 Veronica Zingales
Veronica Zingales Alfio Distefano1
Alfio Distefano1 Marco Raffaele
Marco Raffaele Ignazio Barbagallo
Ignazio Barbagallo Luca Vanella
Luca Vanella- 1Department of Drug Science, Biochemistry Section, University of Catania, Catania, Italy
- 2Department of Surgery, Azienda Ospedaliero Universitaria Policlinico Vittorio Emanuele, Catania, Italy
Prostate cancer (PCa) has become the most frequent type of cancer in men. Recent data suggest that diabetic patients taking metformin have a lower incidence of certain cancer, including PCa. Metformin is the most common drug used in type II diabetes mellitus; its use has been shown to lower the incidence of several cancers, although there are ambiguous data about the anticancer activity of metformin. A large number of studies examined the potential antineoplastic mechanism of metformin although it is not still completely understood. This review summarizes the literature concerning the effects of metformin on PCa cells, highlighting its numerous mechanisms of action through which it can act. We analyze the possible causes of the discordances regarding the impact of metformin on risk of PCa; we discuss the latest findings in this field, suggesting that metformin may have a future role in the management of PCa both as monotherapy and in combination with other drugs.
Introduction
Prostate cancer (PCa) represents one-third of all new cancer cases each year and the second cause of cancer-related death in US (1).
Prostate cancer is the stage subsequent to premalignant lesions due to a progressive transition from normal prostatic epithelial cells to prostatic intraepithelial neoplasia. Over time, most tumors evolve in castration-resistant prostate cancer (CRPC) with development of metastasis (2, 3). Advanced stages of the disease and formation of metastasis are the main causes of most PCa-related death. Multiple treatment strategies exist but the survival rates remain low. The current strategies for the management of PCa include surgery, chemotherapy, radiation, and endocrine therapy.
In the early stage, PCa is characterized by androgen-dependent growth and medical castration through androgen-deprivation therapy (ADT) is the first-line therapy choice for its treatment. This therapy depresses the proliferative function of androgen receptor (AR) but after 12 and 18 months, patients treated with ADT develop resistance to this therapy (4). In fact, over time, androgen-dependent PCa evolves to androgen-independent PCa and ADT is not useful to treat the progressive stage of PCa. CRPC is the most aggressive form of PCa and it shows resistance to current available therapeutic strategies. Recent studies demonstrated the existence of a relationship between diabetes, insulin levels, and risk of cancer, including PCa, but other studies, investigating the association between diabetes mellitus and PCa, have reported inconsistent findings (5). Notably, although the PCa affects a considerable proportion of men, a reduced incidence of this type of cancer has been observed in subject with type II diabetes mellitus (T2DM). Probably, lower hormone levels, such as testosterone, insulin, and insulin-like growth factor-1 (IGF-1) in patients with uncontrolled T2DM induces a lower risk of developing PCa (6). Although pieces of epidemiological evidence showed that diabetes mellitus is inversely associated with the development of PCa (6, 7), other studies proved opposite results (8–10). Circulating insulin levels, higher than average, have been shown to be associated with increased cancer growth and mortality (11–13). This could be explained by analyzing the role of insulin and/or IGF-1 on insulin-like growth factor-1 receptor (IGF-1R). High levels of IGF-1R are associated with invasion, aggressiveness cancer, and poorer prognosis (14–18).
In addition to the escalation of the incidence of PCa, over the last three decades, an ever-going epidemic of diabetes occurred particularly in developed countries. World Health Organization estimated that diabetes will become one of the main causes of death in the world by 2030 (19).
Metformin is an insulin-sensitizing oral biguanide used by diabetic patients every day to maintain their glycemic homeostasis. Metformin is an ideal drug: it is well tolerated and inexpensive. Metformin regulates glucose homeostasis exerting an important control of metabolism. In particular, metformin reduces intestinal absorption of glucose and it increases peripheral glucose uptake and its utilization by adipose tissue and skeletal muscles leading to increased insulin sensitivity. Through AMPK activation, metformin decreases insulin secretion, inhibits gluconeogenesis and energy consuming processes (such as protein and fatty acid synthesis), and stimulates ATP-generating processes (such as glycolysis and fatty acid oxidation). This results in a shift from anabolic to catabolic metabolism and in an inhibition of proliferation. Metformin and a safety lifestyle, the latter based on healthy and well-balanced meals and a regular physical activity, are currently the first-line treatment for the management of T2DM.
The Antineoplastic Effects of Metformin
Recent studies were interested to investigate whether metformin might have potential benefits on other widespread diseases, such as the cancer, suggesting a new potential use of this drug beyond its classical indications. In fact, it seems that metformin owns antineoplastic activity and this ability might be the cause of the inverse relationship between diabetes and risk of developing PCa. This hypothesis remains credible and should continue to be investigated. Recently, metformin has been shown to exert antineoplastic effects in several cancers. Its anticancer activity has been highlighted by diabetes specialists who noticed diabetic patients taking metformin had a lower risk to develop cancer (including PCa) than other diabetic patients. The role of metformin in glucose and fatty acids metabolism is very well known but further studies are needed to explore the cellular mechanism and its cell targets in cancer cells.
In the last years, many researchers focused the attention on the metformin and several studies and meta-analysis were conducted. These studies showed an unclear role of metformin on prevention of PCa: some studies showed that use of metformin is associated with a reduced risk of development of PCa; other studies showed that this association does not exist (20, 21).
A retrospective cohort study showed that metformin use is associated with a reduced risk of PCa in Asian patients with type II diabetes (22). Meta-analysis by Hwang et al. revealed that metformin use causes a 20% reduction in risk of recurrence in PCa patients with T2DM (23).
Another large retrospective cohort study did not find an association between metformin use and risk of PCa in men with diabetes, regardless of cancer grade (high-grade and low-grade cancer) or diagnosis method (24).
The effect of metformin on the prognosis of patients with cancer was also investigated, showing a statistically significant decrease in cancer-related mortality, following metformin use (25). In addition, in patients with PCa, metformin use was associated with a lower risk of death due to PCa, and the benefit increased in a way that is dependent on the duration of its use (26).
Other studies showed different results (27) and these discrepancies could be explained by differences in patient characteristics, different origin, grade, and biological features of PCa and dose and duration of drug treatment (metformin seems to require long-term use to exert its anticancer activity) (28, 29). However, from a large number of studies, metformin results to have anticancer activity, decreasing the incidence of different types of cancer, such as colon, breast, pancreatic, and PCa in vivo and in vitro (30–34).
AMPK-Dependent and Independent Molecular Mechanism
AMPK activation appears the main mechanism through which metformin inhibits cancer growth. AMPK plays a key role in the maintaining of cellular energy homeostasis (35). It is an important sensor of the AMP/ATP ratio. AMPK appears as a potential anticancer agent when it is highly activated, but it may not be critical as inhibitor of cancer growth when it acts at low levels (36).
Metformin primarily acts to directly inhibit the mitochondrial respiratory chain which then reduces the production of ATP resulting in an increase in the ratio of AMP to ATP which then results in activation of AMPK. Under energy stress conditions, the tumor suppressor LKB1 (37) is the major kinase involved in the AMPK activation and mTOR reduction.
Through the mTOR inhibition, metformin arrests cell cycle and cell growth, because mTOR is a downstream effector of PI3K/AKT pathway, a signaling pathway linked to cancer cell growth and proliferation. PI3K/AKT/mTOR signaling pathway leads to an abnormal cells proliferation, inhibition of apoptosis, and carcinogenesis.
Colquhoun et al. showed that metformin owns an anti-proliferative effect in PCa cells through the activation of pAMPK and subsequent inhibition of downstream mTOR signaling and the induction of cell cycle arrest. In this study, metformin was used in combination with bicalutamide, a known agent used in the hormonal therapy of PCa. It acts blocking the AR and inducing a G1/S phase arrest of the cell cycle. Combining metformin with bicalutamide, the authors obtained a reduction of PCa cell survival, especially in cells expressing functional AR (38).
The anti-PCa effect of metformin via AMPK activation was also observed by Tsutsumi et al. They demonstrated, in vitro and in vivo, that metformin induces apoptosis and attenuates PCa cell proliferation. Furthermore, a stronger decrease of PCa growth was achieved when metformin was combined with Exenedin-4, a glucagon-like peptide-1 receptor agonists (39).
A key feature of cancer cells is an increase in dependency on glycolysis for energy production. This is known as “Warburg effect.” 2-Deoxyglucose (2DG) has been considered a potential anticancer agent for its capacity to inhibit glucose metabolism and induce intracellular ATP reduction and autophagy (40). In the study conducted by Ben Sahra et al., the combination of metformin and 2DG resulted in a synergistically effect on cell viability in PCa cells, due to a stronger depletion of intracellular ATP, through the attack to two different sources of energy: mitochondrial complex 1 and glycolysis (41).
Although AMPK activation appears the main mechanism of action through which metformin exerts its anticancer effect, other antineoplastic mechanisms of action AMPK-independent have been shown.
In DU145 LKB1-negative cells, Biernacka et al. did not observe an increased phosphorylation of AMPK after treatment with metformin, suggesting that, in this specific cell line, metformin induces cell death through a LKB1–AMPK-independent pathway (42).
In addition, Ben Sahra et al. reported that REDD1 and cyclin D1, in an AMPK-independent manner, mediate the anti-proliferative effects of metformin in PCa cells (43, 44).
In addition, it was demonstrated that metformin treatment decreases c-MYC protein levels and the incidence of prostate intraepithelial lesions formation in vivo and in vitro (45). The levels of c-MYC protein and mRNA in the metformin-treated PCa cells were much lower than those in control cells. Furthermore, in LNCaP cells, androgen-sensitive human PCa cells are characterized by the presence of high-affinity AR (46); the reduction of c-MYC levels was associated with a significant reduction of AR.
Activated AR binds androgen response element and regulates the expression of genes involved in PCa cell growth.
Metformin is able to downregulate the levels of AR mRNAs, showing another way to repress the cancer cell proliferation (47). Furthermore, through its ability to downregulate AR, metformin inhibited the migration both AR-negative and AR-positive PCa cells, showing more pronounced effect in these last. The authors suggest that the mechanism through which metformin abrogates the upregulation of AR is via enhanced activity of the MID1 translation regulator complex.
In PCa cells, androgens induce a selective upregulation of IGF-1R, with a subsequent increase of cell proliferation and invasiveness (48). Standard anti-androgen drugs are not capable to block the IGF-1R upregulation. Malaguarnera et al. showed that metformin is able to inhibit androgen-dependent IGF-1R upregulation with subsequent reduction of IGF-1R-mediated proliferation in LNCaP cells (49). This study found that the complex more involved in the androgen-dependent IGF-1R upregulation is the mTORC1 complex, whereas AMPK plays a marginal role in this action.
Since androgens increase proliferation and development of PCa cells, ADT is the first line of PCa treatment. However, after several years, many patients do not well response to this therapy, developing castration-resistant prostate cancer (CRPC). Furthermore, ADT can cause metabolic consequences, such as insulin resistance and development of metabolic syndrome (50). A combination of metformin with androgen deprivation might improve treatment efficacy and minimize side effects.
Some studies showed that ADT might provide microenvironments suitable for the differentiation of cancer cells in hormone-independent cancer cells, increasing factors involved in the epithelial–mesenchymal transition (EMT) and exerting a selective pressure toward EMT (51, 52). Understanding the molecular mechanism by which EMT acts in cancer progression, identifying agents capable of stopping or slowing metastasis, may be an essential step for PCa treatment. Some studies highlighted that COX2/PGE2/STAT2/EMT is involved in cancer cell migration and invasion (53–56). Tong et al. showed that metformin is capable to act on this axis, inhibiting it. In fact, their data revealed that metformin has an anti-EMT effect through the inhibition of COX2 or the block of PEG2-mediated STAT3 phosphorylation and the expression of other EMT markers, when metformin is used at higher concentrations (57). They also demonstrated that metformin is capable, by inhibiting EMT, to restore enzalutamide sensitivity in CRPC (58).
Another known factor associated with EMT in lung fibrosis and breast cancer is FoxM1 (59, 60), but its role in PCa cells has not yet been elucidated. FoxM1 plays a key role in cell proliferation, cell cycle regulation, angiogenesis, invasion, and metastasis (61, 62). Wang et al. found that in PCa cells EMT is inhibited by metformin through the downregulation of FoxM1 expression (63).
Metformin has been shown to inhibit EMT by modulating microRNA, such as miR30a (64). In recent years, the involvement of miRNAs in human PCa has been investigated, discovering several miRNAs expressed abnormally (65, 66). Among these, miR-708-5p is a non-coding RNA with a tumor- and metastasis-suppressive role (67, 68). Yang et al. showed that metformin upregulates miR-708-5p in LNCaP and PC3 cells, inducing ER stress and apoptosis (69). During the early and late stages, PCa is characterized by an increase of lipogenesis that it is associated with tumor growth and the most aggressive forms of PCa (70). Metformin is able to alter the activity and the expression of lipogenic enzymes and transcriptional factors, such as SREBP-1c, FAS, and ACC, causing an energy deficiency (71). The capacity of metformin to decrease the expression of FAS and SREBP-1c is linked with its ability to act on AMPK/mTOR pathway.
Therefore, it has been shown that metformin acts on multifold molecular targets and on a myriad of transduction pathway.
In recent experimental studies, metformin is often used at concentrations clearly higher than those observed in diabetic patients, but it has been demonstrated that many organs and tissues are exposed to metformin concentrations appreciably higher than those present in the general circulation (72). Furthermore, metformin has been shown to have irrelevant effects on benignant cells, suggesting that its pro-apoptotic effects are limited to malignant cells (45).
The Combination of Metformin with Other Drugs or Natural Agents
Researchers evaluated the effect of metformin in combination with other drugs or natural agents on PCa cells. Metformin exerts its antineoplastic effect at lower concentrations when it is used in combination with other agents, such as Plk1 inhibitor. Shao et al. suggest that the inhibition of Plk1 promotes the cytotoxicity of metformin in PCa cells through both signaling and metabolic pathways (73).
In addition to metformin, statins are also the most commonly prescribed drugs in western countries. Recent data suggest that statins may have beneficial effects against PCa (74). The role of the combination of metformin and simvastatin was studied by Pennanen et al., showing a synergistically increase of necrotic cell death and autophagy (75).
Moreover, in the last years, natural-derived and medical plants have acquired more and more importance, such as solamargine (76, 77). The capacity of solamargine alone and in combination with metformin on CRPC cells has been explored (78). This study showed that solamargine inhibits the growth of two CRPC cell lines (DU145 and PC3) in the dose-dependent manner; this antineoplastic effect of solamargine is enhanced by metformin. The mechanism through which the combination of two agents reduces the proliferation of CRPC cells consists in the AMPKα-mediated inhibition of p65. A recent study showed that the AMPK-dependent antineoplastic effect of metformin results stronger when it is used in combination with Vitamin D3 (79). This study suggests that vitamin D3 improves metformin’s inhibitory activity due to AMPK activation and G1/S cell cycle arrest. In addition, metformin and vitamin D3 treatment has been shown to reduce anti-apoptotic protein Bcl-2 levels (with subsequent increase of cellular apoptosis) and c-MYC expression in DU145 more than the single treatment, highlighting a synergistic effect of the combination of the two agents.
Conclusion
Overall, metformin may be considered an ideal agent to be used as adjuvant to standard treatments for PCa, both as monotherapy and combined with chemotherapeutics or other drugs (Table 1). Current treatments for PCa result limited because of drug resistance and toxicity that develop over time. Thus, new cellular targets and novel molecular therapeutic agents are needed. To this regard, metformin appears a real candidate more affordable than other expensive therapeutic options: metformin plays a central role as metabolic homeostasis regulator and indirectly as anti-proliferative and anti-carcinogenic agent (Figure 1). Sufficient data from studies on cancer cell lines and animal models suggest that metformin lowers the risk of biochemical recurrence and the rates of mortality in PCa, through its intrinsic proprieties and its pleiotropic effects linked with metformin-mediated fall in plasma glucose and insulin concentrations.
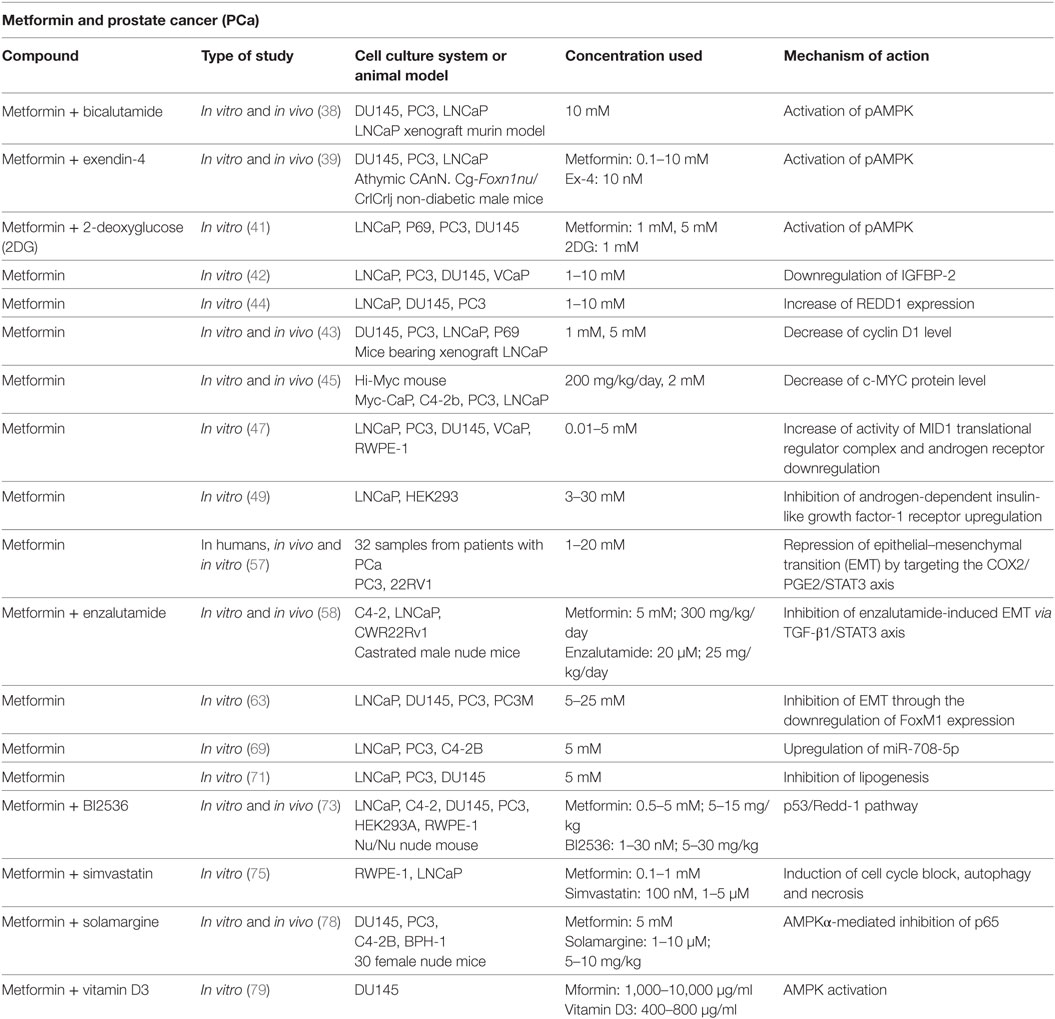
Table 1. Characteristics of selected study.
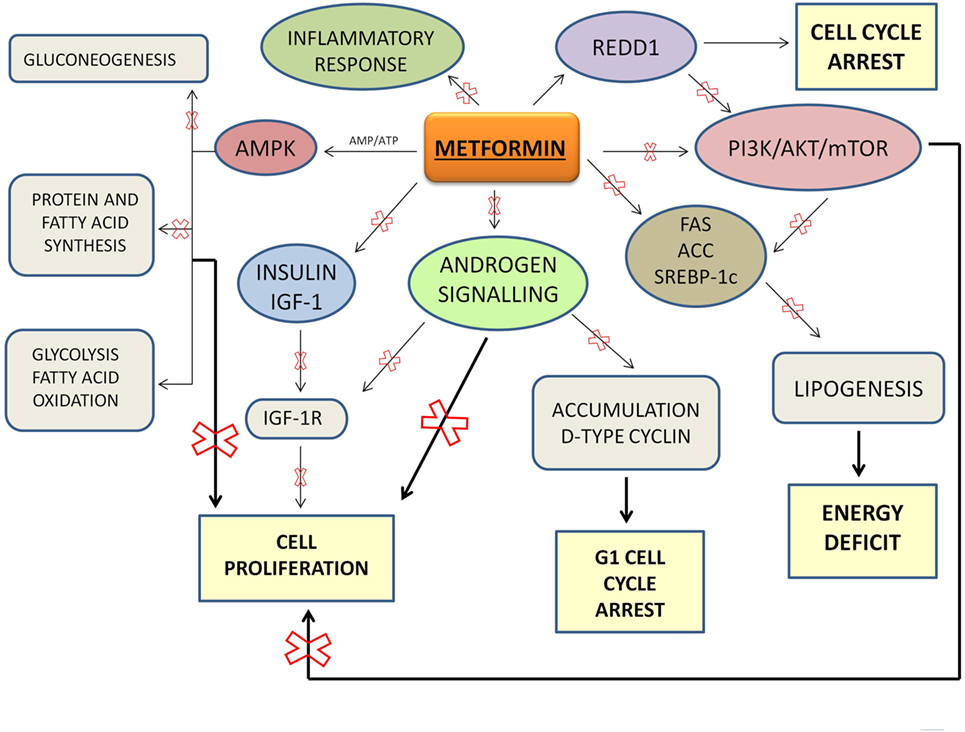
Figure 1. Proposed mechanisms of action for Metformin.
Although the potential mechanism of action of metformin has been largely studied, it is not still completely understood. Other studies are necessary to determine which dose of metformin can cause profound direct antineoplastic effects on cancer cell metabolism and if these doses can be safely administered to patients.
Author Contributions
VZ, AD, and MR collected and analyzed research article; AZ gave conceptual advise; and IB and LV wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gronberg H. Prostate cancer epidemiology. Lancet (2003) 361(9360):859–64. doi:10.1016/S0140-6736(03)12713-4
2. Shin HJ, Ro JY. Prostatic intraepithelial neoplasia: a potential precursor lesion of prostatic adenocarcinoma. Yonsei Med J (1995) 36(3):215–31. doi:10.3349/ymj.1995.36.3.215
3. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin (2010) 60(5):277–300. doi:10.3322/caac.20073
4. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene (2013) 32(49):5501–11. doi:10.1038/onc.2013.206
5. Gacci M, Russo GI, De Nunzio C, Sebastianelli A, Salvi M, Vignozzi L, et al. Meta-analysis of metabolic syndrome and prostate cancer. Prostate Cancer Prostatic Dis (2017) 20(2):146–55. doi:10.1038/pcan.2017.1
6. Kasper JS, Giovannucci E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev (2006) 15(11):2056–62. doi:10.1158/1055-9965.EPI-06-0410
7. Bonovas S, Filioussi K, Tsantes A. Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia (2004) 47(6):1071–8. doi:10.1007/s00125-004-1415-6
8. Tseng CH. Diabetes and risk of prostate cancer: a study using the National Health Insurance. Diabetes Care (2011) 34(3):616–21. doi:10.2337/dc10-1640
9. Tseng CH. Prostate cancer mortality in Taiwanese men: increasing age-standardized trend in general population and increased risk in diabetic men. Ann Med (2011) 43(2):142–50. doi:10.3109/07853890.2010.530683
10. Hsieh MC, Lee TC, Cheng SM, Tu ST, Yen MH, Tseng CH. The influence of type 2 diabetes and glucose-lowering therapies on cancer risk in the Taiwanese. Exp Diabetes Res (2012) 2012:413782. doi:10.1155/2012/413782
11. Hsing AW, Chua S Jr, Gao YT, Gentzschein E, Chang L, Deng J, et al. Prostate cancer risk and serum levels of insulin and leptin: a population-based study. J Natl Cancer Inst (2001) 93(10):783–9. doi:10.1093/jnci/93.10.783
12. Tavani A, Gallus S, Bosetti C, Tzonou A, Lagiou P, Negri E, et al. Diabetes and the risk of prostate cancer. Eur J Cancer Prev (2002) 11(2):125–8. doi:10.1097/00008469-200204000-00003
13. Chien SW, Kuo DY, Liao JM, Wang PS, Yu CH. Growth modulation of diabetic factors and antidiabetic drugs on prostate cancer cell lines. Chin J Physiol (2016) 59(2):109–18. doi:10.4077/CJP.2016.BAE368
14. Liao Y, Abel U, Grobholz R, Hermani A, Trojan L, Angel P, et al. Up-regulation of insulin-like growth factor axis components in human primary prostate cancer correlates with tumor grade. Hum Pathol (2005) 36(11):1186–96. doi:10.1016/j.humpath.2005.07.023
15. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res (2008) 14(20):6364–70. doi:10.1158/1078-0432.CCR-07-4879
16. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8(12):915–28. doi:10.1038/nrc2536
17. Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, et al. Insulin receptor expression by human prostate cancers. Prostate (2009) 69(1):33–40. doi:10.1002/pros.20852
18. Kojima S, Inahara M, Suzuki H, Ichikawa T, Furuya Y. Implications of insulin-like growth factor-I for prostate cancer therapies. Int J Urol (2009) 16(2):161–7. doi:10.1111/j.1442-2042.2008.02224.x
19. Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract (2011) 94(3):311–21. doi:10.1016/j.diabres.2011.10.029
20. Wu GF, Zhang XL, Luo ZG, Yan JJ, Pan SH, Ying XR, et al. Metformin therapy and prostate cancer risk: a meta-analysis of observational studies. Int J Clin Exp Med (2015) 8(8):13089–98.
21. Chen CB, Eurich DT, Majumdar SR, Johnson JA. Metformin and the risk of prostate cancer across racial/ethnic groups: a population-based cohort study. Prostate Cancer Prostatic Dis (2017) 20(1):122–6. doi:10.1038/pcan.2016.65
22. Tseng CH. Metformin significantly reduces incident prostate cancer risk in Taiwanese men with type 2 diabetes mellitus. Eur J Cancer (2014) 50(16):2831–7. doi:10.1016/j.ejca.2014.08.007
23. Hwang IC, Park SM, Shin D, Ahn HY, Rieken M, Shariat SF. Metformin association with lower prostate cancer recurrence in type 2 diabetes: a systematic review and meta-analysis. Asian Pac J Cancer Prev (2015) 16(2):595–600. doi:10.7314/APJCP.2015.16.2.595
24. Margel D, Urbach D, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Association between metformin use and risk of prostate cancer and its grade. J Natl Cancer Inst (2013) 105(15):1123–31. doi:10.1093/jnci/djt170
25. Bo S, Ciccone G, Rosato R, Villois P, Appendino G, Ghigo E, et al. Cancer mortality reduction and metformin: a retrospective cohort study in type 2 diabetic patients. Diabetes Obes Metab (2012) 14(1):23–9. doi:10.1111/j.1463-1326.2011.01480.x
26. Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol (2013) 31(25):3069–75. doi:10.1200/JCO.2012.46.7043
27. Bensimon L, Yin H, Suissa S, Pollak MN, Azoulay L. The use of metformin in patients with prostate cancer and the risk of death. Cancer Epidemiol Biomarkers Prev (2014) 23(10):2111–8. doi:10.1158/1055-9965.EPI-14-0056
28. Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care (2010) 33(6):1304–8. doi:10.2337/dc09-1791
29. Coyle C, Cafferty FH, Vale C, Langley RE. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol (2016) 27(12):2184–95. doi:10.1093/annonc/mdw410
30. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia (2009) 52(9):1766–77. doi:10.1007/s00125-009-1440-6
31. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care (2009) 32(9):1620–5. doi:10.2337/dc08-2175
32. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol (2012) 48(3):R31–43. doi:10.1530/JME-12-0007
33. Hitron A, Adams V, Talbert J, Steinke D. The influence of antidiabetic medications on the development and progression of prostate cancer. Cancer Epidemiol (2012) 36(4):e243–50. doi:10.1016/j.canep.2012.02.005
34. Tseng CH. Diabetes, metformin use, and colon cancer: a population-based cohort study in Taiwan. Eur J Endocrinol (2012) 167(3):409–16. doi:10.1530/EJE-12-0369
35. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab (2005) 1(1):15–25. doi:10.1016/j.cmet.2004.12.003
36. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res (2013) 73(10):2929–35. doi:10.1158/0008-5472.CAN-12-3876
37. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (2005) 310(5754):1642–6. doi:10.1126/science.1120781
38. Colquhoun AJ, Venier NA, Vandersluis AD, Besla R, Sugar LM, Kiss A, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis (2012) 15(4):346–52. doi:10.1038/pcan.2012.16
39. Tsutsumi Y, Nomiyama T, Kawanami T, Hamaguchi Y, Terawaki Y, Tanaka T, et al. Combined treatment with exendin-4 and metformin attenuates prostate cancer growth. PLoS One (2015) 10(10):e0139709. doi:10.1371/journal.pone.0139709
40. Brown J. Effects of 2-deoxyglucose on carbohydrate metablism: review of the literature and studies in the rat. Metabolism (1962) 11:1098–112.
41. Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res (2010) 70(6):2465–75. doi:10.1158/0008-5472.CAN-09-2782
42. Biernacka KM, Persad RA, Bahl A, Gillatt D, Holly JM, Perks CM. Hyperglycaemia-induced resistance to docetaxel is negated by metformin: a role for IGFBP-2. Endocr Relat Cancer (2017) 24(1):17–30. doi:10.1530/ERC-16-0095
43. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene (2008) 27(25):3576–86. doi:10.1038/sj.onc.1211024
44. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res (2011) 71(13):4366–72. doi:10.1158/0008-5472.CAN-10-1769
45. Akinyeke T, Matsumura S, Wang X, Wu Y, Schalfer ED, Saxena A, et al. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis (2013) 34(12):2823–32. doi:10.1093/carcin/bgt307
46. Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res (1983) 43(4):1809–18.
47. Demir U, Koehler A, Schneider R, Schweiger S, Klocker H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer (2014) 14:52. doi:10.1186/1471-2407-14-52
48. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65(5):1849–57. doi:10.1158/0008-5472.CAN-04-1837
49. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, et al. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology (2014) 155(4):1207–21. doi:10.1210/en.2013-1925
50. Braga-Basaria M, Dobs AS, Muller DC, Carducci MA, John M, Egan J, et al. Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J Clin Oncol (2006) 24(24):3979–83. doi:10.1200/JCO.2006.05.9741
51. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res (2012) 72(2):527–36. doi:10.1158/0008-5472.CAN-11-3004
52. Rycaj K, Cho EJ, Liu X, Chao HP, Liu B, Li Q, et al. Longitudinal tracking of subpopulation dynamics and molecular changes during LNCaP cell castration and identification of inhibitors that could target the PSA-/lo castration-resistant cells. Oncotarget (2016) 7(12):14220–40. doi:10.18632/oncotarget.7303
53. Vo BT, Morton D Jr, Komaragiri S, Millena AC, Leath C, Khan SA. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology (2013) 154(5):1768–79. doi:10.1210/en.2012-2074
54. Qiu X, Cheng JC, Chang HM, Leung PC. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr Relat Cancer (2014) 21(4):533–43. doi:10.1530/ERC-13-0450
55. Jorvig JE, Chakraborty A. Zerumbone inhibits growth of hormone refractory prostate cancer cells by inhibiting JAK2/STAT3 pathway and increases paclitaxel sensitivity. Anticancer Drugs (2015) 26(2):160–6. doi:10.1097/CAD.0000000000000171
56. Qin G, Xu F, Qin T, Zheng Q, Shi D, Xia W, et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget (2015) 6(39):41794–808. doi:10.18632/oncotarget.5993
57. Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang Y, et al. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett (2017) 389:23–32. doi:10.1016/j.canlet.2016.12.031
58. Liu Q, Tong D, Liu G, Xu J, Do K, Geary K, et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-beta1/STAT3 axis-regulated EMT. Cell Death Dis (2017) 8(8):e3007. doi:10.1038/cddis.2017.417
59. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J (2013) 32(2):231–44. doi:10.1038/emboj.2012.336
60. Yang C, Chen H, Tan G, Gao W, Cheng L, Jiang X, et al. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett (2013) 340(1):104–12. doi:10.1016/j.canlet.2013.07.004
61. Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res (2007) 67(17):8293–300. doi:10.1158/0008-5472.CAN-07-1265
62. Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D, et al. Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res (2009) 69(8):3501–9. doi:10.1158/0008-5472.CAN-08-3045
63. Wang Y, Yao B, Wang Y, Zhang M, Fu S, Gao H, et al. Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int J Mol Med (2014) 33(6):1514–22. doi:10.3892/ijmm.2014.1707
64. Zhang J, Shen C, Wang L, Ma Q, Xia P, Qi M, et al. Metformin inhibits epithelial-mesenchymal transition in prostate cancer cells: involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem Biophys Res Commun (2014) 452(3):746–52. doi:10.1016/j.bbrc.2014.08.154
65. Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res (2007) 67(13):6130–5. doi:10.1158/0008-5472.CAN-07-0533
66. Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol (2009) 27(34):5848–56. doi:10.1200/JCO.2009.24.0317
67. Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, et al. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res (2011) 71(19):6208–19. doi:10.1158/0008-5472.CAN-11-0073
68. Ryu S, McDonnell K, Choi H, Gao D, Hahn M, Joshi N, et al. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell (2013) 23(1):63–76. doi:10.1016/j.ccr.2012.11.019
69. Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P, et al. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis (2015) 4:e158. doi:10.1038/oncsis.2015.18
70. Swinnen JV, Esquenet M, Goossens K, Heyns W, Verhoeven G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res (1997) 57(6):1086–90.
71. Loubiere C, Goiran T, Laurent K, Djabari Z, Tanti JF, Bost F. Metformin-induced energy deficiency leads to the inhibition of lipogenesis in prostate cancer cells. Oncotarget (2015) 6(17):15652–61. doi:10.18632/oncotarget.3404
72. Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica (1994) 24(1):49–57. doi:10.3109/00498259409043220
73. Shao C, Ahmad N, Hodges K, Kuang S, Ratliff T, Liu X. Inhibition of polo-like kinase 1 (Plk1) enhances the antineoplastic activity of metformin in prostate cancer. J Biol Chem (2015) 290(4):2024–33. doi:10.1074/jbc.M114.596817
74. Zhang Y, Zang T. Association between statin usage and prostate cancer prevention: a refined meta-analysis based on literature from the years 2005–2010. Urol Int (2013) 90(3):259–62. doi:10.1159/000341977
75. Pennanen P, Syvala H, Blauer M, Savinainen K, Ylikomi T, Tammela TL, et al. The effects of metformin and simvastatin on the growth of LNCaP and RWPE-1 prostate epithelial cell lines. Eur J Pharmacol (2016) 788:160–7. doi:10.1016/j.ejphar.2016.06.036
76. Cui CZ, Wen XS, Cui M, Gao J, Sun B, Lou HX. Synthesis of solasodine glycoside derivatives and evaluation of their cytotoxic effects on human cancer cells. Drug Discov Ther (2012) 6(1):9–17. doi:10.5582/ddt.2012.v6.1.9
77. Munari CC, de Oliveira PF, Campos JC, Martins Sde P, Da Costa JC, Bastos JK, et al. Antiproliferative activity of Solanum lycocarpum alkaloidic extract and their constituents, solamargine and solasonine, in tumor cell lines. J Nat Med (2014) 68(1):236–41. doi:10.1007/s11418-013-0757-0
78. Xiang S, Zhang Q, Tang Q, Zheng F, Wu J, Yang L, et al. Activation of AMPKalpha mediates additive effects of solamargine and metformin on suppressing MUC1 expression in castration-resistant prostate cancer cells. Sci Rep (2016) 6:36721. doi:10.1038/srep36721
Keywords: diabetes, metformin, prostate, cancer, apoptosis
Citation: Zingales V, Distefano A, Raffaele M, Zanghi A, Barbagallo I and Vanella L (2017) Metformin: A Bridge between Diabetes and Prostate Cancer. Front. Oncol. 7:243. doi: 10.3389/fonc.2017.00243
Received: 25 July 2017; Accepted: 25 September 2017;
Published: 11 October 2017
Edited by:
Anna Rita Migliaccio, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Victor C. Kok, Asia University, TaiwanJeff M. P. Holly, University of Bristol, United Kingdom
Copyright: © 2017 Zingales, Distefano, Raffaele, Zanghi, Barbagallo and Vanella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignazio Barbagallo, aWduYXppby5iYXJiYWdhbGxvQHVuaWN0Lml0