Zebrafish xenograft models of cancer and metastasis for drug discovery. Expert Opin Drug Discov (2017) 12:379–89.10.1080/17460441.2017.1297416
58
Liu C Zhang Y Lim S Hosaka K Yang Y Pavlova T et al A zebrafish model discovers a novel mechanism of stromal fibroblast-mediated cancer metastasis. Clin Cancer Res (2017) 23(16):4769–79.10.1158/1078-0432.CCR-17-0101
59
Schabet M Herrlinger U . Animal models of leptomeningeal metastasis. J Neurooncol (1998) 38:199–205.10.1023/A:1005936304256
60
Shultz LD Ishikawa F Greiner DL . Humanized mice in translational biomedical research. Nat Rev Immunol (2007) 7:118–30.10.1038/nri2017
61
Hughes P Marshall D Reid Y Parkes H Gelber C . The costs of using unauthenticated, over-passaged cell lines: how much more data do we need?Biotechniques (2007) 43:575, 577–8, 581–572 passim.10.2144/000112598
62
Francia G Cruz-Munoz W Man S Xu P Kerbel RS . Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer (2011) 11:135–41.10.1038/nrc3001
63
Bos PD Zhang XH Nadal C Shu W Gomis RR Nguyen DX et al Genes that mediate breast cancer metastasis to the brain. Nature (2009) 459:1005–9.10.1038/nature08021
64
Fidler IJ . Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2’-deoxyuridine. J Natl Cancer Inst (1970) 45:773–82.
65
Song HT Jordan EK Lewis BK Liu W Ganjei J Klaunberg B et al Rat model of metastatic breast cancer monitored by MRI at 3 tesla and bioluminescence imaging with histological correlation. J Transl Med (2009) 7:88.10.1186/1479-5876-7-88
66
Kircher DA Silvis MR Cho JH Holmen SL . Melanoma brain metastasis: mechanisms, models, and medicine. Int J Mol Sci (2016) 17:E1468.10.3390/ijms17091468
67
Daphu I Sundstrøm T Horn S Huszthy PC Niclou SP Sakariassen PØ et al In vivo animal models for studying brain metastasis: value and limitations. Clin Exp Metastasis (2013) 30:695–710.10.1007/s10585-013-9566-9
68
Martinez-Aranda A Hernandez V Picon C Modolell I Sierra A . Development of a preclinical therapeutic model of human brain metastasis with chemoradiotherapy. Int J Mol Sci (2013) 14:8306–27.10.3390/ijms14048306
69
Zhang Z Hatori T Nonaka H . An experimental model of brain metastasis of lung carcinoma. Neuropathology (2008) 28:24–8.10.1111/j.1440-1789.2007.00826.x
70
Sakamoto S Inoue H Ohba S Kohda Y Usami I Masuda T et al New metastatic model of human small-cell lung cancer by orthotopic transplantation in mice. Cancer Sci (2015) 106:367–74.10.1111/cas.12624
71
Cruz-Munoz W Man S Xu P Kerbel RS . Development of a preclinical model of spontaneous human melanoma central nervous system metastasis. Cancer Res (2008) 68:4500–5.10.1158/0008-5472.CAN-08-0041
72
Marsden CG Wright MJ Carrier L Moroz K Pochampally R Rowan BG . A novel in vivo model for the study of human breast cancer metastasis using primary breast tumor-initiating cells from patient biopsies. BMC Cancer (2012) 12:10.10.1186/1471-2407-12-10
73
Choi SA Kwak PA Kim SK Park SH Lee JY Wang KC et al In vivo bioluminescence imaging for leptomeningeal dissemination of medulloblastoma in mouse models. BMC Cancer (2016) 16:723.10.1186/s12885-016-2742-y
74
Reijneveld JC Taphoorn MJ Voest EE . A simple mouse model for leptomeningeal metastases and repeated intrathecal therapy. J Neurooncol (1999) 42:137–42.10.1023/A:1006237917632
75
Janczewski KH Chalk CL Pyles RB Parysek LM Unger LW Warnick RE . A simple, reproducible technique for establishing leptomeningeal tumors in nude rats. J Neurosci Methods (1998) 85:45–9.10.1016/S0165-0270(98)00115-0
76
Boire A DeAngelis L Massagué J . Development of a mouse model of leptomeningeal metastasis (P7.014). Neurology (2014) 82:7.014.10.1093/neuonc/nou240.6
77
Boire A Zou Y Shieh J Macalinao DG Pentsova E Massagué J . Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell (2017) 168:1101–13.e1113.10.1016/j.cell.2017.02.025
78
Singh M Venugopal C Tokar T Brown KR McFarlane N Bakhshinyan D et al RNAi screen identifies essential regulators of human brain metastasis-initiating cells. Acta Neuropathol (2017).10.1007/s00401-017-1757-z
79
Eberth C . Zur entwick pung des ephlheliomas (cholesteatomas) der pia and der lung. Virchow S Arch (1870) 49:51–63.10.1007/BF02214196
80
Cantillo R Jain J Singhakowinta A Vaitkevicius VK . Blindness as initial manifestation of meningeal carcinomatosis in breast cancer. Cancer (1979) 44:755–7.10.1002/1097-0142(197908)44:2<755::AID-CNCR2820440249>3.0.CO;2-E
81
Ahluwalia MS Vogelbaum MV Chao ST Mehta MM . Brain metastasis and treatment. F1000Prime Rep (2014) 6:114.10.12703/P6-114
82
Franciosi V Cocconi G Michiara M Di Costanzo F Fosser V Tonato M et al Front-line chemotherapy with cisplatin and etoposide for patients with brain metastases from breast carcinoma, nonsmall cell lung carcinoma, or malignant melanoma: a prospective study. Cancer (1999) 85:1599–605.10.1002/(SICI)1097-0142(19990401)85:7<1599::AID-CNCR23>3.3.CO;2-R
83
Rosner D Nemoto T Lane WW . Chemotherapy induces regression of brain metastases in breast carcinoma. Cancer (1986) 58:832–9.10.1002/1097-0142(19860815)58:4<832::AID-CNCR2820580404>3.0.CO;2-W
84
Sevinc A Kalender ME Altinbas M Ozkan M Dikilitas M Camci C et al Irinotecan as a second-line monotherapy for small cell lung cancer. Asian Pac J Cancer Prev (2011) 12:1055–9.10.4172/2476-2253.1000102
85
Zhao KH Zhang C Bai Y Li Y Kang X Chen JX et al Antiglioma effects of cytarabine on leptomeningeal metastasis of high-grade glioma by targeting the PI3K/Akt/mTOR pathway. Drug Des Devel Ther (2017) 11:1905–15.10.2147/DDDT.S135711
86
Niwińska A Rudnicka H Murawska M . Breast cancer leptomeningeal metastasis: the results of combined treatment and the comparison of methotrexate and liposomal cytarabine as intra-cerebrospinal fluid chemotherapy. Clin Breast Cancer (2015) 15:66–72.10.1016/j.clbc.2014.07.004
87
Nagpal S Riess J Wakelee H . Treatment of leptomeningeal spread of NSCLC: a continuing challenge. Curr Treat Options Oncol (2012) 13:491–504.10.1007/s11864-012-0206-4
88
Petrelli F Ghidini M Lonati V Tomasello G Borgonovo K Ghilardi M et al The efficacy of lapatinib and capecitabine in HER-2 positive breast cancer with brain metastases: a systematic review and pooled analysis. Eur J Cancer (2017) 84:141–8.10.1016/j.ejca.2017.07.024
89
Bachelot T Romieu G Campone M Diéras V Cropet C Dalenc F et al Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol (2013) 14:64–71.10.1016/S1470-2045(12)70432-1
90
Ceresoli GL Cappuzzo F Gregorc V Bartolini S Crinò L Villa E . Gefitinib in patients with brain metastases from non-small-cell lung cancer: a prospective trial. Ann Oncol (2004) 15:1042–7.10.1093/annonc/mdh276
91
Koba T Kijima T Takimoto T Hirata H Naito Y Hamaguchi M et al Rapid intracranial response to osimertinib, without radiotherapy, in nonsmall cell lung cancer patients harboring the EGFR T790M mutation: two case reports. Medicine (Baltimore) (2017) 96:e6087.10.1097/MD.0000000000006087
92
Sperduto PW Wang M Robins HI Schell MC Werner-Wasik M Komaki R . A phase 3 trial of whole brain radiation therapy and stereotactic radiosurgery alone versus WBRT and SRS with temozolomide or erlotinib for non-small cell lung cancer and 1 to 3 brain metastases: Radiation Therapy Oncology Group 0320. Int J Radiat Oncol Biol Phys (2013) 85:1312–18.10.1016/j.ijrobp.2012.11.042
93
Lin NU Carey LA Liu MC Younger J Come SE Ewend M et al Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol (2008) 26:1993–9.10.1200/JCO.2007.12.3588
94
Lin NU Diéras V Paul D Lossignol D Christodoulou C Stemmler HJ et al Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res (2009) 15:1452–9.10.1158/1078-0432.CCR-08-1080
95
Dummer R Goldinger SM Turtschi CP Eggmann NB Michielin O Mitchell L et al Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer (2014) 50:611–21.10.1016/j.ejca.2013.11.002
96
Long GV Trefzer U Davies MA Kefford RF Ascierto PA Chapman PB et al Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol (2012) 13:1087–95.10.1016/S1470-2045(12)70431-X
97
Yoshida T Oya Y Tanaka K Shimizu J Horio Y Kuroda H et al Clinical impact of crizotinib on central nervous system progression in ALK-positive non-small lung cancer. Lung Cancer (2016) 97:43–7.10.1016/j.lungcan.2016.04.006
98
Melosky B Agulnik J Albadine R Banerji S Bebb DG Bethune D et al Canadian consensus: inhibition of ALK-positive tumours in advanced non-small-cell lung cancer. Curr Oncol (2016) 23:196–200.10.3747/co.23.3120
99
Gadgeel SM Gandhi L Riely GJ Chiappori AA West HL Azada MC et al Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol (2014) 15:1119–28.10.1016/S1470-2045(14)70362-6
100
Besse B Le Moulec S Mazières J Senellart H Barlesi F Chouaid C et al Bevacizumab in patients with nonsquamous non-small cell lung cancer and asymptomatic, untreated brain metastases (BRAIN): a nonrandomized, phase II study. Clin Cancer Res (2015) 21:1896–903.10.1158/1078-0432.CCR-14-2082
101
Schulz H Pels H Schmidt-Wolf I Zeelen U Germing U Engert A . Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematologica (2004) 89:753–4.
102
Park YH Park MJ Ji SH Yi SY Lim DH Nam DH et al Trastuzumab treatment improves brain metastasis outcomes through control and durable prolongation of systemic extracranial disease in HER2-overexpressing breast cancer patients. Br J Cancer (2009) 100:894–900.10.1038/sj.bjc.6604941
103
Margolin K Ernstoff MS Hamid O Lawrence D McDermott D Puzanov I et al Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol (2012) 13:459–65.10.1016/S1470-2045(12)70090-6
104
Dudnik E Yust-Katz S Nechushtan H Goldstein DA Zer A Flex D et al Intracranial response to nivolumab in NSCLC patients with untreated or progressing CNS metastases. Lung Cancer (2016) 98:114–7.10.1016/j.lungcan.2016.05.031
105
Goldberg SB Gettinger SN Mahajan A Chiang AC Herbst RS Sznol M et al Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol (2016) 17:976–83.10.1016/S1470-2045(16)30053-5
106
Walko CM Lindley C . Capecitabine: a review. Clin Ther (2005) 27:23–44.10.1016/j.clinthera.2005.01.005
107
Zhu W Zhou L Qian JQ Qiu TZ Shu YQ Liu P . Temozolomide for treatment of brain metastases: a review of 21 clinical trials. World J Clin Oncol (2014) 5:19–27.10.5306/wjco.v5.i1.19
108
Gleissner B Chamberlain MC . Neoplastic meningitis. Lancet Neurol (2006) 5:443–52.10.1016/S1474-4422(06)70443-4
109
Shih T Lindley C . Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther (2006) 28:1779–802.10.1016/j.clinthera.2006.11.015
110
Johanns T Waqar SN Morgensztern D . Immune checkpoint inhibition in patients with brain metastases. Ann Transl Med (2016) 4:S9.10.21037/atm.2016.09.40
111
Farber SH Tsvankin V Narloch JL Kim GJ Salama AK Vlahovic G et al Embracing rejection: immunologic trends in brain metastasis. Oncoimmunology (2016) 5:e1172153.10.1080/2162402X.2016.1172153
112
Wu X Northcott PA Dubuc A Dupuy AJ Shih DJ Witt H et al Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature (2012) 482:529–33.10.1038/nature10825
113
Morrissy AS Garzia L Shih DJ Zuyderduyn S Huang X Skowron P et al Divergent clonal selection dominates medulloblastoma at recurrence. Nature (2016) 529:351–7.10.1038/nature16478
114
Redzic Z . Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS (2011) 8:3.10.1186/2045-8118-8-3
115
Banks WA . Characteristics of compounds that cross the blood-brain barrier. BMC Neurol (2009) 9(Suppl 1):S3.10.1186/1471-2377-9-S1-S3
116
Oldendorf WH . Lipid solubility and drug penetration of the blood brain barrier. Proc Soc Exp Biol Med (1974) 147:813–5.10.3181/00379727-147-38444
117
Fauré J Lachenal G Court M Hirrlinger J Chatellard-Causse C Blot B et al Exosomes are released by cultured cortical neurones. Mol Cell Neurosci (2006) 31:642–8.10.1016/j.mcn.2005.12.003
118
Alvarez-Erviti L Seow Y Yin H Betts C Lakhal S Wood MJ . Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol (2011) 29:341–5.10.1038/nbt.1807
Summary
Keywords
leptomeningeal metastasis, brain metastasis, in vivo models, metastasis, brain metastasis therapies
Citation
Singh M, Bakhshinyan D, Venugopal C and Singh SK (2017) Preclinical Modeling and Therapeutic Avenues for Cancer Metastasis to the Central Nervous System. Front. Oncol. 7:220. doi: 10.3389/fonc.2017.00220
Received
23 June 2017
Accepted
01 September 2017
Published
19 September 2017
Volume
7 - 2017
Edited by
David D. Eisenstat, University of Alberta, Canada
Reviewed by
Janusz Rak, McGill University, Canada; Sunit Das, University of Toronto, Canada; Todd Charles Hollon, University of Michigan Health System, United States
Updates
Copyright
© 2017 Singh, Bakhshinyan, Venugopal and Singh.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheila K. Singh, ssingh@mcmaster.ca
Specialty section: This article was submitted to Neuro-Oncology and Neurosurgical Oncology, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.