 Rui Lu1,2
Rui Lu1,2 Gang Greg Wang1,2*
Gang Greg Wang1,2*
- 1Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC, United States
- 2Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Acute myeloid leukemia (AML), a common hematological cancer of myeloid lineage cells, generally exhibits poor prognosis in the clinic and demands new treatment options. Recently, direct sequencing of samples from human AMLs and pre-leukemic diseases has unveiled their mutational landscapes and significantly advanced the molecular understanding of AML pathogenesis. The newly identified recurrent mutations frequently “hit” genes encoding epigenetic modulators, a wide range of chromatin-modifying enzymes and regulatory factors involved in gene expression regulation, supporting aberration of chromatin structure and epigenetic modification as a main oncogenic mechanism and cancer-initiating event. Increasing body of evidence demonstrates that chromatin modification aberrations underlying the formation of blood cancer can be reversed by pharmacological targeting of the responsible epigenetic modulators, thus providing new mechanism-based treatment strategies. Here, we summarize recent advances in development of small-molecule inhibitors specific to chromatin factors and their potential applications in the treatment of genetically defined AMLs. These compounds selectively inhibit various subclasses of “epigenetic writers” (such as histone methyltransferases MLL/KMT2A, G9A/KMT1C, EZH2/KMT6A, DOT1L/KMT4, and PRMT1), “epigenetic readers” (such as BRD4 and plant homeodomain finger proteins), and “epigenetic erasers” (such as histone demethylases LSD1/KDM1A and JMJD2C/KDM4C). We also discuss about the molecular mechanisms underpinning therapeutic effect of these epigenetic compounds in AML and favor their potential usage for combinational therapy and treatment of pre-leukemia diseases.
Introduction
Epigenetic modifications, including DNA methylation and a myriad of post-translational modifications of the DNA-packaging histone proteins, represent a fundamental means for regulating gene expression and other DNA-templated processes (1–4). These modifications of DNA or histones are increasingly appreciated to be dynamically regulated by epigenetic modulators, a broad class of proteins that consist of “epigenetic writer” enzymes catalyzing chromatin modification, “epigenetic eraser” enzymes removing the modification, “epigenetic readers” or “effectors” recognizing the modification to elicit biological consequences, and various other cellular regulators that indirectly influence the level or readout of epigenetic modification (2, 5). While the dynamic regulation of epigenetic modification enables cells to adapt and function differently in response to developmental and environmental cues, their mis-regulation often perturbs gene expression and cellular function leading to pathogenesis of human disease such as cancer. Indeed, recent deep sequencing of human cancer patient samples has identified novel recurrent mutations in genes encoding a wide range of epigenetic modulators and even histones themselves (6–9).
Acute myeloid leukemia (AML), a common malignancy of myeloid-lineage precursor cells in the blood, is characterized by two hallmarks, uncontrolled cell proliferation and impaired differentiation. Previously, progression and characteristics of AML were linked to several key pathways (10, 11), including inactivation of tumor suppressors [such as TP53 and Wilm’s Tumor-1 (WT1)], gain-of-function mutation of oncogenic kinases (such as FLT3, NRAS, and KRAS), and stem cell transcription factors (TFs) [such as rearrangement and/or overexpression of HOX cluster genes and their cofactors such as MEIS1 (12–14)], as well as inactivating mutation of differentiation-promoting TFs (such as PU.1 and CEBP/α). Recently, deep sequencing of samples from human patients with AML and pre-leukemia diseases such as myelodysplastic syndrome (MDS) and clonal hematopoiesis of indeterminate potential (CHIP) additionally revealed frequent somatic mutations of genes involved in epigenetic modulation or RNA splicing (11, 15–26). Among the various affected epigenetic pathway genes include the DNA (cytosine-5)-methyltransferase 3 A (DNMT3A, a DNA methylation “writer”), Tet Methylcytosine dioxygenase 2 (TET2, a DNA methylation “eraser” or demethylase), Enhancer of zeste homolog 2 [EZH2/KMT6A, a “writer” mediating methylation of histone H3, Lys27 (H3K27)], Additional Sex Combs Like 1 and 2 (ASXL1 and ASXL2, an EZH2-associated cofactor family), the Cohesin complex (SMC3-SMC1-RAD21-STAG) genes, and Isocitrate Dehydrogenase 1 and 2 (IDH1 and IDH2). These newly identified somatic mutations of DNA/chromatin modifiers and structural organizers are in agreement with previous karyotyping/FISH-based analyses of AML patients, which already identified recurrent chromosomal translocation or abnormality of genes encoding various members of epigenetic “writers” (MLL/KMT2A, NSD1/KMT3B, NSD3/WHSC1L1/KMT3F) (27–31), “erasers” (JARID1A/KDM5A) (32, 33), and “readers” (PHF23) (32, 34). Importantly, mutations of DNMT3A, TET2, IDH1/2, or ASXL1 were frequently detected among apparently healthy individuals with clonal hematopoiesis or CHIP (22, 24, 35, 36) and in AML patients who received complete disease remission after chemotherapy (26, 35, 37–39), supporting the pivotal roles of epigenetic deregulation in initiation, clonal evolution and relapse of AMLs.
In contrast to significant advances in molecular appreciation of human AML’s mutational landscape and putative “driving” pathways, chemotherapy remains as the frontline treatment for most AML patients, with an exception of all-trans retinoic acid (ATRA) used as targeted therapy of the acute promyelocytic leukemia (APL) subtype. AML patients still suffer from low overall survival and a high rate of recurrence, demanding new treatments to be developed. Recent studies of AML and other tumors have increasingly shown that genetic lesion of epigenetic modulator often induces a subsequent chain reaction leading to aberrations in chromatin modification/remodeling, gene-expression program, and cellular states during tumorigenesis (2, 5, 29, 40–43). Thus, pharmacologic targeting of epigenetic players responsible for the above chromatin/gene mis-regulation shall represent new mechanism-based strategies for therapeutic intervention. This review aims to summarize recent advances in specific inhibition of histone-modifying enzymes and regulatory proteins as potential AML therapeutics, with the already discovered inhibitors sub-grouped into the categories targeting either the “writing,” “reading,” or “erasing” function of epigenetic modulators (Table 1).
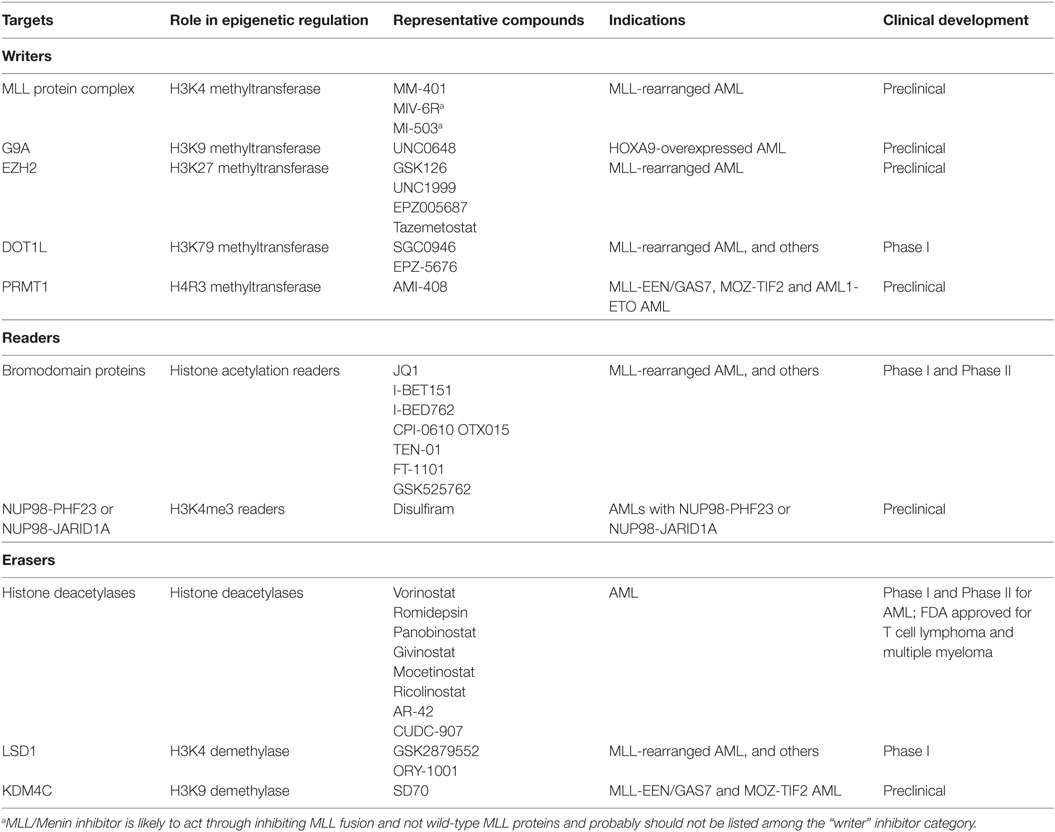
Table 1. Epigenetic therapies in acute myeloid leukemia (AML): targets, compounds, and clinical development.
Targeting Chromatin “Writers”
MLL Inhibitors (MLLi)
The Mixed-Lineage Leukemia gene (MLL/MLL1/KMT2A) encodes one of the KMT2 family of methyltransferase enzymes that contain multiple structural domains, including a C-terminal SET domain catalyzing methylation of histone H3, Lys4 (H3K4) (44–46). MLL rearrangement and translocation, which typically affect one allele, are responsible for about 70% of infant leukemias and 5–10% of childhood and adult AML cases (28, 29). Often, the leukemia-associated MLL gene rearrangement produces the MLL fusion oncoprotein that loses MLL’s C-terminal SET domain and gains a partial sequence from its fusion partner such as AF4, AF9, AF10, or ENL, which recruits the DOT1L-associated transcription elongation complexes. MLL fusion oncoproteins still retain MLL’s N-terminal domains, which mediate chromatin association and interaction with functional cofactors such as Menin. Previously, the remaining wild-type MLL allele in cancer cells was shown to be critical for leukemogenesis induced by MLL fusion (47); however, a recent study reported that MLL2/KMT2B, another trithorax family methyltransferase that is most closely related to MLL/KMT2A (48), sustains growth of MLL-rearranged leukemia and represents a more relevant drug target (49). While the transcription elongation activity acquired by MLL fusion remains as an attractive targeting strategy (see the section of DOT1Li), these studies have justified development of MLL1/2 inhibitors (MLLi) for the treatment of MLL-rearranged leukemias.
Using the structure-guided design, Cao et al. developed an MLLi termed MM-401 (Figure 1A, left and Table 1) to disrupt direct interaction of MLL1 with WDR5, a cofactor associated with the SET domain of MLL/KMT2 enzymes, and thus inhibit MLL1’s methylase or “writer” function (50). In vitro biochemical assays showed that MM-401 specifically targets WDR5 interaction to MLL1, and not other MLL/KMT2 family enzymes. Treatment with MM-401 blocked proliferation and induced myeloid differentiation of MLL-rearranged leukemia cells while not significantly affecting normal blood stem/progenitor cells (50). A recent study reported that MLL2 represents a more relevant therapeutic target in a range of MLL-rearranged leukemia models and that MLL2 and MLL1 collaborate to maintain oncogenesis via regulating distinctive gene-expression pathways (49). Therefore, dual inhibitors of MLL2 and MLL1 or a specific one against MLL2 need to be developed and may provide a more effective treatment strategy.
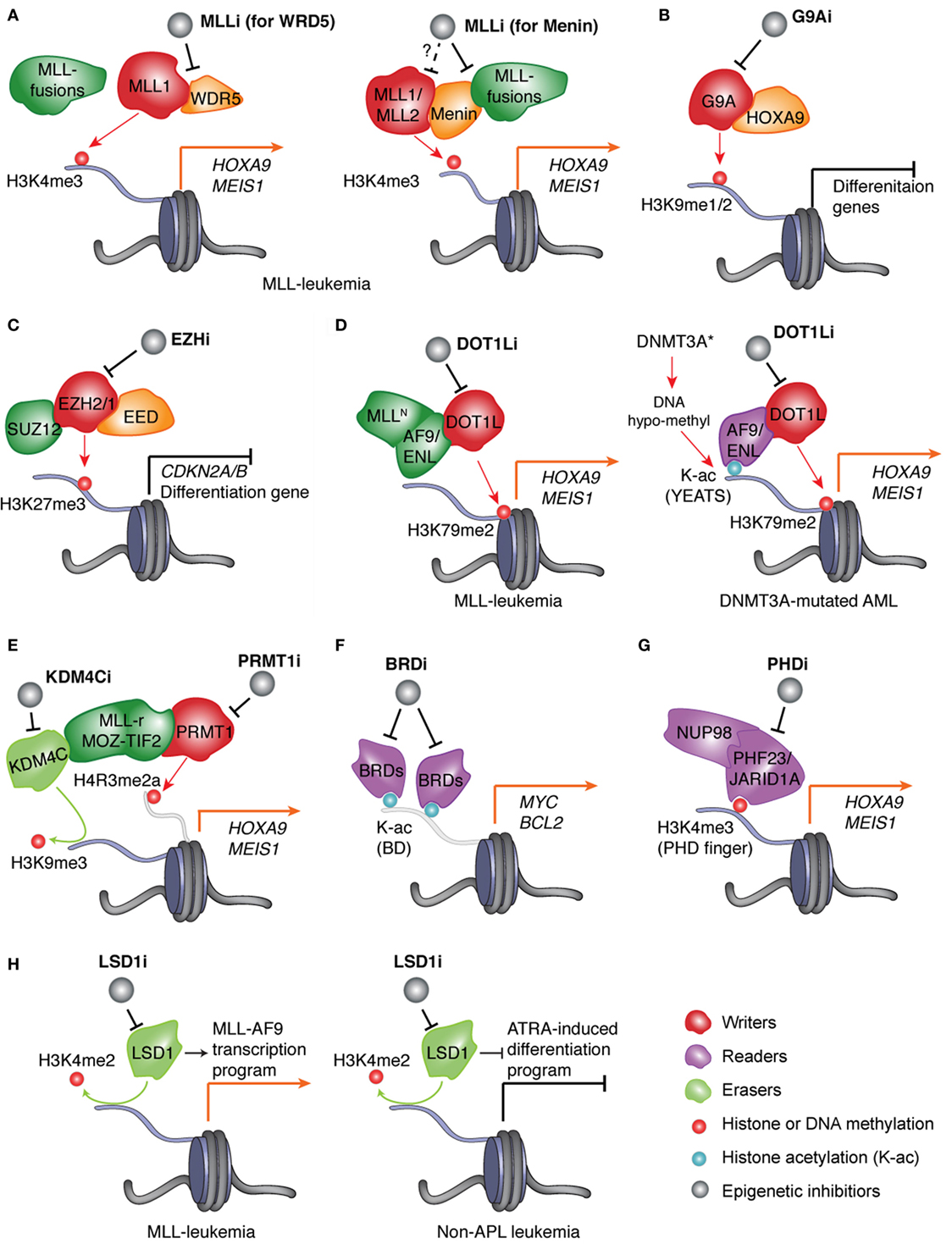
Figure 1. Pharmacological inhibition of the epigenetic “writers,” “readers,” or “erasers” responsible for deregulation of chromatin modification and gene expression in AMLs. (A) In leukemias with MLL rearrangement (MLL-r), protein complexes assembled by the wild-type MLL and aberrant MLL fusion proteins induce H3K4me3 and H3K79me2, respectively, to cooperatively mediate activation of MLL targets such as “stemness” genes HOXA9 and MEIS1. Inhibitor of MLL (MLLi) disrupts physical association of MLL (MLL1 or MLL2) and MLL-fusion to its interacting partner, either WDR5 (left) or Menin (right), thereby preventing target gene activation and AML development. (B) HOXA9, a transcription factor (TF) found overexpressed in ~50–70% of AML patients, promotes leukemogenesis partly through recruiting G9A, an H3K9me1/2-specific “writer” enzyme, to suppress gene-expression programs crucial for myeloid differentiation. Inhibitor of G9A (G9Ai) targets this differentiation-arrest mechanism in AMLs with HOXA9 overexpression. (C) In AMLs, treatment with inhibitor of EZH2 and/or EZH1 (EZHi) results in suppression of H3K27me3 and de-repression of polycomb repressive complex 2 (PRC2) target genes, which include tumor suppressor genes (such as CDKN2A/B) and myeloid differentiation-associated genes. (D) Left panel: in MLL-rearranged leukemias, MLL fusion partners such as AF9 and ENL recruit DOT1L, an H3K79me2-specific “writer” enzyme, to maintain high expression of target genes such as MEIS1 and HOXA9. Right panel: in normal-karyotype AMLs with DNMT3A mutation, focal decrease of DNA methylation (i.e., hypo-methylation) results in increase of histone acetylation (K-ac) and binding of the YEAST domain-containing K-ac “reader” proteins AF9 and ENL, which subsequently recruit DOT1L to promote H3K79me2 and transcriptional activation/elongation of “stemness” genes. In both genetically defined AML subtypes, inhibitor of DOT1L (DOT1Li) blocks the above oncogenic program and leukemia progression. (E) In leukemias with aberrant fusion of MLL or MOZ-TIF2, PRMT1, an H4R3-specific methyltransferase/“writer,” and KDM4C, an H3K9-specific demethylase/“eraser,” are recruited by leukemic fusion oncoproteins to modulate histone methylation and promote target gene activation. Blockage of PRMT1 or KDM4C provides a new treatment strategy. (F) In AMLs, inhibitor of bromodomain (BRD)-containing K-ac “readers” (BRDi) selectively blocks interaction of bromodomain proteins (BRD4 and related BRD2/3) with K-ac and represses expression of vital oncogenes such as MYC and BCL2, thus suppressing leukemic growth. (G) In AML patients, aberrant rearrangement of the gene encoding the H3K4me3-“reading” proteins JARID1A/KDM5A and PHF23 produces the leukemogenic fusion protein NUP98-JARID1A and NUP98-PHF23, respectively, which rely on their H3K4me3-“reading” plant homeodomain (PHD) finger domains to maintain high expression of AML-associated genes. Inhibitor of PHD fingers (PHDi) shall provide an attractive therapeutic method for these AML patients. (H) Left: in MLL-rearranged leukemia, inhibitor of LSD1 (LSD1i) downregulates MLL target genes and inhibits leukemia development. Right: in non-acute promyelocytic leukemia (APL) leukemia, LSD1i promotes all-trans retinoic acid (ATRA)-induced cell differentiation thereby suppressing leukemogenesis.
Menin, a cofactor associated with the N-terminal region of both MLL fusion and wild-type MLL1/2 proteins, is required for MLL- and MLL fusion-mediated target gene activation and for leukemic transformation caused by MLL rearrangement (51–55). Menin is required for association and/or recruitment of MLL and MLL fusion proteins to their gene targets and represents a validated drug target of MLL-rearranged leukemia. Recently, through high-throughput screening and structure-based development, a series of MLLi, including MIV-6R (56), MI-463, and MI-503 (57), were discovered and optimized to disrupt MLL–Menin interaction, with some achieving in vitro inhibition in the nanomole range (Figure 1A, right; Table 1). These MLL–Menin inhibitors efficiently suppressed growth of MLL-rearranged leukemia cells in vitro/vivo and did not affect that of non-MLL-rearranged leukemias. Treatment with these MLLi led to down-regulation of gene-expression programs enforced by MLL fusion, such as HOXA9 and MEIS1, in the leukemia cells. The effect of MLL–Menin inhibitors on steady-state normal hematopoiesis appears to be small (57), suggesting that their anti-leukemia effect is mainly through inhibiting Menin interaction to MLL fusion and not wild-type MLL1 proteins. For this reason, MLL–Menin inhibitors should not be categorized as the “writer” inhibitor. However, it is worthy noting that, besides MLL1/KMT2A, Menin also interacts with MLL2/KMT2B through conserved interfaces (46, 51, 53). It remains to be determined whether the above MLLi also targets MLL2, a recently validated oncoprotein that sustains MLL-rearranged leukemias (49). For convenience, we decide to list the MLL–Menin inhibitors as MLLi and “writer” inhibitors (Table 1).
G9A Inhibitors (G9Ai)
Euchromatic histone lysine methyltransferase 2 (EHMT2, also known as G9A/KMT1C) encodes a methyltransferase that catalyzes mono/di-methylation of histone H3, Lys9 (H3K9me1/2), a histone modification correlated with gene silencing. Knockout of G9A in hematopoietic systems led to decreased proliferation of myeloid progenitors without affecting the function of long-term repopulating hematopoietic stem cells (58). In mouse AMLs induced by HOXA9, a homeodomain TF gene found over-expressed in about 50–70% of human AMLs, loss of G9A suppressed leukemogenesis. Mechanistically, G9A physically interacts with HOXA9. Inhibition of G9A led to de-repression of HOXA9 target genes (58). UNC0638 (59), a recently developed G9Ai, demonstrated similar AML therapeutic effect (Figure 1B; Table 1). While no method is currently available for directly targeting HOXA9 oncoprotein, the above studies provide an alternative strategy.
EZH Inhibitors (EZHi)
EZH2/KMT6A serves as the catalytic subunit of the polycomb repressive complex 2 (PRC2) mediating transcriptional repression through tri-methylation of H3K27 (H3K27me3) (60). EZH1, an EZH2-related methylase, can partially compensate EZH2’s functions on a subset of gene targets when assembled in a separate complex with the same set of PRC2 components such as SUZ12 and EED (60, 61). Genomic deletion and loss-of-function mutations of EZH2/KMT6A were frequently found in MDS and other myeloid malignancies (62), whereas its gain-of-function mutations occur in 10–20% of B-cell lymphoma patients (63–65). Such EZH2/KMT6A somatic mutation is rare among AMLs (66). Recent investigation of animal blood cancer models, however, has shown that complete loss of EZH2 promotes MDS development but prevents AML transformation (67). MDS induced by EZH2 loss requires EZH1 for disease progression (68), indicating a context-dependent role of these PRC2 enzymatic complexes in development of MDS and blood malignancy. Furthermore, several studies demonstrated that the MLL-rearranged leukemias require functionality of EZH2 and/or EZH1 to maintain leukemogenecity (69–74). Mechanistically, these PRC2 enzymes suppress genes related to tumor suppression (such as Cdkn2a/b) and cell differentiation (such as Egr1) through inducing gene-repressive H3K27me3/2 (Figure 1C). Additionally, PRC2 was found to promote expression of MYC-associated gene signatures probably via an indirect mechanism. Furthermore, about 5–10% of AML patients carry the inactivating mutation of the WT1 gene, which was shown to induce a DNA hyper-methylation phenotype through interfering with WT1-mediated recruitment of TET DNA demethylases (75, 76). The induced DNA hyper-methylation sites were found enriched in myeloid differentiation genes and PRC2 targets, and EZH2 is highly expressed in WT1-mutated AMLs to maintain repression of genes with DNA hyper-methylation, leading to cell differentiation block (77). Importantly, in cellular and murine models of MLL-rearranged (69, 70, 72) or WT1-mutated AMLs (77), knockdown or knockout of PRC2 inhibited cell proliferation and restored gene-expression programs involved in myeloid differentiation. These studies unveiled the oncogenic function of PRC2 and EZH2 in these genetically defined AMLs, supporting PRC2 as a drug target of AML.
Due to frequent overexpression and gain-of-function mutation of EZH2 in solid cancer and lymphoma, several pharmaceutical companies have embarked on high-throughput screening campaigns leading to discovery of a series of small-molecule compounds (Table 1) that compete binding of S-adenosyl-methionine (SAM), the methyl donor of PRC2, thereby suppressing PRC2’s methyltransferase activity (78–82). These EZHi compounds demonstrate high selectivity and high potency toward EZH2 and/or EZH1. In MLL-rearranged AML models, dual inhibition of EZH2 and EZH1 by an EZHi, UNC1999, derepressed PRC2 target genes and significantly suppressed AML malignant growth in vitro and in vivo (74) (Figure 1C). Treatment of WT1-mutated AML cells with GSK126 (79), an EZH2-selective inhibitor, had similar anti-cancer effect (77). Currently, several EZHis show drug-like properties and are used in clinical trials of lymphoma treatment. Their potential therapeutic effect in AMLs remains to be determined in clinical settings.
DOT1L Inhibitors (DOT1Li)
Disruptor of telomeric silencing 1-like (DOT1L/KMT4) is a histone H3 Lys79 (H3K79)-specific methyltransferase that regulates gene transcriptional elongation, telomeric silencing, and DNA damage response (83). Biochemical interaction studies found that DOT1L interacts with transcriptional elongation factors including AF4, AF9, AF10, and ENL, which are also common fusion partners of MLL-rearrangement in AMLs (29, 84–86). DOT1L loss-of-function studies in MLL-rearranged leukemias support its crucial role in leukemogeneicity, possibly through maintaining expression of target transcripts of MLL-fusion such as HOX cluster genes and MEIS1 (84, 87–91).
Structure-based design has led to development of several DOT1Li (Table 1) that specifically targets the SAM-binding pocket of DOT1L enzymes (92, 93). Consistent with DOT1L loss-of-function studies, these DOT1Li also selectively inhibited expression of MLL-fusion target genes such as HOXA9 and MEIS1 and selectively killed MLL-rearranged leukemia cells and xenografted tumors (90–92, 94). Furthermore, recent investigation supports that DOT1L can potentially serve as a therapeutic target of other genetically defined AMLs, which include the subtype with translocation of NUP98-NSD1 (95), somatic mutation of DNMT3A (96, 97), NPM1 (98) or IDH1/2 (99), or overexpression of MN1 (100). While NUP98-NSD1 induced leukemic transformation through direct targeting and epigenetic modulation of AML-promoting “stemness” genes (HOX gene clusters and MEIS1) (30), a DNMT3A hotspot mutation (DNMT3AR882H) was recently found to focally suppress DNA methylation at cis-regulatory elements of these genes thereby promoting their transcription activation (96). In addition, aberrant over-expression of HOX cluster genes is a hallmark of AMLs that harbor NPM1 mutation (98), and overexpression of MN1 was found to induce an aggressive myeloid leukemia that strictly relies on the same “stemness” genes-expression program in the leukemia-initiating cells (100). Leukemia cells from the above AML subtypes were found generally sensitive to DOT1Li, and DOT1Li treatments repressed “stemness” gene-expression programs, supporting a broader role of DOT1L and “stemness” TF nodes in AML biology (Figure 1D). EPZ-5676 (94) represents the first DOT1Li used for clinical trials for MLL-rearranged leukemia; however, drug-like properties of the disclosed DOT1Li such as half-life in vivo are generally poor and need to be improved.
PRMT1 Inhibitors (PRMT1i)
Protein arginine methyltransferase 1 (PRMT1) encodes a methyltransferase for histone H4 arginine-3 (H4R3) and associates with gene activation. PRMT1 was shown to interact with AML1-ETO, a gene fusion product defining AML with t(8;21) translocation, activate the downstream target genes of AML1-ETO, and promote progression of AML1-ETO-asssociated leukemia (101). Recent studies have additionally demonstrated specific requirement of PRMT1 in leukemogenesis induced by MLL-rearrangement (such as MLL-GAS7) or the MOZ-TIF2 translocation (102, 103). Similar to what was found in t(8;21) AMLs, PRMT1 physically associates with these leukemic fusion oncoproteins and is required for high expression of their target genes such as HOX and MEIS1, supporting targeting PRMT1 as new AML therapeutics. Indeed, in various leukemia cell lines and animal models with MLL fusion or MOZ-TIF2, AMI-408 (104), a PRMT1i, suppressed AML growth (103) (Figure 1E; Table 1). These works have established a foundation for further validation of PRMT1i’s therapeutic effect in clinical settings.
Targeting Chromatin “Readers”
Epigenetic or chromatin “readers” are a subclass of factors that specifically recognize DNA or histone modification to induce subsequent events and elicit functional readout of the modification (1, 2, 105–107). Compared to a generally high druggability of chromatin-modifying “writer” or “eraser” enzymes, that of various epigenetic “reader” families varies (108). Despite challenges, targeting chromatin “reader” function is increasingly considered as promising partly due to recent success in discovery of bromodomain (BRD) protein inhibitors.
BRD Inhibitors (BRDi)
BRD-containing proteins BRD4 and related BRD2/3 recognize histone lysine acetylations subsequently recruiting pTEFb, a CDK9/Cyclin-T kinase complex, to activate RNA polymerase II and target gene expression (109). Originally, these BRD genes were found aberrantly rearranged in malignant NUT midline carcinomas. A pioneering functional genomics screening of chromatin regulators in MLL-rearranged leukemia unveiled a role for BRD4 in maintenance of C-MYC expression and leukemia oncogenicity (110). Since advent of JQ1, the first highly selective and highly potent BRDi (showing a nano-molar range inhibition of BRDs) that competes BRD4 off acetylated histone ligands (111), multiple BRDis have been developed and their therapeutic effect seen in a wide range of human diseases including AML and other cancers (109). Mechanistically, BRDi such as JQ1 and I-BET151 repressed expression of a number of key oncogenic nodes including C-MYC and BCL2 in mouse and human leukemia models carrying MLL-rearrangement (110, 112) (Figure 1F). BRDi was also found effective in treating non-MLL-rearranged AMLs such as those with NPM1 mutation (113) or deletion of chromosome 7 and 7q [−7/del(7q)] (114), supporting their broader application in AML therapeutics. Even more potent BRDi, including a degrader derivative that can both inhibit BRD’s “reading” function and induce its proteasome-mediated degradation (115), have been developed, with several currently under clinical evaluation for the treatment of refractory AMLs (109). Following these encouraging advances, inhibitors of other RNA Pol-II activators such as the CDK7 and CDK9 kinases are on the horizon becoming a strategy to target transcriptional addiction to vital oncogenes seen in cancer (116, 117).
Plant Homeodomain (PHD) Finger Inhibitors (PHDi)
The PHD finger-containing proteins comprise a large class of chromatin-associated proteins, some of which harbor the “reading” specificity toward H3K4 methylation (2, 106). In human AMLs, genes encoding the PHD finger-containing protein JARID1A (also known as KDM5A, a PHD finger-containing histone demethylase) and PHF23 were altered due to chromosomal abnormalities, resulting in in-frame fusion of their C-terminal H3K4me3-“reading” PHD finger to NUP98, a promiscuous gene translocation partner in human AMLs (32, 106). Despite generally low frequency of these genetic abnormalities in AMLs, the NUP98-JARID1A/KDM5A translocation was reported to be recurrent and detected in ~10% of the pediatric acute megakaryoblastic leukemia subtype (33). The produced NUP98-JARID1A or NUP98-PHF23 oncoproteins were highly potent in inducing AML transformation in vitro/vivo and rely on their H3K4me3-“reading” PHD finger domain to maintain high expression of “stemness” nodes, notably HOX and MEIS1 (32, 118). Disulfiram, a previously FDA-approved drug, was found to carry the ability to inhibit binding of these PHD fingers to H3K4me3 possibly through structural alteration (119) and to selectively kill the leukemic cells transformed by NUP98-PHF23 or NUP98-JARID1A/KDM5A (118) (Figure 1G). However, the potency and selectivity of disulfiram appear poor and the ligand-competitive inhibitors still remain to be developed for these PHD fusion oncoproteins.
Targeting Chromatin “Erasers”
HDAC Inhibitors (HDACi)
Histone deacetylases (HDACs) remove acetylation off histones to influent gene expression. HDACi (Table 1) including Vorinostat (also known as SAHA) and Panobinostat are the earliest inhibitors of epigenetic “erasers” approved by FDA for treatment of cutaneous T cell lymphoma and, recently, multiple myeloma. Currently, HDACi is under phase I/II trials of relapsed AML patients. As HDACs also deacetylate numerous non-histone substrates, effect of HDACi remains controversial as of the detailed mechanisms, especially through targeting histone versus non-histone proteins.
LSD1 Inhibitors (LSD1i)
Lysine-specific demethylase 1 (LSD1/KDM1A) is the first identified histone demethylase with specificity toward H3K4 mono/di-methylation (H3K4me1/2) (120). Several LSD1i have been developed. In the MLL-rearranged leukemias, terminal differentiation arrest was partially enforced by LSD1, and LSD1i treatment induced myeloid differentiation and suppressed leukemogenesis in vivo (Figure 1H) (121). Mechanistically, LSD1i may perturb the H3K4me3/H3K4me2 ratio at MLL target genes thus reducing their transcription (121). Also, therapeutic effect of LSD1i was reported in AMLs without PML-RARA (i.e., non-APL AML), where LSD1i sensitized the pro-differentiation effect of ATRA, an agent only for PML-RARA-positive APLs (Figure 1H) (122). Here, combinational treatment of non-APL human AMLs with ATRA and LSD1i showed a potent anti-leukemic effect, with increased H3K4me2 and expression found at the myeloid differentiation genes (122). Several LSD1i are now in clinical trials in refractory AMLs.
KDM4C Inhibitors
KDM4C (also known as JMJD2C/GASC1) encodes an “eraser” carrying the H3K9-demethylating and gene-activating activities. Like PRMT1, KDM4C was also found to interact with various AML oncoproteins including MLL-GAS7 and MOZ-TIF2 (103). Knockdown of KDM4C partially reversed target gene activation mediated by these AML fusion proteins. Moreover, pharmacological inhibition of KDM4C can be achieved by an inhibitor SD70 and proposed to be a potentially new AML treatment strategy (103) (Figure 1E).
Perspectives
In short, epigenetic modulators emerge rapidly as potential drug targets for the treatment of currently incurable AMLs. With many showing high selectivity, high potency and/or promising drug-like properties, the already developed epigenetic inhibitors shall provide potential alternatives or adjuvants to current therapeutic arsenal that frequently relies on non-specific cytotoxic agents. While the area is in its infancy, we wish to pinpoint several directions that may broaden application of epigenetic inhibitors.
Newly Validated Epigenetic Factors and Cancer Cell Dependency Pathways Remain to Be Targeted
An existing advance in understanding the biology of gene activation is recent identification of a YEATS family of protein domains as a new “reader” class of histone acylation such as acetylation and crotonylation (123, 124). In the MLL-rearranged AML cells, a YEATS domain harbored in ENL was recently shown to be crucial for tethering/stabilizing the MLL fusion proteins at sites with histone acetylation to induce downstream gene activation (125, 126). Similar mechanisms might be also functional among DNMT3A-mutated leukemias (Figure 1D, right) where DNMT3A mutations perturb efficient CpG methylation at cis-regulatory sites leading to elevated histone acetylation and increased binding of DOT1L-associated complexes that harbor YEATS-containing AF9 and ENL (96). Furthermore, LEDGF (lens epithelium-derived growth factor), a protein that mediates chromatin association of the MLL complex, was previously found to be essential for MLL-rearrangement-induced leukemic transformation (55). A recent work reports that the PWWP domain of LEDGF recognizes and “reads” H3K36 methylation added by the ASH1L methyltransferase at proximal promoter chromatin, and this event was found critical for recruiting/stabilizing MLL fusions onto target sites to activate gene expression in leukemia cells (127). Additionally, NSD1 and NSD3, two related H3K36 methyltransferases, were previously found to be aberrantly rearranged in ~15% of pediatric AMLs (31) and their “writing” SET domains represent the validated site that remains to be pharmacologically targeted (30). Thus, these discovered circuits should offer additional therapeutic opportunities, both in the “reading” domains (YEATS of AF9 or ENL; PWWP of LEDGF) and the catalytic “writing” domains (SET of ASH1L and NSD1/3), for AML treatment.
Identification of BRD4 as a novel AML dependency was achieved through shRNA-based functional screening of epigenetic factors (110). Small-guide RNA-based CRISPR/Cas9 technology has provided an alternative system to perform screening in human AML cell lines, which recently led to identification of the histone acetyltransferase KAT2A/GCN5 as an AML dependency gene (128). In future, functional genomics studies using a range of AML cell lines that represent various genetically defined AML subtypes, as well as validation with primary human AML samples, are likely to produce useful information for subtype-specific dependencies on epigenetic modulators, which would guide drug discovery efforts aiming to developing the personalized AML treatment.
Implication in the Treatment of Pre-leukemic Disease
Somatic mutations of several epigenetic modulators (DNMT3A, TET2, IDH1/2) occur frequently among patients with pre-leukemia diseases such as MDS and apparently healthy individuals with clonal hematopoiesis or CHIP, an aging-related phenotype associated with increased risk of AML (21, 22, 25, 26). These mutations and resultant epigenetic deregulations are likely to be the “founder” lesion initiating pre-malignant disease and shaping subsequent malignant formation. Identification of the epigenetic vulnerabilities associated with these gene mutations in the context of AML shall provide useful information on how to treat premalignant diseases. For example, using a murine AML model harboring the coexisting kinase and DNMT3A mutations, a recent study demonstrated that DNMT3A mutation induced epigenetic dysregulation to promote “stemness” gene-expression programs, a process that can be reversed by DOT1L inhibitors (Figure 1D, right) (96). We speculate that the same mechanism/pathways act among premalignant diseases, and if so, the similar epigenetic inhibitors could reverse the premalignant alternations thus preventing malignant development in individuals with MDS or CHIP. In support, the epigenetic inhibitors and hypomethyating agents such as 5-Aza delay malignant transformation of MDS and are FDA-approved drugs for its treatment. However, as a life-threatening disease with a risk of conversion into AML, MDS has additional immediate needs to treat other complications such as anemia and transfusion associated iron overload, bleeding and infectious risk associated with the cytopenias. Currently, the definitive cure of MDS-associated leukemia risk is still allogeneic HSC transplantation. As for CHIP, there is consensus in the field that the relatively low risk of transformation of CHIP does not warrant the targeted therapies. Potential application of targeted epigenetic inhibitors in the treatment of pre-AML diseases such as MDS and myeloproliferative neoplasms warrants further investigation.
Potential Drug Resistance and Combinational Therapy
Resistance to drug remains a challenge in achieving durable remissions in cancer and epigenetically targeted drugs are no exception. The molecular understanding of resistance in epigenetic therapy is just at its beginning. For example, MLL-rearranged leukemias with PRC2 loss, either pre-existing or acquired, are resistant to BRDi presumably due to enhanced transcription of oncogenes such as MYC (129); furthermore, recent reports documented acquisition of somatic mutation by blood cancer cells during resistance to BRDi or EZHi (129, 130). Conceptually, combinational treatment using two or more drugs that target multiple cancer cell dependencies should help overcome treatment resistance. Furthermore, regardless of drug resistance, combinational therapy should improve treatment when their potential toxic effect can be mitigated. As mentioned above, a good example is that LSD1i sensitizes non-APL AML cells to ATRA treatment (122). In addition, DOT1Li and BRDi are shown to be synergistic in treating MLL-rearranged leukemia, possibly due to functional collaboration between DOT1L and BRD4 at the highly transcribed super-enhancer genes (131). Future studies of drug resistance, toxicity, and combinational treatment strategies would be necessary to further develop and optimize the existing leads into those useful compounds for clinical trials.
Author Contributions
RL and GW wrote the manuscript and generated the figures/tables.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
GW is supported by a Kimmel Scholar award and the NCI grants R01CA211336 and R01CA215284.
References
1. Strahl BD, Allis CD. The language of covalent histone modifications. Nature (2000) 403(6765):41–5. doi:10.1038/47412
2. Chi P, Allis CD, Wang GG. Covalent histone modifications – miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer (2010) 10(7):457–69. doi:10.1038/nrc2876
3. Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol (2010) 28(10):1079–88. doi:10.1038/nbt.1684
4. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28(10):1057–68. doi:10.1038/nbt.1685
5. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell (2012) 150(1):12–27. doi:10.1016/j.cell.2012.06.013
6. Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer (2011) 11(10):726–34. doi:10.1038/nrc3130
7. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482(7384):226–31. doi:10.1038/nature10833
8. Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood (2013) 121(18):3563–72. doi:10.1182/blood-2013-01-451781
9. Garraway LA, Lander ES. Lessons from the cancer genome. Cell (2013) 153(1):17–37. doi:10.1016/j.cell.2013.03.002
10. Estey E, Dohner H. Acute myeloid leukaemia. Lancet (2006) 368(9550):1894–907. doi:10.1016/S0140-6736(06)69780-8
11. Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers (2016) 2:16010. doi:10.1038/nrdp.2016.10
12. Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J (2001) 20(3):350–61. doi:10.1093/emboj/20.3.350
13. Wang GG, Pasillas MP, Kamps MP. Meis1 programs transcription of FLT3 and cancer stem cell character, using a mechanism that requires interaction with Pbx and a novel function of the Meis1 C-terminus. Blood (2005) 106(1):254–64. doi:10.1182/blood-2004-12-4664
14. Alharbi RA, Pettengell R, Pandha HS, Morgan R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia (2013) 27(5):1000–8. doi:10.1038/leu.2012.356
15. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med (2010) 363(25):2424–33. doi:10.1056/NEJMoa1005143
16. Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, Mauch J, Kelkenberg-Schade S, Haldemann B, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood (2012) 120(18):e83–92. doi:10.1182/blood-2011-12-401471
17. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med (2012) 366(12):1079–89. doi:10.1056/NEJMoa1112304
18. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer (2012) 12(9):599–612. doi:10.1038/nrc3343
19. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi:10.1056/NEJMoa1301689
20. Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet (2013) 45(10):1232–7. doi:10.1038/ng.2731
21. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371(26):2477–87. doi:10.1056/NEJMoa1409405
22. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi:10.1056/NEJMoa1408617
23. Genovese G, Jaiswal S, Ebert BL, McCarroll SA. Clonal hematopoiesis and blood-cancer risk. N Engl J Med (2015) 372(11):1071–2. doi:10.1056/NEJMc1500684
24. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126(1):9–16. doi:10.1182/blood-2015-03-631747
25. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): an update. Mutat Res Rev Mutat Res (2016) 769:47–62. doi:10.1016/j.mrrev.2016.04.009
26. Koeffler HP, Leong G. Preleukemia: one name, many meanings. Leukemia (2017) 31(3):534–42. doi:10.1038/leu.2016.364
27. Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood (2002) 99(10):3857–60. doi:10.1182/blood.V99.10.3857
28. Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med (2004) 10(10):500–7. doi:10.1016/j.molmed.2004.08.005
29. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer (2007) 7(11):823–33. doi:10.1038/nrc2253
30. Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol (2007) 9(7):804–12. doi:10.1038/ncb1608
31. Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood (2011) 118(13):3645–56. doi:10.1182/blood-2011-04-346643
32. Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature (2009) 459(7248):847–51. doi:10.1038/nature08036
33. de Rooij JD, Hollink IH, Arentsen-Peters ST, van Galen JF, Berna Beverloo H, Baruchel A, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia (2013) 27(12):2280–8. doi:10.1038/leu.2013.87
34. Reader JC, Meekins JS, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia (2007) 21(4):842–4.
35. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature (2014) 506(7488):328–33. doi:10.1038/nature13038
36. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20(12):1472–8. doi:10.1038/nm.3733
37. Wakita S, Yamaguchi H, Omori I, Terada K, Ueda T, Manabe E, et al. Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia (2013) 27(5):1044–52. doi:10.1038/leu.2012.317
38. Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A (2014) 111(7):2548–53. doi:10.1073/pnas.1324297111
39. Ploen GG, Nederby L, Guldberg P, Hansen M, Ebbesen LH, Jensen UB, et al. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br J Haematol (2014) 167(4):478–86. doi:10.1111/bjh.13062
40. Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends Mol Med (2007) 13(9):373–80. doi:10.1016/j.molmed.2007.07.004
41. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483(7390):474–8. doi:10.1038/nature10860
42. Coombs CC, Tallman MS, Levine RL. Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol (2016) 13(5):305–18. doi:10.1038/nrclinonc.2015.210
43. Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer (2017) 3(5):372–86. doi:10.1016/j.trecan.2017.04.004
44. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell (2002) 10(5):1107–17. doi:10.1016/S1097-2765(02)00741-4
45. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol (2006) 13(8):713–9. doi:10.1038/nsmb1128
46. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem (2012) 81:65–95. doi:10.1146/annurev-biochem-051710-134100
47. Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell (2010) 17(2):148–59. doi:10.1016/j.ccr.2009.12.034
48. Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene (2017) 627:337–42. doi:10.1016/j.gene.2017.06.056
49. Chen Y, Anastassiadis K, Kranz A, Stewart AF, Arndt K, Waskow C, et al. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell (2017) 31(6):755–770e756. doi:10.1016/j.ccell.2017.05.002
50. Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell (2014) 53(2):247–61. doi:10.1016/j.molcel.2013.12.001
51. Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell (2004) 13(4):587–97. doi:10.1016/S1097-2765(04)00081-4
52. Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol (2004) 24(13):5639–49. doi:10.1128/MCB.24.13.5639-5649.2004
53. Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell (2005) 123(2):207–18. doi:10.1016/j.cell.2005.09.025
54. Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res (2007) 67(15):7275–83. doi:10.1158/0008-5472.CAN-06-2369
55. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell (2008) 14(1):36–46. doi:10.1016/j.ccr.2008.05.003
56. He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem (2014) 57(4):1543–56. doi:10.1021/jm401868d
57. Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell (2015) 27(4):589–602. doi:10.1016/j.ccell.2015.02.016
58. Lehnertz B, Pabst C, Su L, Miller M, Liu F, Yi L, et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev (2014) 28(4):317–27. doi:10.1101/gad.236794.113
59. Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol (2011) 7(8):566–74. doi:10.1038/nchembio.599
60. Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature (2011) 469(7330):343–9. doi:10.1038/nature09784
61. Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell (2008) 32(4):491–502. doi:10.1016/j.molcel.2008.10.016
62. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet (2010) 42(8):722–U109. doi:10.1038/ng.621
63. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi:10.1038/ng.518
64. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A (2010) 107(49):20980–5. doi:10.1073/pnas.1012525107
65. Wang GG, Konze KD, Tao J. Polycomb genes, miRNA, and their deregulation in B-cell malignancies. Blood (2015) 125(8):1217–25. doi:10.1182/blood-2014-10-606822
66. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi:10.1056/NEJMoa1301689
67. Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun (2014) 5:4177. doi:10.1038/ncomms5177
68. Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood (2015) 126(10):1172–83. doi:10.1182/blood-2015-03-634428
69. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A (2012) 109(13):5028–33. doi:10.1073/pnas.1202258109
70. Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood (2012) 120(5):1107–17. doi:10.1182/blood-2011-11-394932
71. Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol (2013) 9(10):643–50. doi:10.1038/nchembio.1331
72. Shi J, Wang E, Zuber J, Rappaport A, Taylor M, Johns C, et al. The polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9;Nras(G12D) acute myeloid leukemia. Oncogene (2013) 32(7):930–8. doi:10.1038/onc.2012.110
73. Danis E, Yamauchi T, Echanique K, Haladyna J, Kalkur R, Riedel S, et al. Inactivation of Eed impedes MLL-AF9-mediated leukemogenesis through Cdkn2a-dependent and Cdkn2a-independent mechanisms in a murine model. Exp Hematol (2015) 43(11):930–935e936. doi:10.1016/j.exphem.2015.06.005
74. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood (2015) 125(2):346–57. doi:10.1182/blood-2014-06-581082
75. Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep (2014) 9(5):1841–55. doi:10.1016/j.celrep.2014.11.004
76. Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell (2015) 57(4):662–73. doi:10.1016/j.molcel.2014.12.023
77. Sinha S, Thomas D, Yu L, Gentles AJ, Jung N, Corces-Zimmerman MR, et al. Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood (2015) 125(2):316–26. doi:10.1182/blood-2014-03-566018
78. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol (2012) 8(11):890–6. doi:10.1038/nchembio.1084
79. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492(7427):108–12. doi:10.1038/nature11606
80. Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A (2012) 109(52):21360–5. doi:10.1073/pnas.1210371110
81. Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol (2013) 20(11):1329–39. doi:10.1016/j.chembiol.2013.09.013
82. Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol (2013) 8(6):1324–34. doi:10.1021/cb400133j
83. Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev (2011) 25(13):1345–58. doi:10.1101/gad.2057811
84. Okada Y, Feng Q, Lin YH, Jiang Q, Li YQ, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell (2005) 121(2):167–78. doi:10.1016/j.cell.2005.02.020
85. Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev (2010) 24(6):574–89. doi:10.1101/gad.1898410
86. He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, et al. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A (2011) 108(36):E636–45. doi:10.1073/pnas.1107107108
87. Chang M-J, Wu H, Achille NJ, Reisenauer MR, Chou C-W, Zeleznik-Le NJ, et al. Histone H3 lysine 79 methyltransferase dot1 is required for immortalization by MLL oncogenes. Cancer Res (2010) 70(24):10234–42. doi:10.1158/0008-5472.can-10-3294
88. Anh Tram N, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood (2011) 117(25):6912–22. doi:10.1182/blood-2011-02-334359
89. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell (2011) 20(1):66–78. doi:10.1016/j.ccr.2011.06.010
90. Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia (2013) 27(4):813–22. doi:10.1038/leu.2012.327
91. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood (2013) 121(13):2533–41. doi:10.1182/blood-2012-11-465120
92. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell (2011) 20(1):53–65. doi:10.1016/j.ccr.2011.06.009
93. Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun (2012) 3:1288. doi:10.1038/ncomms2304
94. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood (2013) 122(6):1017–25. doi:10.1182/blood-2013-04-497644
95. Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell (2014) 26(6):896–908. doi:10.1016/j.ccell.2014.10.009
96. Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell (2016) 30(1):92–107. doi:10.1016/j.ccell.2016.05.008
97. Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood (2016) 128(7):971–81. doi:10.1182/blood-2015-11-684225
98. Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov (2016) 6(10):1166–81. doi:10.1158/2159-8290.CD-16-0237
99. Sarkaria SM, Christopher MJ, Klco JM, Ley TJ. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia (2014) 28(12):2403–6. doi:10.1038/leu.2014.235
100. Riedel SS, Haladyna JN, Bezzant M, Stevens B, Pollyea DA, Sinha AU, et al. MLL1 and DOT1L cooperate with meningioma-1 to induce acute myeloid leukemia. J Clin Invest (2016) 126(4):1438–50. doi:10.1172/JCI80825
101. Shia W-J, Okumura AJ, Yan M, Sarkeshik A, Lo M-C, Matsuura S, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood (2012) 119(21):4953–62. doi:10.1182/blood-2011-04-347476
102. Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol (2007) 9(10):1208–15. doi:10.1038/ncb1642
103. Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell (2016) 29(1):32–48. doi:10.1016/j.ccell.2015.12.007
104. Dillon MB, Bachovchin DA, Brown SJ, Finn MG, Rosen H, Cravatt BF, et al. Novel inhibitors for PRMT1 discovered by high-throughput screening using activity-based fluorescence polarization. ACS Chem Biol (2012) 7(7):1198–204. doi:10.1021/cb300024c
105. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell (2007) 25(1):15–30. doi:10.1016/j.molcel.2006.12.014
106. Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res (2008) 647(1–2):3–12. doi:10.1016/j.mrfmmm.2008.07.004
107. Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol (2012) 19(12):1218–27. doi:10.1038/nsmb.2436
108. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov (2012) 11(5):384–400. doi:10.1038/nrd3674
109. French CA. Small-molecule targeting of BET proteins in cancer. Adv Cancer Res (2016) 131:21–58. doi:10.1016/bs.acr.2016.04.001
110. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature (2011) 478(7370):524–U124. doi:10.1038/nature10334
111. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature (2010) 468(7327):1067–73. doi:10.1038/nature09504
112. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature (2011) 478(7370):529–33. doi:10.1038/nature10509
113. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia (2014) 28(2):311–20. doi:10.1038/leu.2013.338
114. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell (2014) 25(5):652–65. doi:10.1016/j.ccr.2014.03.016
115. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (2015) 348(6241):1376–81. doi:10.1126/science.aab1433
116. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell (2014) 159(5):1126–39. doi:10.1016/j.cell.2014.10.024
117. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature (2014) 511(7511):616–20. doi:10.1038/nature13393
118. Gough SM, Lee F, Yang F, Walker RL, Zhu YJ, Pineda M, et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov (2014) 4(5):564–77. doi:10.1158/2159-8290.CD-13-0419
119. Wagner EK, Nath N, Flemming R, Feltenberger JB, Denu JM. Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry (2012) 51(41):8293–306. doi:10.1021/bi3009278
120. Shi YJ, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119(7):941–53. doi:10.1016/j.cell.2004.12.012
121. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell (2012) 21(4):473–87. doi:10.1016/j.ccr.2012.03.014
122. Schenk T, Chen WC, Goellner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med (2012) 18(4):605–11. doi:10.1038/nm.2661
123. Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell (2014) 159(3):558–71. doi:10.1016/j.cell.2014.09.049
124. Zhao D, Li Y, Xiong X, Chen Z, Li H. YEATS domain-a histone acylation reader in health and disease. J Mol Biol (2017) 429(13):1994–2002. doi:10.1016/j.jmb.2017.03.010
125. Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature (2017) 543(7644):270–4. doi:10.1038/nature21688
126. Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature (2017) 543(7644):265–9. doi:10.1038/nature21687
127. Zhu L, Li Q, Wong SH, Huang M, Klein BJ, Shen J, et al. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov (2016) 6(7):770–83. doi:10.1158/2159-8290.CD-16-0058
128. Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep (2016) 17(4):1193–205. doi:10.1016/j.celrep.2016.09.079
129. Rathert P, Roth M, Neumann T, Muerdter F, Roe J-S, Muhar M, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature (2015) 525(7570):543–7. doi:10.1038/nature14898
130. Xu B, Konze KD, Jin J, Wang GG. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp Hematol (2015) 43(8):698–712. doi:10.1016/j.exphem.2015.05.001
Keywords: epigenetic modulator, small-molecule inhibitors, acute myeloid leukemia, bromodomain, MLL, EZH2, DNMT3A, DOT1L
Citation: Lu R and Wang GG (2017) Pharmacologic Targeting of Chromatin Modulators As Therapeutics of Acute Myeloid Leukemia. Front. Oncol. 7:241. doi: 10.3389/fonc.2017.00241
Received: 29 July 2017; Accepted: 21 September 2017;
Published: 12 October 2017
Edited by:
Keisuke Ito, Albert Einstein College of Medicine, United StatesReviewed by:
Kathrin Maria Bernt, Children’s Hospital of Philadelphia, United StatesRachel Rau, Baylor College of Medicine, United States
Copyright: © 2017 Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Greg Wang, Z3JlZ193YW5nQG1lZC51bmMuZWR1