 Alia Daoud
Alia Daoud Quincy S. Chu
Quincy S. Chu- Department of Medical Oncology, Cross Cancer Institute, University of Alberta, Edmonton, AB, Canada
Discovery of the epidermal growth factor receptor gene mutation and the anaplastic lymphoma kinase chromosomal translocation in non-small cell lung cancer has prompted efforts around the world to identify many less common targetable oncogenic drivers. Such concerted efforts have been variably successful in both non-squamous and squamous cell carcinomas of the lung. Some of the targeted therapies for these oncogenic drivers have received regulatory approval for clinical use, while others have modest clinical benefit. In this mini-review, several of these targets will be reviewed.
Introduction
Epidermal growth factor receptor (EGFR) activating mutations in exons 18–21 and their exceptional responses to its kinase inhibitors (1, 2) marked the beginning of precision medicine in non-small cell lung cancer (NSCLC). Randomized trials showed treatment naïve, recurrent, or metastatic NSCLC patients harboring these mutations, particularly for exons 19 or 21 (3–10), had improved median progression-free survival (mPFS), tolerability, and quality-of-life from EGFR inhibitors over platinum-based chemotherapy. These studies triggered ongoing research to identify novel targets in both non-squamous (11, 12) and squamous NSCLC (13, 14) (Table 1). Crizotinib for ROS1 and dabrafenib/trametinib for BRAF mutation have received and submitted for regulatory approval, respectively. Selected targets, excluding EGFR and ALK, which will be discussed in separate reviews, will be discussed.
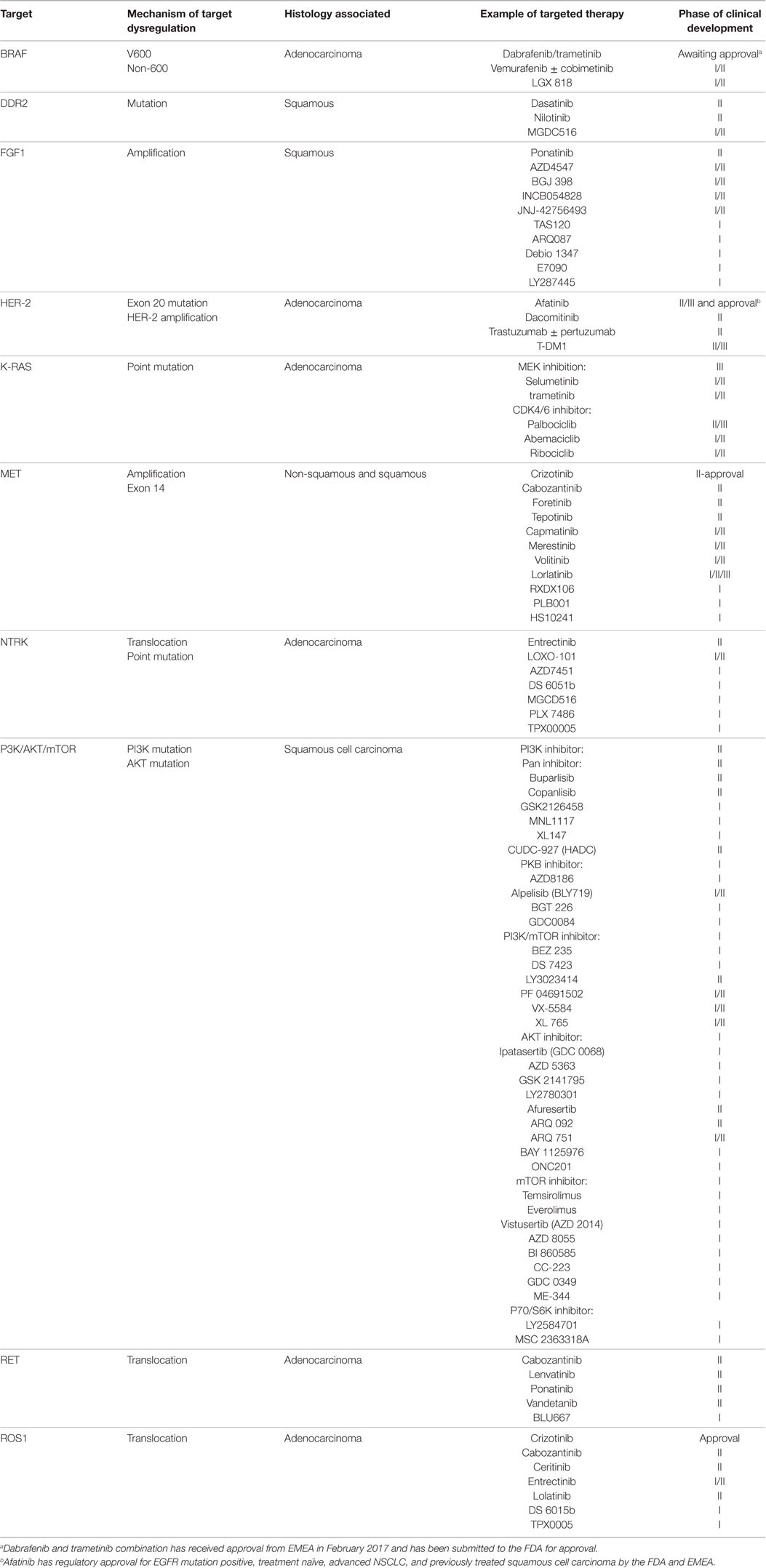
Table 1. Targets, mechanism(s) of target dysregulation, associated histology, and examples of current drugs in development and corresponding phase of clinical development in non-small cell lung cancer.
BRAF Mutations
BRAF is a serine/threonine intracellular kinase and is activated by RAS, subsequently activates mitogen-activated protein kinase (MAPK). Activating BRAF mutations occur in 2–5% of NSCLC (15, 16). It is rare to find concurrent driver mutations, like K-RAS or EGFR (17). Activating BRAF mutations in NSCLC can be categorized into V600 and non-V600, in contrast to the predominance of V600 mutation in melanoma (15, 16). Although non-V600 BRAF mutations are more prevalent in heavy smokers, V600 mutants are found in never or light smokers (17). There are conflicting reports regarding the prognostic difference between the two subtypes (17).
BRAF inhibitors, such as vemurafenib, have shown promising preliminary benefit in V600 BRAF mutant, advanced NSCLC patients with a response rate (RR) of 42% and mPFS of 7.3 months (18). Planchard et al. (19) reported an RR of 33% and mPFS of 5.5 months with dabrafenib. Dual inhibition of BRAF and MEK with dabrafenib and trametinib yielded an RR of 63.2% and mPFS of 9.7 months in 57 evaluable patients (20). Dual inhibition prevents mechanisms leading to MAPK pathway reactivation (21, 22), resulting in more effective growth inhibition. However, resistant mechanisms to dual inhibition may arise as a result of RAS or ERK activation or mutation (23, 24), epigenetic EGFR alteration (25), or overexpression of MCL-1 (26). Non-V600 BRAF mutants may not be as responsive to BRAF inhibition based on BRAF-mutated melanoma data (15, 17, 20). There is no specific targeted therapy developed in this subpopulation.
DDR2 Mutations
DDR2 is a receptor kinase that binds to collagen at the discoidin domain leading to its activation and subsequently to cell migration, proliferation, and survival (27, 28). Activating DDR2 mutations were identified in 4% of squamous NSCLC, with the majority in the kinase domain. Tumor growth inhibition by dasatinib was observed preclinically (29). One partial response (PR), in a patient with S768R DDR2 mutation and wild-type EGFR, of almost 1 year was reported in the Phase II trial of dasatinib and erlotinib in advanced NSCLC (30). Another PR was reported in the Phase II trial of dasatinib in previously treated, advanced NSCLC (31).
A Phase II trial of dasatinib in patients with either inactivating BRAF mutations or DDR2 mutations was conducted. It was terminated prematurely due to intolerable dyspnea, fatigue, and nausea. Patients were on therapy for 9–42 days, with no observed response (32). Trials of dasatinib and MGCD516 in DDR2 mutant solid tumors, including squamous NSCLC, are ongoing. Success in the development of DDR2 inhibitors should modulate the toxicity hindering adequate drug exposure and efficacy by careful dose and schedule selection.
FGFR Pathway Aberrations
The FGF pathway consists of four receptors, FGFR1-4, and 18 ligands. Activation of the pathway leads to downstream activation of the RAS/RAF/MAPK, PI3K/AKT/mTOR, STAT, and PLCγ, which cause cell growth, proliferation, differentiation, migration, and survival (33). Pathway dysregulation can result from overexpression of either FGFs or their receptors, alternative splicing receptor isoforms, impaired downregulation, and degradation of activated FGF signal, FGFR gene amplification, point mutations, or chromosomal translocations (34). FGFR1 amplification is found in 10–25% of squamous NSCLC, commonly in smokers (35, 36). Whether FGFR1 amplification is prognostic remains controversial (36, 37).
An RR of 11.1% and disease-control rate (DCR) of 50% were reported in 36 FGFR1-amplified squamous NSCLC patients treated with BGJ398 (38). In the dose expansion cohort of the Phase 1 erdafitinib (JNJ-42756493) study, no response was observed (39). The criterion for FGFR1 amplification was not specified in either trial (38, 39). Two studies of AZD4547 reported 0/4 and 1/14 PR in evaluable FGFR1-amplified NSCLC, respectively. The responder had high FGFR1 amplification, defined as FGFR1:CEP8 ≥ 2.8 (40, 41).
It is premature to declare that FGFR1 amplification is not a driver mutation. Clinically significant toxicity from FGFR-targeted agents may occur at doses below which adequate growth inhibition of amplified FGFR1 tumors can be achieved. It is still unknown if FGFR1 amplification translates to overexpression or activation of the receptor. The definition of FGFR1 amplification needs to be refined, as in MET amplification and crizotinib efficacy (42).
Chandrani et al. reported that 5.5% of adenocarcinoma NSCLC harbor FGFR3 mutations at S249C, which was previously described in squamous NSCLC, and G691R which are sensitive to FGFR kinase inhibition in preclinical models. The clinical relevance will be established by future clinical trials (43).
HER-2 Mutations and Amplification
HER-2 is a member of the EGFR family. The most common HER-2 mutation is exon 20 in-frame deletion or insertion between codons 776–779 (44, 45), which occur in 1.7–9% of all adenocarcinoma NSCLC (44–47). The length of insertion or deletion is heterogeneous (48). They are most commonly found in females and non-smokers. HER-2 exon 20 mutation or amplification leads to HER-2 phosphorylation, RAS/RAF/MAPK and PI3K/AKT/mTOR activation, and subsequent cell growth, proliferation, survival, and metastasis.
Six patients with HER-2 3+ or amplification had an RR of 83% and mPFS of 8.5 months as compared to an RR of 41% and mPFS of 7.0 months in those without after cisplatin/gemcitabine/trastuzumab treatment (49). A retrospective series of metastatic, HER-2 exon 20 mutant NSCLC reported DCRs of 93 and 100% after trastuzumab (N = 15) and afatinib (N = 3), respectively (46). A Phase II study of dacomitinib in 30 NSCLC with HER-2 aberrations reported an RR of 12% in those with exon 20 mutation and no response in those with amplification (50). The Phase II study of afatinib in 7 exon 20 mutant NSCLC had a DCR of 71% with 1 unconfirmed PR (uPR) (51). The ETOP NICHE trial of afatinib in HER-2 exon 20 mutant NSCLC reported a disappointing DCR at 12-week of 54% and mPFS of 13 weeks (52).
Phase II trials of ado-trastuzumab emtansine in HER-2 exon 20 or point mutations and HER-2 2+/3+ overexpressed NSCLC reported an RR of 6/18 and 10/49 with mPFS of 4 and 2.7 months, respectively (53, 54). The preliminary result of the ongoing MyPathway trial of trastuzumab and pertuzumab, targeting HER-2 dimerization, reported an ORR of 13 and 19% in 16 HER-2-amplified and 12-mutated NSCLC patients, respectively (55).
The benefit of HER-2-targeted therapeutics is modest. It is plausible that HER-2 exon 20 mutation and amplification represent two distinct molecular and therapeutic entities. There may be biological and therapeutic differences to HER-2-targeted agents based on the length of HER-2 exon 20 insertion or deletion (56). The full clinical and molecular data from these trials may help to elucidate the best treatment strategies to these subpopulations of HER-2 gene aberrant NSCLC.
K-RAS Mutations
K-RAS is a member of the guanosine triphosphate gene superfamily. Upon activation by upstream receptors or point mutations at codons 12, 13, 14, or 60/61, K-RAS activates RAF/MAPK and PI3K/AKT/mTOR. These pathways regulate cell proliferation, growth, motility, and apoptosis (57).
K-RAS is mutated in 20–30% of NSCLC, predominantly in adenocarcinoma, non-Asians, and smokers. The incidence of K-RAS mutations may correlate with the amount of cigarettes smoked (11, 57). The majority of K-RAS mutations in NSCLC are at codon 12 (58). In a meta-analysis (59), K-RAS mutation was associated with poorer prognosis (HR = 1.45, 95% CI: 1.29–1.62), particularly in adenocarcinoma and early-stage NSCLC. It remains controversial if K-RAS mutation is predictive of platinum-based palliative chemotherapy efficacy (57), but it is associated with resistance to EGFR inhibitors. It is unclear if K-RAS mutation predicts efficacy to EGFR antibody (60–62), and if K-RAS transversion and transition mutations have different biology and thus therapeutic strategy and outcome (63, 64).
Targeting K-RAS mutation remains elusive. RAS attaches to the cell membrane for activation of downstream pathways via isoprenylation by farnesyltransferase. Alternatively, this is achieved by adding geranyl group by geranylgeranyltransferase I, particularly for K-RAS and H-RAS. Farnesyltransferase inhibitors failed possibly due to this geranylgeranyltransferase pathway (57, 65).
Current therapeutic approaches to K-RAS mutations in NSCLC focus on either the RAF/MAPK pathway or novel K-RAS biology. The MAPK pathway converges at MEK, which in turn activates ERK1/2. Targeting MEK will be expected to be effective in inhibiting the MAPK pathway, regardless of the upstream stimulatory signal. Despite encouraging Phase II results, the Phase III trial of docetaxel/selumetinib, an allosteric MEK1/2 inhibitor, combination over docetaxel alone in platinum-pretreated, advanced K-RAS mutant NSCLC (66), failed to confirm any survival improvement (67).
RAS activation drives G1/S cell cycle transition via cyclin-dependent kinase 2 and 4 (CDK2/4), induces cyclin D1, and downregulates the cdk inhibitor, p27KIP. Cycle D1 activates CDK4/6, which in turn phosphorylates retinoblastoma protein, leading to G1/S transition (68). K-ras mutant NSCLC animal models were particularly sensitive to CDK4/6 inhibition (69, 70). Synergistic antitumor activity was observed with trametinib and CDK4/6 inhibitor because MEK or ERK activation leads to cyclin D1 expression (71). A number of CDK4/6 inhibitors as single agent or in combination with MEK inhibitors are being studied in this population (72).
MET Mutation and Amplification
MET is a receptor kinase and is activated by its ligand, hepatocyte growth factor, which plays a role in cell growth and development. It subsequently activates downstream RAS/RAF/MAPK, PI3K/AKT/mTOR, WNT/β-catenin, and STAT, promoting mitogenesis, motility, invasion, and morphogenesis (73, 74).
MET point mutation is detected in 3–4% of NSCLC. The most common is exon 14 splicing mutation (METex14) in 2–3% of NSCLC, who are older than 70 with non-squamous histology (sarcomatoid > adenosquamous and adenocarcinoma) and smokers. METex14 can have concurrent MET amplification, defined as MET/CEP7 ratio > 5.0 (75, 76). METex14 corresponds to the juxtamembrane domain, which is involved in its degradation by ubiquitin ligase, Cbl, leading to increase in MET activity (74, 77). METex14 alteration is highly variable, making it different to diagnose and predict therapeutic benefit (78). There has been encouraging preliminary antitumor activity of MET inhibitors in METex14 NSCLC (74), like an RR of 44 and 28% uPR after crizotinib (79).
It is challenging to define MET amplification. A recent study suggested the MET/CEP7 ratio > 5 as a sensitive and specific diagnostic test for MET amplification with low oncogenic driver overlap and highly predictive of crizotinib efficacy. These patients were mainly female and ex-smoker. High MET gene copy number was identified in 33% of adenocarcinoma NSCLC, however, none responded to MET inhibitor (80). The Phase II study of crizotinib in advanced NSCLC harboring MET amplification reported RR in low (>1.8–<2.2), intermediate (>2.2–<5) and high (>5) MET/CEP7 ratios of 0, 20, and 50%, respectively (42). It is important to determine MET amplification in non-responding EGFR mutants to EGFR inhibitors, as 2% of them have concurrent MET amplification (81).
Clinical development of MET inhibitors in MET aberration positive and in combination with EGFR inhibitors in EGFR mutant NSCLC is ongoing. This latter strategy may delay the emergence of MET amplification and thus prolong clinical benefit to EGFR inhibitors. Caution should be exercised in patient selection. Onartuzumab, MET antibody, or ARQ-197, MET kinase inhibitor, combined with erlotinib failed to improve survival in either unselected or non-squamous NSCLC with or without wild-type EGFR (82–84). Exploratory analysis found EGFR mutants had a trend toward poorer survival with onartuzumab/erlotinib (82).
NTRK Mutation and Chromosomal Translocation
The NTRK family kinases, NTRK1–3, are activated by ligands from neurotrophin growth factor family. They are involved in neuronal development (85, 86). They subsequently activate downstream PI3K/AKT/mTOR, RAS/RAF/MAPK, PLC-γ, and protein kinase C, leading to cell proliferation, survival, and growth (86, 87). In addition, NTRK overexpression is prognostic (85, 88, 89). NTRK activation can result from translocation of the NTRK kinase to a transcription factor. NTRK1, NTRK2, and NTRK3 translocations account for 3.5, 0.2–1, and 1%, respectively, of adenocarcinoma NSCLC (87). NTRK1 and NTRK2 mutations were identified primarily in large cell carcinoma (85, 87).
Due to the structural similarity in the kinase domain of NTRK, ROS1, and ALK, several pan-inhibitors, such as entrectinib, LOXO101, and TPX-0005, are in clinical investigation. Initial Phase 1 studies reported encouraging preliminary antitumor activity and tolerability (87, 90). Identifying the primary and secondary resistant mechanisms, based on the understanding from ALK and ROS1, will help to improve the efficacy of current inhibitors and identify novel therapeutics, not limited to NTRK inhibitors targeting gatekeeper or solvent front mutation.
PIK3CA/AKT/Phosphatase and Tension Homolog (pTEN)/mTOR Pathway Gene Aberrations
The PI3K/AKT/mTOR pathway is often activated in human cancers, leading to tumor proliferation, growth, and survival (91–93). There are three classes of PI3K. PIK3CA are heterodimers of a single p85 regulatory subunit, and one of the four isoforms of p110 catalytic subunits (α, β, γ, and δ). Different p110 subunit is preferentially expressed in different normal and malignant tissues. PIK3CA can be activated by upstream growth factor receptors, followed by AKT/mTORC1/p70S6K, which exerts a negative feedback on activated PIK3CA. In addition, tumor suppressor pTEN is a key negative regulator to PI3K/AKT/mTOR activation at PIK3CA (91, 94).
Several PI3K pathway activation mechanisms have been documented in NSCLC. Activating mutations in the exon 9 helical and exon 20 kinase domains are uncommon (92, 93, 95, 96). Amplification or polysomy is the predominant mechanism (92, 93). PIK3CA genetic alterations are thought to be more pivotal in squamous NSCLC pathogenesis. A study screening NSCLC, SCLC, extrapulmonary small cell cancer cell lines, and resected NSCLC identified PIK3CA gain in 33.1 and 6.2% of squamous and adenocarcinoma, respectively (92). Squamous NSCLC with PI3K family gene aberrations had inferior median overall survival (mOS) (8.5 versus 19.1 months, p < 0.0001), higher incidence of brain metastases, especially those with truncated pTEN loss (27 versus 11%, p < 0.0001), higher overall disease burden and genomic heterogeneity between the metastatic and primary tumors (37).
AKT consists of three isoforms, AKT1–3. Activating mutation, E17K in exon 4 kinase domain, accounts for 1–7% of all NSCLC (97, 98) with the majority being squamous NSCLC (99). Loss of pTEN expression occurred in up to 75% of NSCLC either by allelic loss (10–20%) (100, 101) or gene methylation (100, 102). It is postulated that pTEN loss leads to PIK3CAβ and downstream pathway activations.
Therapeutics targeting this pathway are currently in progress. Preliminary single agent antitumor activity has been disappointing. Toxicity, including hyperglycemia and GI toxicity, at least in part, limits the delivery of the optimal dose or schedule and thus antitumor activity. Inhibition of specific PIK3CA or AKT isoform leads to compensatory activation of other isoforms, limiting the antitumor activity. Due to extensive negative feedback loops, inhibition of a component leads to rebound activation of the pathway upstream (103). Ongoing studies to fully understand how to best target these genetic alterations, particularly in squamous NSCLC, with single agents, such as the LUNG MAP trial, or in combination with other complementary pathways, such as EGFR, HER-2, BRAF, may help optimize their efficacy.
RET Chromosomal Translocation
RET is a kinase receptor for the giant cell-derived neurotrophic factor ligand. Binding of ligand leads to activation of RAS/RAF/MAPK, PI3K/AKT/mTOR, and PLC-γ, which regulate cell proliferation, migration, and differentiation. RET is important for renal organogenesis and enteric nervous system development (104).
RET was first determined to be oncogenic through the identification of interchromosomal translocation or intrachromosomal inversion in papillary thyroid cancer (105). Subsequently, RET chromosomal rearrangement was identified in NSCLC. The most common 5′ partner of the fusion oncogene is kinesin family member 5B, which is translocated to the kinase domain, leading to activation (106–110).
RET translocation is reported in 1–2% of NSCLC samples and are usually younger than 60, non-smoker, equally distributed in males and females and in mixed or solid adenocarcinoma. Over 30% have signet ring features (106–110).
Preliminary antitumor activity in Phase II trials with cabozantinib (111) and vandetanib (112, 113) demonstrated an RR of 18–47% and mPFS of 4.5–8 months. A global RET inhibitors registry reported an RR of 26% and mPFS of 2.3 months (114). The modest benefit from these multitargeted RET inhibitors may be related to subtherapeutic RET inhibition due to toxicity arising from inhibition of other targets. The heterogeneity of RET fusion partners and concurrent driver mutations may also impact the sensitivity to RET inhibitors. Highly selective RET inhibitors and better understanding of the biological differences in the fusion partners and concurrent mutations may help to improve the outcome of this NSCLC subtype.
ROS-1 Chromosomal Translocation
ROS is a kinase receptor in the insulin receptor superfamily. Rearrangement occurs in 1–2% of non-squamous NSCLC (115, 116). ROS-1 chromosomal rearrangement leads to STAT3, PI3K/AKT/mTOR, and RAS/RAF/MAPK activation, followed by cell growth, proliferation, and survival (117). ROS-1 translocation NSCLC patient is described to be young, female, non-smoker, and with advanced stage adenocarcinoma (115, 117–120). The 5′ partners and the breakpoints of the ROS1 gene are variable (115, 116), which may impact on the biology and benefit to therapy.
The RR of 72%, mPFS of 19.2 months, and 1-year OS rate at 85% in 50 ROS-1 translocation NSCLC patients treated with crizotinib led to recent regulatory approval (121). Based on 77% homology in ALK and ROS-1, especially the kinase domain (121), ALK inhibitors are potentially efficacious. The Phase II study of ceritinib had an RR of 84% and mPFS of 19.3 months (122). In addition, pemetrexed-based chemotherapy may be effective, as ROS1 NSCLC have low thymidylate synthase mRNA levels (123). Further clinical validation is needed.
Overall, ROS1-rearranged NSCLC may have better prognosis with mOS of 36 months after standard chemotherapy and exceeding 5 years with chemotherapy and crizotinib. The incidence of brain metastases may be lower (123). Ongoing development of novel ROS1 inhibitors or combination to improve the benefit and to overcome resistance is important. It is conceivable that the resistant mechanisms to ROS1 inhibition parallel to those to ALK (124), such as secondary kinase domain mutations (125–127), which are sensitive to cabozantinib and lorlatinib, KIT mutation (128), RAS or EGFR pathway activation (129, 130).
Conclusion
Multiple driver mutations have been identified in non-squamous and squamous NSCLC. There is regulatory approval of EGFR-, ALK-, ROS-1-, and BRAF-targeted agents. Benefits from therapies to other targets are preliminary.
To bring targeted therapeutics into the clinic, emphasis should be made on careful selection of true drivers. The criteria remain to be defined (131). Early clinical development efforts to identify and validate the most predictive biomarkers are key. With increasing number of driver mutations and therapies, and limited diagnostic tissues in advanced NSCLC, it is important to optimize diagnostic tissue accruement, minimize unnecessary pathological tests, and implement multiplex mutation analysis. The latter approach and basket trials, such as the LUNG MAP trial, exploring multiple targets simultaneously, can reduce the number and risk of biopsy, increase enrollment, and improve clinical trial efficiency.
Continual basic, translational, and clinical investigations are crucial to understand the targets, their resistance mechanisms, and corresponding therapies. For treatment tumor or plasma biopsies are necessary.
Author Contributions
AD is an internal medicine rotating through medical oncology rotation. This manuscript served as one of her research projects. She was responsible to review the literature on a number of the therapeutic targets reviewed in the manuscript and provided her part of the corresponding manuscript. QC is the corresponding author who reviewed AD’s part of the manuscript, in addition to the review of other therapeutic targets in this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (2004) 304(5676):1497–500. doi:10.1126/science.1099314
2. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med (2004) 350(21):2129–39. doi:10.1056/NEJMoa040938
3. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol (2010) 11(2):121–8. doi:10.1016/S1470-2045(09)70364-X
4. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi:10.1056/NEJMoa0810699
5. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med (2010) 362(25):2380–8. doi:10.1056/NEJMoa0909530
6. Han JY, Park K, Kim SW, Lee DH, Kim HY, Kim HT, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol (2012) 30(10):1122–8. doi:10.1200/JCO.2011.36.8456
7. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol (2011) 12(8):735–42. doi:10.1016/S1470-2045(11)70184-X
8. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi:10.1016/S1470-2045(11)70393-X
9. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol (2013) 31(27):3327–34. doi:10.1200/JCO.2012.44.2806
10. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol (2014) 15(2):213–22. doi:10.1016/S1470-2045(13)70604-1
11. Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, et al. Multi-institutional oncogenic driver mutation analysis in lung adenocarcinoma: the lung cancer mutation consortium experience. J Thorac Oncol (2015) 10(5):768–77. doi:10.1097/JTO.0000000000000516
12. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet (2016) 387(10026):1415–26. doi:10.1016/S0140-6736(16)00004-0
13. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature (2012) 489(7417):519–25. doi:10.1038/nature11404
14. Drilon A, Rekhtman N, Ladanyi M, Paik P. Squamous-cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. Lancet Oncol (2012) 13(10):e418–26. doi:10.1016/S1470-2045(12)70291-7
15. Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res (2002) 62(23):6997–7000.
16. Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res (2002) 62(23):7001–3.
17. Litvak AM, Paik PK, Woo KM, Sima CS, Hellmann MD, Arcila ME, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol (2014) 9(11):1669–74. doi:10.1097/JTO.0000000000000344
18. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med (2015) 373(8):726–36. doi:10.1056/NEJMoa1502309
19. Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol (2016) 17(5):642–50. doi:10.1016/S1470-2045(16)00077-2
20. Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol (2016) 17(7):984–93. doi:10.1016/S1470-2045(16)30146-2
21. Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med (2011) 364(8):772–4. doi:10.1056/NEJMcibr1013704
22. Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov (2014) 4(1):69–79. doi:10.1158/2159-8290.CD-13-0279
23. Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res (2012) 72(4):969–78. doi:10.1158/0008-5472.CAN-11-1875
24. Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res (2014) 74(23):7079–89. doi:10.1158/0008-5472.CAN-14-2073
25. Wang J, Huang SK, Marzese DM, Hsu SC, Kawas NP, Chong KK, et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol (2015) 135(2):532–41. doi:10.1038/jid.2014.418
26. Fofaria NM, Frederick DT, Sullivan RJ, Flaherty KT, Srivastava SK. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget (2015) 6(38):40535–56. doi:10.18632/oncotarget.5755
27. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell (1997) 1(1):13–23. doi:10.1016/S1097-2765(00)80003-9
28. Vogel W. Discoidin domain receptors: structural relations and functional implications. FASEB J (1999) 13:S77–82.
29. Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov (2011) 1(1):78–89. doi:10.1158/2159-8274.CD-11-0005
30. Haura EB, Tanvetyanon T, Chiappori A, Williams C, Simon G, Antonia S, et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(8):1387–94. doi:10.1200/JCO.2009.25.4029
31. Johnson FM, Bekele BN, Feng L, Wistuba I, Tang XM, Tran HT, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(30):4609–15. doi:10.1200/JCO.2010.30.5474
32. Kelley MJ, Jha G, Shoemaker D, Herndon JE II, Gu L, Barry WT, et al. Phase II study of dasatinib in previously treated patients with advanced non-small cell lung cancer. Cancer Invest (2017) 35(1):32–5. doi:10.1080/07357907.2016.1253710
33. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer (2010) 10(2):116–29. doi:10.1038/nrc2780
34. Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR-mediated carcinogenesis. Biochim Biophys Acta (2012) 1823(4):850–60. doi:10.1016/j.bbamcr.2012.01.004
35. Kim HR, Kim DJ, Kang DR, Lee JG, Lim SM, Lee CY, et al. Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer. J Clin Oncol (2013) 31(6):731–7. doi:10.1200/JCO.2012.43.8622
36. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med (2010) 2(62):62ra93. doi:10.1126/scitranslmed.3001451
37. Paik PK, Shen R, Won H, Rekhtman N, Wang L, Sima CS, et al. Next-generation sequencing of stage IV squamous cell lung cancers reveals an association of PI3K aberrations and evidence of clonal heterogeneity in patients with brain metastases. Cancer Discov (2015) 5(6):610–21. doi:10.1158/2159-8290.CD-14-1129
38. Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol (2017) 35(2):157–65. doi:10.1200/JCO.2016.67.2048
39. Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol (2015) 33(30):3401–8. doi:10.1200/JCO.2014.60.7341
40. Paik P, Shen R, Berger M, Ferry D, Soria J, Mathewson A, et al. A phase 1b open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers: preliminary antitumor activity and pharmacodynamic data. J Clin Oncol (2014) 32:A8035.
41. Smyth EC, Turner NC, Peckitt C, Pearson A, Brown G, Chua S, et al. Phase II multicenter proof of concept study of AZD4547 in FGFR amplified tumours. J Clin Oncol (2015) 33:A2508.
42. Camidge DR, Ou S-HI, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA, et al. Efficacy and safety of crizotinib in patients with advanced c-MET amplified NSCLC. Proceedings of the American Society of Clinical Oncology Annual Meeting. Chicago (2014). A8001.
43. Chandrani P, Prabhash K, Prasad R, Sethunath V, Ranjan M, Iyer P, et al. Drug-sensitive FGFR3 mutations in lung adenocarcinoma. Ann Oncol (2017) 28(3):597–603. doi:10.1093/annonc/mdw636
44. Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature (2004) 431(7008):525–6. doi:10.1038/431525b
45. Buttitta F, Barassi F, Fresu G, Felicioni L, Chella A, Paolizzi D, et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer (2006) 119(11):2586–91. doi:10.1002/ijc.22143
46. Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol (2013) 31(16):1997–2003. doi:10.1200/JCO.2012.45.6095
47. Li C, Sun Y, Fang R, Han X, Luo X, Wang R, et al. Lung adenocarcinomas with HER2-activating mutations are associated with distinct clinical features and HER2/EGFR copy number gains. J Thorac Oncol (2012) 7(1):85–9. doi:10.1097/JTO.0b013e318234f0a2
48. Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol (2016) 11(3):414–9. doi:10.1016/j.jtho.2015.10.025
49. Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, et al. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol (2004) 15(1):19–27. doi:10.1093/annonc/mdh031
50. Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O’Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol (2015) 26(7):1421–7. doi:10.1093/annonc/mdv186
51. De Grève J, Moran T, Graas MP, Galdermans D, Vuylsteke P, Canon JL, et al. Phase II study of afatinib, an irreversible ErbB family blocker, in demographically and genotypically defined lung adenocarcinoma. Lung Cancer (2015) 88(1):63–9. doi:10.1016/j.lungcan.2015.01.013
52. Smit EF, Peters S, Dziadziuszko R, Dafni U, Wolf J, Wasąg B, et al. A single-arm phase II trial of afatinib in pretreared patients with advanced NSCLC harboring a HER-2 mutation: the ETOP NICHE trial. J Clin Oncol (2017) 35:A9070.
53. Li BT, Lee A, O’Toole S, Cooper W, Yu B, Chaft JE, et al. HER2 insertion YVMA mutant lung cancer: long natural history and response to afatinib. Lung Cancer (2015) 90(3):617–9. doi:10.1016/j.lungcan.2015.10.025
54. Stinchcombe T, Stahel RA, Bubendorf L, Bonomi P, Villegas AE, Kowalski D, et al. Efficacy, safety and biomarker results of trastuzumab emtansine (T-DM1) in patients (pts) with previously treated HER-2 overexpressing locally advanced or metastatic non-small cell lung cancer (mNSCLC). J Clin Oncol (2017) 35:A8509.
55. Hainsworth JD, Bose R, Sweeney C, Meric-Bernstam I, Huriwitz H, Swanton C, et al. Targeted therapy for non-small cell lung cancer (NSCLC) with HER2, BRAF or hedgehog alterations: interim data from MyPathway. J Clin Oncol (2017) 35:A9073.
56. Gow CH, Liao WY, Lin YN, Shih JY. Discordant HER2 exon 20 mutation status determines a differential sensitivity to afatinib. J Thorac Oncol (2015) 10(7):e58–60. doi:10.1097/JTO.0000000000000518
57. Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-RAS mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer (2006) 8(1):30–8. doi:10.3816/CLC.2006.n.030
58. Piva S, Ganzinelli M, Garassino MC, Caiola E, Farina G, Broggini M, et al. Across the universe of K-RAS mutations in non-small-cell-lung cancer. Curr Pharm Des (2014) 20(24):3933–43. doi:10.2174/13816128113196660761
59. Meng D, Yuan M, Li X, Chen L, Yang J, Zhao X, et al. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis. Lung Cancer (2013) 81(1):1–10. doi:10.1016/j.lungcan.2013.03.019
60. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol (2008) 9(10):962–72. doi:10.1016/S1470-2045(08)70206-7
61. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med (2005) 353(2):133–44. doi:10.1056/NEJMoa050736
62. Khambata-Ford S, Harbison CT, Hart LL, Awad M, Xu LA, Horak CE, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(6):918–27. doi:10.1200/JCO.2009.25.2890
63. Vega F, Iniesta P, Caldes T, Sanchez A, Lopez J, Dejuan C, et al. Association of K-RAS codon 12 transversions with short survival in non-small cell lung cancer. Int J Oncol (1996) 9(6):1307–11.
64. Rosell R, Monzó M, Pifarré A, Ariza A, Sánchez JJ, Moreno I, et al. Molecular staging of non-small cell lung cancer according to K-RAS genotypes. Clin Cancer Res (1996) 2(6):1083–6.
65. Reuter CW, Morgan MA, Bergmann L. Targeting the RAS signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood (2000) 96(5):1655–69.
66. Jänne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol (2013) 14(1):38–47. doi:10.1016/S1470-2045(12)70489-8
67. Jänne PA, van den Heuvel M, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib in combination with docetaxel as second-line treatment for patients with K-RAS mutation advanced non-small cell lung cancer: results from the phase III SELECT-1 trial. European Society of Medical Oncologists Meeting. Copenhagen (2016). Abstract LBA47_PR.
68. Aktas H, Cai H, Cooper GM. RAS links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the CDK inhibitor p27KIP1. Mol Cell Biol (1997) 17(7):3850–7. doi:10.1128/MCB.17.7.3850
69. Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-RAS oncogenes and CDK4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell (2010) 18(1):63–73. doi:10.1016/j.ccr.2010.05.025
70. Mao CQ, Xiong MH, Liu Y, Shen S, Du XJ, Yang XZ, et al. Synthetic lethal therapy for KRAS mutant non-small-cell lung carcinoma with nanoparticle-mediated CDK4 siRNA delivery. Mol Ther (2014) 22(5):964–73. doi:10.1038/mt.2014.18
71. Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang H, Wang P, et al. Coadministration of trametinib and palbociclib radiosensitizes KRAS-mutant non-small cell lung cancers in vitro and in vivo. Clin Cancer Res (2016) 22(1):122–33. doi:10.1158/1078-0432.CCR-15-0589
72. Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov (2016) 6(7):740–53. doi:10.1158/2159-8290.CD-16-0095
73. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol (2017) 12(1):15–26. doi:10.1016/j.jtho.2016.10.014
74. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov (2015) 5(8):850–9. doi:10.1158/2159-8290.CD-15-0285
75. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-MET overexpression. J Clin Oncol (2016) 34(7):721–30. doi:10.1200/JCO.2015.63.4600
76. Tong JH, Yeung SF, Chan AW, Chung LY, Chau SL, Lung RW, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res (2016) 22(12):3048–56. doi:10.1158/1078-0432.CCR-15-2061
77. Awad MM. Impaired c-MET receptor degradation mediated by MET exon 14 mutations in non-small-cell lung cancer. J Clin Oncol (2016) 34(8):879–81. doi:10.1200/JCO.2015.64.2777
78. Drilon A. MET exon 14 alterations in lung cancer: exon skipping extends half-life. Clin Cancer Res (2016) 22(12):2832–4. doi:10.1158/1078-0432.CCR-16-0229
79. Drilon AE, Camidge DR, Ou S-HI, Clark JW, Socinski MA, Weiss J, et al. Efficacy and safety of crizotinib in patients with advanced METex14 altered non-small cell lung cancer. Proceedings of the American Society of Clinical Oncology Annual Meeting. Chicago (2016). Abstract 108.
80. Noonan SA, Berry L, Lu X, Gao D, Barón AE, Chesnut P, et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J Thorac Oncol (2016) 11(8):1293–304. doi:10.1016/j.jtho.2016.04.033
81. Schildhaus HU, Schultheis AM, Rüschoff J, Binot E, Merkelbach-Bruse S, Fassunke J. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin Cancer Res (2015) 21(4):907–15. doi:10.1158/1078-0432.CCR-14-0450
82. Spigel DR, Edelman MJ, O’Byrne K, Paz-Ares L, Mocci S, Phan S, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METLung. J Clin Oncol (2017) 35(4):412–20. doi:10.1200/JCO.2016.69.2160
83. Scagliotti G, von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol (2015) 33(24):2667–74. doi:10.1200/JCO.2014.60.7317
84. Yoshioka H, Azuma K, Yamamoto N, Takahashi T, Nishio M, Katakami N, et al. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-MET inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol (2015) 26(10):2066–72. doi:10.1093/annonc/mdv288
85. Marchetti A, Felicioni L, Pelosi G, Del Grammastro M, Fumagalli C, Sciarrotta M, et al. Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum Mutat (2008) 29(5):609–16. doi:10.1002/humu.20707
86. McDowall J. TRK Receptors. (2005). Available from: https://www.ebi.ac.uk/interpro/potm/2005_8/Page.htm
87. Passiglia F, Caparica R, Giovannetti E, Giallombardo M, Listi A, Diana P, et al. The potential of neurotrophic tyrosine kinase (NTRK) inhibitors for treating lung cancer. Expert Opin Investig Drugs (2016) 25(4):385–92. doi:10.1517/13543784.2016.1152261
88. Eggert A, Grotzer MA, Ikegaki N, Zhao H, Cnaan A, Brodeur GM, et al. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms’ tumor. J Clin Oncol (2001) 19(3):689–96. doi:10.1200/JCO.2001.19.3.689
89. Sclabas GM, Fujioka S, Schmidt C, Li Z, Frederick WA, Yang W, et al. Overexpression of tropomyosin-related kinase B in metastatic human pancreatic cancer cells. Clin Cancer Res (2005) 11(2 Pt 1):440–9.
90. Rolfo C, Ruiz R, Giovannetti E, Gil-Bazo I, Russo A, Passiglia F, et al. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert Opin Investig Drugs (2015) 24(11):1493–500. doi:10.1517/13543784.2015.1096344
91. Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther (2014) 13(5):1021–31. doi:10.1158/1535-7163.MCT-13-0639
92. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res (2008) 68(17):6913–21. doi:10.1158/0008-5472.CAN-07-5084
93. Solomon B, Pearson RB. Class IA phosphatidylinositol 3-kinase signaling in non-small cell lung cancer. J Thorac Oncol (2009) 4(7):787–91. doi:10.1097/JTO.0b013e3181a74dba
94. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol (2010) 28(6):1075–83. doi:10.1200/JCO.2009.25.3641
95. Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist (2011) 16(Suppl 1):12–9. doi:10.1634/theoncologist.2011-S1-12
96. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene (2008) 27(41):5497–510. doi:10.1038/onc.2008.245
97. Do H, Solomon B, Mitchell PL, Fox SB, Dobrovic A. Detection of the transforming AKT1 mutation E17K in non-small cell lung cancer by high resolution melting. BMC Res Notes (2008) 1:14. doi:10.1186/1756-0500-1-14
98. Rekhtman N, Paik PK, Arcila ME, Tafe LJ, Oxnard GR, Moreira AL, et al. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res (2012) 18(4):1167–76. doi:10.1158/1078-0432.CCR-11-2109
99. Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle (2008) 7(5):665–9. doi:10.4161/cc.7.5.5485
100. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol (2005) 36(7):768–76. doi:10.1016/j.humpath.2005.05.006
101. Jin G, Kim MJ, Jeon HS, Choi JE, Kim DS, Lee EB, et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer (2010) 69(3):279–83. doi:10.1016/j.lungcan.2009.11.012
102. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res (2002) 8(5):1178–84.
103. LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol (2016). doi:10.1200/JCO.2014.59.0018
104. Gainor JF, Shaw AT. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist (2013) 18(7):865–75. doi:10.1634/theoncologist.2013-0095
105. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell (1990) 60(4):557–63. doi:10.1016/0092-8674(90)90659-3
106. Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol (2012) 30(35):4352–9. doi:10.1200/JCO.2012.44.1477
107. Sasaki H, Shimizu S, Tani Y, Maekawa M, Okuda K, Yokota K, et al. RET expression and detection of KIF5B/RET gene rearrangements in Japanese lung cancer. Cancer Med (2012) 1(1):68–75. doi:10.1002/cam4.13
108. Yokota K, Sasaki H, Okuda K, Shimizu S, Shitara M, Hikosaka Y, et al. KIF5B/RET fusion gene in surgically-treated adenocarcinoma of the lung. Oncol Rep (2012) 28(4):1187–92. doi:10.3892/or.2012.1908
109. Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res (2012) 22(3):436–45. doi:10.1101/gr.133645.111
110. Platt A, Morten J, Ji Q, Elvin P, Womack C, Su X, et al. A retrospective analysis of RET translocation, gene copy number gain and expression in NSCLC patients treated with vandetanib in four randomized phase III studies. BMC Cancer (2015) 15:171. doi:10.1186/s12885-015-1146-8
111. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol (2016) 17(12):1653–60. doi:10.1016/S1470-2045(16)30562-9
112. Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med (2017) 5(1):42–50. doi:10.1016/S2213-2600(16)30322-8
113. Lee SH, Lee JK, Ahn MJ, Kim DW, Sun JM, Keam B, et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer harboring RET rearrangement: a phase II clinical trial. Ann Oncol (2017) 28(2):292–7. doi:10.1093/annonc/mdw559
114. Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, et al. Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol (2017) 35(13):1403–10. doi:10.1200/JCO.2016.70.9352
115. Zhang L, Jiang T, Zhao C, Li W, Li X, Zhao S, et al. Efficacy of crizotinib and pemetrexed-based chemotherapy in Chinese NSCLC patients with ROS1 rearrangement. Oncotarget (2016) 7(46):75145–54. doi:10.18632/oncotarget.12612
116. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol (2012) 30(8):863–70. doi:10.1200/JCO.2011.35.6345
117. Zhu Q, Zhan P, Zhang X, Lv T, Song Y. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res (2015) 4(3):300–9. doi:10.3978/j.issn.2218-6751.2015.05.01
118. Cao B, Wei P, Liu Z, Bi R, Lu Y, Zhang L, et al. Detection of lung adenocarcinoma with ROS1 rearrangement by IHC, FISH, and RT-PCR and analysis of its clinicopathologic features. Onco Targets Ther (2016) 9:131–8. doi:10.2147/OTT.S94997
119. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med (2014) 371(21):1963–71. doi:10.1056/NEJMoa1406766
120. Warth A, Muley T, Dienemann H, Goeppert B, Stenzinger A, Schnabel PA, et al. ROS1 expression and translocations in non-small-cell lung cancer: clinicopathological analysis of 1478 cases. Histopathology (2014) 65(2):187–94. doi:10.1111/his.12379
121. Shaw AT, Solomon BJ. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med (2015) 372(7):683–4. doi:10.1056/NEJMc1415359
122. Lim S, Cho BC, Kim HR, Lee J-S, Lee KH, Lee Y-G, et al. Ceritinib in ROS1 rearranged non-small cell lung cancer: a Korean nationwide phase II study. European Society of Medical Oncologists Meeting. Copenhagen (2016). Abstract 1205PD.
123. Scheffler M, Schultheis A, Teixido C, Michels S, Morales-Espinosa D, Viteri S, et al. ROS1 rearrangements in lung adenocarcinoma: prognostic impact, therapeutic options and genetic variability. Oncotarget (2015) 6(12):10577–85. doi:10.18632/oncotarget.3387
124. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov (2017) 7(2):137–55. doi:10.1158/2159-8290.CD-16-1123
125. Wu X, Fu Y, Wang Y, Wan S, Zhang J. Gaining insight into crizotinib resistance mechanisms caused by L2026M and G2032R mutations in ROS1 via molecular dynamics simulations and free-energy calculations. J Mol Model (2017) 23(4):141. doi:10.1007/s00894-017-3314-z
126. Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin Cancer Res (2016) 22(24):5983–91. doi:10.1158/1078-0432.CCR-16-0917
127. Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, et al. A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res (2016) 22(10):2351–8. doi:10.1158/1078-0432.CCR-15-2013
128. Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M, et al. An activating KIT mutation induces crizotinib resistance in ROS1-positive lung cancer. J Thorac Oncol (2016) 11(8):1273–81. doi:10.1016/j.jtho.2016.04.001
129. Cargnelutti M, Corso S, Pergolizzi M, Mévellec L, Aisner DL, Dziadziuszko R, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget (2015) 6(7):5182–94. doi:10.18632/oncotarget.3311
130. Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One (2013) 8(12):e82236. doi:10.1371/journal.pone.0082236
Keywords: novel, non-small cell lung cancer, advanced, mutations, chromosomal rearrangement
Citation: Daoud A and Chu QS (2017) Targeting Novel but Less Common Driver Mutations and Chromosomal Translocations in Advanced Non-Small Cell Lung Cancer. Front. Oncol. 7:222. doi: 10.3389/fonc.2017.00222
Received: 14 December 2016; Accepted: 01 September 2017;
Published: 29 September 2017
Edited by:
Stephen V. Liu, Georgetown University, United StatesReviewed by:
Rachel E. Sanborn, Providence Cancer Center, United StatesShadia I. Jalal, Indiana University Bloomington, United States
Copyright: © 2017 Daoud and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quincy S. Chu, cXVpbmN5LmNodUBhbGJlcnRhaGVhbHRoc2VydmljZXMuY2E=