 Martino Marco Gabra
Martino Marco Gabra Leonardo Salmena
Leonardo Salmena- 1Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada
- 2Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
Up until the early 2000s, a functional role for microRNAs (miRNAs) was yet to be elucidated. With the advent of increasingly high-throughput and precise RNA-sequencing techniques within the last two decades, it has become well established that miRNAs can regulate almost all cellular processes through their ability to post-transcriptionally regulate a majority of protein-coding genes and countless other non-coding genes. In cancer, miRNAs have been demonstrated to play critical roles by modifying or controlling all major hallmarks including cell division, self-renewal, invasion, and DNA damage among others. Before the introduction of anthracyclines and cytarabine in the 1960s, acute myeloid leukemia (AML) was considered a fatal disease. In decades since, prognosis has improved substantially; however, long-term survival with AML remains poor. Resistance to chemotherapy, whether it is present at diagnosis or induced during treatment is a major therapeutic challenge in the treatment of this disease. Certain mechanisms such as DNA damage response and drug targeting, cell cycling, cell death, and drug trafficking pathways have been shown to be further dysregulated in treatment resistant cancers. miRNAs playing key roles in the emergence of these drug resistance phenotypes have recently emerged and replacement or inhibition of these miRNAs may be a viable treatment option. Herein, we describe the roles miRNAs can play in drug resistant AML and we describe miRNA-transcript interactions found within other cancer states which may be present within drug resistant AML. We describe the mechanisms of action of these miRNAs and how they can contribute to a poor overall survival and outcome as well. With the precision of miRNA mimic- or antagomir-based therapies, miRNAs provide an avenue for exquisite targeting in the therapy of drug resistant cancers.
Introduction
Despite rapid progress in our understanding of the cellular and molecular etiology of cancer and the development of countless new anticancer agents and therapeutic strategies, little has changed in the treatment of many cancers over the last few decades. For instance, the standard of care for acute myeloid leukemia (AML) which consists of combined cytarabine and anthracycline therapy has been fundamentally unchanged for the past 30 years (1). The long-standing presence of this strategy is owed to its effectiveness with a mean response rate up to 70% and a lack of superior strategies for most AML subtypes (2, 3). New targeted therapy strategies including monoclonal antibodies and small molecule inhibitors are constantly being developed; however to date, none of these targeted therapies have proven more effective than the standard of care with the exception of the use of all-trans retinoic acid (ATRA) in acute promyelocytic leukemia (APL) which has become nearly curable in the majority of cases (4).
Notwithstanding, drug resistance is a major therapeutic challenge in the treatment of AML. Failure of initial therapy can be observed in up to 40% of AML patients, and even when initial therapy is effective, up to 70% of patients eventually succumb to their disease due to aggressive relapse within 5 years (5–7).
The cause of poor long-term survival is primarily drug resistance, which is either intrinsic in patients that fail initial therapy or acquired after chemotherapy through selection or acquisition of mutations (8). Indeed, relapsed AML is often composed of cells that have distinct molecular and cytogenetic characteristics leading to deficiencies or perturbations in various pathways associated with therapeutic resistance including DNA damage response and drug targeting, cell cycling, cell death, and drug trafficking pathways due to increased or altered drug targets are commonly observed (Figure 1) (8–10). Consequently, outcomes of relapsed disease are abysmal, which highlights a desperate need for novel therapeutic approaches with potential to overcome or prevent therapeutic resistance.
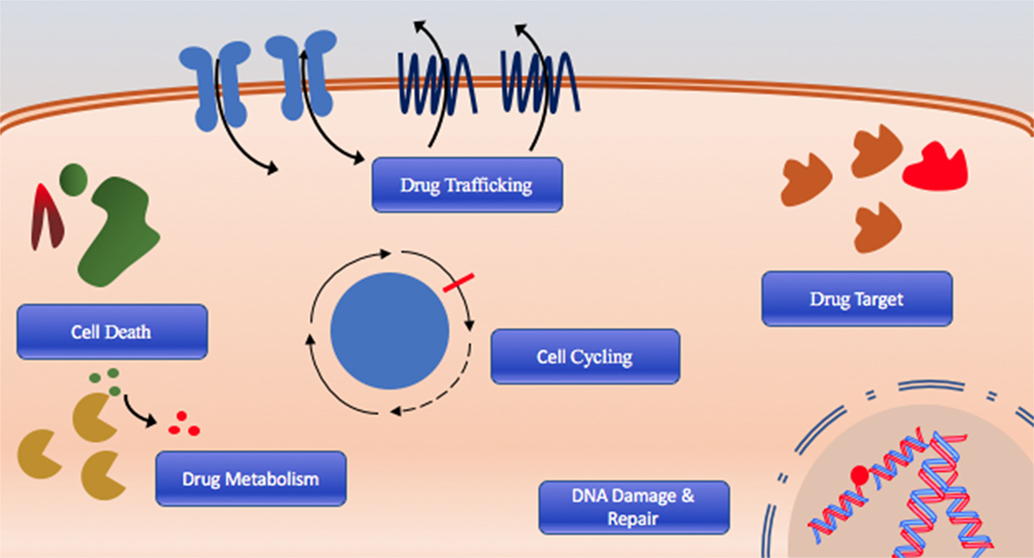
Figure 1. The six hallmarks of drug resistance: DNA damage and repair dysregulation, cell cycle dysregulation, cell death evasion, altered drug metabolism, altered drug target, and dysregulated drug trafficking.
Non-Coding RNAs (ncRNAs) in AML Therapy Resistance
Among several emerging functions, ncRNAs can act as modulators of gene expression through roles in epigenetics, transcription, translation, as well as homology-dependent post-translational regulation of mRNA transcripts (11). The most widely recognized class of ncRNAs are the microRNAs (miRNAs), which are small 18–24 bp dsRNAs that use cellular RNA-interference machinery to suppress protein expression levels by both degrading or blocking translation of mRNA transcripts (12, 13). It has been convincingly demonstrated in numerous cancers that miRNAs can (1) promote or suppress the development of cancer, (2) be of value in prediction of treatment responses and disease prognosis, and (3) be perturbed as a response to chemotherapy (14, 15).
This review is focused on the small ncRNAs, the miRNAs, in drug resistance; however, long non-coding RNAs (lncRNAs) which are typically >200 bp in length and comprise a large proportion of cellular transcribed RNA have numerous emerging functions in AML pathogenesis (16). lncRNA dysregulation in AML have been reported to have consequences for various cellular processes such as proliferation, survival, and migration (17–19) and have been associated with poor clinical outcome (20–23). Furthermore, lncRNAs signatures associated with well-defined cancer types (24). For instance, Homeobox (HOX) transcript antisense RNA (HOTAIR) and HOX antisense intergenic RNA myeloid 1 (HOTAIRM1) are substantially upregulated in AML. It was shown in both cell lines and patient samples that the upregulation of HOTAIR is specifically associated with indirect upregulation of c-kit through sponging of miR-193 (20). Recently, doubt has been raised over the prognostic value of HOTAIR by Sayad et al.; however, in case–control samples, there was a trend toward clinical significance of HOTAIR (25). HOTAIRM1, on the other hand, is thought to behave as an endogenous miRNA-sponge for miR20a, miR-20a/106b, and miR-125b and prevents targeting of ULK1, E2F1, and DRAM2 as demonstrated in luciferase reporter assays (26, 27). In drug resistant AML, however, little is known about the dysregulation of lncRNAs and their respective mechanisms of function.
miRNA Biology
miRNA derive from the transcription of miRNA loci on genomic DNA by RNA polymerases which create a ~80 nt long transcript primary (pri)-miRNA that are then spliced, capped, polyadenylated, and packaged similar to long-stranded transcripts (28). Further splicing and processing by DROSHA and PASHA transform the pri-miRNA into pre-miRNA. When pre-miRNA exits the nucleus through the function of exportin-5, it is folded into a self-bound hairpin secondary structure known as a “stem-loop” (28, 29). At this stage, the 70–100 nt which make up this stem-loop pre-miRNA is cleaved by a cytoplasmic RNase III such as Dicer into a dsRNA dimer which rapidly breaks down into two strands (29). Depending on the stability of the single strand of miRNA either strand can be active (30–32). A functional third miRNA formed from this complex is thought to originate from the loop region, known as loop-miRNA (33, 34). Next, single-stranded mature miRNAs 19–25 nt in length, bind to the argonaute (Ago) proteins which are one member of a complex of proteins collectively known as the RNA-induced silencing complex (RISC) (35, 36).
Guided by miRNAs, Ago and the RISC move to miRNA recognition elements on mRNA which are commonly, but not limited to non-coding 3′-untranslated regions (3′-UTR) (37, 38). Unlike siRNA, miRNA do not require perfect complementary binding; and only binding to the seed-region appears to be a requirement in most cases (39, 40). This comparatively less stringent binding compared to siRNA allows miRNA to regulate the expression levels of multiple RNA transcripts through target promiscuity (39). Once bound to a target, the endonuclease activity of the RISC is activated via the slicer activity of Ago1 (28, 41). Following cleavage, the entire strand is rapidly degraded by endonucleases. Multiple interactions between miRNA and mRNA transcripts are the basis of complex cellular regulatory networks whereby miRNAs control the majority of all protein-coding genes and countless other non-coding genes. In cancer, miRNAs have been demonstrated to play critical roles by modifying or controlling all major hallmarks of cancer including cell division, self-renewal, apoptosis, and DNA damage response among others (42–47).
To date, no comprehensive study examines the role of miRNAs in drug resistant AML. Herein, we describe the miRNAs that have been examined in clinical samples and we highlight miRNA that have been examined mechanistically. Furthermore, we discuss potential miRNA-binding partners of important AML drug resistance machinery found within other cancers to guide future research.
AML Chemotherapy, DNA Damage, and miRNA Dysregulation
The most common treatment for AML includes an anthracycline like daunorubicin and a nucleoside analog like cytarabine in the “7 + 3” regimen where daunorubicin is administered IV for the first 3 days concomitantly to the IV infusion of cytarabine for 7 days (48, 49). The 7 + 3 regimen is termed induction therapy (because of its intent is to induce remission) and has been in place since the 1960s (50). The aim of induction therapy is achieving complete remission (CR), defined clinically as myeloid blast counts in the bone marrow below 5% or minimum residual disease status (49). Efforts to enhance this regimen by escalating dose or adding a third drug has only resulted in increased toxicity with minimal improvement in patient survival. Upon achieving CR, treatment can be consolidated using high doses of cytarabine. Unfortunately, despite undergoing such aggressive chemotherapy regimen with all the associated toxicities and side effect, many patients still relapse within 5 years (48, 49). This is in part due to lack of targeting of leukemic-initiating cells, selection of rare pre-existing resistant AML clones, or the mutagenic effects of the treatments, all of which increase the probability of generating more aggressive AML.
Fundamentally, drug resistance occurs in cells which can evade or withstand treatment. While tumor heterogeneity may explain selection of a pre-existing clone with a favorable mutation, acquired drug resistance is generally defined as the ability of a cell to resist response to the drug to which it was initially responsive. Acquired resistance may be achieved through multiple dosing of the same drug or through as little as a single dose may be explained by the mechanism of drug action.
For instance, anthracyclines used to treat AML such as daunorubicin, doxorubicin, and idarubicin, intercalate DNA, and stall proper DNA replication events (51). Anthracyclines can also target topoisomerase II which normally binds to the scaffold/matrix-associated protein region (S/MAR) to resolve DNA supercoils (52, 53). By binding to topoisomerase II in its open DNA-bound conformation, a stall occurs which can lead to a double-strand break. These double-strand breaks may be fixed aberrantly through non-homologous end joining which can lead to gene mutation. One common mutation in AML, t4:11, occurs at an S/MAR (54–56). This mutation has also been shown to occur in significant proportions in secondary AML patients as well (56). Loss or translocation of the S/MAR may further modulate various miRNAs. As demonstrated by Chavali et al., protein binding to the S/MAR induces histone acetylation that leads to the increased expression of the miR-17-92 cluster and the miRNAs miR-221, miR-93, miR-17, and let-7b (57). As DNA damage is most likely to occur in these regions due to daunorubicin, it is likely that dysregulation of miRNA expression can be due to daunorubicin-induced damage directly.
Cytarabine, on the other hand, is a cytosine analog that terminates translation and replication events. It primarily inhibits cells in S phase (DNA replication) but can also inhibit the progression from G1 phase into S phase (58, 59). It is known that cytarabine is first metabolized into the triphosphate bound product by deoxycytidine kinase (DCK) and other nucleoside analog enzymes whereby it can then incorporate into the DNA. It is shown that its incorporation can often lead to extensive DNA damage including chromatid breaks (60). Stalled replication forks can also lead to bypass mechanisms such as translesion synthesis (61). This method of DNA replication is more error prone and can lead to mutation events as well. Each of these mechanisms can be demonstrated to have direct or indirect consequences for miRNA function.
As described with both drugs, genotoxic effects can lead to breaks that are then repaired using homologous or non-homologous repair mechanisms leading to miRNA alterations and the upregulation of drug resistance mechanisms. Conversely, miRNA which regulate these associated pathways may also contribute to drug resistance when perturbed by increasing tolerance to DNA damage. For instance, ataxia telangiectasia mutated (ATM) is an important DNA damage sensing and DNA damage response protein that has been demonstrated to contribute to chemoresistance (62). In experiments conducted in leukemic HL60, NB4, and K562 cell lines, it was found that the overexpression of miR-181a leads to increased cell proliferation and increased cell cycling through ATM targeting and downregulation (63). Similarly, miR-128 was reported to affect the propensity for DNA damage in AML cells. In a study conducted in HL60 cells, it was observed that the transfection of miR-128 led to increased apoptosis, drug sensitivity, and the amount of DNA damage tolerated; however, the mechanism is yet to be elucidated (64). miR-128 is thought to be upregulated in various cancers, but its levels are reduced in AML cells carrying NPM1 mutations (Figure 2; Table 1) (65, 66).
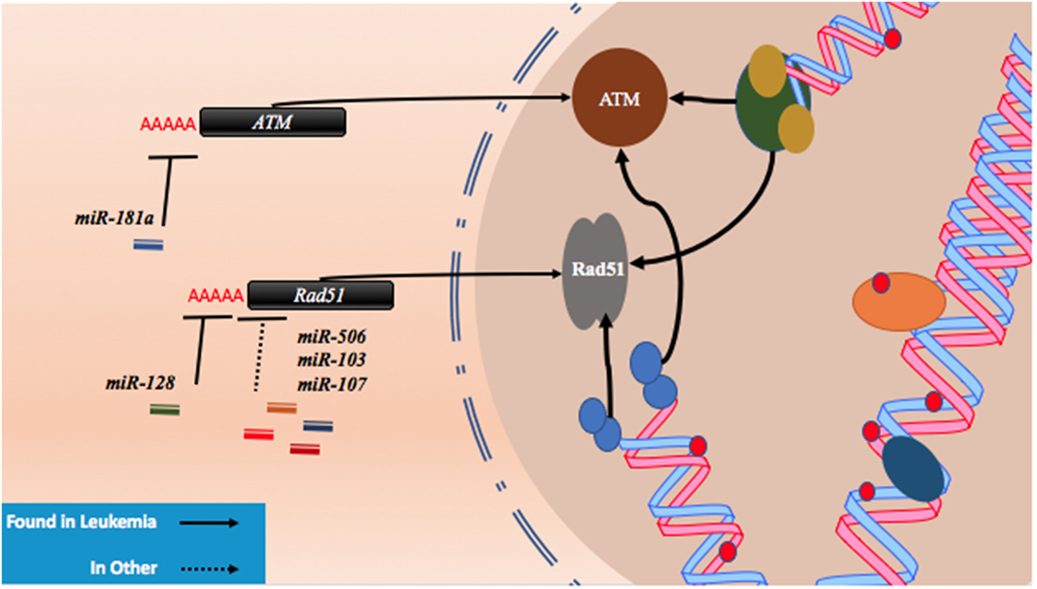
Figure 2. microRNAs (miRNAs) regulate DNA damage response by regulating proteins that behave as DNA damage response elements. In the process of generating DNA damage through genotoxic drugs such as the anthracyclines and the cytosine analogs, the upregulation of effector and response proteins such as ataxia telangiectasia mutated (ATM) and Rad51 is likely to occur. The inhibition of ATM through miR-181a targeting allows tolerance for DNA damage. Reduction of Rad51 through miR-128, miR-506, miR-103, and miR-107 reduces DNA damage response and also contributes to DNA damage tolerance.
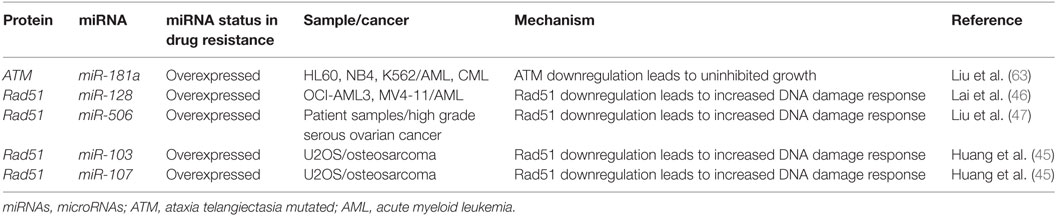
Table 1. miRNAs demonstrated to directly bind to DNA damage regulatory proteins.
Recently, Lai et al. identified a mechanism by which miR-128 is likely targeting Rad51 directly and leading to the increased DNA damage response in OCI-AML3 and MV4-11 AML cell lines. In these experiments, miR-128 led to the sensitization of these cell lines to sapacitabine, a novel oral nucleoside analog prodrug (46). In other cancers, Rad51 has been shown to be a direct target of other miRNAs such as miR-506, miR-103, and miR-107. Clinical significance in chemoresistant high-grade serous ovarian cancers was established for miR-506 while a miRNA mimic library screen revealed miR-103 and miR-107 as strong drug resistance contributors in the U2OS cell line, a model for osteosarcoma (Figure 2; Table 1) (45, 47). To date, proteins that are thought to be integral to the activity of anthracyclines and nucleoside analogs such as topoisomerase II and the DNA polymerases are not known to interact with miRNAs. However, topoisomerase II has been demonstrated to be downregulated in drug resistant subtypes of AML (67, 68). miRNA targeting may prove to be a mechanism of topoisomerase II downregulation, but more research is required to establish important links of miRNA-induced dysregulation of DNA repair machinery in drug resistant AML.
miRNA and Cell Cycling in AML Resistance
The cell cycle represents a series of events that require the input of various checkpoint proteins known as cyclins and cyclin-dependent kinases (CDK) to proceed into division (69). These proteins, in turn, receive input from DNA damage sensing proteins such as ATM/ATR and CHK1/2 (70, 71). The majority of rapidly dividing cancer cells can be found in one of two major phases: the interphase; which consists of G1, S phase (DNA replication) followed by G2; and the M phase, where cells undergo mitosis. Cell cycle manipulation can be a drug resistance mechanism as cell cycle arrest at different phases or quiescence can lead to chemotherapy evasion; however, increased proliferation can also contribute to resistance (72–75).
The process of cell division begins in G1 by the duplication of various proteins, chromatin remodeling, and the verification that the DNA is free of DNA damage. In a healthy cell, if substantial levels of DNA damage are found, ATM/ATR become activated leading to eventual CDK2 inhibition and arrest at the G1/S checkpoint through p21 signaling, where the mechanisms of action of many miRNAs have been elucidated (76). CDK2 has been found to be inhibited by miR-638, where it was demonstrated in HL-60, NB4, and THP-1 that an upregulation of miR-638 leads to a reduction in cell cycling and a differentiation block in APL (77). The differentiation block was found to occur at the G1/S checkpoint and differentiation inducers like ATRA were found to be more effective in cells with miR-638 downregulation (77).
CDK2 has been demonstrated to be a target of various miRNAs in cancer including miR-885-5p, miR-372, and miR-188 (Figure 3; Table 2). In contrast to miR-638 in AML, miR-885-5p was demonstrated to play a tumor suppressive role in neuroblastoma by inhibiting CDK2 and promoting senescence and apoptosis (78). miR-372 demonstrated targeting of both CDK2 and cyclin A1, which is highly expressed during S phase. Like miR-885-5p, miR-372 was demonstrated to play a tumor suppressive role as demonstrated in HeLa cells and tissue samples of cervical cancer (79). miR-188 was demonstrated to directly bind several genes which play a role in cycling such as cyclin D1, cyclin D3, cyclin A2, cyclin E1, CDK2, and CDK4 with varying degrees and it demonstrated modest knockdown of CDK2 relative to the other genes (80). In this study, it was found that the arrest occurs at the G1/S transition and that miR-188 plays a tumor suppressive role (80).
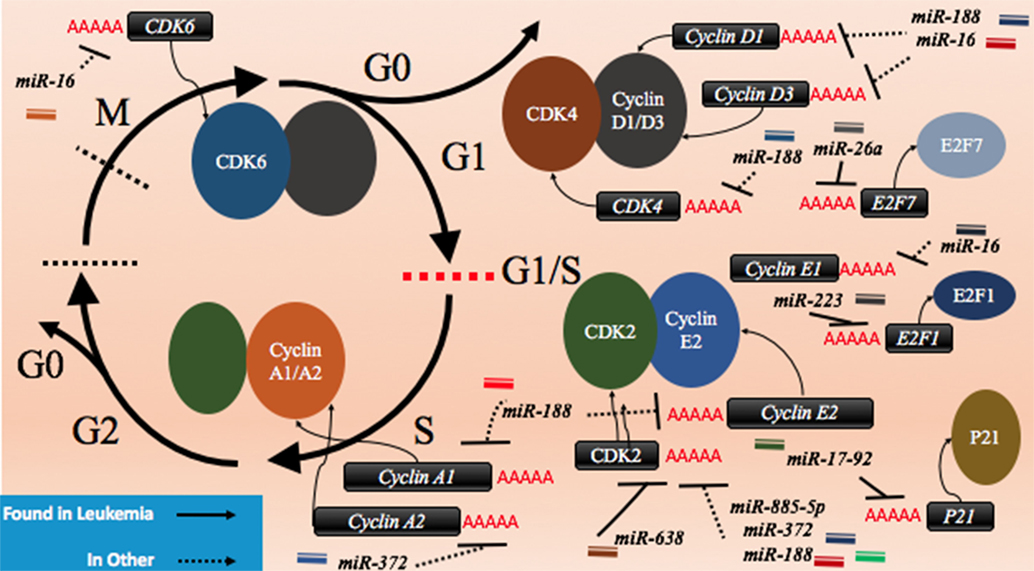
Figure 3. microRNAs (miRNAs) can dysregulate cell cycling mechanisms by dysregulating several phases of the cell cycle, but the majority of known targeting occurs at the G1 and S phases and at the G1/S transition. The downregulation of the cyclins that would normally signal for cell cycling to proceed can be downregulated. Cyclin D1 and cyclin D3 can be dysregulated by miR-188 and miR-16, cyclin E1 can be knocked down by miR-16 while cyclin E2 can be downregulated by miR-17-92 and finally, cyclin A1 and A2 are downregulated by miR-188 and miR-372, respectively. The cyclin-dependent kinases (CDKs) are also adjustable through miRNA targeting and their targeting reduces cycling as well. CDK2 can be downregulated by miR-638, miR-885-5p, miR-372, and miR-188; CDK4 is downregulated by miR-188, and CDK6 is downregulated by miR-16. Effector proteins such as E2F1, E2F7, and p21 can also be downregulated by miRNAs to lead to differentiation blocks. They can be targeted by miR-223, miR-26a, and miR-17-92, respectively.
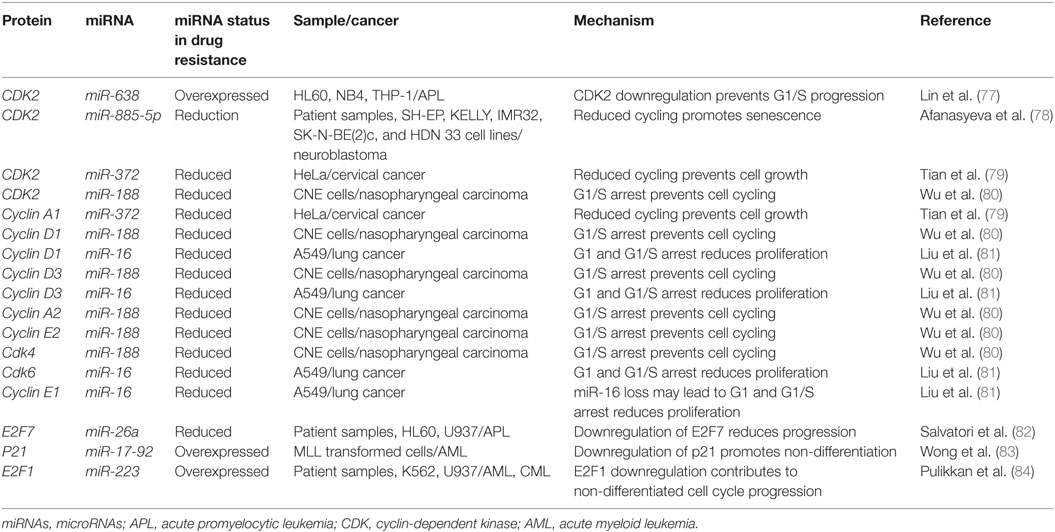
Table 2. Cell cycling gene dysregulations induced by miRNA binding.
Other miRNAs such as the miR-16 family members famously known for downregulation of BCL2 (Figure 4) are also shown to simultaneously directly target several cycling genes such as cyclin D1, cyclin D3, cyclin E1, and CDK6 (Figure 3; Table 2). As demonstrated in the A549 cell line by Liu et al., this targeting and likely the targeting of downstream effectors leads to the arrest in G1 and at G1/S, a phenomenon observed by others (81, 85, 86). The targeting of Cyclin E has since been demonstrated as playing an important role in certain cancers such as cervical cancer and breast cancer (86–89). The miR-15 and miR-16 family may be response elements of E2F1 and as such, may be contributing to a feedback mechanism (90).
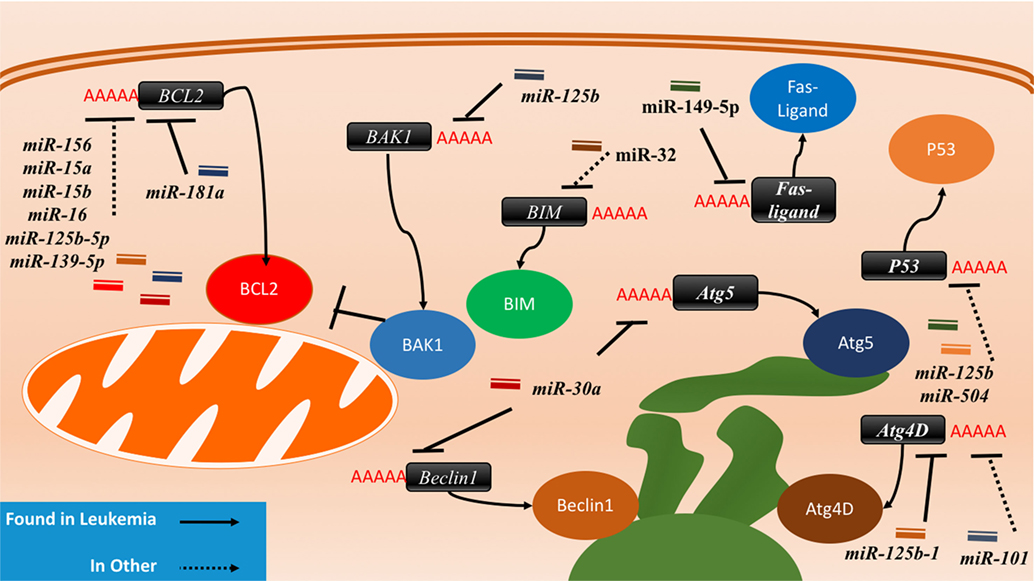
Figure 4. The interactions between microRNAs (miRNAs) and cell death-related proteins in drug resistant cells. Within the apoptosis cell death mechanism, proteins part of the intrinsic or extrinsic pathway can respond to miRNAs to inhibit apoptosis or reduce their regulatory signaling of apoptosis. BCL2, an anti-apoptosis gene, will gain signaling when the associated miRNAs such as miR-156, miR-15a/b, miR-16, miR-125b-5p, and miR-139-5p are lost in the drug resistant cell. The gain of BAK1 miRNA targeting through miR-125b or the gain of BIM targeting through miR-32 will lead to the same effect as well. The Fas-ligand can also be suppressed by miR-149-5p thus ending extrinsic apoptosis signaling. P53 suppression through miR-125b and miR-504 will prevent apoptosis as well. Dysregulating autophagy through increased targeting may increase drug resistance through the binding of miR-125b and miR-101 on Atg4D. miR-30a is known to inversely correlate with Beclin1 and Atg5 in leukemia cell lines, but less is known about the outcome of this interaction.
The transcription factor E2F family may also be a target of miRNAs. E2F7, a transcriptional response element gene implicated in cell cycling, is downregulated by miR-26a in AML (82). This inhibition in turn reduces c-myc transcriptional activation and sequential miR-17-92 reduced transcription, which has previously been implicated in promoting a differentiation block (82, 91, 92). When active, miR-17-92 members may be in part directly targeting p21 and promoting cycling, as demonstrated in MLL transformed leukemic cells by Wong et al. (83). The inhibition of E2F7 may lead to a reduction of miRNAs involved in proliferation such as miR-25, miR-26a, miR-27b, miR-92a, and miR-7 thus behaving as a regulatory mechanism (93).
In other instances, miRNAs can behave as direct inhibitors of their own transcriptional repressor thus behaving as autoregulatory elements. It has been demonstrated by Pulikkan et al. that this is the case for miR-223 and E2F1 regulation (84). E2F1, an important response element in G1/S, can repress transcription of miR-223 which in turn can repress E2F1 (84, 94, 95). The differentiation block observed in APL may be further exacerbated by miRNAs like miR-223 (Figure 3; Table 2). The complexity of interactions within miRNA–mRNA networks demonstrates the need for further analyses elucidating the major pathways of feedback and feedforward signaling.
Cell Death and miRNA
In the majority of blast cells that experience sufficient levels of DNA damage upon chemotherapy, programmed cell death (PCD) will become activated. PCD may take the form of apoptosis or autophagy. Apoptosis is characterized by specific changes in morphology such as cell shrinkage and pyknosis (96). Autophagy, on the other hand, is characterized by cellular degradation and the re-introduction of catabolic products into anabolic processes (97, 98). Autophagy can play both a detrimental and a beneficial role in cancer cells and it can also contribute to the generation of leukemia (98–100). Apoptosis, on the other hand, while it is an essential component of normal cell turnover, only its downregulation will often be a major contributor for aberrant cancer growth and its further suppression can lead to drug resistance.
miRNA and BCL2 Family Members
miRNA-associated dysregulation of apoptosis has been observed in drug resistant AML cells. Given that AML is often characterized by aberrant DNA repair and maintenance, tolerance of these damaged lesions is observed through the downregulation of pro-apoptotic markers and damage sensors, or the upregulation of antiapoptotic factors. Of the apoptosis-related families, the BCL2 protein family is the most well described in miRNA dysregulation driven in AML. The BCL2 protein itself is commonly considered as a crucial anti-apoptosis gene as it inhibits the mitochondrial pro-apoptotic proteins such as Bak and Bax. While it can be dysregulated or mutated in cancers, it is observed that dysregulation may also occur in the development of drug resistance. Many miRNAs including miR-15/miR-16, miR-125b-5p, miR-139-5p, miR-145, and miR-181a have been shown to suppress the translation of BCL2 and decrease the propensity for activation of apoptosis (Figure 4; Table 3).
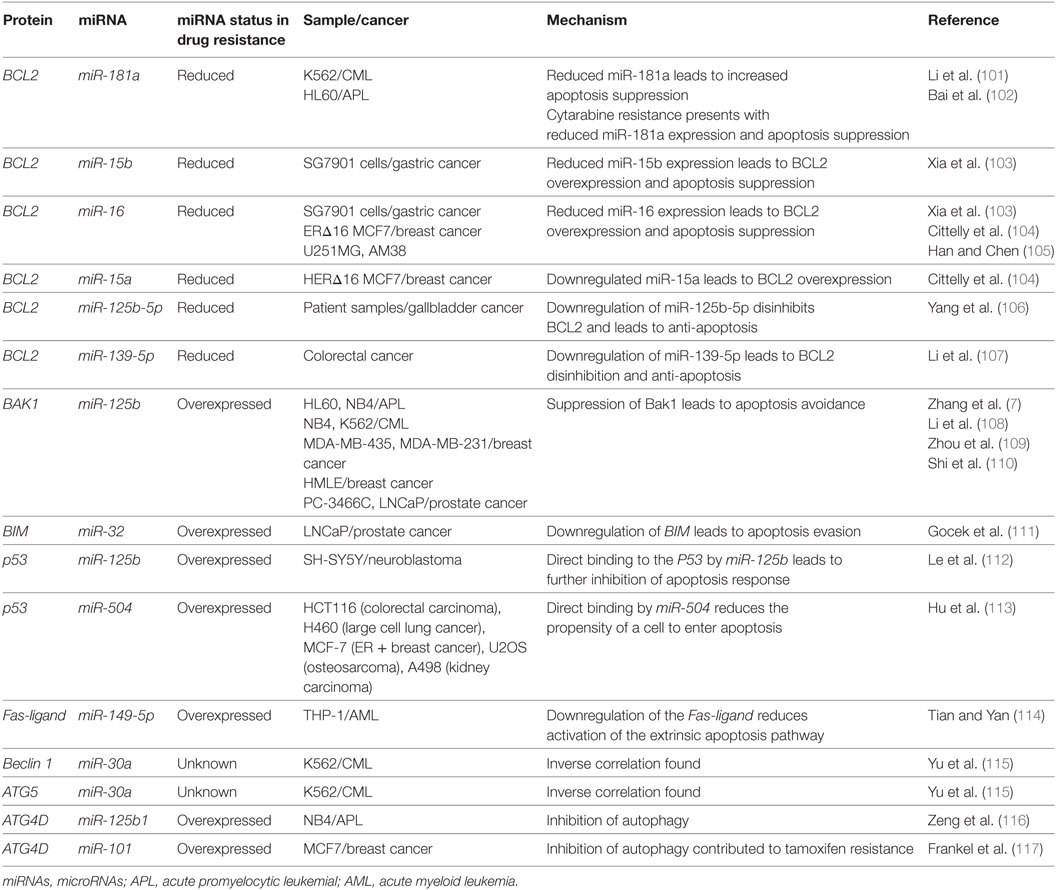
Table 3. The interactions of miRNAs with cell death-related proteins.
Of the BCL2-targeting miRNAs, only miR-181a has been shown to do so in AML cells. In K562 CML cells, it was demonstrated by Li et al. that the drug resistant form had 40% of the miR-181a levels found in the parental cell line. When the parental cells were transfected with a miR-181a inhibitor, resistance developed (101). In a separate study conducted by Bai et al. in cytarabine resistant HL60, it was found that the resistance phenotype can be also be attributed to reduced BCL2 targeting by miR-181a, whereas its ectopic expression sensitizes the cells to treatment to cytarabine (102). Other studies of miR-181a in AML have also demonstrated that it is often downregulated in drug resistant AML, that it can serve as an independent prognostic marker and potentially modulate the interaction with natural killer cells as well (118–122). In molecular poor risk group AML with FLT3-ITD mutations, it was demonstrated that high miR-181a also strongly predicted better survival (123).
The miR-15/16 have been shown to suppress BCL2 in multiple cancers including gastric cancer, breast cancer, and glioma and the loss of this locus has also been observed in CLL (124–127). Xia et al. demonstrated that miR-15b and miR-16 are lost in vincristine resistant SGC7901 cells, a gastric cell line (103). Cittelly et al. later demonstrated that in a common mutation of the HER2 gene, HERΔ16, representative of 30% of HER2 dysregulations in estrogen receptor positive breast cancers, the downregulation of miR-15a and miR-16 is observed (104). In MCF-7 cells ectopically expressing this mutant variant, it was shown that tamoxifen resistance may be in part due to the reduced regulation of BCL2 by miR-15a and miR-16, which leads to apoptosis evasion (104). In glioma cells that are resistant to temozolomide, it was demonstrated that the loss of miR-16 specifically can contribute to resistance in the U251MG/Temozolomide resistant cell line and that the blocking of miR-16 in the temozolomide sensitive AM38 cell line increased resistance by de-repressing BCL2 (105).
In a genome-wide gene expression analysis of gallbladder cancer clinical samples, miR-125b-5p was found to be statistically downregulated in cisplatin resistant patients (N = 6). Analyses demonstrated that this miRNA can directly bind to the 3′UTR of BCL2, contribute to cisplatin desensitization, and increase tumor formation in mice (106). A similar analysis of patient samples conducted in colorectal cancer demonstrated that miR-139-5p inhibits the epithelial-to-mesenchymal transition and contributes to drug resistance by downregulating BCL2 (107). Bioinformatic studies also demonstrate binding of other miRNAs to the BCL2 mRNA as putative mechanisms of miRNA-induced downregulations. For instance, bioinformatic analysis of miR-451 through miRBase and miRanda identified it as an inhibitor of BCL2 (128). Similarly, in paclitaxel-resistant breast cancer, it was demonstrated that miR-451 may also inhibit BCL2.
The BCL2 antagonist/killer 1 (Bak1) protein is upregulated in the progression of apoptosis in normal cells; in drug resistant cancers, however, it is observed that there is Bak1 suppression through miR-125b binding. The binding of miR-125b to the Bak1 transcript was initially examined in the prostate cancer cell lines PC-346C and LNCaP in the context of androgen-independent signaling, but effect on drug resistance was not examined (110). In APL, miR-125b was demonstrated to be clinically relevant, in CML mice models, and it was further demonstrated that direct suppression occurs in the cell lines NB4, HL60, and K562 (7, 108). A similar link between miR-125b and Bak1 was established in MDA-MB-435 and MDA-MB-231 where it was demonstrated that miR-125b is capable of Bak1 suppression in Taxol resistant cells (109). The mechanism of miR-125b upregulation was further elucidated to be through Wnt signaling and specifically through Snail binding; an upregulation thought to also occur in cancer stem cells (129).
The Bcl-2-like protein 11, also known as, BIM, has been demonstrated to be a direct target of miR-32 in a previous study in LNCaP prostate cancer cells. This pro-apoptotic protein can be downregulated by miR-32 and consequently lead to resistance and increased cell proliferation (130). Studies in the AML cell lines HL60 and U937 also demonstrated an inverse correlation between miR-32 and BIM (111).
miRNA and P53 Regulation
The tumor-suppressor protein p53, often referred to as guardian of the genome is dysregulated in 50% of all cancers. In wild-type cells, p53 is often suppressed and destabilized by mdm2, mdm4, and mdmx which behave like E3 ligases, marking P53 by ubiquitination for degradation. Phosphorylation of p53 by ATM leads to its stabilization and release from the mdm protein family. p53 can then behave as a transcription factor by activating apoptosis-related genes (both intrinsic and extrinsic), cell cycle arrest related genes or DNA repair related genes and it can directly bind to the mitochondria to participate in membrane permeabilization (131, 132).
P53 has been identified as a direct target of miRNA binding by miR-125b and miR-504. miR-125b was shown to directly decrease P53 transcript levels and consequently decrease apoptosis response to irradiation in neuroblastoma cells and in lung fibroblasts (Figure 4; Table 3) (112). miR-504 was first computationally predicted and then demonstrated in various cell lines including HCT116 (colorectal carcinoma), H460 (large cell lung cancer), MCF-7 (ER + breast cancer), U2OS (osteosarcoma), and A498 (kidney carcinoma) cells to directly target the 3′UTR of P53 (113). P53 is also importantly downregulated through indirect ways by miR-34a, which is thought to play a crucial role in P53’s pro-apoptotic abilities (133, 134). It has been demonstrated that miR-34a can indirectly increase P53 by inhibiting P53 negative regulators such as SIRT1 in colon cancer as demonstrated by Yamakuchi et al. and likely through binding of mdm4 as well, as predicted bioinformatically (135–137).
Furthermore, it has been demonstrated that P53 transcriptionally activates miR-34a which in turn modulates and fine tunes P53’s signal (134). Consequently, the relationship between miR-34a and P53 is context dependent as the mutation status of P53 can influence the response and outcome of miR-34a activity (138). In the study conducted by Rücker et al., it was found that P53 alterations were the most common molecular lesions which coincided with complex karyotypes in AML (138). Low miR-34a and P53 alterations were shown to have the poorest clinical outcome in terms of drug resistance and survival. The low expression was shown to also correlate with a specific gene expression profile consisting of P53-associated proteins. In complex karyotypes that did not have a P53 alteration, high miR-34a predicted a poor overall survival while loss of P53 and high miR-34a predicted better outcome (138). The interplay between miR-34a and P53 demonstrates that the same miRNA can have opposite effects depending on the mutation status of the associated mRNA and highlights the necessity of describing miRNA activity in relation to the activity of associated mRNA.
Other Apoptosis-Related Proteins
For the apoptotic extrinsic pathway, it was reported by Tian et al. that miR-149-5p can directly downregulate the Fas-ligand and reduce the levels of the apoptosis effector proteins caspase-8, caspase-2, and caspase-3; however, no effect on drug resistance is demonstrated (114). It is possible that miR-181a and miR-21 can suppress the Fas-ligand in cancers as they are shown to interact with the Fas-ligand in bone marrow-derived mesenchymal cells and cardiomyocytes, respectively (139, 140). The binding of miRNAs to caspases has also not been examined closely in cancers, but in an experiment conducted by Zhang et al. in endothelial cells demonstrated caspase-3 downregulation due to let-7g inhibition. As such, this targeting reduced the progression of apoptosis and lead to higher tolerance of oxidative stress (141).
Autophagy and miRNA
Autophagy is regulated by many autophagy related (ATG) proteins which play various roles in the formation of the autophagosome (100, 142). It has been observed that miRNAs can likely play a role in autophagy and that AML cells can have dysregulated autophagy (97). To date, two miRNAs have been found to associate with autophagy in leukemia: miR-30a and miR-125b1 (115, 116). miR-30a is inversely correlated with Beclin1 and ATG5 in K562, but direct binding and relevance to drug resistance is yet to be demonstrated (115). miR-125b1, on the other hand, can bind RAM2, ATG4D, and UVRAG as demonstrated in NB4 cells (116). The activity of miR-125b1 in this circumstance contributed to inhibition of autophagy through ATG4D. In other cancers, ATG4D was found to be a direct target of miR-101 and its inhibition may contribute to 4-hydroxytamoxifen sensitization in the breast cancer lines MCF7 and T47D (117).
Drug Metabolism and Chemoresistance
Drug activation and drug clearance can be altered in cells to reduce the effective dose of the drug. These proteins are highly varied, but can largely be characterized into two major classes: the phase I and the phase II class of enzymes. Phase I enzymes typically perform redox reactions or hydrolysis reactions. While they often precede phase II enzyme activity, this is not always required. Phase II enzymes typically increase the polarity of the molecule through the addition of a sub-group such as UDP-glucoronate, sulfate, methane, acetate, or glutathione (143).
Anthracyclines are active drugs that can carry out their genotoxic effects directly. Their metabolism into the semiquinone form, the hydroxyaglycone form, deoxyaglycone form, or the alcohol form will decrease its likelihood of intercalating DNA as it reduces the anthracycline’s lipophilicity. It is unclear whether the anthracyclines lose efficacy through metabolism. As demonstrated from cardiotoxicity assays in rat and rabbit, the metabolites may have differing effects depending on the organism in question and the rate of metabolism. In rats, the alcohol form may retain some activity, but the effects of the active drug are more pronounced (143, 144). In rabbits, the alcohol derivative is implicated in the cardiotoxic effects of the anthracyclines (143, 145). It is thought that the enzymes CBR1/3 and AKR1A1/C3 can act on the parent drug to form the alcohol form. The hydroxaglycone and the deoxyaglycone forms can be generated in part by certain cytochrome P450 (CYP) enzymes such as CYP3A4/5, CYP2D6, xanthine dehydrogenase (XDH), and NAD(P)H quinone dehydrogenase 1 (NQO1) (146–150). XDH, NQO1 along with nitric oxide synthase can help in generating the semiquinone form (151–153).
Cytarabine and other nucleoside analogs require phosphorylation through DNA/RNA synthesizing enzymes such as the nucleoside kinases to become candidates for incorporation into nascent DNA. Cytarabine requires activation by several enzymes including deoxycytidine monophosphate kinase, nucleoside diphosphate kinase, and the rate limiting DCK (Figure 5) (154, 155). It is then metabolized by various enzymes including CYP3A4, 5′ nucleotidase, cytidine deaminase, and deoxycytidylate deaminase (154, 155).
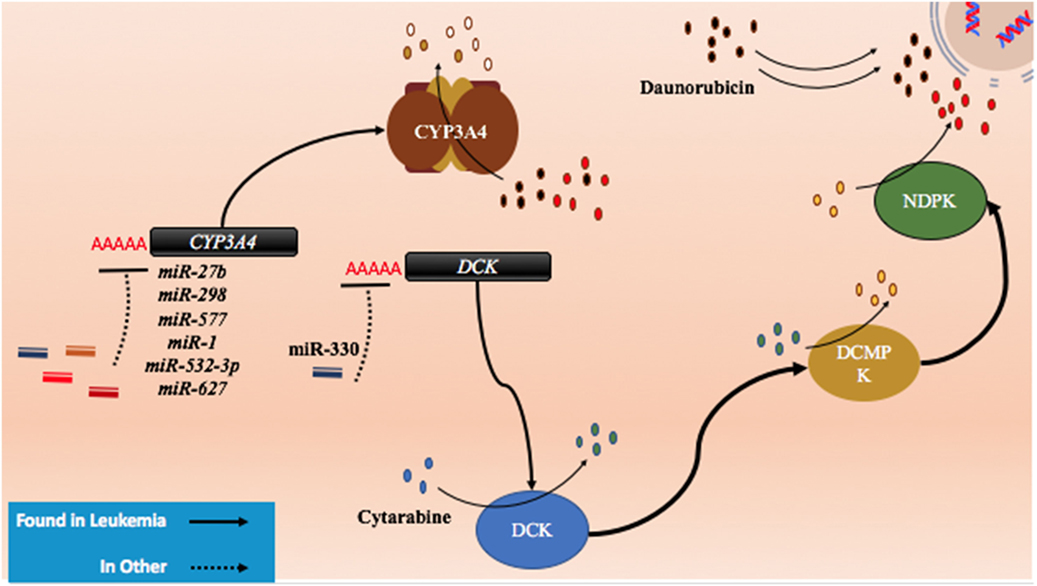
Figure 5. The role of metabolism and microRNA (miRNA) in daunorubicin and cytarabine treatment. While daunorubicin is an active drug, cytosine requires bio-activation. As a cytosine analog, it must undergo three phosphorylation steps to become fully activated and capable of incorporating into the genome. The deactivation of daunorubicin and cytarabine is partially dependent on the cytochrome P450s and they commonly share CYP3A4 in their pathway of degradation. In other cancer, CYP3A4 has been shown to be targeted by miR-27b, miR-298, miR-577, miR-1, miR-532-3p, and miR-627. In the pathway of cytarabine activation, deoxycytidine kinase (DCK) has been shown to be downregulated by miR-330 in other cancers.
Currently, there are few publications that highlight the role of miRNAs in anthracycline and cytosine analog metabolizing enzymes in AML. However, certain miRNAs such as miR-27b and miR-298 have demonstrated direct binding of CYP3A4 in a pancreatic cell line and miR-577, miR-1, miR-532-3p, and miR-627 were found to target CYP3A4 in HEK 293T cells (Figure 5; Table 4) (156, 157). In gemcitabine resistant colon and lung cancer cells, Hodzic et al. established a correlation between miRNA-330 and DCK expression levels (158). Further studies interrogating the role of computationally predicted miRNAs and miRNAs discovered in other cancer subtypes may help establish a role for miRNAs in metabolism in drug resistant AML.
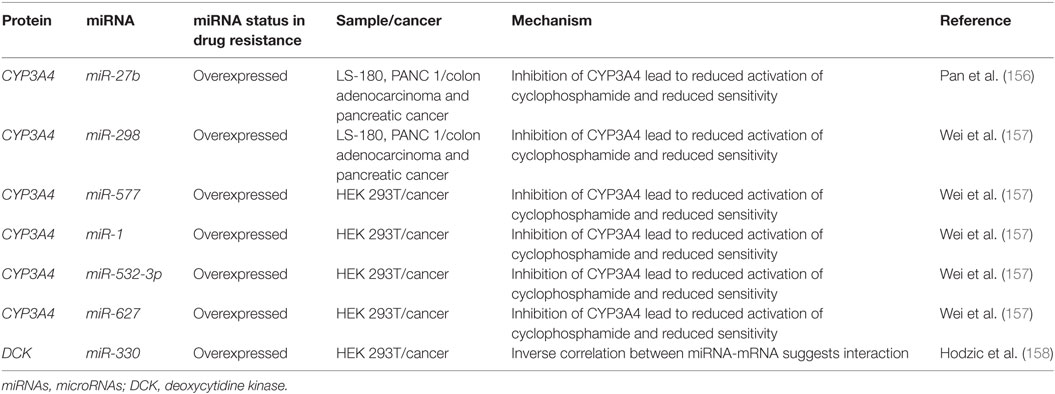
Table 4. miRNA targeting proteins involved in drug metabolism.
Drug Trafficking and miRNA in Chemoresistance
The trafficking of the anticancer drugs can dramatically modulate treatment response as a reduction in influx or an increase in efflux will reduce the effective intracellular concentration of drug. Due to the lipophilicity of the anthracyclines, they can freely diffuse into the cell, but they can also bind to the SLC22A16 solute pump to enter cells (153, 159–161). While there are some reports that suggest the role of SLC22A16 in bleomycin resistance, the role of this transporter in anthracycline resistance is yet to be explored (162, 163). As such, while there are predicted miRNA-binding sites on this protein, none are yet confirmed.
Cytarabine and other cytosine analogs, on the other hand, necessitate the function of nucleoside transporters to enter the cell. The nucleoside transporters are composed of six major protein families: human equilibrative nucleoside transporters (hENTs) and human concentrative nucleoside transporters (hCNTs), organic anion transporters, organic cation transporters, peptide transporters, and the multidrug resistance protein family (MRP), with the hCNTs and hENTs playing the most major role of cytarabine import (164–166). In childhood leukemia, the hENT protein family has demonstrated to correlate with cytarabine resistance, but miRNA-mediated mechanisms are yet to be confirmed (167, 168).
In contrast, many efflux pumps can confer resistance to diverse and seemingly unrelated drugs and the characterization of several of these transporters has been extensive in AML. These ATP-binding cassette (ABC) proteins can be upregulated in the drug resistant forms of cancers and as such, the downregulation of miRNAs that target efflux pumps can contribute to resistance. Within this class, ABCB1 (P-glycoprotein, MDR1), ABCC1 (MRP1), ABCC2 (MRP2), and ABCG2 (BCRP) have been the most extensively examined out of 48 proteins within this functionally similar class (Figure 6) (169, 170). Indeed, previous treatments of drug resistant AML centered on the targeting of P-glycoprotein. It has been clearly demonstrated that the surface expression of P-glycoprotein is inversely proportional to the concentration of intracellular daunorubicin in blast cells and in tissue culture samples; however, blocking of P-glycoprotein did not yield positive results in clinical settings (171).
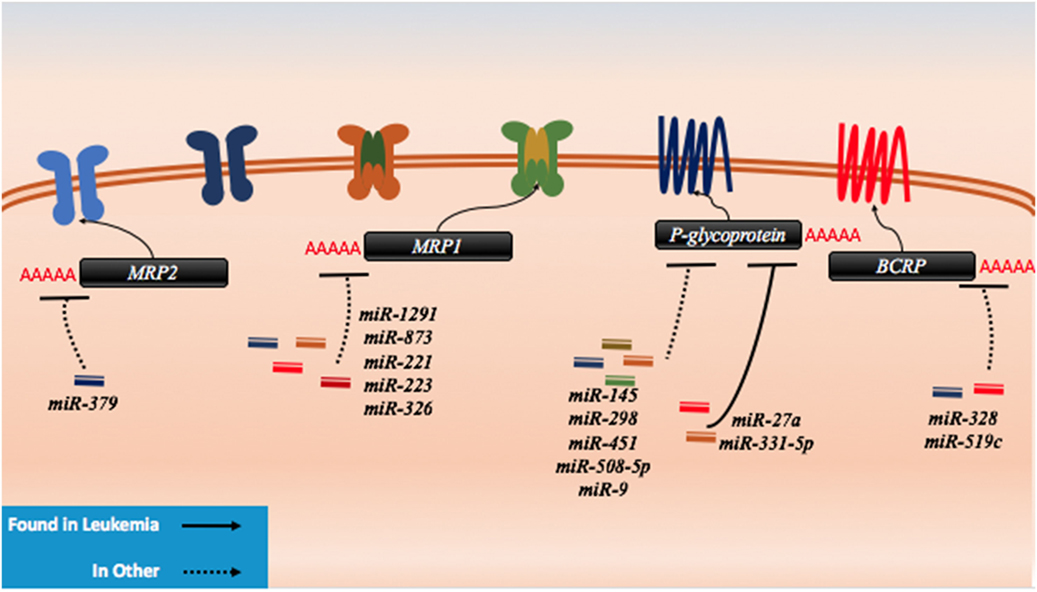
Figure 6. microRNAs (miRNAs) have been shown to dysregulate drug efflux mechanisms in both leukemia and other cancer. There are no known miRNA regulators of the drug influx proteins. In leukemia, P-glycoprotein has been demonstrably targeted by miR-27a and miR-331. In other cancers, P-glycoprotein has been shown to be regulated by miR-145, miR-298, miR-451, miR-508-5p, and miR-9. MRP1 has been targeted by miR-1291, miR-873, miR-221, miR-223, and miR-326, while MRP2 has been shown to be targeted my miR-379. The last protein to exhibit miRNA binding in lab setting is BCRP which has been shown to be a target of miR-328 and miR-519c.
P-glycoprotein can be targeted by several miRNAs including miR-27a, miR-331-5p, miR-145, miR-298, miR-508-5p, miR-9, and miR-451 (Figure 6; Table 5). In leukemia, only miR-27a and miR-331-5p have been demonstrated to bind to P-glycoprotein in the K562 and HL-60 leukemia cell lines (172). In ovarian and cervix cell lines, it was demonstrated that the downregulation of both miR-27a and miR-451 can lead to downregulation of P-glycoprotein; however, in the case of miR-27a, this contradictory effect on P-glycoprotein is likely in part due to targeting of HPK2 upstream (173). This was further phenotypically demonstrated by the reduced uptake of intracellular dyes and by the response to cisplatin and methotrexate (174). In more recent experiments conducted in hepatocellular carcinoma cells, in addition to direct binding to P-glycoprotein and HPK2 binding, it was demonstrated that the inhibitory effect of miR-27a on P-glycoprotein may also be partially attributed to upstream modulation of the β-catenin pathway through direct binding of SFRP1 and potentially through FZD7 as well (172, 175). It is possible and likely that P-glycoprotein is involved in processes that are unrelated to drug trafficking as well such as apoptosis which may explain the contradictory expression in different cancers and the varying predisposition of its mutagenicity in certain cancers; however, its actions remain unclear (176).
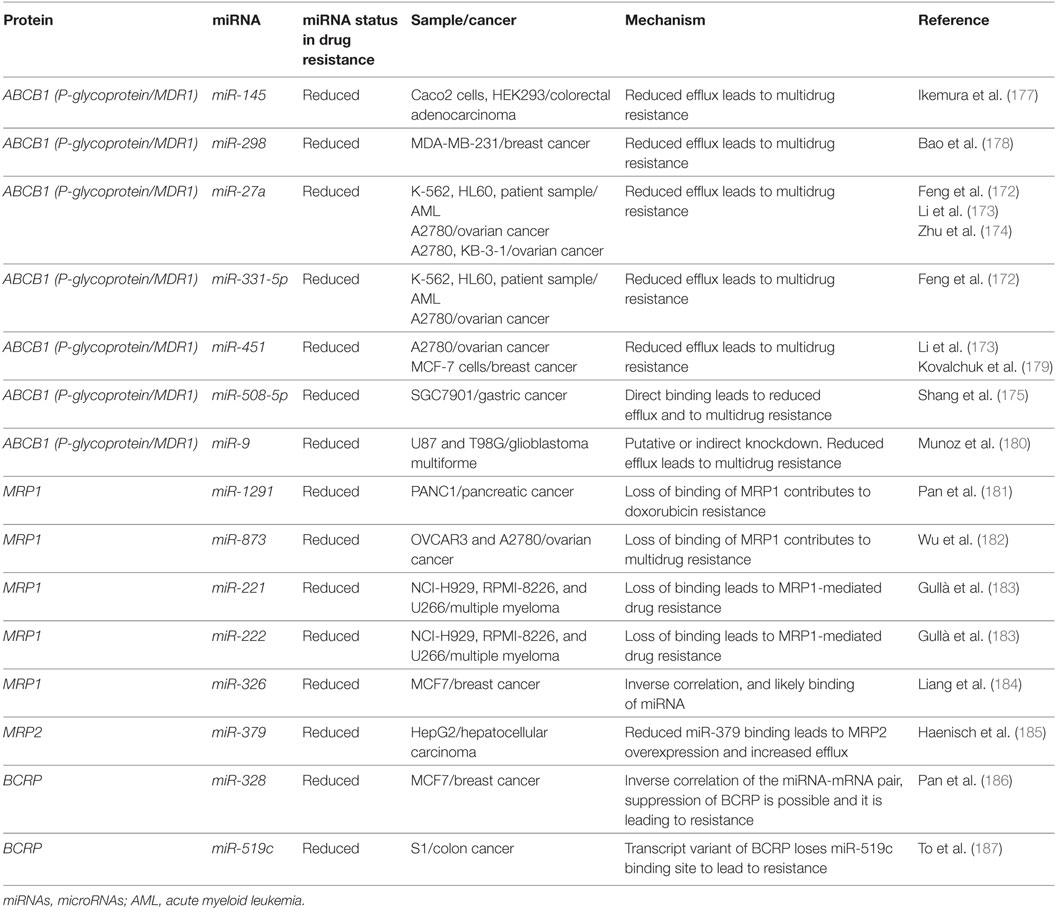
Table 5. Drug trafficking gene disinhibitions caused by loss of miRNAs can lead to drug resistance.
Direct binding of miR-451 to P-glycoprotein transcripts was demonstrated in MCF-7 cells, where it was demonstrated that it could contribute to doxorubicin resistance; however, this has not yet been demonstrated to be clinically significant in cancer patients (179). In colon cancer cell-derived cell lines and HEK293 cells, it was demonstrated that miR-145 can play a role in the repression of P-glycoprotein and increase the efflux of rhodamine 123 (177). miR-298 was demonstrated to directly bind to the transcript in resistant breast cancer cell lines (178). This suggests that it may play a role in patients, but follow-up studies are needed. miR-508-5p was demonstrated to directly bind to P-glycoprotein in gastric cancers and its upregulation was found clinically as well (175). It has also been suggested by Munoz et al. that miR-9 may also target P-glycoprotein and confer resistance to temozolomide in glioblastoma multiforme cells (180). These miRNAs may also prove to be relevant in AML, but no studies have been attempted to date.
While the MRP1 gene has not demonstrated miRNA binding in AML, it was demonstrated in other cancers that the MRP1 gene can also be targeted by miRNAs such as miR-1291, miR-873, miR-221/222, and miR-326 (Figure 6; Table 5). In an analysis conducted by Pan et al., doxorubicin treatment of pancreatic cancer cells demonstrated that miR-1291 will become upregulated and target MRP1 directly (181). MRP1 downregulation contributes to multidrug resistance as well in other cancers such as ovarian cancer (182). It was recently demonstrated through in vivo and in vitro studies that miR-873 can be biologically significant in paclitaxel and cisplatin resistance in ovarian cancer cell lines where it can directly bind to MRP1 (182). Consequently, miR-873 is often downregulated in MRP1-dependent ovarian cancers. In melphalan-refractory multiple myeloma cells, Gulla et al. demonstrated that miR-221/222 may be binding and reducing MRP1 thus contributing to drug resistance (183). Finally, miR-326 was inversely correlated with MRP1 in multidrug resistant MCF7 cell lines (184). Less is known about MRP2 targeting by miRNAs, but in the liver cell line HepG2, miR-379 was demonstrated to be highly upregulated and to target MRP2 directly as a response to Rifampicin resistance (185).
BCRP, in contrast, has been shown to be a target of miR-520h, miR-328, and miR-519c and to potentially play a role in the hematopoietic system (Figure 6; Table 5). In CD34+CD38− hematopoietic stem cells, it was demonstrated that miR-520h is enriched compared to CD34+ cells alone and that it can directly target BCRP in this fraction (188). An examination of miR-520h in leukemic cells and AML may demonstrate a similar trend of upregulation and a contribution of miR-520h to drug resistance, but more experiments are required. In mitoxantrone-resistant MCF-7 cells, Pan et al. showed that the expression of miR-328 is inversely correlated with BCRP and that it is directly suppressing BCRP, leading to resistance (186). To et al. demonstrated that miR-519c may play a role in downregulating BCRP in S1 colon cancer cell lines; however, they demonstrated that binding of miR-519c was limited to a longer form of the transcript only found in their parental cell line compared to their mitoxantrone-resistant counterpart (187, 189). This study highlights the importance of splice variants and how they may gain or lose miRNA-binding sites and thereby contribute to resistance.
Implications in Treatment
Drug resistance is only a single aspect of clinical setbacks; however, it is a major contributor to therapy failure. Although treatment has improved substantially in some cancers in the past few decades, many other cancer types continue to demonstrate substantial patient populations that relapse after an initially successful treatment. While we focused on the regulation of drug resistance-associated miRNAs common between different cancers and drug classes, there are likely various miRNA that are specific to different drug treatments and cancers. However, the miRNA dysregulations discussed may have therapeutic value beyond AML. Furthermore, although we describe several drug resistance proteins, our analysis only focused on miRNA specifically implied in drug resistance where they were demonstrated to have direct activity and as such, the list is not exhaustive (190).
There are also many other molecular changes that occur in the development of drug resistance such as copy number variations, aberrant methylation, and aberrant post-transcriptional and post-translational processing (191, 192). The modulation of miRNAs offers a new perspective on drug resistance as miRNA replacement therapy and miRNA inhibition therapy raises the potential of developing new and effective drug therapies. Subtle miRNA changes can lead to significant changes in protein-coding gene expression and can consequently lead to changes in tumor progression and patient outcome. Experimental success in vitro and in vivo models may point to the likely coming of more miRNA-based clinical trials.
Previously, Mrx34 emerged as a promising therapy for the treatment of unresectable primary liver cancer. Due to multiple immune-related adverse events, this therapy was terminated in phase I although there was evidence of benefit in a subset of patients (193). Its promise came from being a p53-response element that was thought to mediate p53’s antitumor effects and consequently affecting downstream signaling in proliferation arrest and induction of apoptosis by targeting c-MYC, CDK6, and c-MET (194). However, recent research now demonstrates that it may not always behave as a tumor-suppressor either and furthermore, p53 may also be a direct target of miR-34a (138, 195). In liver cancers with β-catenin mutations, it is demonstrated that LNA-34a, a miR-34a inhibitor, displays antitumor effects. This is suggested to occur through blocking HNF-4α targeting which in turn decreases cyclin D1 and inhibits proliferation (196, 197).
A miR-16 mimic has also been recently introduced in patients in an open-label phase I clinical trial for mesothelioma and non-small cell lung cancer (NSLC). miR-16 was shown to be dysregulated in many different cancers (87, 89, 90, 103–105, 124, 198). A directed analysis in mesothelioma showed that miR-16 is reduced in patient samples and that a knock-in of a miR-16 mimic is tumor suppressive (198). This observation was repeated in xenografted mice with high success (198). Currently, there are no miRNA-based therapies for drug resistant AML or AML-related diseases.
Currently, there are two miRNA-based therapies intended to treat different cancers that are on-going or with pending results. MesomiR-1, a miR-16 mimic, was in a multi-center Phase I trial intended to treat mesothelioma and NSLC. This trial has been completed as of January 2017 and the results are currently pending. MRG-106 is a miRNA inhibitor that targets miR-155 that is currently being examined in cutaneous T-cell lymphoma and mycosis fungoides. Like mesomiR-1, it is also currently in phase I. It is thought to block the action of miR-155 from targeting tumor suppressors such as C/EBPβ and altering the TGF-β response (199). This study is currently still recruiting patients. These studies may offer promise of miRNA treatment as therapy and pave the way for future studies similar in nature.
Concluding Statement
Today, the main hurdle for miRNA-based therapies remains to be the method of delivery. Many types of viruses are thought to be potentially useful for treatment and many stabilizing modifications such as phosphorothioate, methyl- and fluoro-substitutions on RNA species may help to overcome this hurdle (200, 201). Given the diverse set of roles that miRNAs play in regular cellular function, it is evident that clear elucidation of specific miRNA mechanisms may be required before their integration into modern cancer therapy (202). In contrast, due to the dependence and overexpression of a few coding mRNA in tumorigenic cells, it is possible that miRNAs may have a higher therapeutic index. miRNAs may prove to be an important addition to treatment in the years to come to treat drug resistant cancers in the future.
Author Contributions
MG contributed to the research, figure design, and writing of manuscript. LS contributed to the research, editing, and overall design of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
LS is a recipient of a Tier II Canada Research Chair. MG is supported by a scholarship from the Centre for Pharmaceutical Oncology at the Leslie Dan Faculty of Pharmacy, University of Toronto.
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373(12):1136–52. doi:10.1056/NEJMra1406184
2. Büchner T, Schlenk RF, Schaich M, Döhner K, Krahl R, Krauter J, et al. Acute myeloid leukemia (AML): different treatment strategies versus a common standard arm – combined prospective analysis by the German AML Intergroup. J Clin Oncol (2012) 30(29):3604–10. doi:10.1200/JCO.2012.42.2907
3. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J (2016) 6(7):e441. doi:10.1038/bcj.2016.50
4. Coombs CC, Tavakkoli M, Tallman MS. Acute promyelocytic leukemia: where did we start, where are we now, and the future. Blood Cancer J (2015) 5:e304. doi:10.1038/bcj.2015.25
5. Crespo-Solis E, Contreras-Cisneros J, Demichelis-Gómez R, Rosas-López A, Vera-Zertuche JM, Aguayo A, et al. Survival and treatment response in adults with acute promyelocytic leukemia treated with a modified international consortium on acute promyelocytic leukemia protocol. Rev Bras Hematol Hemoter (2016) 38(4):285–90. doi:10.1016/j.bjhh.2016.08.002
6. Verma D, Kantarjian H, Faderl S, O’Brien S, Pierce S, Vu K, et al. Late relapses in acute myeloid leukemia: analysis of characteristics and outcome. Leuk Lymphoma (2010) 51(5):778–82. doi:10.3109/10428191003661852
7. Zhang H, Luo X-Q, Feng D-D, Zhang X-J, Wu J, Zheng Y-S, et al. Upregulation of microRNA-125b contributes to leukemogenesis and increases drug resistance in pediatric acute promyelocytic leukemia. Mol Cancer (2011) 10:108. doi:10.1186/1476-4598-10-108
8. Zahreddine H, Borden KLB. Mechanisms and insights into drug resistance in cancer. Front Pharmacol (2013) 4:28. doi:10.3389/fphar.2013.00028
9. Shaffer BC, Gillet J-P, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat (2012) 15(1–2):62–9. doi:10.1016/j.drup.2012.02.001
10. Cree IA, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer (2017) 17(1):10. doi:10.1186/s12885-016-2999-1
11. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell (2014) 157(1):77–94. doi:10.1016/j.cell.2014.03.008
12. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature (2000) 403(6772):901–6. doi:10.1038/35002607
13. Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell (2003) 113(6):673–6. doi:10.1016/S0092-8674(03)00428-8
14. Macha MA, Seshacharyulu P, Krishn SR, Pai P, Rachagani S, Jain M, et al. MicroRNAs (miRNAs) as biomarker(s) for prognosis and diagnosis of gastrointestinal (GI) cancers. Curr Pharm Des (2014) 20(33):5287–97. doi:10.2174/1381612820666140128213117
15. Grady WM, Tewari M. The next thing in prognostic molecular markers: microRNA signatures of cancer. Gut (2010) 59(6):706–8. doi:10.1136/gut.2009.200022
16. Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics (2013) 193(3):651–69. doi:10.1534/genetics.112.146704
17. Huarte M. The emerging role of lncRNAs in cancer. Nat Med (2015) 21(11):1253–61. doi:10.1038/nm.3981
18. Sun L, Xue H, Jiang C, Zhou H, Gu L, Liu Y, et al. LncRNA DQ786243 contributes to proliferation and metastasis of colorectal cancer both in vitro and in vivo. Biosci Rep (2016) 36(3):e00328. doi:10.1042/BSR20160048
19. Zhang Z-L, Zhao L-J, Chai L, Zhou S-H, Wang F, Wei Y, et al. Seven lncRNA-mRNA based risk score predicts the survival of head and neck squamous cell carcinoma. Sci Rep (2017) 7(1):309. doi:10.1038/s41598-017-00252-2
20. Xing C, Hu X, Xie F, Yu Z, Li H, Zhou B, et al. Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett (2015) 589(15):1981–7. doi:10.1016/j.febslet.2015.04.061
21. Wu S, Zheng C, Chen S, Cai X, Shi Y, Lin B, et al. Overexpression of long non-coding RNA HOTAIR predicts a poor prognosis in patients with acute myeloid leukemia. Oncol Lett (2015) 10(4):2410–4. doi:10.3892/ol.2015.3552
22. Hao S, Shao Z. HOTAIR is upregulated in acute myeloid leukemia and that indicates a poor prognosis. Int J Clin Exp Pathol (2015) 8(6):7223–8.
23. Díaz-Beyá M, Brunet S, Nomdedéu J, Pratcorona M, Cordeiro A, Gallardo D, et al. The lincRNA HOTAIRM1, located in the HOXA genomic region, is expressed in acute myeloid leukemia, impacts prognosis in patients in the intermediate-risk cytogenetic category, and is associated with a distinctive microRNA signature. Oncotarget (2015) 6(31):31613–27. doi:10.18632/oncotarget.5148
24. Garzon R, Volinia S, Papaioannou D, Nicolet D, Kohlschmidt J, Yan PS, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci U S A (2014) 111(52):18679–84. doi:10.1073/pnas.1422050112
25. Sayad A, Hajifathali A, Hamidieh AA, Roshandel E, Taheri M. HOTAIR long noncoding RNA is not a biomarker for acute myeloid leukemia (AML) in Iranian patients. Asian Pac J Cancer Prev (2017) 18(6):1581–4. doi:10.22034/APJCP.2017.18.6.1581
26. Chen Z-H, Wang W-T, Huang W, Fang K, Sun Y-M, Liu S-R, et al. The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ (2017) 24(2):212–24. doi:10.1038/cdd.2016.111
27. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell (2011) 146(3):353–8. doi:10.1016/j.cell.2011.07.014
28. Macfarlane L-A, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics (2010) 11(7):537–61. doi:10.2174/138920210793175895
29. Gunaratne PH, Creighton CJ, Watson M, Tennakoon JB. Large-scale integration of microRNA and gene expression data for identification of enriched microRNA-mRNA associations in biological systems. Methods Mol Biol (2010) 667:297–315. doi:10.1007/978-1-60761-811-9_20
30. Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, et al. Differential regulation of microRNA stability. RNA (2010) 16(5):1032–9. doi:10.1261/rna.1851510
31. Okamura K, Liu N, Lai EC. Distinct mechanisms for microRNA strand selection by drosophila argonautes. Mol Cell (2009) 36(3):431–44. doi:10.1016/j.molcel.2009.09.027
32. Li S-C, Liao Y-L, Ho M-R, Tsai K-W, Lai C-H, Lin W. miRNA arm selection and isomiR distribution in gastric cancer. BMC Genomics (2012) 13(Suppl 1):S13. doi:10.1186/1471-2164-13-S1-S13
33. Winter J, Link S, Witzigmann D, Hildenbrand C, Previti C, Diederichs S. Loop-miRs: active microRNAs generated from single-stranded loop regions. Nucleic Acids Res (2013) 41(10):5503–12. doi:10.1093/nar/gkt251
34. Okamura K, Ladewig E, Zhou L, Lai EC. Functional small RNAs are generated from select miRNA hairpin loops in flies and mammals. Genes Dev (2013) 27(7):778–92. doi:10.1101/gad.211698.112
35. Redfern AD, Colley SM, Beveridge DJ, Ikeda N, Epis MR, Li X, et al. RNA-induced silencing complex (RISC) proteins PACT, TRBP, and dicer are SRA binding nuclear receptor coregulators. Proc Natl Acad Sci U S A (2013) 110(16):6536–41. doi:10.1073/pnas.1301620110
36. Klusmann J-H, Li Z, Böhmer K, Maroz A, Koch ML, Emmrich S, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev (2010) 24(5):478–90. doi:10.1101/gad.1856210
37. Chen K, Rajewsky N. Deep conservation of microRNA-target relationships and 3’UTR motifs in vertebrates, flies, and nematodes. Cold Spring Harb Symp Quant Biol (2006) 71:149–56. doi:10.1101/sqb.2006.71.039
38. Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature (2008) 455(7216):1124–8. doi:10.1038/nature07299
39. Lam JKW, Chow MYT, Zhang Y, Leung SWS. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids (2015) 4:e252. doi:10.1038/mtna.2015.23
40. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature (2005) 433(7027):769–73. doi:10.1038/nature03315
41. Nakanishi K, Ascano M, Gogakos T, Ishibe-Murakami S, Serganov AA, Briskin D, et al. Eukaryote-specific insertion elements control human ARGONAUTE slicer activity. Cell Rep (2013) 3(6):1893–900. doi:10.1016/j.celrep.2013.06.010
42. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
43. Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the microRNA pathway. Nature (2005) 435(7044):974–8. doi:10.1038/nature03816
44. Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells (2011) 29(11):1661–71. doi:10.1002/stem.741
45. Huang J-W, Wang Y, Dhillon KK, Calses P, Villegas E, Mitchell PS, et al. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol Cancer Res (2013) 11(12):1564–73. doi:10.1158/1541-7786.MCR-13-0292
46. Lai T-H, Ewald B, Zecevic A, Liu C, Sulda M, Papaioannou D, et al. HDAC inhibition induces microRNA-182, which targets RAD51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin Cancer Res (2016) 22(14):3537–49. doi:10.1158/1078-0432.CCR-15-1063
47. Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst (2015) 107(7):djv108. doi:10.1093/jnci/djv108
48. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood (2010) 115(3):453–74. doi:10.1182/blood-2009-07-235358
49. Estey E. AML in older patients: are we making progress? Best Pract Res Clin Haematol (2009) 22(4):529–36. doi:10.1016/j.beha.2009.08.007
51. Barone G, Guerra CF, Gambino N, Silvestri A, Lauria A, Almerico AM, et al. Intercalation of daunomycin into stacked DNA base pairs. DFT study of an anticancer drug. J Biomol Struct Dyn (2008) 26(1):115–30. doi:10.1080/07391102.2008.10507229
52. D’Ugo E, Bruni R, Argentini C, Giuseppetti R, Rapicetta M. Identification of scaffold/matrix attachment region in recurrent site of woodchuck hepatitis virus integration. DNA Cell Biol (1998) 17(6):519–27. doi:10.1089/dna.1998.17.519
53. Sumer H, Craig JM, Sibson M, Choo KHA. A rapid method of genomic array analysis of scaffold/matrix attachment regions (S/MARs) identifies a 2.5-Mb region of enhanced scaffold/matrix attachment at a human neocentromere. Genome Res (2003) 13(7):1737–43. doi:10.1101/gr.1095903
54. Heidenreich E, Novotny R, Kneidinger B, Holzmann V, Wintersberger U. Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. EMBO J (2003) 22(9):2274–83. doi:10.1093/emboj/cdg203
55. Hensel JP, Gillert E, Fey GH, Marschalek R. Breakpoints of t(4;11) translocations in the human MLL and AF4 genes in ALL patients are preferentially clustered outside of high-affinity matrix attachment regions. J Cell Biochem (2001) 82(2):299–309. doi:10.1002/jcb.1161
56. Domer PH, Head DR, Renganathan N, Raimondi SC, Yang E, Atlas M. Molecular analysis of 13 cases of MLL/11q23 secondary acute leukemia and identification of topoisomerase II consensus-binding sequences near the chromosomal breakpoint of a secondary leukemia with the t(4;11). Leukemia (1995) 9(8):1305–12.
57. Chavali PL, Funa K, Chavali S. Cis-regulation of microRNA expression by scaffold/matrix-attachment regions. Nucleic Acids Res (2011) 39(16):6908–18. doi:10.1093/nar/gkr303
58. Fathi AT, Karp JE. New agents in acute myeloid leukemia: beyond cytarabine and anthracyclines. Curr Oncol Rep (2009) 11(5):346–52. doi:10.1007/s11912-009-0047-x
59. Seedhouse C, Grundy M, Shang S, Ronan J, Pimblett H, Russell N, et al. Impaired S-phase arrest in acute myeloid leukemia cells with a FLT3 internal tandem duplication treated with clofarabine. Clin Cancer Res (2009) 15(23):7291–8. doi:10.1158/1078-0432.CCR-09-1222
60. Prakasha Gowda AS, Polizzi JM, Eckert KA, Spratt TE. Incorporation of gemcitabine and cytarabine into DNA by DNA polymerase beta and ligase III/XRCC1. Biochemistry (2010) 49(23):4833–40. doi:10.1021/bi100200c
61. Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem (2005) 74:317–53. doi:10.1146/annurev.biochem.74.082803.133250
62. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer (2012) 12(9):587–98. doi:10.1038/nrc3342
63. Liu X, Liao W, Peng H, Luo X, Luo Z, Jiang H, et al. miR-181a promotes G1/S transition and cell proliferation in pediatric acute myeloid leukemia by targeting ATM. J Cancer Res Clin Oncol (2016) 142(1):77–87. doi:10.1007/s00432-015-1995-1
64. Seca H, Lima RT, Almeida GM, Sobrinho-Simoes M, Bergantim R, Guimaraes JE, et al. Effect of miR-128 in DNA damage of HL-60 acute myeloid leukemia cells. Curr Pharm Biotechnol (2014) 15(5):492–502. doi:10.2174/1389201015666140519122524
65. Volinia S, Calin GA, Liu C-G, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A (2006) 103(7):2257–61. doi:10.1073/pnas.0510565103
66. Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci U S A (2008) 105(10):3945–50. doi:10.1073/pnas.0800135105
67. Chikamori K, Hill JE, Grabowski DR, Zarkhin E, Grozav AG, Vaziri SAJ, et al. Downregulation of topoisomerase IIbeta in myeloid leukemia cell lines leads to activation of apoptosis following all-trans retinoic acid-induced differentiation/growth arrest. Leukemia (2006) 20(10):1809–18. doi:10.1038/sj.leu.2404351
68. Hermanson DL, Das SG, Li Y, Xing C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIβ downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Mol Pharmacol (2013) 84(2):236–43. doi:10.1124/mol.113.086140
69. Lim S, Kaldis P. CDKs, cyclins and CKIs: roles beyond cell cycle regulation. Development (2013) 140(15):3079–93. doi:10.1242/dev.091744
70. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol (2013) 14(4):197–210. doi:10.1038/nrm3546
71. Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J (2003) 22(20):5612–21. doi:10.1093/emboj/cdg541
72. Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res (2001) 7(8):2168–81.
73. Beaumont KA, Hill DS, Daignault SM, Lui GYL, Sharp DM, Gabrielli B, et al. Cell cycle phase-specific drug resistance as an escape mechanism of melanoma cells. J Invest Dermatol (2016) 136(7):1479–89. doi:10.1016/j.jid.2016.02.805
74. Kaaijk P, Kaspers GJL, Van Wering ER, Broekema GJ, Loonen AH, Hählen K, et al. Cell proliferation is related to in vitro drug resistance in childhood acute leukaemia. Br J Cancer (2003) 88(5):775–81. doi:10.1038/sj.bjc.6600787
75. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) (2012) 7(4):597–615. doi:10.2217/nnm.12.22
76. Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta (2011) 1812(5):592–601. doi:10.1016/j.bbadis.2011.02.002
77. Lin Y, Li D, Liang Q, Liu S, Zuo X, Li L, et al. miR-638 regulates differentiation and proliferation in leukemic cells by targeting cyclin-dependent kinase 2. J Biol Chem (2015) 290(3):1818–28. doi:10.1074/jbc.M114.599191
78. Afanasyeva EA, Mestdagh P, Kumps C, Vandesompele J, Ehemann V, Theissen J, et al. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ (2011) 18(6):974–84. doi:10.1038/cdd.2010.164
79. Tian R-Q, Wang X-H, Hou L-J, Jia W-H, Yang Q, Li Y-X, et al. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human cervical cancer, which may contribute to tumorigenesis. J Biol Chem (2011) 286(29):25556–63. doi:10.1074/jbc.M111.221564
80. Wu J, Lv Q, He J, Zhang H, Mei X, Cui K, et al. MicroRNA-188 suppresses G1/S transition by targeting multiple cyclin/CDK complexes. Cell Commun Signal (2014) 12:66. doi:10.1186/s12964-014-0066-6
81. Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, et al. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res (2008) 36(16):5391–404. doi:10.1093/nar/gkn522
82. Salvatori B, Iosue I, Mangiavacchi A, Loddo G, Padula F, Chiaretti S, et al. The microRNA-26a target E2F7 sustains cell proliferation and inhibits monocytic differentiation of acute myeloid leukemia cells. Cell Death Dis (2012) 3:e413. doi:10.1038/cddis.2012.151
83. Wong P, Iwasaki M, Somervaille TCP, Ficara F, Carico C, Arnold C, et al. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res (2010) 70(9):3833–42. doi:10.1158/0008-5472.CAN-09-3268
84. Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Müller-Tidow C, Bohlander SK, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood (2010) 115(9):1768–78. doi:10.1182/blood-2009-08-240101
85. Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol (2007) 27(6):2240–52. doi:10.1128/MCB.02005-06
86. Wang F, Fu X-D, Zhou Y, Zhang Y. Down-regulation of the cyclin E1 oncogene expression by microRNA-16-1 induces cell cycle arrest in human cancer cells. BMB Rep (2009) 42(11):725–30. doi:10.5483/BMBRep.2009.42.11.725
87. Zubillaga-Guerrero MI, Alarcón-Romero LDC, Illades-Aguiar B, Flores-Alfaro E, Bermúdez-Morales VH, Deas J, et al. MicroRNA miR-16-1 regulates CCNE1 (cyclin E1) gene expression in human cervical cancer cells. Int J Clin Exp Med (2015) 8(9):15999–6006.
88. Guo X, Connick MC, Vanderhoof J, Ishak M-A, Hartley RS. MicroRNA-16 modulates HuR regulation of cyclin E1 in breast cancer cells. Int J Mol Sci (2015) 16(4):7112–32. doi:10.3390/ijms16047112
89. Rivas MA, Venturutti L, Huang Y-W, Schillaci R, Huang TH-M, Elizalde PV. Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development. Breast Cancer Res (2012) 14(3):R77. doi:10.1186/bcr3187
90. Ofir M, Hacohen D, Ginsberg D. miR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res (2011) 9(4):440–7. doi:10.1158/1541-7786.MCR-10-0344
91. Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, et al. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol (2007) 9(7):775–87. doi:10.1038/ncb1613
92. Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A, et al. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. EMBO J (2011) 30(21):4450–64. doi:10.1038/emboj.2011.317
93. Mitxelena J, Apraiz A, Vallejo-Rodríguez J, Malumbres M, Zubiaga AM. E2F7 regulates transcription and maturation of multiple microRNAs to restrain cell proliferation. Nucleic Acids Res (2016) 44(12):5557–70. doi:10.1093/nar/gkw146
94. Eyholzer M, Schmid S, Schardt JA, Haefliger S, Mueller BU, Pabst T. Complexity of miR-223 regulation by CEBPA in human AML. Leuk Res (2010) 34(5):672–6. doi:10.1016/j.leukres.2009.11.019
95. Rodriguez-Ubreva J, Ciudad L, van Oevelen C, Parra M, Graf T, Ballestar E. C/EBPa-mediated activation of microRNAs 34a and 223 inhibits Lef1 expression to achieve efficient reprogramming into macrophages. Mol Cell Biol (2014) 34(6):1145–57. doi:10.1128/MCB.01487-13
96. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi:10.1080/01926230701320337
97. Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov (2015) 1:15008. doi:10.1038/cddiscovery.2015.8
98. Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM, et al. Autophagy in acute leukemias: a double-edged sword with important therapeutic implications. Biochim Biophys Acta (2015) 1853(1):14–26. doi:10.1016/j.bbamcr.2014.09.023
99. Zhang S-P, Niu Y-N, Yuan N, Zhang A-H, Chao D, Xu Q-P, et al. Role of autophagy in acute myeloid leukemia therapy. Chin J Cancer (2013) 32(3):130–5. doi:10.5732/cjc.012.10073
100. Gozuacik D, Akkoc Y, Ozturk DG, Kocak M. Autophagy-regulating microRNAs and cancer. Front Oncol (2017) 7:65. doi:10.3389/fonc.2017.00065
101. Li H, Hui L, Xu W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Chin J Biochem Biophys (2012) 44(3):269–77. doi:10.1093/abbs/gmr128
102. Bai H, Cao Z, Deng C, Zhou L, Wang C. miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C by inducing apoptosis. J Cancer Res Clin Oncol (2012) 138(4):595–602. doi:10.1007/s00432-011-1137-3
103. Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer (2008) 123(2):372–9. doi:10.1002/ijc.23501
104. Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2{Delta}16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis (2010) 31(12):2049–57. doi:10.1093/carcin/bgq192
105. Han J, Chen Q. miR-16 modulate temozolomide resistance by regulating BCL-2 in human glioma cells. Int J Clin Exp Pathol (2015) 8(10):12698–707.
106. Yang D, Zhan M, Chen T, Chen W, Zhang Y, Xu S, et al. miR-125b-5p enhances chemotherapy sensitivity to cisplatin by down-regulating Bcl2 in gallbladder cancer. Sci Rep (2017) 7:43109. doi:10.1038/srep43109
107. Li Q, Liang X, Wang Y, Meng X, Xu Y, Cai S, et al. miR-139-5p inhibits the epithelial-mesenchymal transition and enhances the chemotherapeutic sensitivity of colorectal cancer cells by down regulating BCL2. Sci Rep (2016) 6:27157. doi:10.1038/srep27157
108. Li Q, Wu Y, Zhang Y, Sun H, Lu Z, Du K, et al. miR-125b regulates cell progression in chronic myeloid leukemia via targeting BAK1. Am J Transl Res (2016) 8(2):447–59.
109. Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem (2010) 285(28):21496–507. doi:10.1074/jbc.M109.083337
110. Shi X-B, Xue L, Yang J, Ma A-H, Zhao J, Xu M, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A (2007) 104(50):19983–8. doi:10.1073/pnas.0706641104
111. Gocek E, Wang X, Liu X, Liu C-G, Studzinski GP. MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of AraC-induced apoptosis. Cancer Res (2011) 71(19):6230–9. doi:10.1158/0008-5472.CAN-11-1717
112. Le MTN, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev (2009) 23(7):862–76. doi:10.1101/gad.1767609
113. Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song JS, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell (2010) 38(5):689–99. doi:10.1016/j.molcel.2010.05.027
114. Tian P, Yan L. Inhibition of microRNA-149-5p induces apoptosis of acute myeloid leukemia cell line THP-1 by targeting Fas ligand (FASLG). Med Sci Monit (2016) 22:5116–23. doi:10.12659/MSM.899114
115. Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, et al. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia (2012) 26(8):1752–60. doi:10.1038/leu.2012.65
116. Zeng C-W, Chen Z-H, Zhang X-J, Han B-W, Lin K-Y, Li X-J, et al. MIR125B1 represses the degradation of the PML-RARA oncoprotein by an autophagy-lysosomal pathway in acute promyelocytic leukemia. Autophagy (2014) 10(10):1726–37. doi:10.4161/auto.29592
117. Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. MicroRNA-101 is a potent inhibitor of autophagy. EMBO J (2011) 30(22):4628–41. doi:10.1038/emboj.2011.331
118. Nanbakhsh A, Visentin G, Olive D, Janji B, Mussard E, Dessen P, et al. miR-181a modulates acute myeloid leukemia susceptibility to natural killer cells. Oncoimmunology (2015) 4(12):e996475. doi:10.1080/2162402X.2014.996475
119. Mrózek K, Radmacher MD, Bloomfield CD, Marcucci G. Molecular signatures in acute myeloid leukemia. Curr Opin Hematol (2009) 16(2):64–9. doi:10.1097/MOH.0b013e3283257b42
120. Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJM, Löwenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood (2008) 111(10):5078–85. doi:10.1182/blood-2008-01-133355
121. Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol (2008) 26(31):5078–87. doi:10.1200/JCO.2008.17.5554
122. Volinia S, Galasso M, Costinean S, Tagliavini L, Gamberoni G, Drusco A, et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res (2010) 20(5):589–99. doi:10.1101/gr.098046.109
123. Schwind S, Maharry K, Radmacher MD, Mrózek K, Holland KB, Margeson D, et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol (2010) 28(36):5257–64. doi:10.1200/JCO.2010.29.2953
124. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A (2002) 99(24):15524–9. doi:10.1073/pnas.242606799
125. Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin Y-C, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood (2007) 109(12):5079–86. doi:10.1182/blood-2007-02-071225
126. Balatti V, Pekarky Y, Croce CM. Role of microRNA in chronic lymphocytic leukemia onset and progression. J Hematol Oncol (2015) 8:12. doi:10.1186/s13045-015-0112-x
127. Allegra D, Bilan V, Garding A, Döhner H, Stilgenbauer S, Kuchenbauer F, et al. Defective DROSHA processing contributes to downregulation of miR-15/-16 in chronic lymphocytic leukemia. Leukemia (2014) 28(1):98–107. doi:10.1038/leu.2013.246
128. Gu X, Li J-Y, Guo J, Li P-S, Zhang W-H. Influence of miR-451 on drug resistances of paclitaxel-resistant breast cancer cell line. Med Sci Monit (2015) 21:3291–7. doi:10.12659/MSM.894475
129. Liu Z, Liu H, Desai S, Schmitt DC, Zhou M, Khong HT, et al. miR-125b functions as a key mediator for snail-induced stem cell propagation and chemoresistance. J Biol Chem (2013) 288(6):4334–45. doi:10.1074/jbc.M112.419168
130. Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res (2008) 68(15):6162–70. doi:10.1158/0008-5472.CAN-08-0144
131. Jamil S, Lam I, Majd M, Tsai S-H, Duronio V. Etoposide induces cell death via mitochondrial-dependent actions of p53. Cancer Cell Int (2015) 15:79. doi:10.1186/s12935-015-0231-z
132. Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature (1994) 370(6486):220–3. doi:10.1038/370220a0
133. Goeman F, Strano S, Blandino G. Micrornas as key effectors in the p53 network. Int Rev Cell Mol Biol (2017) 333:51–90. doi:10.1016/bs.ircmb.2017.04.003
134. Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell (2007) 26(5):745–52. doi:10.1016/j.molcel.2007.05.010
135. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature (2007) 447(7148):1130–4. doi:10.1038/nature05939
136. Hünten S, Siemens H, Kaller M, Hermeking H. The p53/microRNA network in cancer: experimental and bioinformatics approaches. Adv Exp Med Biol (2013) 774:77–101. doi:10.1007/978-94-007-5590-1_5
137. Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A (2008) 105(36):13421–6. doi:10.1073/pnas.0801613105
138. Rücker FG, Russ AC, Cocciardi S, Kett H, Schlenk RF, Botzenhardt U, et al. Altered miRNA and gene expression in acute myeloid leukemia with complex karyotype identify networks of prognostic relevance. Leukemia (2013) 27(2):353–61. doi:10.1038/leu.2012.208
139. Shao B, Liao L, Yu Y, Shuai Y, Su X, Jing H, et al. Estrogen preserves Fas ligand levels by inhibiting microRNA-181a in bone marrow-derived mesenchymal stem cells to maintain bone remodeling balance. FASEB J (2015) 29(9):3935–44. doi:10.1096/fj.15-272823
140. Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, et al. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem (2010) 285(26):20281–90. doi:10.1074/jbc.M110.109207
141. Zhang Y, Chen N, Zhang J, Tong Y. Hsa-let-7g miRNA targets caspase-3 and inhibits the apoptosis induced by ox-LDL in endothelial cells. Int J Mol Sci (2013) 14(11):22708–20. doi:10.3390/ijms141122708
142. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi:10.1146/annurev-cellbio-092910-154005
143. Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub (2010) 154(2):103–16. doi:10.5507/bp.2010.017
144. Platel D, Bonoron-Adèle S, Robert J. Role of daunorubicinol in daunorubicin-induced cardiotoxicity as evaluated with the model of isolated perfused rat heart. Pharmacol Toxicol (2001) 88(5):250–4. doi:10.1111/j.1600-0773.2001.880505.x
145. Cusack BJ, Mushlin PS, Voulelis LD, Li X, Boucek RJ, Olson RD. Daunorubicin-induced cardiac injury in the rabbit: a role for daunorubicinol? Toxicol Appl Pharmacol (1993) 118(2):177–85. doi:10.1006/taap.1993.1023
146. Huang Z, Wang J, Qian J, Li Y, Xu Z, Chen M, et al. Effects of cytochrome P450 family 3 subfamily A member 5 gene polymorphisms on daunorubicin metabolism and adverse reactions in patients with acute leukemia. Mol Med Rep (2017) 15(6):3493–8. doi:10.3892/mmr.2017.6470
147. Colburn DE, Giles FJ, Oladovich D, Smith JA. In vitro evaluation of cytochrome P450-mediated drug interactions between cytarabine, idarubicin, itraconazole and caspofungin. Hematology (2004) 9(3):217–21. doi:10.1080/10245330410001701585
148. Yee SB, Pritsos CA. Comparison of oxygen radical generation from the reductive activation of doxorubicin, streptonigrin, and menadione by xanthine oxidase and xanthine dehydrogenase. Arch Biochem Biophys (1997) 347(2):235–41. doi:10.1006/abbi.1997.0340
149. Gustafson DL, Swanson JD, Pritsos CA. Role of xanthine oxidase in the potentiation of doxorubicin-induced cardiotoxicity by mitomycin C. Cancer Commun (1991) 3(9):299–304.
150. Jamieson D, Cresti N, Bray J, Sludden J, Griffin MJ, Hawsawi NM, et al. Two minor NQO1 and NQO2 alleles predict poor response of breast cancer patients to adjuvant doxorubicin and cyclophosphamide therapy. Pharmacogenet Genomics (2011) 21(12):808–19. doi:10.1097/FPC.0b013e32834b6918
151. Deng S, Kruger A, Schmidt A, Metzger A, Yan T, Gödtel-Armbrust U, et al. Differential roles of nitric oxide synthase isozymes in cardiotoxicity and mortality following chronic doxorubicin treatment in mice. Naunyn Schmiedebergs Arch Pharmacol (2009) 380(1):25–34. doi:10.1007/s00210-009-0407-y
152. Oktem G, Bilir A, Selvi N, Yurtseven ME, Vatansever S, Ates U, et al. Chemotherapy influences inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) activity on 3D breast cancer cell line. Oncol Res (2006) 16(4):195–203.
153. Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics (2011) 21(7):440–6. doi:10.1097/FPC.0b013e32833ffb56
154. Lamba JK. Genetic factors influencing cytarabine therapy. Pharmacogenomics (2009) 10(10):1657–74. doi:10.2217/pgs.09.118
155. Liu X, Zhou B, Mi S, Xue L, Shih J, Lee J, et al. An increase of cytochrome C oxidase mediated disruption of gemcitabine incorporation into DNA in a resistant KB clone. Biochem Pharmacol (2007) 73(12):1927–38. doi:10.1016/j.bcp.2007.03.014
156. Pan Y-Z, Gao W, Yu A-M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab Dispos (2009) 37(10):2112–7. doi:10.1124/dmd.109.027680
157. Wei Z, Jiang S, Zhang Y, Wang X, Peng X, Meng C, et al. The effect of microRNAs in the regulation of human CYP3A4: a systematic study using a mathematical model. Sci Rep (2014) 4:4283. doi:10.1038/srep04283
158. Hodzic J, Giovannetti E, Diosdado B, Adema AD, Peters GJ. Regulation of deoxycytidine kinase expression and sensitivity to gemcitabine by micro-RNA 330 and promoter methylation in cancer cells. Nucleosides Nucleotides Nucleic Acids (2011) 30(12):1214–22. doi:10.1080/15257770.2011.629271
159. Armenian SH, Ding Y, Mills G, Sun C, Venkataraman K, Wong FL, et al. Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br J Haematol (2013) 163(2):205–13. doi:10.1111/bjh.12516
160. Szachowicz-Petelska B, Figaszewski Z, Lewandowski W. Mechanisms of transport across cell membranes of complexes contained in antitumour drugs. Int J Pharm (2001) 222(2):169–82. doi:10.1016/S0378-5173(01)00713-X
161. El-Kareh AW, Secomb TW. Two-mechanism peak concentration model for cellular pharmacodynamics of doxorubicin. Neoplasia (2005) 7(7):705–13. doi:10.1593/neo.05118
162. Aouida M, Poulin R, Ramotar D. The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J Biol Chem (2010) 285(9):6275–84. doi:10.1074/jbc.M109.046151
163. Aouida M, Ramotar D. A new twist in cellular resistance to the anticancer drug bleomycin-A5. Curr Drug Metab (2010) 11(7):595–602. doi:10.2174/138920010792927307
164. Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, et al. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene (2003) 22(47):7524–36. doi:10.1038/sj.onc.1206952
165. Molina-Arcas M, Pastor-Anglada M. Role of nucleoside transporters in nucleoside-derived drug sensitivity. Nucleosides Nucleotides Nucleic Acids (2010) 29(4–6):335–46. doi:10.1080/15257771003729823
166. Pastor-Anglada M, Cano-Soldado P, Molina-Arcas M, Lostao MP, Larráyoz I, Martínez-Picado J, et al. Cell entry and export of nucleoside analogues. Virus Res (2005) 107(2):151–64. doi:10.1016/j.virusres.2004.11.005
167. Hubeek I, Stam RW, Peters GJ, Broekhuizen R, Meijerink JPP, van Wering ER, et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer (2005) 93(12):1388–94. doi:10.1038/sj.bjc.6602881
168. Galmarini CM, Thomas X, Calvo F, Rousselot P, El Jafaari A, Cros E, et al. Potential mechanisms of resistance to cytarabine in AML patients. Leuk Res (2002) 26(7):621–9. doi:10.1016/S0145-2126(01)00184-9
169. Leslie EM, Deeley RG, Cole SPC. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol (2005) 204(3):216–37. doi:10.1016/j.taap.2004.10.012
170. Cole SPC. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem (2014) 289(45):30880–8. doi:10.1074/jbc.R114.609248
171. Greenberg PL, Lee SJ, Advani R, Tallman MS, Sikic BI, Letendre L, et al. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995). J Clin Oncol (2004) 22(6):1078–86. doi:10.1200/JCO.2004.07.048
172. Feng D-D, Zhang H, Zhang P, Zheng Y-S, Zhang X-J, Han B-W, et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J Cell Mol Med (2011) 15(10):2164–75. doi:10.1111/j.1582-4934.2010.01213.x
173. Li Z, Hu S, Wang J, Cai J, Xiao L, Yu L, et al. miR-27a modulates MDR1/P-glycoprotein expression by targeting HIPK2 in human ovarian cancer cells. Gynecol Oncol (2010) 119(1):125–30. doi:10.1016/j.ygyno.2010.06.004
174. Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu C-G, et al. Role of microRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem Pharmacol (2008) 76(5):582–8. doi:10.1016/j.bcp.2008.06.007
175. Shang Y, Feng B, Zhou L, Ren G, Zhang Z, Fan X, et al. The miR27b-CCNG1-P53-miR-508-5p axis regulates multidrug resistance of gastric cancer. Oncotarget (2016) 7(1):538–49. doi:10.18632/oncotarget.6374
176. Lopes-Rodrigues V, Seca H, Sousa D, Sousa E, Lima RT, Vasconcelos MH. The network of P-glycoprotein and microRNAs interactions. Int J Cancer (2014) 135(2):253–63. doi:10.1002/ijc.28500
177. Ikemura K, Yamamoto M, Miyazaki S, Mizutani H, Iwamoto T, Okuda M. MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol Pharmacol (2013) 83(2):399–405. doi:10.1124/mol.112.081844
178. Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol (2012) 180(6):2490–503. doi:10.1016/j.ajpath.2012.02.024
179. Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryndyak VP, Chekhun VF, et al. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther (2008) 7(7):2152–9. doi:10.1158/1535-7163.MCT-08-0021
180. Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol Ther Nucleic Acids (2013) 2:e126. doi:10.1038/mtna.2013.60
181. Pan Y-Z, Zhou A, Hu Z, Yu A-M. Small nucleolar RNA-derived microRNA hsa-miR-1291 modulates cellular drug disposition through direct targeting of ABC transporter ABCC1. Drug Metab Dispos (2013) 41(10):1744–51. doi:10.1124/dmd.113.052092
182. Wu D, Li X-S, Meng X-N, Yan J, Zong Z-H. MicroRNA-873 mediates multidrug resistance in ovarian cancer cells by targeting ABCB1. Tumour Biol (2016) 37(8):10499–506. doi:10.1007/s13277-016-4944-y
183. Gullà A, Di Martino MT, Gallo Cantafio ME, Morelli E, Amodio N, Botta C, et al. A 13 mer LNA-i-miR-221 inhibitor restores drug sensitivity in melphalan-refractory multiple myeloma cells. Clin Cancer Res (2016) 22(5):1222–33. doi:10.1158/1078-0432.CCR-15-0489
184. Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol (2010) 79(6):817–24. doi:10.1016/j.bcp.2009.10.017
185. Haenisch S, Laechelt S, Bruckmueller H, Werk A, Noack A, Bruhn O, et al. Down-regulation of ATP-binding cassette C2 protein expression in HepG2 cells after rifampicin treatment is mediated by microRNA-379. Mol Pharmacol (2011) 80(2):314–20. doi:10.1124/mol.110.070714
186. Pan Y-Z, Morris ME, Yu A-M. MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol Pharmacol (2009) 75(6):1374–9. doi:10.1124/mol.108.054163
187. To KKW, Zhan Z, Litman T, Bates SE. Regulation of ABCG2 expression at the 3’ untranslated region of its mRNA through modulation of transcript stability and protein translation by a putative microRNA in the S1 colon cancer cell line. Mol Cell Biol (2008) 28(17):5147–61. doi:10.1128/MCB.00331-08
188. Liao R, Sun J, Zhang L, Lou G, Chen M, Zhou D, et al. MicroRNAs play a role in the development of human hematopoietic stem cells. J Cell Biochem (2008) 104(3):805–17. doi:10.1002/jcb.21668
189. Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res (1999) 59(1):8–13.
190. Yeh C-H, Moles R, Nicot C. Clinical significance of microRNAs in chronic and acute human leukemia. Mol Cancer (2016) 15(1):37. doi:10.1186/s12943-016-0518-2
191. Varma S, Pommier Y, Sunshine M, Weinstein JN, Reinhold WC. High resolution copy number variation data in the NCI-60 cancer cell lines from whole genome microarrays accessible through cell miner. PLoS One (2014) 9(3):e92047. doi:10.1371/journal.pone.0092047
192. de Necochea-Campion R, Shouse GP, Zhou Q, Mirshahidi S, Chen C-S. Aberrant splicing and drug resistance in AML. J Hematol Oncol (2016) 9(1):85. doi:10.1186/s13045-016-0315-9
193. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang Y-K, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs (2017) 35(2):180–8. doi:10.1007/s10637-016-0407-y
194. Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, et al. miR-34: a new weapon against cancer? Mol Ther Nucleic Acids (2014) 3:e194. doi:10.1038/mtna.2014.47
195. Navarro F, Lieberman J. miR-34 and p53: new insights into a complex functional relationship. PLoS One (2015) 10(7):e0132767. doi:10.1371/journal.pone.0132767
196. Takagi S, Nakajima M, Kida K, Yamaura Y, Fukami T, Yokoi T. MicroRNAs regulate human hepatocyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. J Biol Chem (2010) 285(7):4415–22. doi:10.1074/jbc.M109.085431
197. Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat Commun (2015) 6:7466. doi:10.1038/ncomms8466
198. Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol (2013) 24(12):3128–35. doi:10.1093/annonc/mdt412
199. Johansson J, Berg T, Kurzejamska E, Pang MF, Tabor V, Jansson M, et al. miR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene (2013) 32(50):5614–24. doi:10.1038/onc.2013.322
200. Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol (2012) 19(1):60–71. doi:10.1016/j.chembiol.2011.12.008
201. Mali S. Delivery systems for gene therapy. Indian J Hum Genet (2013) 19(1):3–8. doi:10.4103/0971-6866.112870
Keywords: microRNA, acute myeloid leukemia, drug resistance, RNA therapy, daunorubicin, cytarabine, chemotherapy
Citation: Gabra MM and Salmena L (2017) microRNAs and Acute Myeloid Leukemia Chemoresistance: A Mechanistic Overview. Front. Oncol. 7:255. doi: 10.3389/fonc.2017.00255
Received: 17 August 2017; Accepted: 11 October 2017;
Published: 30 October 2017
Edited by:
Angela Re, Istituto Italiano di Tecnologia, ItalyReviewed by:
Bernd Groner, Georg Speyer Haus, GermanyGabriele Multhoff, Technische Universität München, Germany
Copyright: © 2017 Gabra and Salmena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo Salmena, bGVvbmFyZG8uc2FsbWVuYUB1dG9yb250by5jYQ==