 Pravanya Nandakumar
Pravanya Nandakumar Alireza Mansouri2,3
Alireza Mansouri2,3 Sunit Das
Sunit Das- 1Division of Neurosurgery, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada
- 2Center for Cancer Research, Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 3Division of Neuro-Oncology, Johns Hopkins University, Baltimore, MD, United States
- 4The Arthur and Sonia Labatt Brain Tumour Centre, Hospital for Sick Kids, University of Toronto, Toronto, ON, Canada
The current World Health Organization classification of CNS tumors has made a tremendous leap from past editions by incorporating molecular criteria in addition to the pre-existing histological parameters. The revised version has had a particular impact on the classification of diffuse low-grade gliomas and their high-grade variants. The ATRX status is one of the critical markers that define the molecular classification of gliomas. In this review, we will first provide an overview of the role of ATRX in regular cell biology. Furthermore, the role of ATRX in tumorigenesis, specifically gliomas, is comprehensively elucidated. The possible correlation of ATRX status with other genetic/epigenetic modifications is also presented. We conclude by discussing some of the challenges associated with incorporating ATRX status assessment into routine clinical practice while also exploring opportunities for future diagnostics/therapeutics in gliomas based on ATRX status.
The World Health Organization (WHO) Classification of CNS Tumors
The latest version of the WHO classification scheme of the central nervous system tumors has undergone an iterative process of transformation since its inception in 1979 (1), reflecting the emergence of the novel innovative technology and clinical findings of a particular era. Initially based on pure histological features (2, 3), immunohistochemical features were incorporated into the armamentarium of diagnosis in 1993 (2, 4). The year 2000 saw the integration of genetic profiles of tumors, in addition to immunohistochemical information; furthermore, salient features of the epidemiology of each tumor subgroup were also incorporated (5). The grouping of tumors based on histological phenotype was initiated in the 2007 version, wherein diffuse gliomas were subcategorized as astrocytomas, oligodendrogliomas, or oligoastrocytomas and each subcategory was further recognized by its grade of malignancy (2). Although at the time, characteristic genetic alterations within histological categories were starting to emerge, it was not until 2016 that they were implemented into the classification scheme. The current WHO classification of CNS tumors released in 2016 is a revised version of the 4th edition rather than a 5th edition (1) (Table 1). It has made a tremendous leap from past editions by incorporating molecular criteria in addition to the pre-existing histological parameters (1). One of the most important contributions of incorporating molecular features is envisioned to be the minimization of interobserver variability and, thus, striving toward improving diagnostic reproducibility (6). This has enabled a more robust approach toward diagnosis, prognostication, and management of CNS tumors (1).
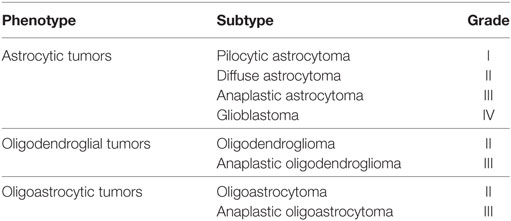
Table 1. 2007 WHO classification scheme of diffuse glioma according to histology and grade.
The revised version has had a particular impact on the classification of diffuse low-grade gliomas and their high-grade variants (7). IDH mutation and 1p/19q codeletion statuses have captured the biological characteristics (DNA methylation, mRNA, DNA copy number, and microRNA) of lower-grade gliomas (LGGs) with a greater reliability than histological classes (8). In this regard, the four molecular parameters utilized for diffuse gliomas are absence/presence of IDH mutations, 1p/19q chromosomes codeletion, TP53 mutation, and ATRX loss (1). Figure 1 provides a graphical overview of the phylogeny of gliomas within the low- to high-grade spectrum. As shown in Figure 1, astrocytomas are either IDH mutants with ATRX loss and TP53 mutations or are IDH wild-type, whereas oligodendrogliomas are IDH mutants with 1p/19q codeletion. The diagnosis and management of diffuse low-grade gliomas based on these subcategories is a step toward the emergence of precision medicine. Therefore, it is critical for specialists involved in the field of neuro-oncology to have a thorough understanding of the particular molecular/genetic components of interest. To this end, detailed knowledge about the role of each component within regular cell metabolism and tumorigenesis is paramount. Furthermore, a fundamental understanding can outline avenues of therapeutic potential and future possibilities in the management of these complex tumors. In the following review, we will focus on the central role of the ATRX gene and its product in regular cell biology while also elucidating its role in tumorigenesis. Furthermore, we will highlight its potential role in clinical practice, aside from mere diagnostics. We conclude by addressing some of the challenges associated with the incorporation of such molecular markers in the future care of patients with gliomas.
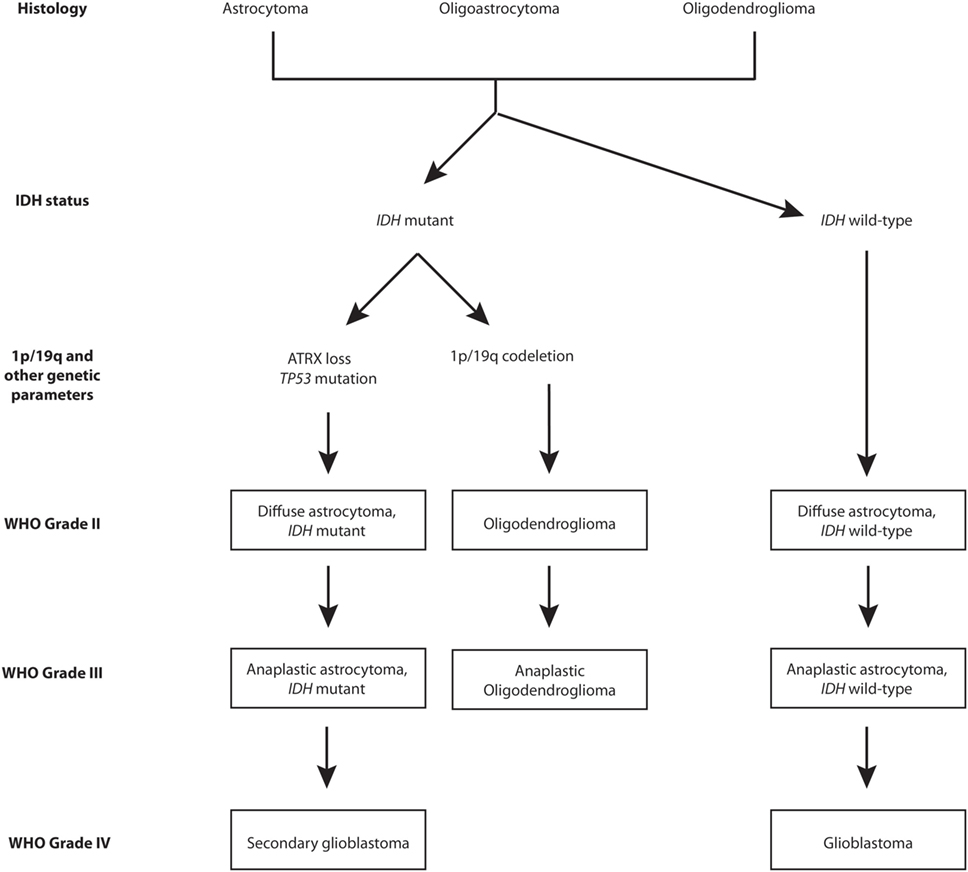
Figure 1. Schema of molecular alterations in various grades of gliomas.
ATRX in Normal Biology
Chromatin Remodeling
The ATRX gene was first discovered through a study assessing patients with the x-linked mental retardation (MR) syndrome (ATRX syndrome) presenting with α-thalassemia, severe psychomotor impairments, urogenital abnormalities, and patterns of characteristic facial dysmorphism (9). The ATRX protein exists as two isoforms (180 and 280 kDa) and is highly enriched at GC-rich and repetitive sequences (10, 11). The C-terminus of the ATRX protein harbors the helicase/ATPase domain, classifying ATRX as part of the SNF2 (SWI/SNF2) family of chromatin-remodeling proteins (12, 13). At the N-terminus of the ATRX protein lies the ATRX–DNMT3–DNMT3L (ADD) domain, receiving its name from having cysteine-rich motifs with similar features to the DNMT3 proteins involved in DNA methylation (14, 15). The ADD domain comprises a GATA-like zinc finger, a plant homeodomain (PHD)-like zinc finger, and a C-terminal α-helix (15, 16). The presence of GATA-like zinc finger suggests a DNA/chromatin-binding role for ATRX (15), whereas the PHD-like zinc finger implies a function in chromatin regulation/transcription (14, 17). Further support of ATRX’s role in mediating chromatin remodeling and potential link with DNA methylation and gene expression has been elucidated by Gibbons and colleagues (18) who have found varied DNA methylation patterns on ATRX syndrome patients on repetitive sequences including rDNA arrays, Y-specific repeat DYZ2, and in a family of subtelomeric repeats (TelBam3.4).
Further confirming the function of ATRX as a regulator of chromatin remodeling and transcription is evidence of the formation of an ATP-dependent complex with transcription cofactor DAXX. In addition, ATRX also alters the DNase I digestion pattern and triple helix displacement activity (19). ATRX has been found to localize at pericentromeric heterochromatin (PCH), ribosomal DNA arrays on acrocentric chromosomes, telomeres, and promyelocytic leukemia (PML) nuclear bodies within mouse and human cells (13, 19, 20). Therefore, ATRX likely plays a key role in gene expression regulation. The amphipathic α-helices located on the N-terminal of DAXX can bind to one of the two regions on ATRX; the first region, located on amino acids 1,189–1,326, binds to DAXX more strongly than the second region between amino acids 321–865 (13). Although DAXX has no effect on the ATPase activity of ATRX, DAXX does attenuate its transcription repression activity and plays a role in recruiting ATRX to PML nuclear bodies (13). PML nuclear bodies have been implicated as tumor suppressors, possessing antiviral functions, and possible regulators of DNA replication and transcription (21, 22).
Interactions with Histone Variants
The ATRX–DAXX complex plays a key role in maintaining genomic stability through its deposition of H3.3 at telomeres and PCH; this function is independent of the H3.3 deposition at regulatory elements and the histone cell cycle regulator (HIRA) complex-mediated H3.3 deposition at euchromatic regions (16, 23–25). Rather, the ADD domain of ATRX interacts with the N-terminal tail of H3.3 through its two binding pockets that are sensitive to the methylation states of specific lysine (lys) residues located on H3 tails (16, 25). One binding pocket is sensitive to unmodified Lys4 (H3K4me0) and the other is responsive to di-/tri-methylated Lys 9 (H3K9me3) (25); this readout of histone H3 modifications and interactions with heterochromatin protein 1 and MeCP2 protein allows ATRX to be recruited to heterochromatin for H3.3 deposition (16, 25, 26). In addition, ATRX conserves genomic stability by aiding in the formation of heterochromatin at intracisternal-A particle retrotransposons (27).
Within rodents, ATRX prevents transcriptional activation and accessibility to DNA-damaging elements. Although DAXX is necessary for this function, this is not linked to H3.3 deposition (27). Within mouse embryonic stem (mES) cells, the ATRX/DAXX complex protects repetitive sequences during DNA hypomethylation and imprinted loci from aberrant transcription and recombination through silencing achieved by H3K9 trimethylation; this serves to conserve genomic integrity (28, 29). In another study, it was found that the sensitivity of ATRX to histone modification H3K9 trimethylation and serine 10 phosphorylation in postmitotic neurons allowed ATRX to maintain the transcription of heterochromatin/silenced repetitive sequences during increased activity, suggesting that irregular repetitive element transcription may occur in the absence of ATRX (30). Additional evidence suggests that H3.3 is not the only histone that ATRX interacts with as parts of its regular function.
Ratnakumar and colleagues (31) observed that ATRX associates with another histone variant, macroH2A1, independent of its interaction with DAXX and H3.3. ATRX acts as a negative regulator of macroH2A1 deposition at telomeres and α-globin cluster and dysfunctional regulation is linked to the α-thalassemia phenotype in ATRX syndrome patients (31). As well, ATRX controls the binding of macroH2A1 with poly (ADP-ribose) polymerase tankyrase 1 and ATRX loss has been shown to prevent tankyrase 1 from resolving telomere sister cohesion in alternative lengthening telomere (ALT) cells (32). This cohesion is a favorable factor in tumors presenting ALT that overcome the gradual loss of telomeric DNA after each cell division; the cohesion promotes recombination between sister telomeres, which is crucial for DNA repair and telomere maintenance, and restricts recombination between non-sisters that would affect cell growth (32). These studies highlight ATRX’s role in maintaining the integrity of the genome through modifications of heterochromatin.
Cell Cycle Regulation
Aside from histone deposition, ATRX also partakes the responsibility of activities regulating cell cycle and maintaining the stability of the genome. ATRX depletion within HeLa cells has been shown to induce lobulated nuclei and intranuclear bridges during interphase, poor cell proliferation and viability, lengthened transition between pro-metaphase and metaphase, abnormal chromosome congression, and reduced sister chromatid cohesion (10). ATRX absence has been strongly linked to DNA damage and replicative stress (33, 34). Conte and colleagues (35) discovered that multiple cell types of ATRX-null mice are sensitized to agents that cause DNA damage and apoptosis through intrinsic pathways dependent on the DNA damage checkpoint p53, suggesting that ATRX normally inhibits p53-mediated apoptosis. In another in vivo study, rodent GBM models exhibiting ATRX and p53 loss demonstrated an impairment in non-homologous end joining (NHEJ) DNA repair, had decreased amounts of NHEJ-related proteins pDNA-PKcs, and were more responsive to double-stranded DNA-damaging therapy than controls with p53 loss alone but ATRX maintained (33). In vitro analysis of mES and human cell lines revealed that ATRX associates at DNA damage sites and interacts with the Mre11–Rad50–Nbs1 protein complex, which is involved in repairing double-strand breaks and restarting stalled replication forks (34, 36). Watson and authors (37) observed that ATRX depletion within mouse neuroprogenitor cells (NPCs) had augmented replicative stress-induced DNA damage that was amplified by p53 loss at PCH and telomeres, as well as enhanced telomeric defects including telomeric fusions. Moreover, they identified that ATRX-null NPCs were sensitive to DNA damage caused by secondary DNA structure G4-stabilizing ligand telomestatin, which suggests a role in G4 replication for ATRX (37). Through work on ATRX-null mouse embryos, Seah and colleagues (38) demonstrated an absence of dentate granule cells within the neocortex and hippocampus; this was attributed to an increased rate of p53-mediated apoptosis in the hippocampus and the basal telencephalon. This was accompanied by an increased expression of Cyclin G1 and p21, both of which are known targets of p53. Finally, increased apoptotic death rate was not observed in p53 mutant ATRX-null mice (38). These studies present a critical function for ATRX for regulating cell cycle-related activities and preserving the stability of the genome.
Implications of ATRX Mutation in ATRX Syndrome
Mutations linked to the ATRX syndrome mostly cluster within the helicase and PHD domains of ATRX (39). Although mutations within both domains present similar clinical phenotypes and although there seems to exist a clinical spectrum and severity of symptoms, including MR, gross motor ability, genital abnormality, and α-thalassemia, studies have correlated high psychomotor impairments to mutations within the PHD domain (39, 40). However, there still remains uncertainty in regard to whether mutations in specific domains define symptoms as one study has associated severity of urogenital abnormality with mutations within the PHD domain (39), whereas another, based on a larger cohort of patients, has linked it to mutations within the C-terminus (9). Even within related individuals with the same mutation, severity of MR can differ (41). Patients with ATRX syndrome have also presented white matter abnormalities, delayed myelination, and non-specific brain atrophy (42, 43). In fact, ATRX-null mice express significant neuronal loss within the neocortex and hippocampus and decreased forebrain size due to heightened apoptosis during early stages of corticogenesis; this implies that ATRX may be required for cell survival during early (E11-13.5) and postnatal neuronal differentiation and may be linked to the MR seen in human patients (44). Future studies will shed more light on the pathogenesis of the ATRX syndrome.
ATRX in Gliomas
Although significant advances have been made in the molecular aspects of brain tumors, deciphering a comprehensive role for ATRX in gliomas is still in its infancy. So far, studies have found a strong association of IDH canonical mutations and ATRX mutation (45, 46), whereas cooccurrence of 1p/19q codeletion and ATRX loss have been nearly non-existent; enabling neuropathologists to be able to make determine whether a tumor is of astrocytic or oligodendrocytic lineage without requiring both studies (8, 45). ATRX inactivation within gliomas can be due to mutations, deletions, gene fusions, or an amalgam of these causes (8). Ikemura and colleagues have demonstrated the feasibility of detecting ATRX protein expression using immunohistochemistry and correlating these expression levels with mutation status (47); this significantly simplifies the incorporation of ATRX status detection in clinical practice, given that standard sequencing methods would be difficult to apply for a large gene such as ATRX. Furthermore, ATRX mutations correlate with other prominent features including the ALT phenotype, TP53 mutations, and occur most often in astrocytic tumors (45, 48, 49). Platelet-derived growth factor receptor alpha gene (PDGFRA) amplification has also been shown to be significantly associated with ATRX loss and the ALT phenotype; future studies should look in whether possible inhibition of the PDGFRA signaling cascade may serve as a specialized therapeutic intervention within these subset of glioma patients (49, 50). Interestingly, Kannan and colleagues (45) reported that within their cohort, mutations related to ATRX cofactor DAXX were not found in LGGs and therefore in these tumors, interactions with histone is not as important perhaps. In terms of prognostication, low-grade glioma patients with ATRX retention and IDH mutations have lower progression-free survival and overall survival (OS) than tumors with 1p/19q codeletion and IDH mutations and longer time to treatment failure than those patients with IDH mutation and wild-type ATRX (55.6 vs. 31.8 months, respectively) (46, 51). This disparity aligns well with the astrocytic vs. oligodendrocytic lineage of these tumors.
In glioblastomas of young adults and pediatric patients, some studies have identified a small percentage of patients who are IDH wild-type and have a loss of ATRX expression (52–54). Ebrahimi and colleagues found these patients to have H3F3A G34 or K27 mutations, which is concordant with Ikemura and colleagues’ finding of ATRX-loss glioblastomas in younger patients being most commonly non-hemispheric in location (47, 52). On the other end of the age spectrum, the authors also identified loss of ATRX in 26 patients above the age of 55 and in 22 of these an IDH1 mutation was identified. Furthermore, contrary to some other studies, the authors showed that in IDH-mutated tumors ATRX retention is not mutually exclusive of 1p/19q codeletion (55). Together, these findings culminated in the authors recommending assessment of both IDH and ATRX status and sequencing for both IDH1/2 and H3F3A for any age group when a tumor is found to have ATRX loss and lacking IDH1/2 mutations by immunohistochemistry.
In regard to glioblastomas, reports of ATRX deficiency seem to vary; Liu and colleagues (56) observed absence of ATRX within secondary glioblastomas and more in younger patients, whereas Cai and colleagues (57) have observed lower ATRX expression more prominently in primary GBM and anaplastic gliomas than grade II gliomas and have suggested it as a malignancy marker. Therefore, it appears that additional mechanisms of tumorigenesis are involved in higher-grade gliomas and the role of ATRX is not specific in this category of tumors.
Furthermore, DNA methylation and genetic expression profiles have been found to differ among tumors with high- and low-ATRX mRNA expression; the low-ATRX subgroup had augmented methylation levels at chromatin ends (57). Cai and colleagues (57) also found that tumors with low-ATRX expression levels overexpressed genes involved in the transport, modification, and ubiquitination of proteins, in addition to (signal transduction, including GTP-related signal transduction and positive regulation of GTPase). In these tumors, transcription regulation and chromatin modification were downregulated. In addition, in vitro analysis of ATRX knockdown in glioma cells inhibited cell migration, increased cell death, and reduced cell viability (57). Overall, these studies highlight some important characteristics of ATRX mutations within gliomas that will aid in their detection. However, most of these studies are correlation-based and have not addressed the functionality of ATRX and its impacts when it is lost within gliomas; future studies should tailor their experiments to address the role of ATRX in the pathogenesis of gliomas so a better understanding can be acquired to implement novel, specialized treatments.
Possible Therapeutic Interventions for ATRX-Deficient Gliomas
A substantial contribution of the inclusion of molecular parameters in glioma diagnosis has been the refinement of diagnostic and prognostic regimens. Numerous studies have focused their research on the ALT phenotype that allows cancer cells to escape replicative senescence and has definitive features including the presence of extrachromosomal C-circles, PML nuclear bodies (APBs), and telomeric sister chromatid exchange (TSCE) (55, 56). As discussed earlier, ATRX plays a pivotal role in the interaction of macroH2A1 and tankyrase (32) and highlights the ATRX–macroH2A1–tankyrase axis as a potential therapeutic target within ALT-positive, ATRX-mutant/loss tumors. Furthermore, Ramamoorthy and Smith (32) found forced sister cohesion promotes TSCE and that tankyrase overexpression resulted in the resolution of telomere cohesion, decreased recombination events, and increased copying of non-homologous telomeres, thereby impeding cell growth. Forced resolution of sister telomere cohesion was recommended as a plausible treatment target for ALT-positive tumors within ATRX-mutant/loss gliomas; identifying small peptides/molecules that can bind to macroH2A1 to release tankyrase or interrupting the interaction between macroH2A1 and PARsylated tankyrase 1 through PAR-binding domain of macroH2A1 was a goal the authors suggested for new research (32). Another study found that ectopic ATRX expression within telomerase-deficient, ALT-positive osteosarcoma epithelial (U-2 OS) cells led to DAXX-dependent reduction of several features of the ALT phenotype (58), signifying that ATRX loss is imperative for the maintenance of the ALT phenotype (58). Clynes and colleagues’ (58) study highlights that another route that can be taken toward targeting the ALT pathway may be to reduce replicative stress, possibly through nucleoside supplementation. However, ATRX deficiency alone may not trigger the ALT phenotype (59), therefore, caution should be exercised with implementing treatment strategies targeting ATRX-mutant/loss tumors.
Flynn and authors (59) observed aberrant levels of telomeric repeat-containing RNA and prolonged association of replication protein A (RPA) with telomeric ssDNA in ATRX-mutant/loss, ALT-positive cells, whereas under normal circumstances this DNA replication intermediate is released from the telomeres during the S phase. With the inhibition of DNA damage response (DDR) kinase ATR through VE-821, a regulator of the recombination carried out by RPA, chromosome destabilization, and cell death occurred in ALT-positive cancer cells; it should be noted, however, that ATRX knockdown did not induce cells to be hypersensitive to the serine/threonine protein kinase ATR treatment (59). In contrast, Deeg and colleagues (60) have reported that ATR inhibition alone is not adequate to treat ALT-positive tumors; sensitivity to ATR inhibition did not rely on the presence of ALT activity but rather was dependent on specific cell line lineage and other factors. Interestingly, another study has found ATM- (involved in DDR as like ATR) or p53- (effector protein of the ATM pathway) deficient cancer cells are more responsive to ATR inhibition treatment and result in apoptosis than normal cells (61). This potency of reduced cell survival was increased in most cancer cell lines when the ATR inhibitor VE-821 was paired with genotoxic agents, especially cisplatin; a significant synergy between VE-821 and cisplatin was also seen within ATM-/p53-deficient cells (61). The authors suggested ATR inhibition coupled with cisplatin as a treatment option for p53-deficient tumors that increase their cell survival by reducing DNA damage through ATR (61). Therefore, ATR inhibition may serve as a plausible solution to control tumor progression in ATRX-deficient tumors displaying p53 loss as well.
Future Directions
While the identification of the role of ATRX in normal cell function and tumorigenesis has been a key step forward, additional research is warranted.
Diagnostic Challenges
Optimal Tissue Collection, Storage, and Analysis
Practical hurdles are foreseeable with the use of molecular diagnostics. Although with its updated classification of CNS tumors WHO has tried to implement a consensus on diagnostic practices worldwide, there still remains a lack of guidelines on which molecular techniques to use (62). With the addition of many clinically relevant biomarkers, a major challenge is the limited amount of tissue available for molecular tests; therefore, there lies a need for the advancement of technology that can perform multiplexed/global profiling for simultaneous tests of various biomarkers (63). Formalin-fixed paraffin-embedded tissue (FFPE) remains as a popular method of tissue processing; however, biosample processing also needs to be updated to adhere for future applications of molecular diagnostics as currently FFPE degrades proteins for standard analysis (63). A survey among members of the European Confederation of Neuropathological Societies revealed heterogeneity within and between countries with regard to access to molecular diagnostics, molecular techniques, and laboratory practices, with low-income countries reporting to use fewer biomarkers in diagnostic tests (62).
Logistic Barriers
The newly proposed integrative approach includes other technical challenges as well, including training laboratory personnel with molecular testing, acquiring skills to interpret the combined information from traditional tests and molecular pathology, assuring reliability, reproducibility, and quality control of molecular testing, treatment difficulties with those patients who have been diagnosed with uncertain NOS (not otherwise specified) gliomas and increased costs for the new generation of technology (64, 65). Hence, huge changes to existing infrastructures and resources are foreseeable to bring about equal access to molecular diagnostics worldwide, which includes creating new regulating and accreditation agencies to ensure quality and competence of laboratories, setting up standardized procedures, references, and testing software, generation of health-care schemes and insurance policies for equal coverage of these tests, determining ethical bindings and a need for higher understanding of pathology informatics so that the multitude of data generated from molecular tests can be transformed into pathology reports relevant for clinical needs (62, 65, 66).
Opportunities in Future Clinical Trials
As part of the spectrum of therapeutic opportunities, controlling PDGFRA amplification may be another route that can be explored in managing tumors with ATRX loss (49, 50) as well elucidating the connection between ALT and ATRX loss. However, understanding the pathogenesis of gliomas lacking ATRX is more crucial before specific treatment regimens can be developed; there still exists a large knowledge gap in how these subset of gliomas behave differently from the others and what underlies their tumor progression.
Traditional approaches toward diffuse LGGs has been the “wait-and-see” method (63). A recent survey among Canadian neurologists also demonstrated a high variability in the clinical management of LGGs (67). The ability to categorize LGG patients into molecular-based, prognostically relevant subgroups can help refine clinical practice and trial design (63). This presents an opportunity for further exploration.
Author Contributions
PN and AM performed literature review and analysis and were involved in the writing of the manuscript. SD performed critical analysis and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SD is supported by awards from the Canadian Cancer Society Research Institute and Canadian Institutes of Health Research.
References
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131:803–20. doi:10.1007/s00401-016-1545-1
2. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol (2007) 114(2):97–109. doi:10.1007/s00401-007-0243-4
3. Mennel HD. Grading of intracranial tumors following the WHO classification. Neurosurg Rev (1991) 14(4):249–60. doi:10.1007/BF00383256
4. Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumors. Brain Pathol (1993) 3(3):255–68. doi:10.1111/j.1750-3639.1993.tb00752.x
5. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol (2002) 61(3):215–25. doi:10.1093/jnen/61.3.215
6. Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer (1997) 79(7):1381–93. doi:10.1002/(SICI)1097-0142(19970401)79:7<1381::AID-CNCR16>3.0.CO;2-W
7. Tabouret E, Nguyen AT, Dehais C, Carpentier C, Ducray F, Idbaih A, et al. Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol (2016) 132(4):625–34. doi:10.1007/s00401-016-1611-8
8. Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med (2015) 372(26):2481–98. doi:10.1056/NEJMoa1402121
9. Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with α-thalassemia (ATR-X syndrome). Cell (1995) 80(6):837–45. doi:10.1016/0092-8674(95)90287-2
10. Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Bérubé NG. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol (2008) 180(2):315–24. doi:10.1083/jcb.200706083
11. Law MJ, Lower KM, Voon HPJ, Hughes JR, Garrick D, Viprakasit V, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell (2010) 143(3):367–78. doi:10.1016/j.cell.2010.09.023
12. Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, Gibbons RJ. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet (1996) 5(12):1899–907. doi:10.1093/hmg/5.12.1899
13. Tang J, Wu S, Liu H, Stratt R, Barak OG, Shiekhattar R, et al. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J Biol Chem (2004) 279(19):20369–77. doi:10.1074/jbc.M401321200
14. Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene (1999) 236:87–95. doi:10.1016/S0378-1119(99)00252-8
15. Argentaro A, Yang JC, Chapman L, Kowalczyk MS, Gibbons RJ, Higgs DR, et al. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc Natl Acad Sci U S A (2007) 104(29):11939–44. doi:10.1073/pnas.0704057104
16. Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat Struct Mol Biol (2011) 18(7):769–76. doi:10.1038/nsmb.2062
17. Aasland R, Gibson TJ, Stewart AF. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci (1995) 20(2):56–9. doi:10.1016/S0968-0004(00)88957-4
18. Gibbons RJ, TL M, Raman S, DM O, Garrick D, Ayyub H, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet (2000) 24(4):368–71. doi:10.1038/74191
19. Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A (2003) 100(19):10635–40. doi:10.1073/pnas.1937626100
20. McDowell TL, Gibbons RJ, Sutherland H, O’Rourke DM, Bickmore WA, Pombo A, et al. Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc Natl Acad Sci U S A (1999) 96(24):13983–8. doi:10.1073/pnas.96.24.13983
21. Borden KL. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol Cell Biol (2002) 22(15):5259–69. doi:10.1128/MCB.22.15.5259-5269.2002
22. Sahin U, Lallemand-Breitenbach V, de Thé H. PML nuclear bodies: regulation, function and therapeutic perspectives. J Pathol (2014) 234(3):289–91. doi:10.1002/path.4426
23. Wong LH, Mcghie JD, Sim M, Anderson MA, Ahn S, Hannan RD, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res (2010) 20(3):351–60. doi:10.1101/gr.101477.109
24. Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A (2010) 107(32):14075–80. doi:10.1073/pnas.1008850107
25. Eustermann S, Yang J-C, Law MJ, Amos R, Chapman LM, Jelinska C, et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol (2011) 18(7):777–83. doi:10.1038/nsmb.2070
26. Nan XS, Hou JH, Maclean A, Nasir J, Lafuente MJ, Shu XH, et al. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A (2007) 104(8):2709–14. doi:10.1073/pnas.0608056104
27. Sadic D, Schmidt K, Groh S, Kondofersky I, Ellwart J, Fuchs C, et al. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep (2015) 16(7):836–50. doi:10.15252/embr.201439937
28. He Q, Kim H, Huang R, Lu W, Tang M, Shi F, et al. The DAXX/ATRX complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell (2015) 17(3):273–86. doi:10.1016/j.stem.2015.07.022
29. Voon HPJ, Hughes JR, Rode C, DeLaRosa-Velàzquez IA, Jenuwein T, Feil R, et al. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep (2015) 11(3):405–18. doi:10.1016/j.celrep.2015.03.036
30. Noh K-M, Maze I, Zhao D, Xiang B, Wenderski W, Lewis PW, et al. ATRX tolerates activity-dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc Natl Acad Sci U S A (2014) 112(22):6820–7. doi:10.1073/pnas.1411258112
31. Ratnakumar K, Duarte LF, LeRoy G, Hasson D, Smeets D, Vardabasso C, et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev (2012) 26(5):433–8. doi:10.1101/gad.179416.111
32. Ramamoorthy M, Smith S. Loss of ATRX suppresses resolution of telomere cohesion to control recombination in ALT cancer cells. Cancer Cell (2015) 28(3):357–69. doi:10.1016/j.ccell.2015.08.003
33. Koschmann C, Calinescu A-A, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med (2016) 8(328):328ra28. doi:10.1126/scitranslmed.aac8228
34. Leung JWC, Ghosal G, Wang W, Shen X, Wang J, Li L, et al. Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem (2013) 288(9):6342–50. doi:10.1074/jbc.M112.411603
35. Conte D, Huh M, Goodall E, Delorme M, Parks RJ, Picketts DJ. Loss of Atrx sensitizes cells to DNA damaging agents through p53-mediated death pathways. PLoS One (2012) 7(12):e52167. doi:10.1371/journal.pone.0052167
36. Clynes D, Jelinska C, Xella B, Ayyub H, Taylor S, Mitson M, et al. ATRX dysfunction induces replication defects in primary mouse cells. PLoS One (2014) 9(3):e92915. doi:10.1371/journal.pone.0092915
37. Watson LA, Solomon LA, Li JR, Jiang Y, Edwards M, Shin-Ya K, et al. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J Clin Invest (2013) 123(5):2049–63. doi:10.1172/JCI65634
38. Seah C, Levy MA, Jiang Y, Mokhtarzada S, Higgs DR, Gibbons RJ, et al. Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. J Neurosci (2008) 28(47):12570–80. doi:10.1523/JNEUROSCI.4048-08.2008
39. Badens C, Lacoste C, Philip N, Martini N, Courrier S, Giuliano F, et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin Genet (2006) 70(1):57–62. doi:10.1111/j.1399-0004.2006.00641.x
40. Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP, et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat (2008) 29(6):796–802. doi:10.1002/humu.20734
41. Guerrini R, Shanahan JL, Carrozzo R, Bonanni P, Higgs DR, Gibbons RJ. A nonsense mutation of the ATRX gene causing mild mental retardation and epilepsy. Ann Neurol (2000) 47(1):117–21. doi:10.1002/1531-8249(200001)47:1<117::AID-ANA20>3.0.CO;2-A
42. Lee JS, Lee S, Lim BC, Kim KJ, Hwang YS, Choi M, et al. Alpha-thalassemia X-linked intellectual disability syndrome identified by whole exome sequencing in two boys with white matter changes and developmental retardation. Gene (2015) 569(2):318–22. doi:10.1016/j.gene.2015.04.075
43. Wada T, Ban H, Matsufuji M, Okamoto N, Enomoto K, Kurosawa K, et al. Neuroradiologic features in X-linked α-thalassemia/mental retardation syndrome. AJNR Am J Neuroradiol (2013) 34(10):2034–8. doi:10.3174/ajnr.A3560
44. Bérubé NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest (2005) 115(2):258–67. doi:10.1172/JCI22329
45. Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget (2012) 3(10):1194–203. doi:10.18632/oncotarget.689
46. Leeper HE, Caron AA, Decker PA, Jenkins RB, Lachance D, Giannini C. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget (2015) 6(30):30295–305. doi:10.18632/oncotarget.4497
47. Ikemura M, Shibahara J, Mukasa A, Takayanagi S, Aihara K, Saito N, et al. Utility of ATRX immunohistochemistry in diagnosis of adult diffuse gliomas. Histopathology (2016) 69(2):260–7. doi:10.1111/his.12927
48. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol (2012) 124(5):615–25. doi:10.1007/s00401-012-1031-3
49. Abedalthagafi M, Phillips JJ, Kim GE, Mueller S, Haas-Kogen DA, Marshall RE, et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: a multi-institutional study of 214 astrocytomas. Mod Pathol (2013) 26(10):1425–32. doi:10.1038/modpathol.2013.90
50. Purkait S, Miller CA, Kumar A, Sharma V, Pathak P, Jha P, et al. ATRX in diffuse gliomas with its mosaic/heterogeneous expression in a subset. Brain Pathol (2017) 27(2):138–45. doi:10.1111/bpa.12364
51. Wiestler B, Capper D, Holland-Letz T, Korshunov A, Von Deimling A, Pfister SM, et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol (2013) 126(3):443–51. doi:10.1007/s00401-013-1156-z
52. Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Mühleisen H, Franziska E, et al. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun (2016) 4:60. doi:10.1186/s40478-016-0331-6
53. Wiestler B, Capper D, Sill M, Jones DT, Hovestadt V, Sturm D, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol (2014) 128(4):561–71. doi:10.1007/s00401-014-1315-x
54. Pathak P, Jha P, Purkait S, Sharma V, Suri V, Sharma MC, et al. Altered global histone-trimethylation code and H3F3A-ATRX mutation in pediatric GBM. J Neurooncol (2015) 121(3):489–97. doi:10.1007/s11060-014-1675-z
55. Hewer E, Vajtai I, Dettmer MS, Berezowska S, Vassella E. Combined ATRX/IDH1 immunohistochemistry predicts genotype of oligoastrocytomas. Histopathology (2016) 68(2):272–8. doi:10.1111/his.12743
56. Liu N, Wang P, Song H, Kong L, Yao K, Qi X, et al. Immunostaining of IDH-1 R132H and ATRX proteins in the classification of adult glioblastomas. Int J Clin Exp Pathol (2016) 9(12):12849–54.
57. Cai J, Chen J, Zhang W, Yang P, Zhang C, Li M, et al. Loss of ATRX, associated with DNA methylation pattern of chromosome end, impacted biological behaviors of astrocytic tumors. Oncotarget (2015) 6(20):18105–15. doi:10.18632/oncotarget.3906
58. Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, et al. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun (2015) 6:7538. doi:10.1038/ncomms8538
59. Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science (2015) 347(6219):273–7. doi:10.1126/science.1257216
60. Deeg KI, Chung I, Bauer C, Rippe K. Cancer cells with alternative lengthening of telomeres do not display a general hypersensitivity to ATR inhibition. Front Oncol (2016) 6:186. doi:10.3389/fonc.2016.00186
61. Reaper PM, Griffiths MR, Long JM, Charrier J-D, MacCormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol (2011) 7(7):428–30. doi:10.1038/nchembio.573
62. Woehrer A, Kristensen BW, Vital A, Hainfellner JA. Patterns of diagnostic marker assessment in adult diffuse glioma: a survey of the European Confederation of Neuropathological Societies (Euro-CNS). Clin Neuropathol (2017) 36(1):5–14. doi:10.5414/NP301009
63. Huse JT, Wallace M, Aldape KD, Berger MS, Bettegowda C, Brat DJ, et al. Where are we now? And where are we going? A report from the accelerate brain cancer cure (ABC2) low-grade glioma research workshop. Neuro Oncol (2014) 16(2):173–8. doi:10.1093/neuonc/not229
64. Malzkorn B, Reifenberger G. Practical implications of integrated glioma classification according to the World Health Organization classification of tumors of the central nervous system 2016. Curr Opin Oncol (2016) 28(6):494–501. doi:10.1097/CCO.0000000000000327
65. Salto-Tellez M, James JA, Hamilton PW. Molecular pathology – the value of an integrative approach. Mol Oncol (2014) 8(7):1163–8. doi:10.1016/j.molonc.2014.07.021
66. Dubbink HJ, Deans ZC, Tops BBJ, van Kemenade FJ, Koljenović S, van Krieken HJM, et al. Next generation diagnostic molecular pathology: critical appraisal of quality assurance in Europe. Mol Oncol (2014) 8(4):830–9. doi:10.1016/j.molonc.2014.03.004
Keywords: glioma, oligodendroglioma, astrocytoma, IDH, ATRX
Citation: Nandakumar P, Mansouri A and Das S (2017) The Role of ATRX in Glioma Biology. Front. Oncol. 7:236. doi: 10.3389/fonc.2017.00236
Received: 27 July 2017; Accepted: 14 September 2017;
Published: 29 September 2017
Edited by:
Pamela Stuart Jones, University of California, San Diego, United StatesReviewed by:
Vinesh Puliyappadamba, University of Texas Southwestern Medical Center, United StatesSeunggu Jude Han, Oregon Health & Science University, United States
Copyright: © 2017 Nandakumar, Mansouri and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunit Das, c3VuaXQuZGFzQHV0b3JvbnRvLmNh