 Fatma E. El-Khouly1,2*
Fatma E. El-Khouly1,2* Dannis G. van Vuurden1
Dannis G. van Vuurden1 Thom Stroink3Esther Hulleman1Gertjan J. L. Kaspers1,4
Thom Stroink3Esther Hulleman1Gertjan J. L. Kaspers1,4 N. Harry Hendrikse2,5
N. Harry Hendrikse2,5 Sophie E. M. Veldhuijzen van Zanten1
Sophie E. M. Veldhuijzen van Zanten1
- 1Department of Pediatric Oncology – Hematology, VU University Medical Center, Amsterdam, Netherlands
- 2Department of Clinical Pharmacology and Pharmacy, VU University Medical Center, Amsterdam, Netherlands
- 3Department of Pharmaceutical Sciences, Utrecht University, Utrecht, Netherlands
- 4Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands
- 5Department of Radiology and Nuclear Medicine, VU University Medical Center, Amsterdam, Netherlands
Despite decades of clinical trials for diffuse intrinsic pontine glioma (DIPG), patient survival does not exceed 10% at two years post-diagnosis. Lack of benefit from systemic chemotherapy may be attributed to an intact bloodbrain barrier (BBB). We aim to develop a theoretical model including relevant physicochemical properties in order to review whether applied chemotherapeutics are suitable for passive diffusion through an intact BBB or whether local administration via convection-enhanced delivery (CED) may increase their therapeutic potential. Physicochemical properties (lipophilicity, molecular weight, and charge in physiological environment) of anticancer drugs historically and currently administered to DIPG patients, that affect passive diffusion over the BBB, were included in the model. Subsequently, the likelihood of BBB passage of these drugs was ascertained, as well as their potential for intratumoral administration via CED. As only non-molecularly charged, lipophilic, and relatively small sized drugs are likely to passively diffuse through the BBB, out of 51 drugs modeled, only 8 (15%)—carmustine, lomustine, erlotinib, vismodegib, lenalomide, thalidomide, vorinostat, and mebendazole—are theoretically qualified for systemic administration in DIPG. Local administration via CED might create more therapeutic options, excluding only positively charged drugs and drugs that are either prodrugs and/or only available as oral formulation. A wide variety of drugs have been administered systemically to DIPG patients. Our model shows that only few are likely to penetrate the BBB via passive diffusion, which may partly explain the lack of efficacy. Drug distribution via CED is less dependent on physicochemical properties and may increase the therapeutic options for DIPG.
Background
Diffuse intrinsic pontine glioma (DIPG) is a rare, aggressive childhood malignancy of the brainstem with a 2-year survival rate of 10% (1, 2). Unlike the spectacular increase in survival of childhood leukemia patients from <10% to over 80% in the last 50 years, the prospect for patients suffering from DIPG has not changed (3–5), likely due to a lack of success from (chemo)therapeutic strategies. Historically, this was blamed on the supposed resistance of DIPG tumor cells to cytotoxic agents. Preclinical studies have, however, recently shown that primary cultures derived from DIPG patients, are actually not resistant to a number of traditionally used cytotoxic drugs and novel targeted chemotherapeutics (5, 6). These results from pre-clinical studies contrasting with disappointing outcomes of clinical studies led to a shifting paradigm toward the hypothesis of a possible delivery problem of chemotherapeutics over the blood-brain barrier (BBB). This supposedly prevents drugs from reaching the tumor properly (7). In many brain tumors, the integrity of the BBB is affected by the formation of disordered and highly permeable tumor neovasculature. Infiltrating tumors, such as DIPG, however, make use of the existing brain vasculature with normal BBB integrity (8). Noteworthy in this respect, especially early-disease DIPGs show limited contrast enhancement after intravenous administration of gadolinium compared to glioblastoma multiforme tumors that harbor highly neovascular regions. As gadolinium has an average molecular weight of 545 kDa, which largely exceeds the penetration cut-off of the BBB (e.g., 400–600 Da), limited contrast enhancement in these tumors is suggestive of a largely intact BBB (9–11). Furthermore, to illustrate, studies investigating biopsy-derived intratumoral drug concentrations of systemically delivered drugs in adults with high-grade glioma show low, potentially sub-therapeutic local drug concentrations, especially in non-enhancing tumor regions (12–14).
To overcome the BBB, novel drug delivery techniques, such as convection-enhanced delivery (CED), have been developed. With CED, chemotherapeutic agents are administered directly into the tumor microenvironment via a highly controlled positive pressure gradient and a constant flow induced by a pump. This enables homogeneous distribution of high drug concentrations over an easy-to-define distance, presumably increasing the therapeutic potential and avoiding systemic toxicities (14, 15).
In this study, we aim to review all chemotherapeutic drugs (previously) administered systemically to DIPG patients by means of a theoretical model including all physicochemical properties that influence passive diffusion, to indicate their likeliness of passage over an intact BBB in DIPG. Furthermore, we aim to indicate whether local administration of these drugs via convection-enhanced delivery (CED) may increase their therapeutic potential.
Model Design
An extensive search of the literature and trial databases was performed to identify all chemotherapeutics historically employed in DIPG patients. Databases of Medline/PubMed and The Cochrane Library were searched for potentially relevant articles. The search strategy combined controlled and free text words for the target population (e.g., children), the tumor type (e.g., DIPG or pontine glioma) and the application of chemotherapeutic drugs. The reference lists of all included articles were searched for additional studies. In addition, trial registries (www.clinicaltrials.gov, www.clinicaltrialsregister.eu) as well as websites from consortia treating children with brain tumors (www.itcc-consortium.org, www.pbtc.org, www.childrensoncologygroup.org) were searched for clinical trials in DIPG. The complete search strategy can be found in the supplementary data.
Subsequently, drug simulations were performed for both systemic administration to predict passive diffusion over an intact BBB and for local intratumoral drug delivery via CED, to predict convection-distribution efficacy, using relevant physicochemical properties (molecular weight, lipophilicity, and molecular charge). To this aim, physicochemical property data were extracted from various chemical databases: PubChem chemistry database, Drugbank.ca and Clarke’s Analysis of Drugs and Poisons. The molecular charge in physiologic environment was simulated by MarvinSketch® (Chemaxon), an advanced chemical editor for drawing chemical structures and calculating basic physicochemical properties (e.g., molecular charge, log P), using specific algorithms.
Drug Simulation for Systemic Drug Administration
The BBB, formed by tightly interconnected endothelial cells in the capillary walls of the brain vasculature, protects the brain by limiting the inter- and paracellular transport of foreign substances from the systemic blood flow (16). Systemically applied chemotherapeutics enter the brain via passive diffusion or active transport mechanisms. The balance between in- and outflow depends both on the physicochemical properties of the drug itself and on its affinity for drug in- and efflux transporters and receptors expressed in the BBB.
The Lipinski Rule of 5 is used to determine a drug’s permeability. According to this rule good permeability is likely if: (i) the molecular weight is ≤500 Da, (ii) the lipophilicity, measured by the partition coefficient (log P), is ≤5 (optimal value of 2.0–3.5), (iii) the structure has no more than 5 hydrogen bond donors, and (iv) no more than 10 hydrogen bond donor acceptors (16–18). Taking these rules into consideration, the physicochemical properties that determine passive diffusion through the BBB, are molecular weight, lipophilicity, and molecular charge. For every drug, molecular weight and Log P were included in the model and subsequently, the chemical structure was used to simulate the molecular charge of a drug in physiological environment (pH 7.4). Drugs with a molecular (positive or negative) charge of ≤10% were considered to be able to passively diffuse through the BBB. Drugs with a (positive or negative) molecular charge of ≥90% are considered to have a higher affinity for the hydrophilic environment of the blood and are therefore not likely to passively diffuse through the BBB. Drugs with a molecular charge between 10 and 90% are partly able to diffuse through the BBB, but are likely not to reach their therapeutic concentration after systemic administration. In case of prodrugs (i.e., inactive compounds that require metabolization into a pharmacologically active form), the physicochemical properties of the active metabolites were evaluated in the model.
Drug Simulation for Local Administration via CED
For CED, drug distribution over the tumor volume mainly depends on two determinants: positive pressure gradient created by the drug infusion system and the molecular charge of a drug: positively charged molecules tend to form complexes with negatively charged cell membrane components, leading to lower distribution volumes (19–21). Mackay et al. demonstrated that drugs with a positive charge of 10% show a significantly lower distribution than neutral drugs (20). Neutral or negatively charged drugs seem to optimally convect and distribute via CED. However, as negatively charged molecules have previously only been studied up to a charge of 10%, evidence of better convection and distribution of more negatively charged molecules is lacking. In addition, since CED circumvents the systemic circulation and thus the first pass effect, prodrugs are not suitable for local administration.
Efficacy Simulation for Drug Delivery in DIPG
Table 1 shows the variables (i.e., the relevant physicochemical properties discussed above) included in the theoretical model and all drugs found in the literature search. The drugs were grouped based on their mechanisms of action (e.g., alkylating agents, topoisomerase inhibitors, signal transduction inhibitors, cytostatic antibiotics, antimitotic agents, antimetabolites, platinum-containing cytotoxics, antihormones, and others). An “efficacy simulation” was performed including the physicochemical properties of each chemotherapeutic agent and the environmental properties (i.e., pH 7.4). In this efficacy simulation every property was scored to be either optimal (1) or poor (0) based on the “Lipinski rule of five.” The sum of the scores for each variable was used to determine the likelihood of the drug to penetrate the BBB after systemic administration, or the likeliness of drug distribution over the tumor volume using CED. The colors in Table 1 indicate which chemotherapeutic agents are theoretically well suited for systemic and/or local administration via CED (score 3: marked green), or which chemotherapeutic agents are not (score 0/1: marked red). Chemotherapeutic agents with intermediate BBB penetration (score 2) have the potential to passively diffuse through the BBB, but likely in such low concentrations that they presumably do not reach therapeutic concentrations. These drugs are therefore marked yellow.
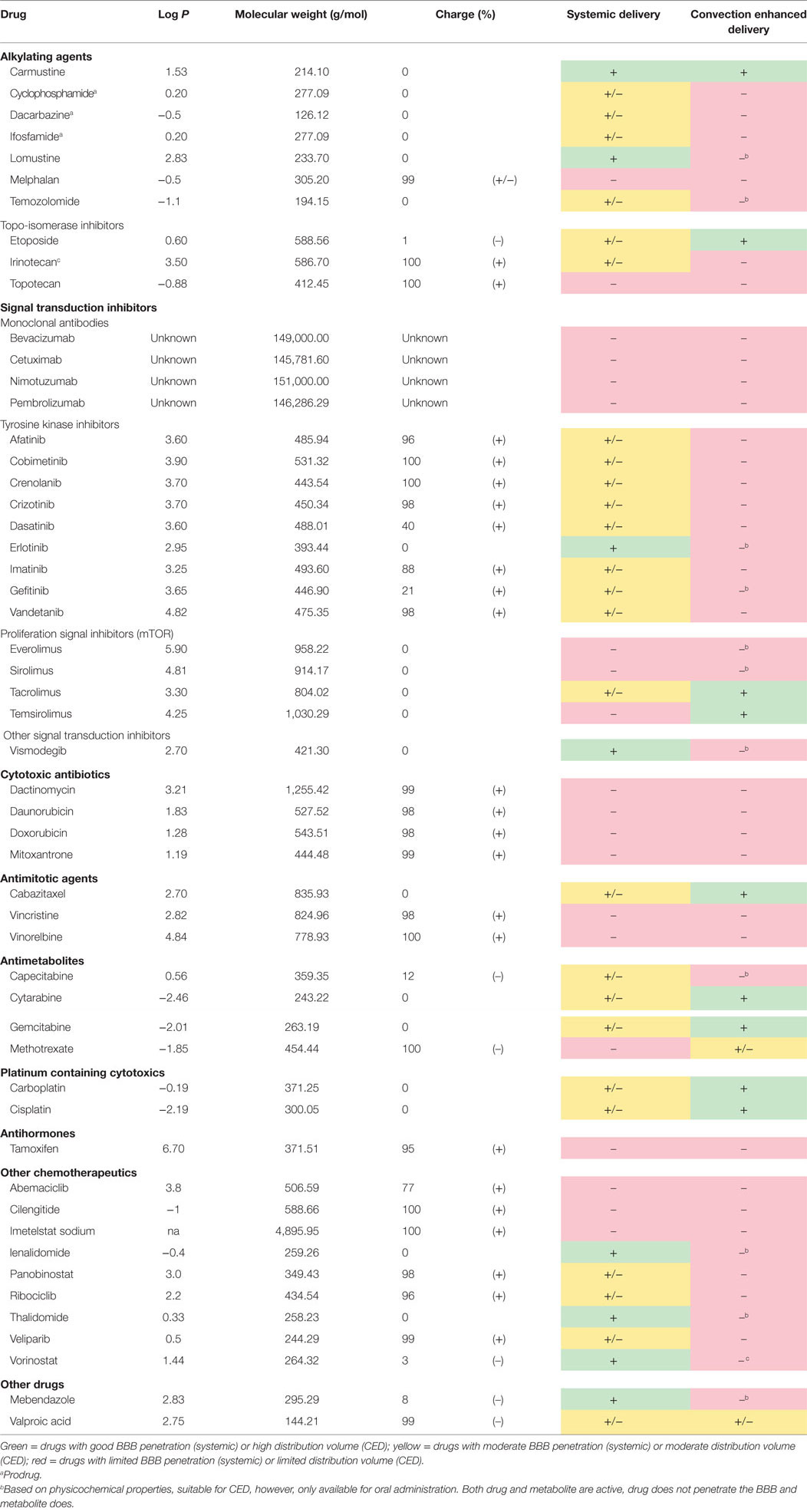
Table 1. Overview of the physicochemical properties of all chemotherapeutic drugs historically applied to DIPG patients.
Drug affinity for efflux transporters (ATP-binding cassette (ABC) transporters) is an important factor in the prediction of ultimate brain uptake. These transporters include P-glycoprotein (P-gp/MDR1/ABCB1), breast cancer-resistant protein (BCRP/ABCG2), and multidrug resistance protein 1 (MRP1/ABCC1) (16, 17, 22). Drug affinity for these transporters has not been investigated for the majority of chemotherapeutics. Besides, as efflux transporter affinity strongly depends on the concentration of a drug, it is difficult to give uniform values on these transporters’ affinity. Therefore, the influence of ABC transporters could not be included in the model. For completeness, Table 2 was designed to summarize all known drug affinities for the efflux transporters located in the BBB.
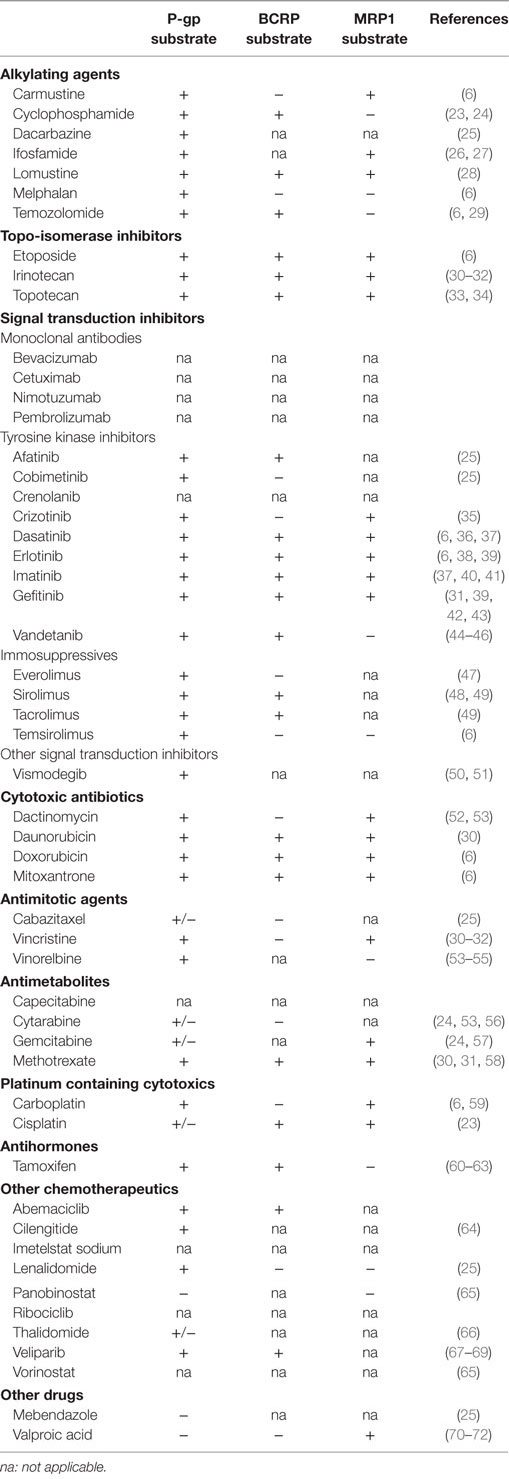
Table 2. Overview of efflux transporter affinity of all chemotherapeutic drugs historically applied to DIPG patients.
Based on our model, including 51 drugs, only 8 (15%)—carmustine, lomustine, erlotinib, vismodegib, lenalidomide, thalidomide, vorinostat, and mebendazole—appear to be of use for systemic administration (green cells in Table 1 column 5), presumably resulting in adequate brain uptake in case of an intact BBB. These drugs will very likely have good BBB passage due to their relatively small molecular weight, lipophilicity and limited molecular charge. Drugs expected to have limited BBB passage (marked in yellow) are either (partly) charged (e.g., dasatinib) or too hydrophilic (e.g., temozolomide), limiting their passive diffusion. Drugs that are marked in red are unlikely to penetrate an intact BBB, mostly due to their molecular charge in physiological environment (up to 100%).
Potential candidates for CED, listed green in Table 1 column 6, are carmustine, etoposide, tacrolimus, temsirolimus, cabazitaxel, cytarabine, gemcitabine, carboplatin, and cisplatin. These drugs have a neutral charge in physiological environment. Methotrexate and valproic acid (in yellow), being negatively charged, might also be suitable for CED, but this is speculative, as only molecules with a negative charge up to 10% have been investigated (20). The other drugs, marked in red, are theoretically not suitable for CED, mainly due to their positive charge (e.g., topotecan). Furthermore, prodrugs such as cyclophosphamide (indicated with superscript a) are not suitable for CED since these drugs require to be metabolized into their active (effective) metabolite. Additionally, drugs indicated with superscript b are currently only available for oral administration and not in liquid form. These drugs however are theoretically suitable for CED based on their favorable neutral charge. It might be worth to investigate reformulation of these drugs into liquid formula for local brain delivery applications.
Discussion
In this study, we developed a theoretical model to review whether chemotherapeutics are suitable for passive diffusion through an intact BBB (after systemic administration) or whether local administration via convection-enhanced delivery (CED) may increase their therapeutic potential. We demonstrated that most systemically (intravenously or orally) administered chemotherapeutic drugs thus far investigated in clinical trials in DIPG are not likely to reach adequate intratumoral concentrations in case of intact BBB, rendering most of these therapies likely ineffective for use in DIPG.
To date, only few studies investigated the actual brain concentration of chemotherapeutic drugs after systemic administration. In 2007, Muldoon et al. reviewed these studies and showed that (from our list of chemotherapeutic drugs historically administered to DIPG patients) there was no brain uptake of doxorubicin, vincristine, and methotrexate after systemic administration in healthy and glioma-bearing dogs and rats (8). Cytarabine and etoposide showed low brain concentrations in these preclinical models. In adults, on-therapy biopsies showed low brain concentrations of cisplatin, imatinib gemcitabine, and methotrexate after systemic administration (8, 12–14). These results are in line with the results of our theoretical review.
Muldoon et al. also showed that the brain:plasma ratio found in preclinical and clinical pharmacology studies was variable, mainly due to the inconsistent interstitial fluid pressure and location of the samples taken, indicating heterogeneous drug distribution (8). Recently, we introduced PET imaging of zirconium-89 (89Zr)-labeled bevacizumab in children with DIPG and demonstrated considerable heterogeneity in drug delivery between patients and within DIPG tumors (73). PET technology enables imaging of radiolabeled drugs, especially monoclonal antibodies and tyrosine kinase inhibitors (74). By this non-invasive in patient quantification of tumor uptake and drug distribution not only therapeutic potential but also toxicity can be predicted. We advocate the development of molecular drug imaging studies additional or parallel to clinical trials because especially in children with cancer, drugs without therapeutic effect (based on a lack of drug-uptake in the tumor), may only cause (life-long) side effects.
In addition to the potential to reach therapeutic concentrations, a drug’s maximum tolerated dose and toxicity profile could be a limiting factor, even when a drug has proven to penetrate the BBB. Recent clinical studies in DIPG have, therefore, started focusing on alternative routes of drug delivery. Alternatives include CED, for which we here show that it theoretically increases the therapeutic potential of suited drugs previously administered systemically in DIPG patients without effect. It should be noted, however, that CED targets only the primary tumor site and will not target disseminated disease. As 13–17% of DIGP patients show distant parenchymal, subependymal, and leptomeningeal metastases in the brain and/or spine, CED should very likely be complemented with “whole-brain therapy” (75). Other alternative drug delivery techniques that are currently being investigated include (i) encapsulation of cationic substances into liposomes (micro- and nanoparticles) to decrease their tissue affinity and thus increase their volume of distribution and (ii) temporary BBB disruption techniques, such as focused or unfocused ultrasound-mediated drug delivery, to enhance uptake of systemically delivered drugs (19–21, 76). Since the variables that determine local drug concentrations in these techniques are heterogeneous and not widely investigated yet, these alternative techniques could not be taken into account in our model design and review. The exact extent and timing of radiotherapy-induced BBB disruption in DIPG is unknown and warrants further investigation.
In conclusion, this review raises awareness for the impact of physicochemical properties of anticancer drugs that influence their passive diffusion through an intact BBB after systemic administration. In diffuse gliomas such as DIPG, in which the BBB is largely intact in large parts of the tumor, during most time of the disease course, one must critically weigh drug candidates that a priori are unlikely to pass the BBB and thus are unlikely to have therapeutic effect in patients suffering from these tumors. This might also be valid for other diffuse growing brain tumors in child- and adulthood that show areas of tumor infiltration behind an intact BBB. We furthermore postulate that a novel drug delivery technique, such as CED theoretically increases the therapeutic potential of some of the drugs previously administered systemically. This might require repositioning of these drugs and reformulation to render them suitable for local delivery strategies.
Future Prospects
Our review calls for further preclinical BBB drug delivery models, as well as clinical research on actual intratumoral drug uptake and local drug delivery techniques such as CED. The model itself may be helpful in the design of future treatment regimens in which the combination of systemic administration and local or alternative delivery of different chemotherapeutics is explored. It aims to provide a first selection of drugs that have the highest potential to penetrate the BBB. Ultimately, best (combinations) of potentially effective drugs against DIPG can be sought combining these data with IC50 data from preclinical studies (77), and information on drug efflux mechanisms. Ideally, this theoretical BBB-passage model needs (pre)clinical validation. Although research into intratumoral drug concentrations remains challenging, especially in DIPG, a preclinical validation study of our model is currently being developed. Clinical validation using on-therapy tumor biopsy studies (i.e., to directly measure intratumoral drug uptake after systemic administration of chemotherapeutic agents), or less invasively with PET imaging (73) are currently emerging and will provide further information on drug uptake in these detrimental diseases.
Author Contributions
SVZ and FE contributed to the design of the work, data collection and interpretation, and wrote the main body of the manuscript. DV contributed to the interpretation of the data and critically revised the manuscript. TS contributed to the interpretation of the data. EH assisted in data collection. NH contributed to the interpretation of the data and revised the manuscript. GK critically revised the manuscript and assisted in writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fonc.2017.00254/full#supplementary-material.
References
1. Veldhuijzen van Zanten SE, Jansen MH, Sanchez Aliaga E, van Vuurden DG, Vandertop WP, Kaspers GJ. A twenty-year review of diagnosing and treating children with diffuse intrinsic pontine glioma in The Netherlands. Expert Rev Anticancer Ther (2015) 15:157–64. doi:10.1586/14737140.2015.974563
2. Veldhuijzen van Zanten SE, Baugh J, Chaney B, De Jongh D, Sanchez Aliaga E, Barkhof F, et al. Development of the SIOPE DIPG network, registry and imaging repository: a collaborative effort to optimize research into a rare and lethal disease. J Neurooncol (2017) 132:255–66. doi:10.1007/s11060-016-2363-y
3. Thomas A. How can we improve on the already impressive results in pediatric ALL? Hematology Am Soc Hematol Educ Program (2015) 2015:414–9. doi:10.1182/asheducation-2015.1.414
4. Hargrave D, Bartels U, Bouffet E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol (2006) 7:241–8. doi:10.1016/S1470-2045(06)70615-5
5. Jansen MHA, van Vuurden DG, Vandertop WP, Kaspers GJL. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev (2012) 38:27–35. doi:10.1016/j.ctrv.2011.06.007
6. Veringa SJ, Biesmans D, van Vuurden DG, Jansen MH, Wedekind LE, Horsman I, et al. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS One (2013) 8:e61512. doi:10.1371/journal.pone.0061512
7. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol (2012) 2:205. doi:10.3389/fonc.2012.00205
8. Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol (2007) 25:2295–305. doi:10.1200/JCO.2006.09.9861
9. Bartels U, Hawkins C, Vézina G, Kun L, Souweidane M, Bouffet E. Proceedings of the diffuse intrinsic pontine glioma (DIPG) Toronto Think Tank: advancing basic and translational research and cooperation in DIPG. J Neurooncol (2011) 105:119–25. doi:10.1007/s11060-011-0704-4
10. Grimm SA, Chamberlain MC. Brainstem glioma: a review. Curr Neurol Neurosci Rep (2013) 13:346. doi:10.1007/s11910-013-0346-3
11. Warren KE. Novel therapeutic delivery approaches in development for pediatric gliomas. CNS Oncol (2013) 2:427–35. doi:10.2217/cns.13.37
12. Holdhoff M, Supko JG, Gallia GL, Hann CL, Bonekamp D, Ye X, et al. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme. J Neurooncol (2010) 97:241–5. doi:10.1007/s11060-009-0008-0
13. Sigmond J, Honeywell RJ, Postma TJ, et al. Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann Oncol (2009) 20:182–7. doi:10.1093/annonc/mdn543
14. Blakeley JO, Olson J, Grossman SA, He X, Weingart J, Supko JG, et al. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J Neurooncol (2009) 91:51–8. doi:10.1007/s11060-008-9678-2
15. Allard E, Passirani C, Benoit J-P. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials (2009) 30:2302–18. doi:10.1016/j.biomaterials.2009.01.003
16. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis (2010) 37:13–25. doi:10.1016/j.nbd.2009.07.030
17. Löscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol (2005) 76:22–76. doi:10.1016/j.pneurobio.2005.04.006
18. Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci (2009) 30:431–40. doi:10.1016/j.tips.2009.05.005
19. Barua NU, Gill SS, Love S. Convection-enhanced drug delivery to the brain: therapeutic potential and neuropathological considerations. Brain Pathol (2013) 24:117–27. doi:10.1111/bpa.12082
20. MacKay JA, Deen DF, Szoka FC. Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res (2005) 1035:139–53. doi:10.1016/j.brainres.2004.12.007
21. Saito R, Krauze MT, Noble CO, Tamas M, Drummond DC, Kirpotin DB, et al. Tissue affinity of the infusate affects the distribution volume during convection-enhanced delivery into rodent brains: implications for local drug delivery. J Neurosci Methods (2006) 154:225–32. doi:10.1016/j.jneumeth.2005.12.027
22. Deeken JF, Löscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res (2007) 13:1663–74. doi:10.1158/1078-0432.CCR-06-2854
23. Pawłowski KM, Mucha J, Majchrzak K, Motyl T, Król M. Expression and role of PGP, BCRP, MRP1 and MRP3 in multidrug resistance of canine mammary cancer cells. BMC Vet Res (2013) 9:119. doi:10.1186/1746-6148-9-119
24. Takara K, Sakaeda T, Yagami T, Kobayashi H, Ohmoto N, Horinouchi M, et al. Cytotoxic effects of 27 anticancer drugs in HeLa and MDR1-overexpressing derivative cell lines. Biol Pharm Bull (2002) 25:771–8. doi:10.1248/bpb.25.771
25. Drugbank Canada. Drug & Drug Target Database. Version 5.0. (2015). Available from: http://www.drugbank.ca
26. Haimeur A, Conseil G, Deeley RG, Cole SPC. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab (2004) 5:21–53. doi:10.2174/1389200043489199
27. Munoz M, Henderson M, Haber M, Norris M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life (2007) 59:752–7. doi:10.1080/15216540701736285
28. Flory AB, Rassnick KM, Al-Sarraf R, Bailey DB, Balkman CE, Kiselow MA, et al. Combination of CCNU and DTIC chemotherapy for treatment of resistant lymphoma in dogs. J Vet Intern Med (2008) 22:164–71. doi:10.1111/j.1939-1676.2007.0005.x
29. Schaich M, Kestel L, Pfirrmann M, Robel K, Illmer T, Kramer M, et al. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann Oncol (2009) 20:175–81. doi:10.1093/annonc/mdn548
30. Nakanishi T. Drug transporters as targets for cancer chemotherapy. Cancer Genomics Proteomics (2007) 4:241–54.
31. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics (2008) 9:105–27. doi:10.2217/14622416.9.1.105
32. Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer (2011) 11:352–63. doi:10.1038/nrc3053
33. Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J Cell Sci (2000) 113:2011–21.
34. Cihalova D, Staud F, Ceckova M. Interactions of cyclin-dependent kinase inhibitors AT-7519, flavopiridol and SNS-032 with ABCB1, ABCG2 and ABCC1 transporters and their potential to overcome multidrug resistance in vitro. Cancer Chemother Pharmacol (2015) 76:105–16. doi:10.1007/s00280-015-2772-1
35. Zhou WJ, Zhang X, Cheng C, Wang F, Wang XK, Liang YJ, et al. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br J Pharmacol (2012) 166:1669–83. doi:10.1111/j.1476-5381.2012.01849.x
36. Tang SC, de Vries N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) gene dosage on plasma pharmacokinetics and brain accumulation of dasatinib, sorafenib, and sunitinib. J Pharmacol Exp Ther (2013) 346:486–94. doi:10.1124/jpet.113.205583
37. Robey RW, Ierano C, Zhan Z, Bates SE. The challenge of exploiting ABCG2 in the clinic. Curr Pharm Biotechnol (2011) 12:595–608. doi:10.2174/138920111795163913
38. de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs (2012) 30:443–9. doi:10.1007/s10637-010-9569-1
39. Lainey E, Sébert M, Thépot S, Scoazec M, Bouteloup C, Leroy C, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle (2012) 11:4079–92. doi:10.4161/cc.22382
40. Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos (2009) 37:946–55. doi:10.1124/dmd.108.024489
41. Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos (2010) 38:1371–80. doi:10.1124/dmd.109.031302
42. Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med (2011) 13:e17. doi:10.1017/S1462399411001888
43. Ozvegy-Laczka C, Hegedus T, Várady G, Ujhelly O, Schuetz JD, Váradi A, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol (2004) 65:1485–95. doi:10.1124/mol.65.6.1485
44. Jovelet C, Bénard J, Forestier F, Farinotti R, Bidart JM, Gil S. Inhibition of P-glycoprotein functionality by vandetanib may reverse cancer cell resistance to doxorubicin. Eur J Pharm Sci (2012) 46:484–91. doi:10.1016/j.ejps.2012.03.012
45. Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int J Pharm (2012) 434:306–14. doi:10.1016/j.ijpharm.2012.05.028
46. Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS One (2009) 4:e5172. doi:10.1371/journal.pone.0005172
47. Food and Drug Administration (FDA). Pharmacologic Class Indexing. Everolimus. (2015). Available from: http://www.accessdata.fda.gov/spl/data/54044090-93f7-4108-b422-3c2996f167ec/54044090-93f7-4108-b422-3c2996f167ec.xml
48. Pop IV, Pop LM, Ghetie M-A, Vitetta ES. Targeting mammalian target of rapamycin to both downregulate and disable the P-glycoprotein pump in multidrug-resistant B-cell lymphoma cell lines. Leuk Lymphoma (2009) 50:1155–62. doi:10.1080/10428190903046722
49. Pawarode A, Shukla S, Minderman H, Fricke SM, Pinder EM, O’Loughlin KL, et al. Differential effects of the immunosuppressive agents cyclosporin A, tacrolimus and sirolimus on drug transport by multidrug resistance proteins. Cancer Chemother Pharmacol (2007) 60:179–88. doi:10.1007/s00280-006-0357-8
50. Sandhiya S, Melvin G, Kumar SS, Dkhar SA. The dawn of hedgehog inhibitors: vismodegib. J Pharmacol Pharmacother (2013) 4:4–7. doi:10.4103/0976-500X.107628
51. Zhang Y, Laterra J, Pomper MG. Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/Pgp. Neoplasia (2009) 11:96–101. doi:10.1593/neo.81264
52. Hill CR, Jamieson D, Thomas HD, Brown CD, Boddy AV, Veal GJ. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem Pharmacol (2013) 85:29–37. doi:10.1016/j.bcp.2012.10.004
53. Prenkert M, Uggla B, Tina E, Tidefelt U, Strid H. Rapid induction of P-glycoprotein mRNA and protein expression by cytarabine in HL-60 cells. Anticancer Res (2009) 29:4071–6.
54. Bessho Y, Oguri T, Ozasa H, Uemura T, Sakamoto H, Miyazaki M, et al. ABCC10/MRP7 is associated with vinorelbine resistance in non-small cell lung cancer. Oncol Rep (2009) 21:263–8.
55. Attaoua C, Vincent LA, Abdel Jaoued A, Hadj-Kaddour K, Baï Q, De Vos J, et al. Differential involvement of glutathione S-transferase mu 1 and multidrug resistance protein 1 in melanoma acquired resistance to vinca alkaloids. Fundam Clin Pharmacol (2015) 29:62–71. doi:10.1111/fcp.12093
56. Stam RW, van den Heuvel-Eibrink MM, den Boer ML, Ebus ME, Janka-Schaub GE, Allen JD, et al. Multidrug resistance genes in infant acute lymphoblastic leukemia: Ara-C is not a substrate for the breast cancer resistance protein. Leukemia (2004) 18:78–83. doi:10.1038/sj.leu.2403168
57. Kohan HG, Boroujerdi M. Time and concentration dependency of P-gp, MRP1 and MRP5 induction in response to gemcitabine uptake in Capan-2 pancreatic cancer cells. Xenobiotica (2015) 45:642–52. doi:10.3109/00498254.2014.1001809
58. Peng X-X, Tiwari AK, Wu H-C, Chen Z-S. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin J Cancer (2012) 31:110–8. doi:10.5732/cjc.011.10327
59. Ceckova M, Vackova Z, Radilova H, Libra A, Buncek M, Staud F. Effect of ABCG2 on cytotoxicity of platinum drugs: interference of EGFP. Toxicol In Vitro (2008) 22:1846–52. doi:10.1016/j.tiv.2008.09.001
60. Iusuf D, Teunissen SF, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. P-glycoprotein (ABCB1) transports the primary active tamoxifen metabolites endoxifen and 4-hydroxytamoxifen and restricts their brain penetration. J Pharmacol Exp Ther (2011) 337:710–7. doi:10.1124/jpet.110.178301
61. Callaghan R, Higgins CF. Interaction of tamoxifen with the multidrug resistance P-glycoprotein. Br J Cancer (1995) 71:294–9. doi:10.1038/bjc.1995.59
62. Sugimoto Y, Tsukahara S, Ishikawa E, Mitsuhashi J. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci (2005) 96:457–65. doi:10.1111/j.1349-7006.2005.00081.x
63. Filipits M, Pohl G, Rudas M, Dietze O, Lax S, Grill R, et al. Clinical role of multidrug resistance protein 1 expression in chemotherapy resistance in early-stage breast cancer: the Austrian Breast and Colorectal Cancer Study Group. J Clin Oncol (2005) 23:1161–8. doi:10.1200/JCO.2005.03.033
64. MacDonald TJ, Stewart CF, Kocak M, Goldman S, Ellenbogen RG, Phillips P, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: pediatric brain tumor consortium study PBTC-012. J Clin Oncol (2008) 26:919–24. doi:10.1200/JCO.2007.14.1812
65. Basseville A, Tamaki A, Ierano C, Trostel S, Ward Y, Robey RW, et al. Histone deacetylase inhibitors influence chemotherapy transport by modulating expression and trafficking of a common polymorphic variant of the ABCG2 efflux transporter. Cancer Res (2012) 72:3642–51. doi:10.1158/0008-5472.CAN-11-2008
66. Zimmermann C, Gutmann H, Drewe J. Thalidomide does not interact with P-glycoprotein. Cancer Chemother Pharmacol (2006) 57:599–606. doi:10.1007/s00280-005-0087-3
67. Li X, Delzer J, Voorman R, de Morais SM, Lao Y. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos (2011) 39:1161–9. doi:10.1124/dmd.110.037820
68. Hauswald S, Duque-Afonso J, Wagner MM, Schertl FM, Lübbert M, Peschel C, et al. Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin Cancer Res (2009) 15:3705–15. doi:10.1158/1078-0432.CCR-08-2048
69. Chao KC, Chang CC, Yen MS, Wang PH. Anti-tumor activity of histone deacetylase inhibitors and the effect on ATP-binding cassette in ovarian carcinoma cells. Eur J Gynaecol Oncol (2010) 31:402–10.
70. Baltes S, Fedrowitz M, Tortós CL, Potschka H, Löscher W. Valproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J Pharmacol Exp Ther (2007) 320:331–43. doi:10.1124/jpet.106.102491
71. Cerveny L, Pavek P, Malakova J, Staud F, Fendrich Z. Lack of interactions between breast cancer resistance protein (bcrp/abcg2) and selected antiepileptic agents. Epilepsia (2006) 47:461–8. doi:10.1111/j.1528-1167.2006.00453.x
72. Eyal S, Lamb JG, Smith-Yockman M, Yagen B, Fibach E, Altschuler Y, et al. The antiepileptic and anticancer agent, valproic acid, induces P-glycoprotein in human tumour cell lines and in rat liver. Br J Pharmacol (2006) 149:250–60. doi:10.1038/sj.bjp.0706830
73. Jansen M, Veldhuijzen van Zanten SE, Van Vuurden DG, Huisman MC, Vugts DJ, Hoekstra OS, et al. Molecular drug imaging: 89Zr-bevacizumab PET in children with diffuse intrinsic pontine glioma. J Nucl Med (2016) 58:711–6. doi:10.2967/jnumed.116.180216
74. Van Dongen GAMS, Poot AJ, Vugts DJ. PET imaging with radiolabeled antibodies and tyrosine kinase inhibitors: immuno-PET and TKI-PET. Tumour Biol (2012) 33:607–15. doi:10.1007/s13277-012-0316-4
75. Gururangan S, McLaughlin CA, Brashears J, Watral MA, Provenzale J, Coleman RE, et al. Incidence and patterns of neuraxis metastases in children with diffuse pontine glioma. J Neurooncol (2006) 77:207–12. doi:10.1007/s11060-005-9029-5
76. Saito R, Tominaga T. Convection-enhanced delivery: from mechanisms to clinical drug delivery for diseases of the central nervous system. Neurol Med Chir (Tokyo) (2012) 52:531–8. doi:10.2176/nmc.52.531
Keywords: diffuse intrinsic pontine glioma, blood–brain barrier, chemotherapy, convection-enhanced delivery, drug delivery
Citation: El-Khouly FE, van Vuurden DG, Stroink T, Hulleman E, Kaspers GJL, Hendrikse NH and Veldhuijzen van Zanten SEM (2017) Effective Drug Delivery in Diffuse Intrinsic Pontine Glioma: A Theoretical Model to Identify Potential Candidates. Front. Oncol. 7:254. doi: 10.3389/fonc.2017.00254
Received: 14 August 2017; Accepted: 11 October 2017;
Published: 30 October 2017
Edited by:
Kerrie Leanne McDonald, University of New South Wales, AustraliaReviewed by:
Han Shen, Westmead Institute for Medical Research, AustraliaSeunggu Jude Han, Oregon Health & Science University, United States
Copyright: © 2017 El-Khouly, van Vuurden, Stroink, Hulleman, Kaspers, Hendrikse and Veldhuijzen van Zanten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatma E. El-Khouly, Zi5lbC1raG91bHlAdnVtYy5ubA==