 S. R. Priya
S. R. Priya Chandra Shekhar Dravid
Chandra Shekhar Dravid Raghunadharao Digumarti2,3
Raghunadharao Digumarti2,3 Mitali Dandekar
Mitali Dandekar- 1Head Neck Surgery, Homi Bhabha Cancer Hospital and Research Centre, Visakhapatnam, India
- 2Tata Memorial Centre, Mumbai, India
- 3Medical Oncology, Homi Bhabha Cancer Hospital and Research Centre, Visakhapatnam, India
- 4Head Neck Surgery, Paras Cancer Centre, Patna, India
Medullary thyroid cancers (MTCs) constitute between 2 and 5% of all thyroid cancers. The 10-year overall survival (OS) rate of patients with localized disease is around 95% while that of patients with regional stage disease is about 75%. Only 20% of patients with distant metastases at diagnosis survive 10 years which is significantly lower than for differentiated thyroid cancers. Cases with regional metastases at presentation have high recurrence rates. Adjuvant external radiation confers local control but not improved OS. The management of residual, recurrent, or metastatic disease till a few years ago was re-surgery with local measures such as radiation. Chemotherapy was used with marginal benefit. The development of targeted therapy has brought in a major advantage in management of such patients. Two drugs—vandetanib and cabozantinib—have been approved for use in progressive or metastatic MTC. In addition, several drugs acting on other steps of the molecular pathway are being investigated with promising results. Targeted radionuclide therapy also provides an effective treatment option with good quality of life. This review covers the rationale of targeted therapy for MTC, present treatment options, drugs and methods under investigation, as well as an outline of the adverse effects and their management.
Background
Medullary thyroid cancer (MTC) which arises from parafollicular cells is less common than differentiated thyroid cancer (DTC) constituting between 2 (1) and 5% (2) of all thyroid malignancies. 13–15% of patients of MTC present with distant metastasis (DM) and have a 10-year survival of approximately 20% (3, 4). As with DTC, surgery is the treatment of choice (1). Unlike DTC, where the iodine avidity of follicular cells makes even metastatic DTC amenable to treatment, parafollicular cells do not concentrate iodine. Management options for residual, recurrent, or metastatic disease till a few years ago were re-surgery and/or radiotherapy (5, 6). Chemotherapy has been used with only marginal benefit (1). The quest to improve survival and quality of life (QOL) of patients with inoperable and/or metastatic MTC has resulted in significant research for other treatment modalities. The development of targeted therapy has brought a major advantage in the management of such patients. Two drugs, Vandetanib and Cabozantinib, have been approved for use in progressive or metastatic MTC (1). In addition, several drugs acting on other steps of the molecular pathway to MTC are being investigated with promising results. The application of targeted radionuclide therapy also provides an effective treatment modality with good QOL (1).
This review covers the rationale of targeted therapy for MTC, presents treatment options, drugs, and methods under investigation, and outlines the adverse effects and their management.
Disease Outcomes
At presentation, 35–50% of patients with MTC have regional metastasis while 13–15% have DM mainly to the lung, bone, and liver (4, 7). An analysis of population-based (Surveillance, Epidemiology, and End Results or SEER) data revealed that among patients with tumors confined to the gland, the 10-year survival rate was 95.6%, whereas patients with regional stage disease had an overall survival (OS) rate of 75.5% (4). Patients presenting with DM at diagnosis had a 10-year survival of approximately 20% (4, 7). Machens et al. (8) corroborated calcitonin levels with prognosis and observed that calcitonin levels of over 150 pg/ml were associated with DM and extrathyroidal extension (in both familial and sporadic cases). Preoperative calcitonin levels of 500 pg/ml, nodal metastasis, and reoperative status best predicted failure to achieve biochemical cure (8). In high risk patients (residual disease, extrathyroidal extension, or nodal involvement), Brierley et al. (9) found that the locoregional recurrence free rate was 86% at 10 years with postoperative external beam radiation therapy (EBRT) as compared to 52% in those without adjuvant EBRT (p = 0.049). However, radiation may not confer survival advantage (6).
Management of Recurrent or Metastatic Disease
Locoregional Recurrence
Surgery remains the mainstay for operable locoregional recurrences (10, 11). At least a third of such patients achieve normalization of calcitonin levels (12) with long-term eradication of biochemical and radiological disease (13).
Metastatic Disease
The liver is a major site for DM of MTC. Such patients are often symptomatic with pain and diarrhea. Surgical resection should be considered for amenable lesions to provide significant symptom-free survival (14, 15). For widespread unresectable lesions, radiofrequency ablation is an effective option with low morbidity (16). For diffuse lesions, transarterial chemoembolization of the liver achieves symptom control and partial response (PR) in a third of patients who have less than 30% of the organ involvement with intact liver and renal function (1, 17).
Lung metastases with associated mediastinal nodal disease should be evaluated for surgery if symptomatic and resectable (14). Alternatively, radiofrequency ablation maybe employed. Bone metastases usually occur in the vertebrae, pelvis, ribs, femur, skull, and humerus. If feasible, bony lesions should be resected for long-term symptomatic cure; postoperative radiation to the site of metastasis helps to ensure durable control (18). Radiofrequency ablation, vertebroplasty, or kyphoplasty along with judicious use of bisphosphonates are useful tools for palliation (19–21).
Single brain metastasis maybe resected if possible for palliation and to prolong survival (22). Multiple brain lesions not amenable to surgery must be considered for EBRT (1, 22).
Soft tissue metastases to sites, such as breast (23), testes (24), and skin (25), though usually amenable to excision or radiation, suggest disseminated disease and such patients usually succumb to disease in less than a year (1, 23–25).
In summary, small isolated metastases may be observed especially if the calcitonin has stabilized (26). For symptomatic progressive metastases not amenable local measures or where a single modality is insufficient for symptomatic relief, systemic therapy is preferred (11).
Rationale for Targeted Therapy in MTC: Why and When
Molecular Basis
Medullary thyroid cancer, whether inherited (MEN2 and FMTC) or sporadic, has a detectable association with mutations of the rearranged during transfection (RET) proto-oncogene which was described first in 1993 (27, 28). MTC is hereditary in 25% cases and occurs as a component heritable syndrome while it is sporadic in about 75% (29). RET is mutated in virtually all familial cases and in roughly 50% of sporadic cases (30, 31). Mutations usually occur in the cysteine-rich areas of exon 10 and 11 but may also occur in the tyrosine kinase domain (32).
Rearranged during transfection is classified as a proto-oncogene because a single activating RET mutation of one allele can lead to neoplastic transformation. Once activated, it transmits mitogenic and survival signals (33) via two major signaling cascades, namely RAS (34, 35) and the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway (36). RAS and PI3K activate other signaling effectors proteins and secondary messengers influencing cell division, growth, and cell death (36), which in turn promote cell proliferation, invasion, and survival, eventually leading to MTC formation (36).
Rearranged during transfection encodes a plasma-membrane-bound receptor tyrosine kinase that is expressed in cells of neural crest and endodermal origin (37). Tyrosine kinases are enzymes that catalyze transfer of a phosphate residue from ATP to tyrosine residues in other proteins (substrates). Tyrosine kinases, which number about 90 in the human genome (38), play central roles in transducing extracellular signals (growth factors and cytokines) into activation of signaling pathways that regulate cell growth. Since a majority of oncoproteins are tyrosine kinases, the study of their inhibition is crucial (39, 40). Most inhibitors of tyrosine kinases receptors (TKRs) act at the intracellular ATP binding site and are low molecular weight inhibitors (in contrast to extracellular acting large molecular weight monoclonal antibodies), thus preventing phosphorylation and further action of the enzyme. Since TKRs are homologous, tyrosine kinase inhibitors (TKIs) inhibit several TKRs, and are, therefore, multikinase inhibitors, as opposed to certain monodrugs that focus on a single pathway such as mTOR/Akt, as described later.
Epidermal growth factor receptor (EGFR)-dependent RET activation: an interaction between EGFR and RET was recently described, with EGFR reported to be over-expressed in thyroid cancer cells (41). EGFR is a transmembrane TKR involved in the activation of MAPK and the PI3K/Akt pathway (42).
Recent lineage tracing (37) reveals that C-cell may have a shared origin from the primitive endoderm, a fact that may explain much of tumor behavior and secretory activity of MTCs. Consequently, expression of Fox-1 (forkhead box protein) is seen in invasive MTC as well as embryonal C-cells but not in differentiated normal C-cells or follicular cells. Thus, Fox-1 promoter can be a target for future research and targeted therapy (37).
Non-RET mutations: RAS family gene mutations (HRAS, KRAS, and NRAS) have been identified in 10–17% of cases of MTC and may be associated with a less aggressive behavior (43). RAS mutations may be exclusive of RET abnormalities (44). Genetic abnormalities found in other cancers, such as TP53, RB1, PIK3CA, and BRAF mutations, are rare in MTC (45).
Overexpression of vascular endothelial growth factor (VEGF)-2 receptors is described in MTC and is associated with increased metastasis (46, 47). Mutations in MET, a proto-oncogene encoding the receptor for hepatocyte growth factor have been reported in MTC (48). Another entity is the fibroblast growth factor receptor (FGFR), which is overexpressed in MTC; its inhibition is seen to decrease the proliferation of MTC cells (49).
All these pathways are potential targets for treatment of MTC. Drugs thus developed, focus on specific oncogenic proteins present in malignant but not normal cells, in contrast to chemotherapeutic agents (50). Further targeting of treatment as per the specific mutational profile may be possible, since there is mutational heterogeneity in MTC, not only between patients but also between tumor subpopulations as well as between primary and metastatic cells in a patient (51).
Clinical Indications
Targeted therapy should be considered where other better tolerated and more accessible local treatments have been exhausted. Assessment of molecular markers calcitonin and carcinoembryonic antigen along with clinical and radiological evaluation helps to decide about when to consider use of these agents. In case of impending compression of specific sites such as the central nervous system or the airway, where a smaller disease volume may be critical, targeted therapy may be considered sooner (52). TKIs also help significantly in palliation of distressing secretory symptoms such as diarrhea and Cushing’s syndrome (53, 54) where they should be considered in adjunct to other palliative measures.
TKIs for MTC
“First Line” TKIs for MTC
Cabozantinib and Vandetinib are considered first line TKIs (Table 1). These drugs have been approved for use in MTC (11).
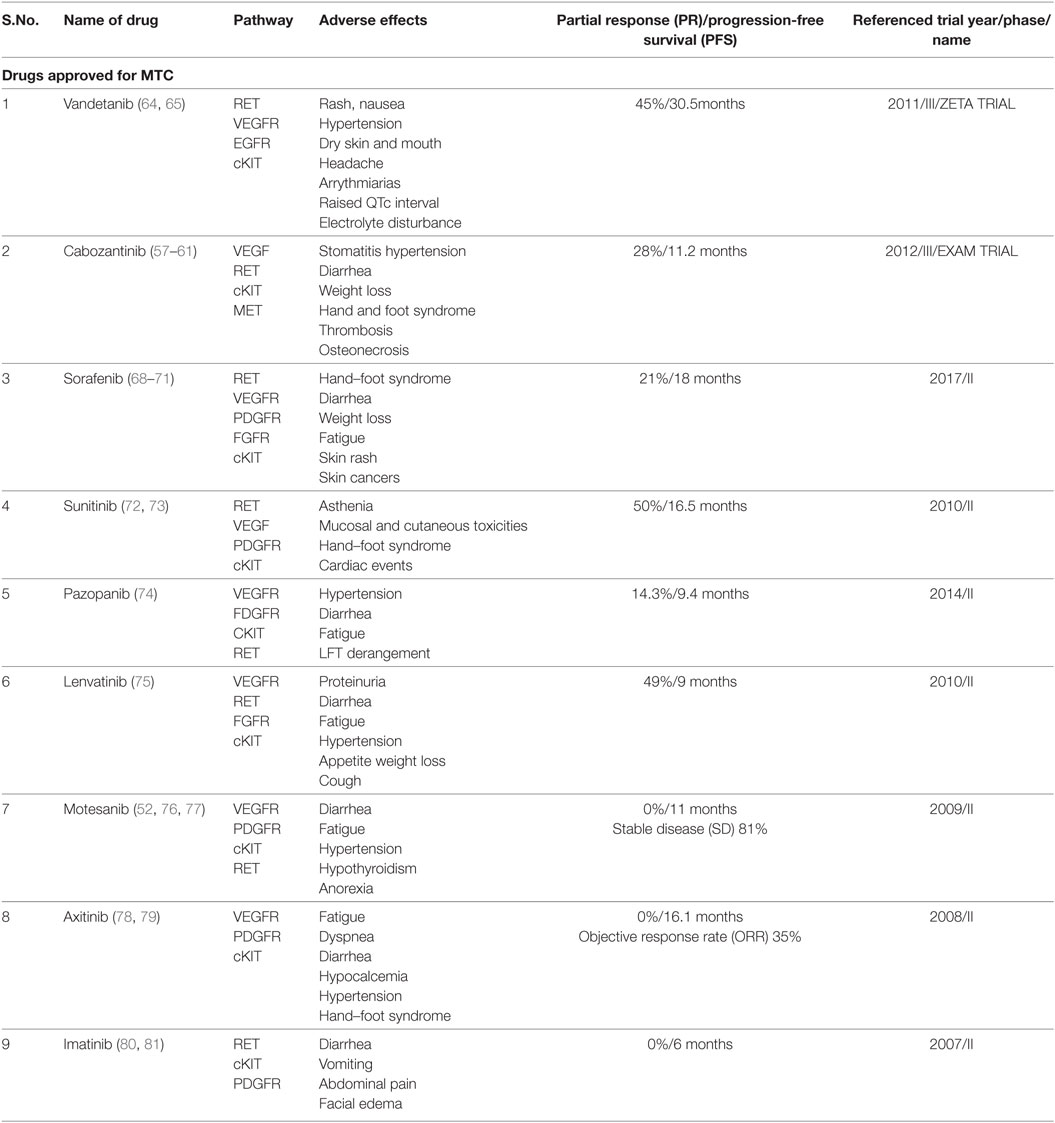
Table 1. Targeted drugs in use for medullary thyroid cancer (MTC).
Cabozantinib
Cabozantinib (XL 184) is an oral, small molecule pantyrosine kinase (RET, VEGFR, and cKIT) inhibitor (55). It was approved by the US Food and Drug Administration (USFDA) in November 2012 for metastatic MTC (56). In a randomized phase III trial (EXAM) (57, 58), 330 patients with progressive metastatic MTC were assigned to either cabozantinib (140 mg) or placebo once daily. A statistically significant prolongation in progression-free survival (PFS) was observed in the cabozantinib group. There was no difference in OS between the arms. The recommended dose is 140 mg daily (adjusted for tolerability). Adverse events (AEs) include stomatitis, hypertension and diarrhea, fatigue, weight loss, palmar-plantar erthyrodysesthesia syndrome (58, 59), uncommonly venous thrombosis and arterial thrombosis (59), rarely osteonecrosis (60), and fistula formation (61). Evaluation of a lower starting dose (60 mg/day) in a phase IV trial is ongoing (EXAMINER, NCT01896479) (62) along with trials for use in patients with renal (XL184-017) (63) and hepatic impairment (XL184-003) (63).
Vandetanib
This is a multikinase inhibitor, acting against RET, VEGFR, and EGFR. The U.S.FDA approved vandetanib for use in MTC in 2011, followed by approval in Europe in 2012. In a phase III (ZETA) trial (64), the efficacy and safety of vandetanib were compared with placebo in patients with locally advanced or metastatic MTC; 331 patients were randomized to receive vandetanib (300 mg/day oral) or placebo. The median PFS was 30.5 months in the vandetanib group vs 19.3 months in the placebo group (P < 0.001). No complete response (CR) was observed; however, PR was observed in 45% of the vandetanib group. Vandetanib was superior in all assessed subgroups (hereditary vs sporadic/unknown disease, prior systemic therapy vs no prior therapy). Frequent AEs were diarrhea, rash, nausea, hypertension, dry skin, dry mouth, and headache. QTc interval was prolonged in 15% subjects. Occasionally, cardiac arrhythmias, corneal opacities, hypocalcemia, hypokalemia, hypomagnesemia, and hypothyroidism were seen. Potentially treatment-related fatality occurred in five patients (2%) treated with vandetanib. An ongoing Phase IV trial is comparing 300 mg daily with 150 mg daily to understand the impact of dosing on response (NCT01496313) (65).
The decision between choice of drug, cabozantinib or vandetanib, must be made after considering the patient’s co-morbodities and the drug’s potential toxicities. A direct comparison between the two agents is not possible presently since the designs of the EXAM and the ZETA trials were different (66). It may be possible in the future to predict response to a TKI according to existing mutations to further individualize treatment (67). An outline of ongoing research on this aspect can be read later in this paper.
“Second Line” TKIs for MTC
Other multikinase inhibitors active against MTC are described below (Table 1). These TKIs showed efficacy in Phase II trials and are considered as second line in patients where cabozantinib or vandetanib are not feasible (11).
Sorafenib
Sorafenib, a multitargeted kinase inhibitor targeting VEGFR, PDGFR, FGFR, c-KIT, BRAF, and RET, is approved for advanced radioiodine-refractory DTC in the USA and Europe. In a phase II trial (68), of 8 patients with MTC, an objective response rate (ORR) of 25% and disease control rate of 75% was observed with toxicities consistent with the known profile of the drug. A systematic review (69) examined the results of 8 phase II studies on the efficacy of sorafenib for all advanced thyroid cancers. The authors concluded that sorafenib at the starting dose of 400 mg twice daily led to PR in 22% of MTC patients which was more than that for differentiated and anaplastic cancers (21 and 6%). PD occurred in 6.5% of MTC patients (vs 21% of DTC). The commonest AEs seen with sorafenib are hand–foot syndrome, diarrhea, weight loss, and fatigue (69). An unusual adverse effect reported (though not in thyroid cancers) is sorafenib-induced skin cancers (70). A small study with DTCs by Chen et al. found that halving the dose of sorafenib reduces the rate of drug discontinuation due to AEs without affecting response (71).
Sunitinib
In a phase II study (72), 26 patients of advanced/metastatic MTC were administered 50 mg/day of sunitinib for 4–6 weeks. Median PFS and OS were 16.5 and 29.4 months. The AEs were asthenia, mucosal and cutaneous toxicities, hand–foot syndrome, and cardiac events (14%). A phase II trial (73) with a smaller dose (37.5 mg) resulted in 50% of patients showing PR but with significant AEs necessitating dose reduction in over half the subjects. Thus, existing data suggest that sunitinib has efficacy in MTC though not without adverse effects.
Pazopanib
Pazopanib, a multikinase inhibitor (potent action on VEGF 1–3, PGDF, FGFR, and milder action on RET) found to have activity against MTCs in preclinical studies, was studied in a multicentre international phase II trial in patients with advanced progressive MTC (74). Of 35 patients, PR was seen in 14.3% with a median PFS of 9.4 months and OS of 19.9 months. Nearly half the patients had received prior therapy with chemoradiation and or other TKIs (vandetanib, cabozantinib, and sunitinib). AEs were hypertension, fatigue, diarrhea, and deranged liver tests, necessitating discontinuation in three subjects. One therapy-related death was reported. Overall, Pazopanib may be considered in MTC patients refractory to other TKIs.
Lenvatinib
Lenvatinib is a multikinase inhibitor most active on FGFR, thus, targeting a pathway involved in resistance to VEGF inhibitors. In a phase II trial (75) enrolling 59 patients with advanced progressive MTC, an ORR of 36% was seen, both in patients with and without prior anti-VEGF therapy/conventional chemotherapy; the 6-month PFS was 67%. The commonest AEs were diarrhea, fatigue, hypertension, decreased appetite, weight loss, and cough; grade V toxicity occurred in 8.5% cases. AEs led to treatment withdrawal in a quarter of the patients. At present, its PFS and toxicity seems similar to the other agents described previously.
Multikinase Inhibitors with Limited Efficacy
These are targeted agents that showed promise in preclinical or phase I or II trials but were not found efficacious enough to be used clinically for MTC.
Motesanib
Motesanib was the first TKI to be tested for MTCs alone. This is an oral small molecule TKI which is specific against VEGF 1, 2, and 3, besides wild-type RET and kit. The use of motesanib is based on the fact that VEGF is elevated upto 20 times in upto 75% of metastatic MTC (76). In a multicentre single arm phase II trial of 91 patients with advanced/metastatic MTC, the ORR was 2%, though 48% of the patients experienced prolonged stable disease (SD). The AEs though frequent were tolerable (52, 77).
Axitinib
Axitinib is a second generation VEGFR1–3 inhibitor and was evaluated in a phase II trial in 2008 (78), at a dose of 5 mg twice daily with 60 patients of advanced thyroid cancer including 11 MTC patients. PR was seen in two patients. On the other hand, a phase II trial (79) of patients with metastatic or unresectable TC, including six MTC patients recorded no PR to Axitinib, though SD was achieved in five cases for more than 4 months. The AEs of Locati et al. (79) Axitinib occur more often in the first year of treatment and rate of discontinuation due to AEs is comparable to that of other TKIs.
Imatinib
This prototype TKI was found to have limited inhibitory activity against RET; two trials included a total of 24 MTC patients who received imatinib. In these trials (80, 81), no objective responses were observed. Furthermore, treatment incurred significant toxicity.
Geftinib
Geftinib has not been found to be of appreciable efficacy for MTC. In a phase II trial for locally advanced or metastatic thyroid cancers, including MTC (82), there were no objective responses. There was tumor reduction in a third of the subjects though not enough to qualify as PR.
Ponatinib
Ponatinib is a newer multikinase inhibitor with exponentially higher activity against RET than other TKIs. Studies have proved its potential in MTC especially in cases where other treatment options had failed (83). However, observations in patients of chronic myeloid leukemia where it is used with good effect, showed a high risk of cardiovascular and peripheral vascular thrombosis (84, 85); this has prevented further development of this drug for MTC (86).
Targeted Drugs in Development for MTC
Other Multikinase Inhibitors
Nintedanib (87), anlotinib (88), regorafenib (89), and sulfatinib (90) all multikinase inhibitors are being investigated in several phase II studies for use in MTC. Dinaciclib (91), a cyclin-dependent kinase inhibitor, has been seen to decrease thyroid cancer proliferation both in vitro and in vivo. It has not yet been investigated for MTC.
Farnesyl Transferase Inhibitors or FTIs (Tipifarnib)
A subset of MTC shows Ras mutations (43, 44). Inhibitors of farnesylation of Ras proteins lead to inhibition of coupling of these proteins with an isoprenyl group, blocking signal transduction and, thus, to cessation of growth (92). Of the three functional Ras genes, H-Ras, N-Ras, and K-Ras, the latter two can alternatively be geranylgeranylated, thereby escaping the inhibition of Ras activation (92). FTIs also exert their effect by inhibiting RET via the downstream MAPK pathway (92). The premise of their efficacy has been upheld in preclinical studies but has not been borne out in phase I, II, and III studies. In two Phase I studies (93, 94), tipifarnib (the most studied FTI) showed synergism when combined with sorafenib and showed sustained PR for patients of thyroid cancers including MTC. Tipifarnib as single agent is currently being investigated in a phase 2 trial in HRAS-mutated tumors, including MTC (NCT02383927) (95). Phase I studies showed myelotoxicity and neurotoxicity as rate limiting effects with other effects being rash, hand/foot skin reaction, fatigue, hypertension, anorexia, and diarrhea (96).
Repositioned Drugs
Cholesterol lowering agents (statins) such as lovastatin have anti-tumor efficacy in K-ras dependent thyroid tumors (97). This “repositioned” drug may be one more therapeutic option for MTC.
Histone deacetylase inhibitors (HDACIs) such as valproic acid (VA): histone proteins surround the DNA strands. Post-translational modifications occurring on the amino acid residues of these proteins modify chromatin structure and, thus, alter gene expression. Histone acetylation and deacetylation are the most well understood modifications. Histone deacetylases (HDACs) exert a pro-oncogenic effect by keeping genes involved in apoptosis in a transcriptionally quiescent state (98). HDAC inhibitors also induce G2/M arrest in neoplastic cells that do not have a functional G2 checkpoint and these cells, thus, undergo apoptosis. Since this effect is not seen in normal cells, HDACIs cause selective cell death of tumor cells (99). Though several HDACIs have not yet shown clinical benefit, these small molecule inhibitors are good potential therapeutic agents and under trial for all advanced thyroid cancers (100, 101). VA inhibits HDAC via its effect on Notch-1 signalling (102). Notch is an EGFR-like transmembrane receptor and an important regulator of ASCL1 gene expression and activity which in turn is essential for the normal and malignant development of thyroid C-cells (103). VA has efficacy on MTC cell lines as it restores Notch function, thus impeding tumor growth (104). Despite the strong preclinical evidence, VA has not been found to have in vivo efficacy against MTC except in combination with other drugs, such as lithium chloride (105).
Monotherapy Agents and Epigenetic Therapy
Nelfinavir (NFV)
The heat shock protein (HSP) 90 chaperone is required for folding and stability of RET mutants (106). HSP90 overexpression is found in a significant proportion of MTCs and may correlate with metastases and RET mutations (107). HSP90 is a molecular target for the HIV protease inhibitor NFV. It has been shown to have wide spectrum activity in vivo against MTC cells and may emerge as an important therapeutic option. The adverse effects of NFV are seen to be similar to those of PI3–AKt pathway inhibitors such as everolimus (108).
mTOR Inhibitors
Everolimus is an mTOR inhibitor presently approved for treatment of neuroendocrine tumors and renal cell carcinoma. Since RET and RAS utilize the mTOR pathway, it follows that it is a potential subject for investigation for MTC. Two phase II trials (109, 110) conducted in patients with progressive metastatic or inoperable thyroid cancer, including MTC showed SD without any objective responses. However, combination of everolimus with sorafenib has shown promising results in the form of PR in 40% of the patients studied (111).
Targeting Other Pathways
MKT-077, a rhodacyanin dye, accumulates in the cellular mitochondria of MTC cells and downregulates RET by direct and indirect pathways making it a potential therapeutic agent (112). cAMP analogs (8-Cl-cAMP) suppress in vitro MTC cell proliferation and may be of clinical use in the treatment of advanced MTC (113).
Combined Targeted Therapy
Certain small molecule inhibitors possess efficacy against MTC in vivo in combination with drugs of other groups (111). Methylating agent temozolomide along with chemotherapeutic agent capecitabine have a beneficial effect on metastatic MTC (114). Sunitinib and cisplatin cooperatively interfere with the autophagic lysosomal pathway and this combination appears to be a promising treatment option for metastatic or progressive MTC (115).
Certain combinations may be more potent than the individual agents alone (101, 105). Acquired resistance to targeted therapy, one of the mechanisms of which is aberrant DNA methylation, can possibly be overcome by employing a combination of targets such as with Everolimus plus 5-aza-2′-deoxycytidine (AZA), proven to be a highly synergistic combination even in Everolimus-resistant cell lines (116).
In a recent meta-analysis (117), the combination of cytotoxic chemotherapy and TKIs (vandetanib, cabozantinib, sorafenib, and sunitinib) was shown to have higher improvement in PFS (but not in OS) than chemotherapy alone in a variety of malignancies. This benefit, however, has to be weighed against AEs associated with this treatment.
Pre-Empting and Managing Adverse Effects and Drug Interactions
Most series have documented significant AEs with targeted therapy (59, 64, 70, 85, 118, 119). Dose-dependent hypertension is a common occurence (120, 121) and guidelines (122) have been developed for its management including pretreatment monitoring of blood pressure, treatment of hypertension, and, if required, dose reduction. Cardiac effects (123–125) include prolonged QT interval (vandetanib, pazopanib), left ventricular dysfunction (pazopanib, axitinib), and congestive heart failure (sunitinib, pazopanib). Cardiac toxicity is often not predicted in preclinical studies (126), possibly due to unintended secondary targets of TKIs, toxic drug metabolites, and drug accumulation in the heart. The reversibility of trastuzumab-induced cardiac toxicity and the possibility of re-administering it to the patient have been mentioned (127). Though the same reversibility has not been described for TKIs in use for MTC, dose reduction, avoiding cardiotoxic drugs and reviewing drug combinations is important (128). Arterial thromboembolic events, such as cerebral infarction, cerebral ischemia, cerebrovascular accidents, myocardial infarction, and myocardial ischemia, are seen especially with panotinib (85) and cabozantinib (59) (upto 2%). Revascularization interventions may be indicated in severe cases (129).
Skin care in the form of protective shoes and use of urea-based cream prevents hand and foot syndrome. Avoiding exposure to soaps and direct sunlight is recommended. Supportive therapy, such as multivitamins, anti-diarrhoeals, dietary advice, and steroids for anorexia, prevents further complications. In view of TKI-induced hepatic AEs (130), pre-treatment baseline liver functions are mandated (131). TKIs are contraindicated in patients with transaminases more than 2.5 times normal in absence of liver metastases and five times in the presence of liver metastases. Development of grade III or IV hepatic toxicity during targeted therapy indicates the need for dose reduction or therapy discontinuation (131).
Drug interactions between TKIs and cytochrome enzyme inducers, such as ketoconazole (132) (alterations in either drug’s levels) and paracetamol (133) (additive hepatotoxicity), have been noted and accordingly the interacting drugs should be stopped or changed.
In view of risk of gastro-intestinal bleed and fistulization especially with VEGF inhibitors, it is important to avoid, stop, or change the therapeutic agent in patients with gastrointestinal bleed, immediately after injury or surgery, after radiation, in cases of impending fistulization or esophageal or tracheal rupture (134, 135). It is also a prudent precaution to avoid TKI treatment in cases of impending retinal bleed (131).
Squamous cancers of the skin which occasionally develop after TKIs (70) may be treated with wide excision (136). As these are unlikely to recur or metastasize, treatment discontinuation is not recommended (137). Sunitinib and sorafenib can lead to hypothyroidism due to decreased gastrointestinal absorption or due to diarrhea causing increased clearance. Monitoring of thyroid levels is, thus, recommended (138). Electrolyte imbalances also should be managed pre-emptively (137). Most of the AEs seen with targeted agents are class effects, that is, due to effect on target tissues, and thus may be unavoidable. There may also be a direct correlation between efficacy and AEs (139). Grade I or II AEs (as per Common terminology criteria—CTCAE) which can be managed with best symptomatic care do not necessitate dose reduction or discontinuation. However, in case of recurrent grade I or II and in case of more severe AEs (grade III or more), discontinuation ought to be considered (131).
Improving Drug Selectivity—Individualization of Targeted Therapy
Consistent efforts are ongoing to correlate mutational status to drug efficacy and treatment decisions. Though important conclusions have been arrived at by different workers, there is still some way to go before definite algorithms can be formulated. The ZETA trial for Vandetanib in MTC (64) showed that patients with sporadic MTCs harboring a somatic M918T mutation had a higher response rate to vandetanib (54.5%; 55 of 101) than those without a somatic M918T mutation (32%; 33 of 103). In another study (140), 5 of 10 vandetanib-treated MTC patients showed PR, all harboring the RET918 mutation; also, mRNA expression of VEGFR was significantly higher in therapy responders. In another study, Kurzrock et al. found that there was no strict correlation between the RET/MET mutational status and clinical response of MTC to Cabozantinib (55). In a phase II trial of Sunitinib in MTC (141), PR or SD greater than 24 weeks was observed in 7 of 9 MTC pts with a M918T RET mutation. Three cell lines (142), respectively, expressing a C634W RET mutation, a M918T RET mutation, and a RET/PTC-1 rearrangement (in a case of DTC) were studied for response to 4TKIs; Cabozantinib was found to be the most efficient inhibitor for MEN2A derived cell lines, whereas vandetanib proved to be the most potent inhibitor for MEN2B. Carlomagno et al. found that the presence of Valine 804 mutation of RET as a structural determinant mediating resistance (143) while sorafenib has been found to have activity in vitro against the same V804 mutant (144). In vitro studies using cell lines with acquired resistance to vandetanib have shown persistent activation of the Ras/Raf/MEK pathway, which can be partly abrogated by sorafenib (145). In patients with RET wild-type tumors, it remains to be seen whether Ras mutations, identified in 60–80% of RET-negative sporadic MTC (35) have an impact on disease progression and if the therapeutic agent used needs to be one targeting Ras as well. In a phase II trial of lenvantinib, there was no difference in treatment response according to RET mutation status; however, high baseline levels of VEGF correlated with greater tumor shrinkage, and low levels of ANG2, sTie-2, HGF, and interleukin (IL)-8 were associated with tumor shrinkage and prolonged PFS (75). This distinction is important as the main serious AEs associated with the current RET TKIs are related to anti-VEGFR or anti-EGFR effects.
Thus, several possibilities for patients with different genetic profiles are gradually being discovered; and various workers continue to study and correlate genetic mutation status so as to determine the therapeutic option best suited to the individual patient.
Resistance
Resistance to targeted therapy is well known and is also called the Escape phenomenon. Resistance may be innate or intrinsic or primary i.e., already present before targeted treatment is begun. In vitro studies show that the RET V804M (valine substituted with methionine) and V804L (valine substituted with leucine) gatekeeper mutations confer resistance to vandetanib (143). Although rare, these could result clinically in primary resistance to vandetanib. In patients with Ras mutations, identified in 60–80% of RET-negative sporadic MTC, intrinsic clinical resistance to vandetanib may exist (35). Intrinsic resistance also includes the ability of the tumor to take alternate and downstream signal pathways, such as Ras/MEK/EGFR (146). Secondary resistance develops after a variable period of definite response to TKI and it may be an On target or an Off target resistance.
On-target resistance is usually secondary but may be an unrecognized rapid cause of primary resistance. There is an alteration in structure or in activity of the target protein that results in reduced drug efficacy. This may be achieved by steric conformational change blocking access to the drug (147); by alteration in the topography of target site (148); or by alteration in ATP affinity (149) and last but not least by gene amplification (150), by which the number of copies of the target gene is increased many times over to enable the malignant cell proliferation to continue despite maximal binding of drug to target. Off target drug resistance is seen when the target oncoprotein stays inhibited and the cancer develops alternative sources of growth. It is termed as evasive or adaptive resistance when alternate pathways further downstream are activated such as FGRF/IL-8 (151); or other pro-angiogenic signaling agents are upregulated such as PGF/GCSF (152) or via negative feedback loops and paradoxical activation (153). Evasive resistance also occurs when there is recruitment of pro-angiogenic cells from the bone marrow; and when the tumor develops the capability to invade in absence of angiogenesis (in case of VEGF TKIs) (154). Finally, tumor cell signaling networks alterations and tumor microenvironment change which occur over time, influence disease progression as well as a drug’s effectiveness, thus resulting in secondary drug resistance (155).
Predicting or Detecting Resistance
Pre-treatment prediction of efficacy of and resistance to a particular TKI, though not yet in routine practice, has been shown to be possible and feasible using cell lines (156) and patient-derived models (157). However, there are several caveats to these procedures and cautious interpretations have to be made. Serum calcitonin may act as a surrogate indicator of the development of resistance to the TKI being used with a study (158) demonstrating that an increase in serum calcitonin levels to greater than 40% may be used reliably as an early indicator of tumor progression or drug resistance and as a guide to consider change of treatment.
Overcoming Resistance
Using a TKI that acts via more than one pathway is one way of countering resistance. This can be seen with cabozantinib that inhibits MET and VEGFR2 and effectively blocks the development of MET-driven evasive resistance, as opposed to agents targeting the VEGF pathway alone. Thus cabozantinib may provide a more sustained therapeutic effect (116, 159). Addition of an additional synergistic agent is another way of evading resistance. For example, cabozantinib induced upregulation of downstream factors, such as VEGF and MET, can be neutralized by adding 2-ME2 (2-methoxyestradiol) to MTC cell lines. This agent downregulates HIF 1α (hypoxia-inducible factor) expression and, thus, inhibits the abovementioned pathways (160). Similarly, chloroquine potentiates action of sunitinib (161) in cell lines by interrupting sunitinib-induced autophagic flux, thus causing further apoptosis in cancer cells.
Conventional Cytotoxic Chemotherapy for MTC
Results with chemotherapy in MTC have been reported as equivocal, whether done with a single agent (dacarbazine or doxorubicin) (162) or as combination (usually dacarbazine along with 5FU/vincristine/doxorubicin/cyclophosphamide) (163, 164). Capecitabine, an oral prodrug of 5-fluorouracil and a reasonably safe option for disease stabilization in MTC (165) has been applied with good benefit in combination with another oral chemotherapeutic agent temozolomide (114). Conventional chemotherapy (with dacarbazine and its combinations) is currently recommended only for cases not eligible for, or resistant to tyrosine kinases (vandetanib or cabozantinib) (11). Cytotoxic agents have been found to act synergistically with small molecule TKIs for metastatic MTCs (115) and their role may re-emerge if these findings are substantiated.
Targeted Radionuclide Therapy or Peptide Receptor Radionuclide Therapy (PRRT)
The molecular rationale for use of PRRT in MTC (or in other well differentiated neuroendocrine tumors) is the over expression of somatostatin type 2 receptors (SSTR) (166). Radiolabeled somatostatin analogs are agents used in diagnosis as well as therapeutics. Such an agent consists of a peptide somatostatin analog with high affinity to SSTR2 receptors (viz. octreotide), a radioactive component [yttrium (Y) or lutetium (Lu)] and an intervening chelator such as DTPA (diethylenetriaminepentaacetic acid). Y and Lu have properties suited to ablation of such tumors and act by direct effect and by anti-angiogenic effect (167). In earlier pilot studies (168) and in more recent trials (169), PRRT was found to be a viable therapeutic option with a beneficial effect on QOL (169). A phase II trial evaluating systemic 90Y-DOTATOC (tetraazacyclododecane-tetraacetic acid Phe-Tyr octreotide) treatment in patients with advanced MTC showed response in 9 out of 31 (29%) patients; there was improved survival in responders to treatment as compared with non-responders (i.e., 74.5 vs 10.8 months); however, 12.9% of patients developed hematological toxicity and 22.6% developed nephrotoxicity (170). Salavati et al. (171) treated over a period of 8 years, a cohort of 28 patients with recurrent and metastatic MTC. All patients had undergone previous treatment in the form of surgery, radiotherapy, conventional chemotherapy, and local palliative measures. Following confirmation of somatostatin expression using 68Ga-DOTATOC PET/CT, patients were treated with PRRT with 90Y or 177Lu-DOTATATE (DOTA-Tyr-octreotate). 17 patients (60.7%) showed SD, 5 of 28 (17.7%) showed PR and 6 (21.4%) showed disease progression. The median survival of patients with SD was 36 months, that of patients with PR was 72 months, and the median survival for patients with PD was 24 months. In a series of 265 patients (172) who underwent LuDOTATATE therapy for neuroendocrine malignancies, significant improvement in QOL scores was seen even in patients who showed no significant disease resolution; there was no case where QOL deteriorated to pre-treatment levels. PRRT has also been used in other neuroendocrine malignancies in the form of an infusion to selectively ablate liver metastases (173). Combination of PRRT with conventional chemotherapy has been found to provide benefit in neuroendocrine tumors (174, 175). Thus, PRRT promises to provide disease outcome benefits with improved QOL scores though the toxicity benefit ratio remains to be quantified (176).
A novel gene therapy technique has shown that MTC cells can be induced to take up iodine through calcitonin promoter-directed human sodium iodide symporter (hNIS) expression and, thus, can be ablated with Radioiodine (177).
Conclusion
Inoperable recurrent and metastatic MTC needs intervention when symptomatic. Solitary, symptomatic metastases are best treated with surgery or radiofrequency ablation for clinical and biochemical benefit. The development of targeted therapy is the logical step forward when the disease becomes unapproachable with conventional treatment modalities. Evolution of knowledge about other molecular pathways that may be targeted and efforts to counter resistance by combining agents acting on separate points on the pathway is the rational next step. Though “personalization” of treatment according to the specific mutation or absence of it is still in the realm of investigation, categorization of patients as per treatment history, disease location, rate of progression and performance status is feasible; the emergence of guidelines concerning several aspects of treatment is evidence of effort in this direction.
Author Contributions
SP, CD, and MD—design, collection of data, manuscript, editing, approval of final version, and accountability. RD—expert comments and approval of final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
DM, distant metastasis; AE, adverse events; HDACI, histone deacetylase inhibitor; VA, valproic acid; QOL, quality of life.
References
1. Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma: the American Thyroid Association Guidelines Task Force on medullary thyroid carcinoma. Thyroid (2015) 25(6):567–610. doi:10.1089/thy.2014.0335
2. Randle RW, Balentine CJ, Leverson GE, Havlena JA, Sippel RS, Schneider DF, et al. Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 Years. Surgery (2017) 161(1):137–46. doi:10.1016/j.surg.2016.04.053
3. Scollo C, Baudin E, Travagli JP, Caillou B, Bellon N, Leboulleux S, et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab (2003) 88(5):2070–5. doi:10.1210/jc.2002-021713
4. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma. Demographic, clinical and pathologic predictors of survival in 1252 cases. Cancer (2006) 107(9):2134–42. doi:10.1002/cncr.22244
5. Call JA, Caudill JS, McIver B, Foote RL. A role for radiotherapy in the management of advanced medullary thyroid carcinoma: the mayo clinic experience. Rare Tumors (2013) 5(3):37. doi:10.4081/rt.2013.e37
6. Youngwirth L, Adam M, Scheri R, Roman SA. External beam radiation is not associated with improved survival in patients with locally advanced medullary thyroid cancer. In thyroid neoplasia (posters). Endocr Soc (2016):SAT–307.
7. Leboulleux S, Baudin E, Travagli JP, Schlumberger M. Medullary thyroid carcinoma. Clin Endocrinol (2004) 61(3):299–310. doi:10.1111/j.1365-2265.2004.02037.x
8. Machens A, Schneyer U, Holzhausen HJ, Dralle H. Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab (2005) 90(4):2029–34. doi:10.1210/jc.2004-1836
9. Brierley J, Tsang R, Simpson WJ, Gospodarowicz M, Sutcliffe S, Panzarella T. Medullary thyroid cancer: analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid (1996) 6(4):305–10. doi:10.1089/thy.1996.6.305
10. Cappagli V, Bottici V, Elisei R. Clinical management of a patient with a locally recurrent medullary thyroid cancer and asymptomatic slowly progressing distant metastases. In: Cooper D, Durante C editors. Thyroid Cancer. Springer International Publishing (2016). p. 347–54. doi:10.1007/978-3-319-22401-5_40
11. National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology (NCCN Guidelines). Thyroid Carcinoma. Version 1.2016. Fort Washington, PA: NCCN (2016).
12. Moley JF, Wells SA, Dilley WG, Tisell LE. Reoperation for recurrent or persistent medullary thyroid cancer. Surg Saint Louis (1993) 114(1):1090–5.
13. Fialkowski E, DeBenedetti M, Moley J. Long-term outcome of reoperations for medullary thyroid carcinoma. World J Surg (2008) 32(5):754–65. doi:10.1007/s00268-007-9317-7
14. Chen H, Roberts JR, Ball DW, Eisele DW, Baylin SB, Udelsman R, et al. Effective long-term palliation of symptomatic, incurable metastatic medullary thyroid cancer by operative resection. Ann Surg (1998) 227(6):887. doi:10.1097/00000658-199806000-00012
15. Harrison LE, Brennan MF, Newman E, Fortner JG, Picardo A, Blumgart LH, et al. Hepatic resection for noncolorectal, nonneuroendocrine metastases: a fifteen-year experience with ninety-six patients. Surgery (1997) 121(6):625–32. doi:10.1016/S0039-6060(97)90050-7
16. Berber E, Flesher N, Siperstein A. Laparoscopic radiofrequency ablation of neuroendocrine liver metastases. World J Surg (2002) 26:985. doi:10.1007/s00268-002-6629-5
17. Fromigué J, De Baere T, Baudin E, Dromain C, Leboulleux S, Schlumberger M. Chemoembolization for Liver metastases from medullary thyroid carcinoma. J Clin Endocrinol Metab (2006) 91(7):2496–9. doi:10.1210/jc.2005-2401
18. Munoz-Bendix C, Santacroce A, Gierga K, Floeth FW, Steiger HJ, Penalonzo MA, et al. Recurrent spinal metastasis of a sporadic medullary carcinoma of the thyroid after radiation therapy: a case report and review of the literature. Clin Case Rep (2016) 4(1):9–18. doi:10.1002/ccr3.409
19. Wexler JA. Approach to the thyroid cancer patient with bone metastases. J Clin Endocrinol Metab (2011) 96:2296–307. doi:10.1210/jc.2010-1996
20. Quan GM, Pointillart V, Palussiere J, Bonichon F. Multidisciplinary treatment and survival of patients with vertebral metastases from thyroid carcinoma. Thyroid (2012) 22:125–30. doi:10.1089/thy.2010.0248
21. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res (2003) 145:S158–64. doi:10.1097/01.blo.0000093057.96273.fb
22. McWilliams RR, Giannini C, Hay ID, Atkinson JL, Stafford SL, Buckner JC. Management of brain metastases from thyroid carcinoma. Cancer (2003) 98:356–62. doi:10.1002/cncr.11488
23. Mandanas S, Margaritidou E, Christoforidou V, Karoglou E, Geranou C, Chrisoulidou A, et al. Breast metastasis from medullary thyroid carcinoma in a male patient: case report and review of the literature. Rare Tumors (2015) 7(2):5765. doi:10.4081/rt.2015.5765
24. Appetecchia M, Barnabei A, Pompeo V, Sentinelli S, Baldelli R, Corsello SM, et al. Testicular and inguinal lymph node metastases of medullary thyroid cancer: a case report and review of the literature. BMC Endocr Disord (2014) 14(1):84. doi:10.1186/1472-6823-14-84
25. Nashed C, Sakpal SV, Cherneykin S, Chamberlain RS. Medullary thyroid carcinoma metastatic to skin. J Cutan Pathol (2010) 37:1237–40. doi:10.1111/j.1600-0560.2009.01365.x
26. Schlumberger M, Bastholt L, Dralle H, Jarzab B, Pacini F, Smit JW. European thyroid association guidelines for metastatic medullary thyroid cancer. Eur Thyroid J (2012) 1(1):5–14. doi:10.1159/000336977
27. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet (1993) 2(7):851–6. doi:10.1093/hmg/2.7.851
28. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature (1993) 363(6428):458. doi:10.1038/363458a0
29. Cakir M, Grossman AB. Medullary thyroid cancer: molecular biology and novel molecular therapies. Neuroendocrinology (2009) 90(4):323–48. doi:10.1159/000220827
30. Dvořáková Š, Vaclavikova E, Sýkorová V, Dušková J, Vlček P, Ryška A, et al. New multiple somatic mutations in the RET proto-oncogene associated with a sporadic medullary thyroid carcinoma. Thyroid (2006) 16(3):311–6. doi:10.1089/thy.2006.16.311
31. de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumor. Endocr Rev (2006) 27:535–60. doi:10.1210/er.2006-0017
32. Castellone MD, Santoro M. Dysregulated RET signaling in thyroid cancer. Endocrinol Metab Clin North Am (2008) 37:363–74. doi:10.1016/j.ecl.2008.02.006
33. Kodama Y, Asai N, Kawai K, Jijiwa M, Murakumo Y, Ichihara M, et al. The RET proto-oncogene: a molecular therapeutic target in thyroid cancer. Cancer Sci (2005) 96(3):143–8. doi:10.1111/j.1349-7006.2005.00023.x
34. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer (2007) 7:295–308. doi:10.1038/nrc2175
35. Moura MM, Cavaco BM, Pinto AE, Leite V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab (2011) 96:E863–8. doi:10.1210/jc.2010-1921
36. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18:1926–45. doi:10.1101/gad.1212704
37. Nilsson M, Williams D. On the origin of cells and derivation of thyroid cancer: C cell story revisited. Eur Thyroid J (2016) 5(2):79–93. doi:10.1159/000447333
38. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science (2002) 298:1912–34. doi:10.1126/science.1075762
39. Pestell KE. Paul workman on the challenges of cancer drug development. Drug Discov Today (2003) 8(17):775–7. doi:10.1016/S1359-6446(03)02838-1
40. Sawyers CL. Rational therapeutic intervention in cancer: kinases as drug targets. Curr Opin Gen Develop (2002) 12:111–5. doi:10.1016/s0959-437x(01)00273-8
41. Croyle M, Akeno N, Knauf JA, Fabbro D, Chen X, Baumgartner JE, et al. RET/PTC induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Res (2008) 68:4183–91. doi:10.1158/0008-5472.CAN-08-0413
42. Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res (2003) 284:31–53. doi:10.1016/S0014-4827(02)00098-8
43. Ciampi R, Mian C, Fugazzola L, Cosci B, Romei C, Barollo S, et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid (2013) 23:50–7. doi:10.1089/thy.2012.0207
44. Barollo S, Pezzani R, Cristiani A, Bertazza L, Rubin B, Bulfone A. Functional significance of the novel H-RAS gene mutation M72I in a patient with medullary thyroid cancer. Exp Clin Endocrinol Diabetes (2013) 121:546–50. doi:10.1055/s-0033-1351299
45. Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab (2013) 98:E1852–60. doi:10.1210/jc.2013-2292
46. Capp C, Wajner SM, Siqueira DR, Brasil BA, Meurer L, Maia AL. Increased expression of vascular endothelial growth factor and its receptors, VEGFR-1 and VEGFR-2, in medullary thyroid carcinoma. Thyroid (2010) 20:863–71. doi:10.1089/thy.2009.0417
47. Rodriguez-Antona C, Pallares J, Montero-Conde C, Inglada-Perez L, Castelblanco E, Landa I, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer (2010) 17:7–16. doi:10.1677/ERC-08-0304
48. Papotti M, Olivero M, Volante M, Negro F, Prat M, Comoglio PM, et al. Expression of hepatocyte growth factor (HGF) and its receptor (MET) in MTC of the thyroid. Endocr Pathol (2000) 11:19–30. doi:10.1385/EP:11:1:19
49. Ezzat S, Huang P, Dackiw A, Asa SL. Dual inhibition of RET and FGFR4 restrains medullary thyroid cancer cell growth. Clin Cancer Res (2005) 11:1336–41.
50. Wells SA Jr, Santoro M. Update: the status of clinical trials with kinase inhibitors in thyroid cancer. J Clin Endocrinol Metab (2014) 99(5):1543–55. doi:10.1210/jc.2013-2622
51. Rodriguez-Antona C, Munoz-Repeto I, Inglada-Perez L, de Cubas AA, Mancikova V, Canamero M, et al. 2013 Influence of RET mutations on the expression of tyrosine kinases in medullary thyroid carcinoma. Endocr Relat Cancer (2013) 20:611–9. doi:10.1530/ERC-12-0316
52. Viola D, Valerio L, Molinaro E, Agate L, Bottici V, Biagini A, et al. Treatment of advanced thyroid cancer with targeted therapies: ten years of experience. Endocr Relat Cancer (2016) 23(4):R185–205. doi:10.1530/ERC-15-0555
53. Nella AA, Lodish MB, Fox E, Balis FM, Quezado MM, Whitcomb PO, et al. Vandetanib successfully controls medullary thyroid cancer-related Cushing syndrome in an adolescent patient. J Clin Endocrinol Metab (2014) 99(9):3055–9. doi:10.1210/jc.2013-4340
54. Barroso-Sousa R, Lerario AM, Evangelista J, Papadia C, Lourenço DM Jr, Lin CS, et al. Complete resolution of hypercortisolism with sorafenib in a patient with advanced medullary thyroid carcinoma and ectopic ACTH (adrenocorticotropic hormone) syndrome. Thyroid (2014) 24(6):1062–6. doi:10.1089/thy.2013.0571
55. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol (2011) 29(19):2660–6. doi:10.1200/JCO.2010.32.4145
56. FDA Approves Cometriq to Treat Rare Type of Thyroid Cancer. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm381452.htm
57. Schoffski P, Elisei R, Müller S, Brose MS, Shah MH, Licitra LF, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients with documented RECIST progression at baseline. J Clin Oncol (2012) 30(15 Suppl):5508–5508. doi:10.1200/jco.2012.30.15_suppl.5508
58. Schlumberger M, Elisei R, Müller S, Schöffski P, Brose MS, Shah MH, et al. Final overall survival analysis of EXAM, an international, double-blind, randomized, placebo-controlled phase III trial of cabozantinib (Cabo) in medullary thyroid carcinoma (MTC) patients with documented RECIST progression at baseline. J Clin Oncol (2015) 33(15 Suppl):6012–6012. doi:10.1200/jco.2015.33.15_suppl.6012
59. European Medicines Agency. COMETRIQ (cabozantinib): Summary of Product Characteristics. (2014). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002640/WC500163703.pdf
60. Marino R, Orlandi F, Arecco F, Gandolfo S, Pentenero M. Osteonecrosis of the jaw in a patient receiving cabozantinib. Aust Dent J (2015) 60(4):528–31. doi:10.1111/adj.12254
61. Blevins DP, Dadu R, Hu M, Baik C, Balachandran D, Ross W, et al. Aerodigestive fistula formation as a rare side effect of antiangiogenic tyrosine kinase inhibitor therapy for thyroid cancer. Thyroid (2014) 24(5):918–22. doi:10.1089/thy.2012.0598
62. Viola D, Cappagli V, Matrone A, Mazzeo S, Elisei R. Cabozantinib: an orphan drug for thyroid cancer. Expert Opin Orphan Drugs (2015) 3(12):1469–77. doi:10.1517/21678707.2015.1112789
63. Spitzweg C, Morris JC, Bible KC. New drugs for medullary thyroid cancer: new promises? Endocr Relat Cancer (2016) 23(6):R287–97. doi:10.1530/ERC-16-0104
64. Wells SA Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol (2012) 30:134–41. doi:10.1200/JCO.2011.35.5040
65. An International, Randomised, Double-Blind, Two-Arm Study To Evaluate The Safety And Efficacy Of Vandetanib 150 And 300mg/Day In Patients With Unresectable Locally Advanced Or Metastatic Medullary Thyroid Carcinoma With Progressive Or Symptomatic Disease (2016). Available from: https://clinicaltrials.gov/ct2/show/NCT01496313
66. Ernani V, Kumar M, Chen AY, Owonikoko TK. Systemic treatment and management approaches for medullary thyroid cancer. Cancer Treat Rev (2016) 50:89–98. doi:10.1016/j.ctrv.2016.09.006
67. Sherman SI, Clary DO, Elisei R, Schlumberger MJ, Cohen EE, Schöffski P, et al. Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer (2016) 122(24):3856–64. doi:10.1002/cncr.30252
68. Ito Y, Onoda N, Ito KI, Sugitani I, Takahashi S, Yamaguchi I, et al. Sorafenib in Japanese patients with locally advanced or metastatic medullary thyroid carcinoma and anaplastic thyroid carcinoma. Thyroid (2017) 27(9):1142–8. doi:10.1089/thy.2016.0621
69. Thomas L, Lai SY, Dong W, Feng L, Dadu R, Regone RM, et al. Sorafenib in metastatic thyroid cancer: a systematic review. Oncologist (2014) 19(3):251–8. doi:10.1634/theoncologist.2013-0362
70. Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol (2011) 50(4):396–402. doi:10.1111/j.1365-4632.2010.04822.x
71. Chen L, Shen Y, Luo Q, Yu Y, Lu H, Zhu R. Response to sorafenib at a low dose in patients with radioiodinerefractory pulmonary metastases from papillary thyroid carcinoma. Thyroid (2011) 21:119–24. doi:10.1089/thy.2010.0199
72. Ravaud A, de la Fouchardière C, Caron P, Doussau A, Do Cao C, Asselineau J, et al. A multicenter phase II study of sunitinib in patients with locally advanced or metastatic differentiated, anaplastic or medullary thyroid carcinomas: mature data from the THYSU study. Eur J Cancer (2017) 76:110–7. doi:10.1016/j.ejca.2017.01.029
73. Carr LL, Mankoff DA, Goulart BH, Eaton KD, Capell PT, Kell EM, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res (2010) 16(21):5260–8. doi:10.1158/1078-0432.CCR-10-0994
74. Bible KC, Suman VJ, Molina JR, Smallridge RC, Maples WJ, Menefee ME, et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab (2014) 99(5):1687–93. doi:10.1210/jc.2013-3713
75. Schlumberger M, Jarzab B, Cabanillas ME, Robinson BG, Pacini F, Ball DW, et al. A phase 2 trial of the multi-targeted tyrosine kinase inhibitor lenvatinib (E7080) in advanced medullary thyroid cancer (MTC). Clin Cancer Res (2016) 22(1):44–53. doi:10.1158/1078-0432.CCR-15-1127
76. Bunone G, Vigneri P, Mariani L, Butó S, Collini P, Pilotti S, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol (1999) 155(6):1967–76. doi:10.1016/S0002-9440(10)65515-0
77. Schlumberger MJ, Elisei R, Bastholt L, Wirth LJ, Martins RG, Locati LD, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol (2009) 27(23):3794–801. doi:10.1200/JCO.2008.18.7815
78. Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clini Oncol (2008) 26(29):4708–13. doi:10.1200/JCO.2007.15.9566
79. Locati LD, Licitra L, Agate L, Ou SH, Boucher A, Jarzab B, et al. Treatment of advanced thyroid cancer with axitinib: phase 2 study with pharmacokinetic/pharmacodynamic and quality-of-life assessments. Cancer (2014) 120(17):2694–703. doi:10.1002/cncr.28766
80. Frank-Raue K, Fabel M, Delorme S, Haberkorn U, Raue F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur J Endocrinol (2007) 157:215–20. doi:10.1530/EJE-06-0695
81. de Groot JWB, Zonnenberg BA, van Ufford-Mannesse PQ, de Vries MM, Links TP, Lips CJM, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab (2007) 92(9):3466–9. doi:10.1210/jc.2007-0649
82. Pennell NA, Daniels GH, Haddad RI, Ross DS, Evans T, Wirth LJ, et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid (2008) 18:317–23. doi:10.1089/thy.2007.0120
83. Mologni L, Redaelli S, Morandi A, Plaza-Menacho I, Gambacorti-Passerini C. Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol Cell Endocrinol (2013) 377(1):1–6. doi:10.1016/j.mce.2013.06.025
84. Cortes JE, Kim DW, Pinilla-Ibarz J, Le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias. N Engl J Med (2013) 369(19):1783–96. doi:10.1056/NEJMoa1306494
85. Mayer K, Gielen GH, Willinek W, Müller MC, Wolf D. Fatal progressive cerebral ischemia in CML under third-line treatment with ponatinib. Leukemia (2014) 28:976–7. doi:10.1038/leu.2013.320
86. Maxwell JE, Sherman SK, O’dorisio TM, Howe JR. Medical management of metastatic medullary thyroid cancer. Cancer (2014) 120(21):3287–301. doi:10.1002/cncr.28858
87. Study of Nintedanib in Thyroid Cancer (BIBF1120) (2016). Available from: http://clinicaltrials.gov/ct2/show/NCT01788982
88. Study of Anlotinib in Patients with Medullary Thyroid Carcinoma (ALTER01031) (2016). Available from: https://clinicaltrials.gov/ct2/show/NCT02586350
89. A Study Using Regorafenib as Second or Third Line Therapy in Metastatic Medullary Thyroid Cancer (2017). Available from: https://clinicaltrials.gov/ct2/show/NCT02657551
90. Chen J, Ji Q, Cao J, Ji D, Bai C, Lin Y, et al. A phase II multicenter trial of the multitargeted kinase inhibitor sulfatinib in advanced medullary thyroid cancer (MTC) and radioiodine (RAI)-refractory differentiated thyroid cancer (DTC). Hypertension (2017) 12:60.
91. iLin S-F, Lin J-D, Hsueh C, Chou T-C, Wong RJ. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS One (2017) 12(2):e0172315. doi:10.1371/journal.pone.0172315
92. Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist (2005) 10(8):565–78. doi:10.1634/theoncologist.10-8-565
93. Hong D, Ye L, Gagel R, Chintala L, El Naggar AK, Wright J, et al. Medullary thyroid cancer: targeting the RET kinase pathway with sorafenib/tipifarnib. Mol Cancer Ther (2008) 7(5):1001–6. doi:10.1158/1535-7163.MCT-07-2422
94. Hong DS, Cabanillas ME, Wheler J, Naing A, Tsimberidou AM, Ye L, et al. Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. J Clin Endocrinol Metab (2011) 96:997–1005. doi:10.1210/jc.2010-1899
95. Ho AL, Chau NG, Wong DJ, Cabanillas ME, Bauman JR, Bible KC, et al. An open-label, phase II study of tipifarnib for the treatment of HRAS mutant solid tumors, including squamous cell carcinomas of the head and neck. J Clin Oncol (2017) 35:TS2618.
96. Punt CJ, van Maanen L, Bol CJ, Seifert WF, Wagener DJ. Phase I and pharmacokinetic study of the orally administered farnesyl transferase inhibitor R115777 in patients with advanced solid tumors. Anticancer Drugs (2001) 12:193–7. doi:10.1097/00001813-200103000-00003
97. Wang CY, Zhong WB, Chang TC, Lai SM, Tsai YF. Lovastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, induces apoptosis and differentiation in human anaplastic thyroid carcinoma cells. J Clin Endocrinol Metab (2003) 88:3021–6. doi:10.1210/jc.2002-021834
98. Brest P, Lassalle S, Hofman V, Bordone O, Tanga VG, Bonnetaud C, et al. miR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr Relat Cancer (2011) 18(6):711–9. doi:10.1530/ERC-10-0257
99. Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol (2012) 90(1):85–94. doi:10.1038/icb.2011.100
100. Catalano MG, Pugliese M, Gallo M, Brignardello E, Milla P, Orlandi F, et al. Valproic acid, a histone deacetylase inhibitor, in combination with paclitaxel for anaplastic thyroid cancer: results of a multicenter randomized controlled phase II/III trial. Int J Endocrinol (2016) 27:2016. doi:10.1155/2016/2930414
101. Pozo K, Zahler S, Tan C, Ishimatsu K, Takahashi M, Bibb J. Targeting tumor growth in a medullary thyroid carcinoma mouse model using a combinatorial treatment with a tyrosine kinase inhibitor and histone deacetylase inhibitor. Cancer Res (2016) 76(14 Suppl):4741–4741. doi:10.1158/1538-7445.AM2016-4741
102. Stockhausen MT, Sjolund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer (2005) 92:751–9. doi:10.1038/sj.bjc.6602309
103. Cook M, Yu XM, Chen H. Notch in the development of thyroid C-cells and the treatment of medullary thyroid cancer. Am J Transl Res (2010) 2(1):119–25.
104. Greenblatt DY, Cayo MA, Adler JT, Ning L, Haymart MR, Kunnimalaiyaan M, et al. Valproic acid activates Notch1 signaling and induces apoptosis in medullary thyroid cancer cells. Ann Surg (2008) 247(6):1036. doi:10.1097/SLA.0b013e3181758d0e
105. Adler JT, Hottinger DG, Kunnimalaiyaan M, Chen H. Inhibition of growth in medullary thyroid cancer cells with histone deacetylase inhibitors and lithium chloride. J Surg Res (2010) 159:640–4. doi:10.1016/j.jss.2008.08.004
106. Alfano L, Guida T, Provitera L, Vecchio G, Billaud M, Santoro M, et al. RET is a heat shock protein 90 (HSP90) client protein and is knocked down upon HSP90 pharmacological block. J Clin Endocrinol Metab (2010) 95(7):3552–7. doi:10.1210/jc.2009-2315
107. Kushchayeva Y, Jensen K, Recupero A, Costello J, Patel A, Klubo-Gwiezdzinska J, et al. The HIV protease inhibitor nelfinavir down-regulates RET signaling and induces apoptosis in medullary thyroid cancer cells. J Clin Endocrinol Metab (2014) 99(5):E734–45. doi:10.1210/jc.2013-3369
108. Valea A, Georgescu CE. A perspective on the current medical approach of advanced medullary thyroid carcinoma. Thyroid Cancer (2016):193–213. doi:10.5772/63650
109. Lim SM, Chang H, Yoon MJ, Hong YK, Kim H, Chung WY, et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol (2013) 24:3089–94. doi:10.1093/annonc/mdt379
110. Schneider TC, de Wit D, Links TP, van Erp NP, van der Hoeven JJ, Gelderblom H, et al. Beneficial effects of the mTOR inhibitor everolimus in patients with advanced medullary thyroid carcinoma: subgroup results of a phase II trial. Int J Endocrinol (2015) 2015:348124. doi:10.1155/2015/348124
111. Sherman E, Ho AL, Fury MG, Baxi SS, Dunn L, Lee JS, et al. Combination of everolimus and sorafenib in the treatment of thyroid cancer: update on phase II study. J Clini Oncol (2015) 33(Suppl):Abstract6069.
112. Starenki D, Park J-I. Selective mitochondrial uptake of MKT-077 can suppress medullary thyroid carcinoma cell survival in vitro and in vivo. Endocrinol Metab (2015) 30(4):593–603. doi:10.3803/EnM.2015.30.4.593
113. Dicitore A, Grassi ES, Caraglia M, Borghi MO, Gaudenzi G, Hofland LJ, et al. The cAMP analogs have potent anti-proliferative effects on medullary thyroid cancer cell lines. Endocrine (2016) 51(1):101–12. doi:10.1007/s12020-015-0597-7
114. Sahin Lacin EE, Karakas Y, Yalcin S. Metastatic medullary thyroid cancer: a dramatic response to a systemic chemotherapy (temozolomide and capecitabine) regimen. Onco Targets Ther (2015) 8:1039. doi:10.2147/OTT.S82906
115. Lopergolo A, Nicolini V, Favini E, Dal Bo L, Tortoreto M, Cominetti D, et al. Synergistic cooperation between sunitinib and cisplatin promotes apoptotic cell death in human medullary thyroid cancer. J Clin Endocrinol Metab (2013) 99(2):498–509. doi:10.1210/jc.2013-2574
116. Vitale G, Dicitore A, Pepe D, Gentilini D, Grassi ES, Borghi MO, et al. Synergistic activity of everolimus and 5-aza-2′-deoxycytidine in medullary thyroid carcinoma cell lines. Mol Oncol (2017). doi:10.1002/1878-0261.12070
117. Funakoshi T, Latif A, Galsky MD. Safety and efficacy of addition of VEGFR and EGFR-family oral small-molecule tyrosine kinase inhibitors to cytotoxic chemotherapy in solid cancers: a systematic review and meta-analysis of randomized controlled trials. Cancer Treat Rev (2014) 40(5):636–47. doi:10.1016/j.ctrv.2014.02.004
118. Sivendran S, Liu Z, Portas LJ, Yu M, Hahn N, Sonpavde G, et al. Treatment-related mortality with vascular endothelial growth factor receptor tyrosine kinase inhibitor therapy in patients with advanced solid tumors: a meta-analysis. Cancer Treat Rev (2012) 38(7):919–25. doi:10.1016/j.ctrv.2012.05.001
119. Schutz FA, Je Y, Richards CJ, Choueiri TK. Meta-analysis of randomized controlled trials for the incidence and risk of treatment-related mortality in patients with cancer treated with vascular endothelial growth factor tyrosine kinase inhibitors. J Clin Oncol (2012) 30(8):871–7. doi:10.1200/JCO.2011.37.1195
120. Shah R. Cardiovascular safety of protein kinase inhibitors: putting their “QT-phobia” in perspective. ADMET DMPK (2016) 4(3):212–31. doi:10.5599/admet.4.3.295
121. Abdel-Rahman O, Fouad M. Risk of cardiovascular toxicities in patients with solid tumors treated with sunitinib, axitinib, cediranib or regorafenib: an updated systematic review and comparative meta-analysis. Crit Rev Oncol Hematol (2014) 92(3):194–207. doi:10.1016/j.critrevonc.2014.06.003
122. Maitland ML, Bakris GL, Black HR, Chen HX, Durand JB, Elliott WJ, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst (2010) 102(9):596–604. doi:10.1093/jnci/djq091
123. Orphanos GS, Ioannidis GN, Ardavanis AG. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol (2009) 48(7):964–70. doi:10.1080/02841860903229124
124. Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation (2008) 118(1):84–95. doi:10.1161/CIRCULATIONAHA.108.776831
125. Qi WX, Shen Z, Tang LN, Yao Y. Congestive heart failure risk in cancer patients treated with VEGFR-TKIs: a systematic review and meta-analysis of 36 clinical trials. Br J Clin Pharmacol (2014) 78(4):748–62. doi:10.1111/bcp.12387
126. Yang B, Papoian T. Tyrosine kinase inhibitor (TKI)-induced cardiotoxicity: approaches to narrow the gaps between preclinical safety evaluation and clinical outcome. J Appl Toxicol (2012) 32(12):945–51. doi:10.1002/jat.2813
127. Ewer MS, Vooletich MT, Durand JB, Woods ML, Davis JR, Valero V, et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J Clin Oncol (2005) 23(31):7820–6. doi:10.1200/JCO.2005.13.300
128. Lenihan DJ, Kowey PR. Overview and management of cardiac adverse events associated with tyrosine kinase inhibitors. Oncologist (2013) 18(8):900–8. doi:10.1634/theoncologist.2012-0466
129. Ponatinib (Iclusig). Ponatinib (Iclusig) National Drug Monograph October 2015 VA Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives REMS RR. Available from: https://www.pbm.va.gov/PBM/clinicalguidance/drugmonographs/Ponatinib_Monograph.pdf
130. Teo YL, Ho HK, Chan A. Risk of tyrosine kinase inhibitors-induced hepatotoxicity in cancer patients: a meta-analysis. Cancer Treat Rev (2013) 39(2):199–206. doi:10.1016/j.ctrv.2012.09.004
131. Carhill AA, Cabanillas ME, Jimenez C, Waguespack SG, Habra MA, Hu M, et al. The noninvestigational use of tyrosine kinase inhibitors in thyroid cancer: establishing a standard for patient safety and monitoring. J Clin Endocrinol Metab (2012) 98(1):31–42. doi:10.1210/jc.2012-2909
132. van Leeuwen RW, van Gelder T, Mathijssen RH, Jansman FG. Drug–drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol (2014) 15(8):e315–26. doi:10.1016/S1470-2045(13)70579-5
133. Liu Y, Ramírez J, Ratain MJ. Inhibition of paracetamol glucuronidation by tyrosine kinase inhibitors. Br J Clin Pharmacol (2011) 71(6):917–20. doi:10.1111/j.1365-2125.2011.03911.x
134. Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer (2007) 96(12):1788–95. doi:10.1038/sj.bjc.6603813
135. Je Y, Schutz FA, Choueiri TK. Risk of bleeding with vascular endothelial growth factor receptor tyrosine-kinase inhibitors sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. Lancet Oncol (2009) 10(10):967–74. doi:10.1016/S1470-2045(09)70222-0
136. Segaert S, Tabernero J, Chosidow O, Dirschka T, Elsner J, Mancini L, et al. The management of skin reactions in cancer patients receiving epidermal growth factor receptor targeted therapies. J Dtsch Dermatol Ges (2005) 3(8):599–606. doi:10.1111/j.1610-0387.2005.05058.x
137. Cabanillas ME, Hu MI, Durand JB, Busaidy NL. Challenges associated with tyrosine kinase inhibitor therapy for metastatic thyroid cancer. J Thyroid Res (2011) 4:2011. doi:10.4061/2011/985780
138. Torino F, Corsello SM, Longo R, Barnabei A, Gasparini G. Hypothyroidism related to tyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat Rev Clin Oncol (2009) 6(4):219–28. doi:10.1038/nrclinonc.2009.4
139. Petrelli F, Borgonovo K, Cabiddu M, Lonati V, Barni S. Relationship between skin rash and outcome in non-small-cell lung cancer patients treated with anti-EGFR tyrosine kinase inhibitors: a literature-based meta-analysis of 24 trials. Lung Cancer (2012) 78(1):8–15. doi:10.1016/j.lungcan.2012.06.009
140. Tiedje V, Ting S, Walter RF, Herold T, Worm K, Badziong J, et al. Prognostic markers and response to vandetanib therapy in sporadic medullary thyroid cancer patients. Eur J Endocrinol (2016) 175(3):173–80. doi:10.1530/EJE-16-0252
141. De Souza JA, Busaidy N, Zimrin A, Seiwert TY, Villaflor VM, Poluru KB, et al. Phase II trial of sunitinib in medullary thyroid cancer (MTC). J Clin Oncol (2010) 28(15_suppl):5504. doi:10.1200/jco.2010.28.15_suppl.5504
142. Verbeek HH, Alves MM, de Groot JW, Osinga J, Plukker JT, Links TP, et al. The effects of four different tyrosine kinase inhibitors on medullary and papillary thyroid cancer cells. J Clin Endocrinol Metab (2011) 96(6):E991–5. doi:10.1210/jc.2010-2381
143. Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene (2004) 23(36):6056–63. doi:10.1038/sj.onc.1207810
144. Carlomagno F, Anaganti S, Guida T, Salvatore G, Troncone G, Wilhelm SM, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst (2006) 98:326–34. doi:10.1093/jnci/djj069
145. Morgillo F, Martinelli E, Troiani T, Orditura M, De Vita F, Ciardiello F. Antitumor activity of sorafenib in human cancer cell lines with acquired resistance to EGFR and VEGFR tyrosine kinase inhibitors. PLoS One (2011) 6:e28841. doi:10.1371/journal.pone.0028841
146. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature (2012) 483(7387):100. doi:10.1038/nature10868
147. Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev (2004) 3:1001–10. doi:10.1038/nrd1579
148. Yoshida T, Zhang G, Haura EB. Targeting epidermal growth factor receptor: central signaling kinase in lung cancer. Biochem Pharmacol (2010) 80:613–23. doi:10.1016/j.bcp.2010.05.014
149. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A (2008) 105:2070–5. doi:10.1073/pnas.0709662105
150. Cepero V, Sierra JR, Corso S, Ghiso E, Casorzo L, Perera T, et al. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res (2010) 70(19):7580–90. doi:10.1158/0008-5472.CAN-10-0436
151. Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1 α-deficient colon cancer cells. Nat Med (2005) 11:992–7. doi:10.1038/nm1294
152. Fernando NT, Koch M, Rothrock C, Gollogly LK, D’Amore PA, Ryeom S, et al. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin Cancer Res (2008) 14:1529–39. doi:10.1158/1078-0432.CCR-07-4126
153. Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal (2013) 6(294):re6. doi:10.1126/scisignal.2004652
154. Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia (2000) 2(4):306–14. doi:10.1038/sj.neo.7900102
155. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8(8):592. doi:10.1038/nrc2442
156. Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res (2014) 74:2377–84. doi:10.1158/0008-5472.CAN-13-2971
157. Rosfjord E, Lucas J, Li G, Gerber HP. Advances in patient-derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem Pharmacol (2014) 91:135–43. doi:10.1016/j.bcp.2014.06.008
158. Werner RA, Schmid JS, Muegge DO, Lückerath K, Higuchi T, Hänscheid H, et al. Prognostic value of serum tumor markers in medullary thyroid cancer patients undergoing vandetanib treatment. Medicine (2015) 94(45):e2016. doi:10.1097/MD.0000000000002016
159. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther (2011) 10(12):2298–308. doi:10.1158/1535-7163.MCT-11-0264
160. Rey S, Schito L, Wouters BG, Eliasof S, Kerbel RS. Targeting hypoxia-inducible factors for antiangiogenic cancer therapy. Trends Cancer (2017) 3(7):529–41. doi:10.1016/j.trecan.2017.05.002
161. Abdel-Aziz AK, Shouman S, El-Demerdash E, Elgendy M, Abdel-Naim AB. Chloroquine as a promising adjuvant chemotherapy together with sunitinib. Sci Proc (2014) 1:1. doi:10.14800/sp.384
162. Husain M, Alsever RN, Lock JP, George WF, Katz FH. Failure of medullary carcinoma of the thyroid to respond to doxorubicin therapy. Horm Res (1978) 9(1):22–5. doi:10.1159/000178893
163. Orlandi F, Caraci P, Berruti A, Puligheddu B, Pivano G, Dogliotti L, et al. Chemotherapy with dacarbazine and 5-fluorouracil in advanced medullary thyroid cancer. Ann Oncol (1994) 5(8):763–5. doi:10.1093/oxfordjournals.annonc.a058984
164. Nocera M, Baudin E, Pellegriti G, Cailleux AF, Mechelany-Corone C, Schlumberger M, et al. Treatment of advanced medullary thyroid cancer with an alternating combination of doxorubicin-streptozocin and 5 FU-dacarbazine. Br J Cancer (2000) 83(6):715. doi:10.1054/bjoc.2000.1314
165. Paiva CE, Michelin OC. Use of capecitabine in refractory metastatic medullary thyroid carcinoma. Thyroid (2008) 18(5):587. doi:10.1089/thy.2007.0220
166. Reubi JC, Schaer JC, Waser B, Mengod G. Expression and localization of somatostatin receptor SSTR1, SSTR2, and SSTR3 messenger RNAs in primary human tumors using in situ hybridization. Cancer Res (1994) 54:3455–9.
167. Reubi JC, Horisberger U, Laissue J. High density of somatostatin receptors in veins surrounding human cancer tissue: role in tumor-host interaction? Int J Cancer (1994) 56:681–8. doi:10.1002/ijc.2910560513
168. Waldherr C, Schumacher T, Pless M, Crazzholara A, Maecke H, Nitzche EU, et al. Pilot study of radionuclide in MTC radiopeptide transmitted internal irradiation of non-iodophil thyroid cancer and conventionally untreatable medullary thyroid cancer using [90Y]-DOTA-D-Phe1Tyr3-octreotide: a pilot study. Nucl Med Commun (2001) 22(6):673–8. doi:10.1097/00006231-200106000-00011
169. Vaisman F, de Castro PH, Lopes FP, Kendler DB, Pessoa CH, Bulzico DA, et al. Is there a role for peptide receptor radionuclide therapy in medullary thyroid cancer? Clin Nucl Med (2015) 40(2):123–7. doi:10.1097/RLU.0000000000000628
170. Iten F, Müller B, Schindler C, Rochlitz C, Oertli D, Mäcke HR, et al. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: a phase II clinical trial. Clin Cancer Res (2007) 13(22):6696–702. doi:10.1158/1078-0432.CCR-07-0935
171. Salavati A, Puranik A, Kulkarni HR, Budiawan H, Baum RP. Peptide Receptor Radionuclide Therapy (PRRT) of medullary and nonmedullary thyroid cancer using radiolabeled somatostatin analogues. Semin Nucl Med (2016) 46(3):215–24. doi:10.1053/j.semnuclmed.2016.01.010
172. Khan S, Krenning EP, van Essen M, Kam BL, Teunissen JJ, Kwekkeboom DJ. Quality of life in 265 patients with gastroenteropancreatic or bronchial neuroendocrine tumors treated with [177Lu-DOTA0,Tyr3]octreotate. J Nucl Med (2011) 52:1361–8. doi:10.2967/jnumed.111.087932
173. Limouris GS, Chatziioannou A, Kontogeorgakos D, Mourikis D, Lyra M, Dimitriou P, et al. Selective hepatic arterial infusion of In-111-DTPA-Phe1-octreotide in neuroendocrine liver metastases. Eur J Nucl Med Mol Imaging (2008) 35:1827–37. doi:10.1007/s00259-008-0779-0
174. Kong G, Johnston V, Ramdave S, Lau E, Rischin D, Hicks RJ. High-administered activity In-111 octreotide therapy with concomitant radiosensitizing 5FU chemotherapy for treatment of neuroendocrine tumors: preliminary experience. Cancer Biother Radiopharm (2009) 24:527–33. doi:10.1089/cbr.2009.0644
175. Claringbold PG, Brayshaw PA, Price RA, Turner JH. Phase II study of radiopeptide 177Lu-octreotate and capecitabine therapy of progressive disseminated neuroendocrine tumours. Eur J Nucl Med Mol Imaging (2011) 38:302–11. doi:10.1007/s00259-010-1631-x
176. Van Der Zwan WA, Bodei L, Mueller-Brand J, De Herder WW, Kvols LK, Kwekkeboom DJ. GEP–NETs UPDATE: radionuclide therapy in neuroendocrine tumors. Eur J Endocrinol (2015) 172(1):R1–8. doi:10.1530/EJE-14-0488
Keywords: medullary thyroid cancer, targeted therapy thyroid cancer, vandetanib medullary thyroid, cabozantinib medullary thyroid, targeted radionuclide medullary thyroid, peptide receptor radionuclide therapy medullary thyroid, thyroid cancer recurrence, small molecule thyroid
Citation: Priya SR, Dravid CS, Digumarti R and Dandekar M (2017) Targeted Therapy for Medullary Thyroid Cancer: A Review. Front. Oncol. 7:238. doi: 10.3389/fonc.2017.00238
Received: 16 July 2017; Accepted: 19 September 2017;
Published: 06 October 2017
Edited by:
Vincent Vander Poorten, KU Leuven, BelgiumReviewed by:
Massimo Bongiovanni, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandJeffrey Knauf, Memorial Sloan-Kettering Cancer Center, United States
Copyright: © 2017 Priya, Dravid, Digumarti and Dandekar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitali Dandekar, bWl0YWxpZGFuZGVrYXJAZ21haWwuY29t