 Milica Vučetić
Milica Vučetić Yann Cormerais
Yann Cormerais Scott K. Parks
Scott K. Parks Jacques Pouysségur
Jacques Pouysségur- 1Medical Biology Department, Centre Scientifique de Monaco (CSM), Monaco, Monaco
- 2Institute for Research on Cancer and Aging (IRCAN), CNRS, INSERM, Centre A. Lacassagne, Université Côte d’Azur, Nice, France
A fine balance in reactive oxygen species (ROS) production and removal is of utmost importance for homeostasis of all cells and especially in highly proliferating cells that encounter increased ROS production due to enhanced metabolism. Consequently, increased production of these highly reactive molecules requires coupling with increased antioxidant defense production within cells. This coupling is observed in cancer cells that allocate significant energy reserves to maintain their intracellular redox balance. Glutathione (GSH), as a first line of defense, represents the most important, non-enzymatic antioxidant component together with the NADPH/NADP+ couple, which ensures the maintenance of the pool of reduced GSH. In this review, the central role of amino acids (AAs) in the maintenance of redox homeostasis in cancer, through GSH synthesis (cysteine, glutamate, and glycine), and nicotinamide adenine dinucleotide (phosphate) production (serine, and glutamine/glutamate) are illustrated. Special emphasis is placed on the importance of AA transporters known to be upregulated in cancers (such as system xc-light chain and alanine-serine-cysteine transporter 2) in the maintenance of AA homeostasis, and thus indirectly, the redox homeostasis of cancer cells. The role of the ROS varies (often described as a “two-edged sword”) during the processes of carcinogenesis, metastasis, and cancer treatment. Therefore, the context-dependent role of specific AAs in the initiation, progression, and dissemination of cancer, as well as in the redox-dependent sensitivity/resistance of the neoplastic cells to chemotherapy are highlighted.
Introduction
The potential of targeting redox homeostasis for both cancer prevention and development of novel anticancer treatments has been recognized during past decades. However, despite intensive efforts, development of an effective redox-based therapy remains challenging. A main reason for this is cancer cell plasticity but also our inability to adequately perceive the complexity of redox homeostasis. Namely, antioxidant prophylaxis led to the “antioxidant paradox” (1, 2), while use of chemotherapeutics that compromise the oxidative status of cancer cells encountered resistance (3) and the ability of some cancer cells to upregulate antioxidant protective mechanisms (4). Currently, most attention on targeting redox homeostasis focuses on the attack and downregulation of endogenous antioxidant tumor cell defense mechanisms (5). In this review, we approach cancer redox balance from a different perspective with the main players involving amino acids (AAs).
Although the idea of AA dependency of cancer antioxidant defense (AOD) has received more attention recently, a unified review on this subject is lacking. In 2015, Jones and Sies (6) labeled the nicotinamide adenine dinucleotide (NAD, NADP) and thiol/dysulfide [glutathione (GSH)/glutathione oxidized (GSSG) in the first place] systems together with thiol redox proteome as carriers of the cellular “Redox Code.” According to this principle, spatiotemporal organization of these systems is fundamental for physiology, while its disruption inevitably leads to pathology. Interestingly, accumulating literature indicates that AA availability and metabolism are upstream and superior to these systems, especially in cancer cells. Our review will address this particular aspect of redox regulation in tumors. However, before considering the involvement of AA homeostasis in cancer redox balance, it is necessary to point out some important findings, as well as delusions, that exist in the complex cancer redox field.
Partially Reduced Oxygen—“Activated” Oxygen
The first steps in understanding oxygen toxicity occurred in the mid-twentieth century when Gerschman et al. (7–9) proposed that the damaging effects of oxygen could be attributed to the formation of oxygen radicals. At approximately the same time, research with [18O2] and mass spectrometry showed that oxygen atoms from molecular oxygen [O2] could be introduced into biomolecules (10, 11). The susceptibility of biomolecules to oxidation gave a biological frame to oxygen toxicity, and together with the discovery of superoxide dismutase [SOD; (12)] fueled research in the field of oxidative damage in biological systems. The term “oxidative stress” was introduced into scientific literature for the first time in 1985 (13).
Now it is clear that the oxidative capacity of molecular oxygen in vivo is minimal, but that is not the case for its partially reduced counterparts known as “reactive oxygen species—ROS.” ROS is a term widely used to describe a number of reactive molecules and free radicals derived from molecular oxygen. However, we feel obliged to emphasize the generic nature of this term. ROS includes both radical (superoxide anion radical, ; hydroxyl radical, [HO⋅]; peroxyl radicals, [ROO⋅]; nitric oxide, [NO⋅]) and non-radical (hydrogen peroxide, [H2O2]; hydroxyl anion, [HO−]; singlet oxygen, [1O2]; organic hydroperoxides, [ROOH]) species, which differ significantly in terms of half-life, water/lipid solubility and reactivity. For example, the cellular half-life of lipophobic [HO⋅] is only ~10−9 s because of its reactivity, compared to ~1 ms for [H2O2], which also can diffuse through lipid cellular compartments (14). However, use of the common term ROS is sometimes unavoidable (15) due to the complex nature of biological systems, an inability to exactly measure the species generated in a spatiotemporal manner in addition to the so-called theory of “kindling radicals” by which a few primary ROS “inflame” a cascade of ROS amplification by stimulating the sources of secondary ROS (16).
ROS in Cancer
The terms “ROS” and “cancer” cover a wide range of molecules and diseases, which makes broad generalizations almost impossible. Is it possible, however, to conceptualize some common denominators of the cancer redox state? Widespread opinion is that virtually all malignant cells are in a pro-oxidative state, mostly due to oncogene-driven altered and/or intensified cell metabolism [reviewed in Ref. (17–21)]. However, Halliwell (20) raised important questions regarding ROS measurement in malignant (and other) cells in classical culture conditions that include 21% oxygen and media that is usually deficient in antioxidants/antioxidant precursors and contains free iron ions. These conditions, per se, favor ROS generation, and thus special attention should be paid in extrapolating results obtained in vitro to the in vivo state. Considering this point in combination with current advances in the cancer redox field, a major conclusion that can be drawn is that cancer cells indeed experience mild oxidative pressure in comparison to normal cells (Figure 1) that can help them to exhibit characteristic cancer hallmarks [for detailed review refer to Hornsveld and Dansen (22)].
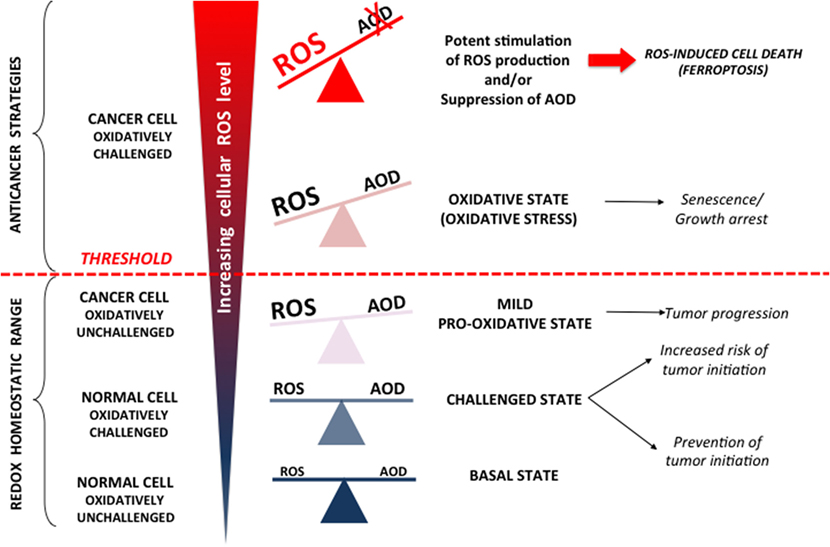
Figure 1. Reactive oxygen species (ROS) can (i) promote cancer, (ii) cause growth arrest, and (iii) be cytotoxic. In normal cells, increased (endogenous or exogenous) oxidative pressure leads to adequate upregulation of cellular antioxidant defense (AOD), which prevent mutagenic events and initiation of cancer formation. However, AOD is not 100% efficient, and thus, these “challenging states” also represent well-known risk factors for cancer development. Once formed, cancer progression seems to be further stimulated by a mild pro-oxidative state due to intensified metabolism, ROS-producing foci, etc. Importantly, this state is still maintained within “redox homeostatic range” thanks to strongly upregulated AOD of cancer cells. However, due to maximized AOD, cancer cells do not support further increase in ROS levels and thus cross the threshold into the state of “oxidative stress.” If ROS level increase further (e.g., due to chemotherapy), the only way for cancer cells to prevent further damage is by decreasing ROS production via cell-cycle arrest to repair damage and prevent cell death (cytostatic effects of ROS). However, if ROS burst induces irreversible damage and/or there is not enough components required for repair systems (e.g., glutathione), cancer cells experience programmed cell death or necrosis (cytotoxic effects of ROS).
According to the previous paragraph, it seems that a pro-oxidative state could facilitate initiation and progression of tumorigenesis. However, when reactive and very short living species such as ROS are considered, the situation is not so clear. Accordingly, studies on the effects of antioxidant supplements for cancer prevention and treatment showed opposed and mainly unpromising results, giving rise to confusion and the “antioxidant paradox” (1). Another redox consideration for cancer treatment includes increased ROS levels in cancer cells that already “walk on the edge of oxidative abyss” (23–25). This stand point arises from the very well know concept of hormesis that has been recognized since the XVI century by Paracelsus’s—“Die Dosis macht das Gift” or “the dose makes the poison” (26). The concept of hormesis, which revolutionized modern toxicology, claims that the dose–response curve is U-shaped, generally meaning that a drug/stimulus can have opposite effects in small and large doses [for more details refer to Calabrese and Baldwin (27) and papers stemming from it]. Indeed, it has been shown that a mild oxidative state promotes all hallmarks of cancer cells; however, if the threshold is exceeded (reaching the top of the arm of the U-shaped curve), influence of the oxidative environment can easily become anti-carcinogenous, promoting cell-cycle arrest, senescence, programmed cell death, or necrosis (Figure 1). Thus, it has been shown that increased oxidative pressure in the blood, if not adequately balanced by internal AOD, may limit the efficiency of melanoma cells to form distant tumors (28). These results are a textbook example of the antioxidant paradox suggesting how dietary supplementation of antioxidants may promote the metastatic potential of the cancer cells.
The anticancer effects of many conventional therapies, including irradiation and DNA-damaging chemotherapeutics (cisplatin, doxorubicin, gemcitabine, and 5-fluorouracil), rely mostly or partially on increased ROS production, due to mitochondria damage and dysfunction, as well as activation of NADPH oxidase (NOX) enzymes (29–33). However, these treatments encounter resistance with initial response being followed by the development of protective mechanisms against these oxidative/genotoxic insults. The mechanisms of resistance are complex involving drug modification, inhibition, degradation, and/or efflux [for further readings refer to Housman et al. (3)]. In spite of this complexity, the central role that AOD plays in these processes provided the rational for developing anticancer therapies targeting this aspect of cancer redox balance.
AOD in Cancer
As mentioned previously, oncogenic mutations lead to a pro-oxidative state of cancer cells. However, these cells are still required to maintain ROS levels below the threshold that would become detrimental (Figure 1). Indeed, antioxidant pathways known to respond to increased oxidative pressure in normal cells are constitutively activated in some cancers. The best example is the nuclear factor (erythroid-derived-2)-like 2 (NRF2)-signaling pathway [reviewed elsewhere in great detail (34, 35)]. NRF2 is the main transcription factor regulating expression of AOD enzymes. Under normal conditions, NRF2 is constantly ubiquitinated by Kelch-like ECH-associated protein 1 (KEAP1) and degraded by the proteasome. Oxidants/electrophiles inactivate Keap1 and stabilize NRF2, which then translocates into the nucleus, binds to the antioxidant response element, and activates the transcription of many cytoprotective genes that encode detoxifying enzymes and antioxidant proteins. Constitutive activation of NRF2, due to gain-of-function mutations in NRF2 (36), or loss-of-function mutation in its negative regulator KEAP1, was observed in different types of cancers (37–41). In addition, several tumor-suppressor genes act to repress tumor cell proliferation or cause cells to enter permanent cell-cycle arrest in response to ROS overproduction. These include retinoblastoma, p16INK4A, JNK, p38, p53, and forkhead box O. Most of these tumor-suppressor proteins sense changes in the cellular oxidative status and respond accordingly by inhibiting the cell cycle, and thus allowing cells time to recover after oxidative stress, and/or to induce expression of AOD enzymes (22).
Antioxidant defense is divided into enzymatic and non-enzymatic parts. Enzymatic AOD includes enzymes such as SODs, catalases, gluthatione peroxidases (GSH-Px), and glutathione S-transferases, as well as redox proteins such as thioredoxins (TRXs), peroxiredoxins, and glutaredoxins. Non-enzymatic AOD components are low-molecular weight compounds such as the key AOD tripeptide glutathione (GSH), vitamins (vitamins C and E), β-carotene, and uric acid. Complementary to these AOD components is the reducing equivalent NADPH that maintains catalases in active forms, serves as a cofactor for TRX and glutathione reductase [which converts oxidized glutathione (GSSG) into its reduced state (GSH)], and acts as a reducing agent for regeneration of glutaredoxins.
The concept of the Redox Code proposed recently by Jones and Sies (6) secludes GSH and NADH/NADPH as main determinants of the dynamic nature of redox signaling and control in multidimensional biological systems. This is even more pronounced in cancer cells due to increased and imbalanced metabolism, mutation accumulation during tumor progression and activated ROS-producing foci (such as defected mitochondria or NOX enzymes). The main reason why GSH and nicotinamide adenine dinucleotide (phosphate) are in the spotlight is the fact that these are the ultimate reducing factors of the cell.
Glutathione
Glutathione, a tripeptide γ-glutamyl-cysteinyl-serine, appears in two forms: the predominant reduced form (GSH), which reaches millimolar concentrations in the cell, and the minor oxidized form (GSSG), which is estimated to be less than 1% of the total GSH (42). The bulk of GSH is found in the cytosol (~90%), while the rest is localized mainly in mitochondria and the endoplasmic reticulum (ER) (43). GSH functions to detoxify electrophilic compounds including xenobiotics, which makes it central to cellular anticancer drug resistance (44). Owing to the sulfhydryl (−SH) group of cysteine, GSH can serve as an electron donor for reduction of peroxides (reactions catalyzed by GSH-Px) or disulfides. GSH can also directly react with various oxidants in a non-enzymatic manner, although these reaction kinetics are generally very slow (45). In addition, GSH is important in its cysteine-storage function (γ-glutamyl cycle).
Similar to ROS, GSH effects can be pro- or antitumorigenic (46). Although it is important in carcinogen detoxification, increased GSH levels and GSH-dependent biotransformation in many tumors may increase resistance to chemotherapy and radiotherapy (47–50). In addition, high GSH levels are associated with cancer hallmarks such as genomic instability, suppression of apoptosis, invasion, and metastatic activity [for further reading refer to Balendiran et al. (46)].
NADPH/NADP+ Couple
Antioxidant defense is completely ineffective without the NADPH/NADP+ cofactor, which serves as a main electron donor for both antioxidant enzymes and catabolic reactions. NADPH supplies reducing equivalents to maintain vital AOD components including the maintenance of active catalase and the regeneration of glutathione, TRX, and glutaredoxin. The NADH/NAD+ system is also involved in reversible 2-electron transfer catalysis and is connected with the NADPH/NADP+ system by activity of mitochondrial energy-linked transhydrogenase (NNT) (51). However, these two nicotinamide nucleotide systems have somewhat different roles in metabolism. Namely, while NADH/NAD+ is involved in catabolism and energy supply, NADPH/NADP+ is central for anabolism, defense, and redox homeostasis [reviewed in Ref. (6)]. The redox potential of these two systems also differs significantly in cells. Namely, the cytosolic redox potential of NADH/NAD+ is more oxidized (−241 mV) (52, 53) while in mitochondria, it operates at a more negative redox potential (−318 mV) (54), providing reductive force for ATP synthesis. Meanwhile, NADPH/NADP+ operates at more negative redox potential than the NAD system both in cytosol (−393 mV) and mitochondria (−415 mV) (53).
The energy-linked mitochondrial enzyme NNT that transfers electrons from NADH to NADPH thus connecting the two systems is of utmost importance in cancers containing mutations in the tricarboxylic acid (TCA) cycle (fumarate hydratase or succinate dehydrogenase) or the electron transport chain (ETC, complex I or III), which have been shown to promote utilization of glutamine by reductive carboxylation (55, 56). Namely, adequate citrate production in these conditions requires high NADPH/NADP+ ratios (57), which are achieved by the activity of the NNT (58).
NADPH production occurs via the pentose phosphate pathway (PPP), folate metabolism, and malic enzymes (MEs). The importance of AAs for NADPH-producing pathways, especially in cancer cells, is discussed below.
AAs Sensing from a Redox Perspective
Glucose, AAs, and fatty acids are the crucial building blocks of cellular biomolecules. Tight regulatory mechanisms have evolved to maintain the level of each within homeostatic range. The two main protein kinases involved in sensing and regulation of AA homeostasis are the mechanistic target of rapamycin complex 1 (mTORC1) and general control non-derepressable 2 (GCN2) [for an extensive reviews refer to Bar-Peled and Sabatini (59), Efeyan et al. (60), and Broer and Broer (61)]. Briefly, mTORC1 is a major sensor of specific AAs (Leu, Arg, and Lys), which also receives integrated, growth factors, hormonal, environmental and stress signals regulating growth, and proliferation. Although mechanisms of mTORC1 activation have progressed considerably in the past 20 years, the precise effects of individual AAs on mTORC1 activation have remained elusive. Sabatini’s group has illuminated AA sensing by demonstrating that mTORC1 translocation to lysosomes, is critical for its activation (59). Interestingly, recent studies revealed that this lysosomal localization allows mTORC1 sensing of AA levels (Arg and Gln), not only in cytoplasm but also in lysosomal compartement via the lysosomal membrane-resident transport protein SLC38A9 that constitutes a physical and functional part of the AA-sensing machinery (62, 63). Conversely, GCN2-kinase senses AA-uncharged tRNA, resulting in a general suppression of protein translation, paralleled by induction of the mechanisms to increase the cellular AA pool. Data regarding redox dependency of these pathways are still scarce and mechanically unclear.
Earlier studies showed that UV radiation activates mTORC1 signaling through MAP kinase activation by promoting phosphorylation of its downstream target p70S6k in an [H2O2] concentration and time-dependent manner (64, 65). mTORC1 activation was also observed when cells were treated with oxidizing agents, and surprisingly, even in AA-depleted conditions (66, 67). By contrast, subcellular localization of the mTORC1-interacting protein complex tuberous sclerosis complex at the peroxisome is responsible for mTORC1 repression and autophagy induction in response to ROS (68). Also, the tumor-suppressor ataxia telangiectasia mutated gene, appears to regulate autophagy through repression of mTORC1 in response to oxidative stress (69, 70). Thus, it seems that net effects of ROS on mTORC1 activity are context, time, and dose dependent. However, it should be emphasized that although the AAs leucine, arginine, and lysine are identified as key stimuli for mTORC1 activation, recent work on hepatoma HepG2 cells revealed significant sensitivity of both mTORC1 and GCN2 kinases to cysteine depletion (71). Prompt (within 60 min) inhibition of mTORC1 upon cysteine removal was observed. Considering that the Cys proteome coevolved with advanced [O2] sensing and [H2O2] signaling systems (72–74), this effect of cysteine on mTORC1 from a redox perspective may be of higher importance than the effects of ROS, per se.
The main downstream target of activated GCN2 is the eukaryotic initiation factor 2α (eIF2α), whose phosphorylation results in a general reduction of translation initiation, while specific mRNAs containing upstream open-reading frames (e.g., ATF4) are actively translated. However, it has been recognized that GCN2 can be activated by a number of different stresses [osmotic, UV, oxidative (such as [H2O2]), and ER] independently of AA depletion/imbalance (75–77). Interestingly, although the mechanisms are not yet known, it is recognized that the response of GCN2 to stressors such as [H2O2] or UV radiation are very fast in comparison to the gradual accumulation of uncharged tRNAs.
In turn, the AA-sensing pathways also influence cellular redox balance. Namely, ATF4, an effector molecule of the GCN2-pathway, also serves as a dimerization partner of the cap “n” collar transcription factor NRF2 (78, 79) promoting resistance to oxidative stress (79, 80). Consistently, it has been shown that mouse fibroblasts lacking Atf4 depend on supplemental reducing substances, such as glutathione, N-acetyl cysteine, or β-mercaptoethanol in their growth media (81). Recent work on HT1080 and A549 tumor cells showed the phosphorylation of eIF2 by protein kinase RNA-like endoplasmic reticulum kinase increases the ability of these cells to cope with increased oxidative pressure in an ATF4-independent manner by activating Akt (82). The importance of the GCN2 kinase in maintaining redox balance was also proved in vivo. Mice lacking GCN2 exhibited an increase in protein carbonylation in response to a leucine-imbalanced diet (83).
As for the effect of mTOR on redox homeostasis, a recent study showed that mTORC1 controls ATF4 activity by regulating the translation and stability of its mRNA (84). These results indicate that mTORC1, besides promoting anabolism and consequently increased ROS production, may also contribute to maintenance of the cellular redox equilibrium through “antioxidant properties” of ATF4.
The results listed earlier favor the hypothesis that redox and AA balance are tightly intertwined. How AAs specifically influence the cellular “Redox Code” (GSH and NADPH levels) will be discussed below with special attention placed on the pathways that might represent “vulnerability points” for design of novel anticancer therapeutics.
Cysteine Levels Determines GSH Levels
Two cytosolic ATP-dependent enzymes are involved in GSH synthesis: glutamate–cysteine ligase (GCL), which catalyzes formation of a particular gamma-peptidic bond between Glu and Cys, and glutathione synthetase. The rate-limiting step in GSH synthesis is the reaction catalyzed by GCL (85). Genetic deletion of the GCL catalytic subunit was lethal in the mouse embryo, while knockout mice for the modifier subunit of the enzyme, although viable and fertile, show a significant decrease of tissue GSH levels (9–16% of wt) (86). The Km of mouse GCL for cysteine is estimated at ~0.2 mM (87), which is near the upper limit of typical cellular cysteine concentrations, while the Km for glutamate is at or below the cellular glutamate concentration for Drosophila, mouse, or human GCLholo enzymes (88–90). Hence, it is not surprising that cysteine is the main regulator of GCL activity, and thus GSH synthesis (Figure 2).
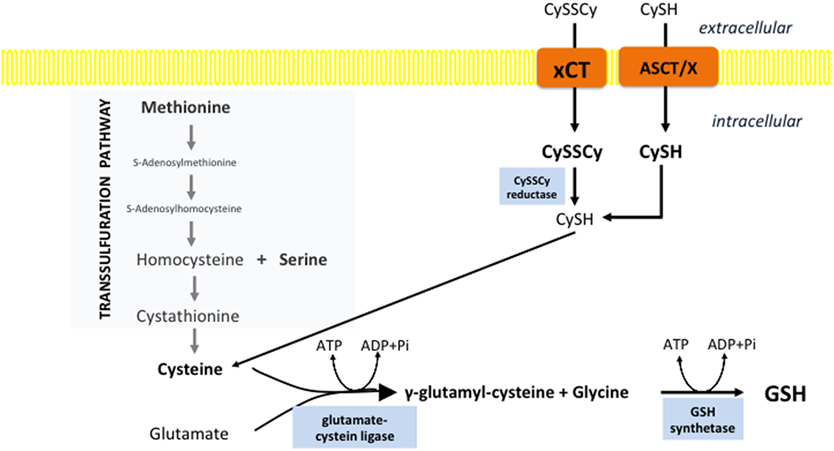
Figure 2. Cystine import is the rate-limiting step in glutathione biosynthesis. Cysteine can be synthesized within the cell through the trans-sulfuration pathway. However, this pathway is often insufficient in cancer cells and therefore cysteine must be imported. Different transporters are involved in the import of the reduced, cysteine (CySH), and oxidized, cystine (CySSCy) form of this semi-essential AA. The heavy-chain transporter subunit of system xc-light chain (xCT) seems to play a pivotal role in the import of CySSCy, the predominant form of cysteine in circulation. After import, CySSCy is reduced by cystine reductase and used for different purposes including GSH biosynthesis. Import of cysteine can occur via ASCT (alanine/serine/cysteine transporter) and other transporters (x).
In physiological conditions, cysteine is not an essential AA as it can be synthetized through trans-sulfuration pathways (TSP) from methionine, mainly in the liver. Approximately 50% of the cysteine in hepatic GSH is derived from methionine via TSP (91). However, high demand for cysteine in cancer cells, make TSP insufficient (Figure 2). Furthermore, some tumors have shown significantly lower expression of TSP enzymes mostly due to transcriptional silencing (92, 93). Consequently, Cramer and coworkers (94) showed that depletion of cyst(e)ine with pharmacologically optimized cyst(e)inase enzymes induced cell-cycle arrest and cancer cell death due to GSH depletion and ROS accumulation, both in vitro and in vivo.
xc-Transport System
Multiple tissue-specific transporters are responsible for the import of cystine (CySSCy), the oxidized and predominant form of the AA in circulation (40–50 µM), and/or cysteine, which is present at substantially lower concentrations (8–10 µM) (95–97). However, increasing data in the literature points toward the xc-system as being crucial for CySSCy import in cancer cells (Figure 2). The system xc- acts as a Na+-independent and Cl−-dependent antiporter of the anionic forms of cystine and glutamate and is composed of the transporter light-chain (xCT, encoded by SLC7A11 gene) and a chaperone heavy-chain (CD98hc aka 4F2hc, encoded by SLC3A2 gene) subunit [for a comprehensive review, see Lewerenz et al. (98)]. Interestingly, although the system xc- seems to be a ubiquitous marker of almost all cells cultured in vitro, its in vivo distribution in humans appears restricted mainly to the CNS, pancreas, fibroblasts, and immune cells (99–105). According to Bannai et al. (106), this induction of the system xc- in culture conditions is caused by the high partial pressure of oxygen. Consistent with this hypothesis, prolonged cultivation of fibroblasts in reduced oxygen partial pressure caused a significant decrease in the system xc-activity (106).
Considering that AA transporters are necessary for tumor cell proliferation, it is not surprising that xCT is upregulated in many patient samples and tumor cell lines including hepatoma, lymphoma, glioma, colon, breast, prostate, and pancreatic (95, 101, 107–113). Expression of the xCT subunit seems to be under direct control of oncogenes including NRF2 and Ets-1 (114–116). In addition, the promoter region of the SLC7A11 gene contains an AA response element, which allows the transcription factor ATF4 to enhance expression of xCT in response to AA depletion and/or oxidative stress (115, 117).
System xc-light chain mediates import of cystine into cells thus regulating GSH levels (118, 119). Since GSH is the most abundant non-enzymatic antioxidant within the cell, upregulation of xCT satisfies the highly proliferative phenotype of cancer cells. This is supported by complete growth inhibition of lymphoma cells and certain glioma, breast, prostate, lung, and pancreatic cancer cells upon pharmacological inhibition of xCT by sulfasalazine or by the cyclic glutamate analog (109, 111). Besides its role in tumor growth, knockdown or pharmacological inhibition of xCT increased adhesion and inhibited tumor cell invasion in vitro and decreased metastases in vivo (120). In addition, xCT was shown to associate with CD44v, a major adhesion molecule for the extracellular matrix, which is involved in tumor invasion and metastasis in lethal gastrointestinal tumors (121) along with the metabolic interplay between tumors and host tissue (122). Furthermore, xCT plays a pivotal role in the chemoresistance of tumor cells (123–125), particularly to anticancer drugs that produce high amounts of ROS, such as geldanamycin and celastrol (126, 127).
The importance of the cystine/glutamate antiporter in redox regulation was further implicated in the newly described type of cell death—ferroptosis (128, 129). Ferroptosis is described as an iron-dependent, programmed form of cell death driven by loss of activity of the lipid repair enzyme glutathione peroxidase 4 and subsequent accumulation of membrane lipid peroxides (130). The first described inducer of ferroptosis in Ras-mutated human foreskin fibroblasts was the xCT inhibitor erastin (131). Depletion of intracellular GSH levels due to inhibition of xCT and subsequent increase of ROS levels seems to be sufficient to trigger erastin-dependent cell death. The same results were observed with sulfasalazine, which is another inhibitor of xCT (109, 132). Interestingly, it has been shown that a loss of cysteinyl-tRNA synthetase might prevent erastin-induced cell death by inducing the TSP (133), suggesting that trans-sulfuration can contribute to resistance to inhibition of xCT and ferroptosis induction.
Serine/Folate Pathway and NADPH Production
Textbooks have stated for years that the main cellular NADPH-producing system is the PPP. Surprisingly, a recent comprehensive study (134) showed that serine-driven one-carbon metabolism (folate cycle) gives almost the same contribution in the NADPH production as the PPP and MEs in proliferating cells. It is also interesting to note that enzymes of both PPP and the serine synthesis pathway (SSP, from which the folate cycle streams out) are induced by NRF2 (135, 136). The function of the folate cycle is ascribed to the collection of one-carbon units from AAs, and subsequent incorporation of these moieties into biomolecules in biosynthetic or methylation reactions. One of the major branching points of the folate cycle is 10-formyl-tetrahydrofolate (10-formyl-THF), which in mitochondria may be used for ATP regeneration [methylene tetrahydrofolate dehydrogenase (MTHFD) reaction], formylation of the mitochondrial initiator N-formylmethionine-tRNA or metabolized to [CO2], generating NADPH (10-formyl-THF dehydrogenase reaction). On the other side, in cytosol, 10-formyl-THF can be used for purine or NADPH synthesis, while its counterpart 5,10-methylene-THF is used for thymidylate synthesis and homocysteine remethylation in the methionine cycle. In cancer, mitochondrial 10-formyl-THF is mainly used for NADPH production due to overexpression of corresponding enzyme, while in citosol, this reaction is prevented so one-carbon unit, required for purine synthesis, would not be wasted (137, 138). Default mitochondria-to-cytosol directionality of the folate cycle is achived by different expression of enzymes in these compartments, as well as more reductive, i.e., oxidative environment in cytosol and mitochondria respectively (139).
Two mitochondrial reactions of the folate cycle contribute to NADPH production; one is catalyzed by MTHFD, and the other is catalyzed by 10-formyl-THF dehydrogenase (ALDH1L2) (Figure 3). Fan et al. showed that depletion of either of these enzymes decreased NADPH/NADP+ and consequently GSH/GSSG ratios and impaired cellular resistance to imposed oxidative stress (134). Similarly, Piskounova et al. showed that redox balancing effects of these enzymes is fundamental for metastatic potential of melanoma cells in vivo (28). Namely, this study showed that knockdown of either MTHFD or ALDH1L2 prevents distant metastasis of melanoma cells that encounter high-oxidative pressure in the blood and visceral organs. Besides, it was reported that the first mitochondrial enzyme of the folate cycle, termed serine hydroxymethyl transferase 2 (SHMT2) is essential for maintaining mitochondrial NADPH and GSH level during hypoxia in neuroblastoma cell lines. This study detected a correlation between high expression of SHMT2 and poor prognosis in neuroblastoma patients (140). Expression of SHMT2 in neuroblastoma cells seems to be controlled by the collaborative action of c-Myc and HIF1α. However, numerous oncogenes are reported to affect enzymes of the folate cycle. For example, it is shown that common KRAS mutation associates with increased expression of MTHFD2 in non-small cell lung cancer cell lines (141), while mTORC1-dependent induction of MTHFD2 is reported in both normal and cancer cells (142).
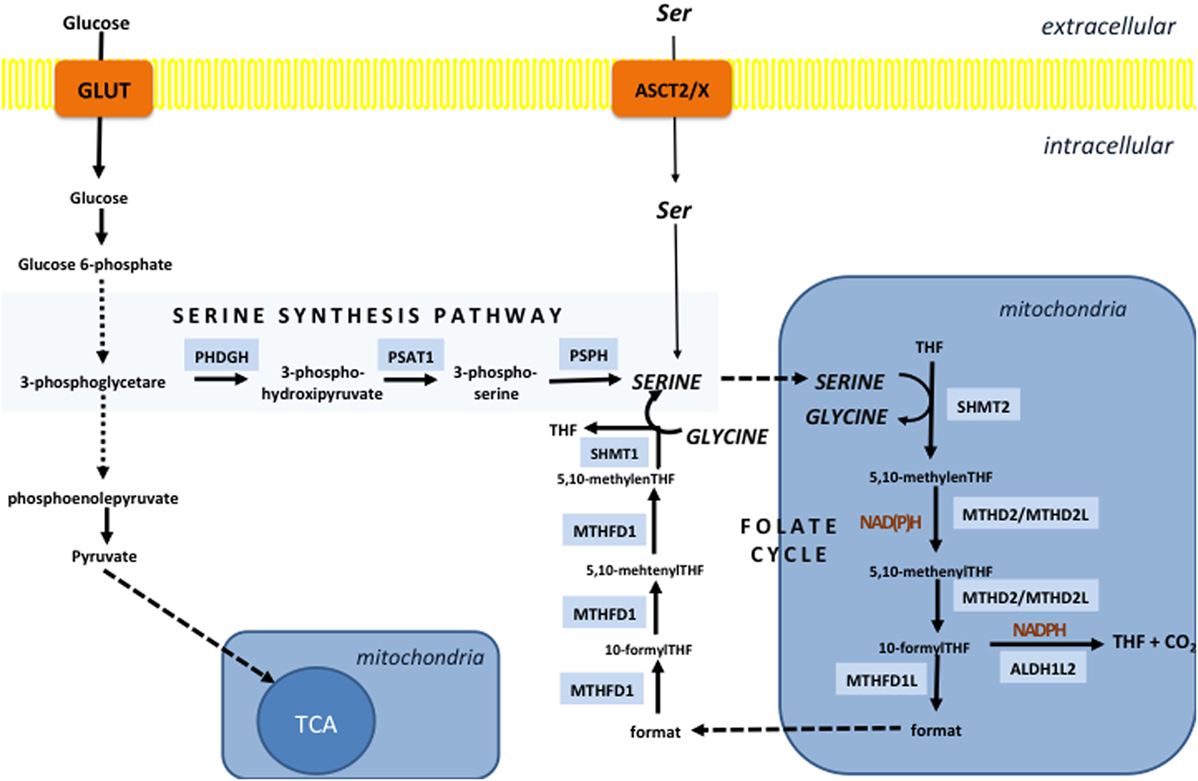
Figure 3. The folate cycle is fueled by the serine synthesis pathway (SSP) and extracellular serine. SSP diverges from glycolysis at the level of 3-phosphoglycerate, which is converted into 3-phospho-hydroxipyruvate by the action of the enzyme phosphoglycerate dehydrogenase (PHDGH) and ultimately to serine following further enzymatic steps. This pathway is of great importance in cancers with mutated or overexpressed PHDGH, while serine import plays a pivotal role in maintenance of the serine cellular balance in cells with unaltered PHDGH activity. The folate cycle in the vast majority of the cells starts in mitochondria by the action of serine hydroxymethyl transferase 2 (SHMT2) which generates glycine and 5,10-methylene-tetrahydrofolate (5,10-methylene-THF). The next reaction can produce NADH or NADPH depending if methenyltetrahydrofolate dehydrogenase 2 (MTHD2) or MTHD2-like (MTHD2L) is used to convert 5,10-methylene-THF into 5,10-methenyl-THF. The same enzyme than generate one-carbon unit—10-formyl-THF, which can be used for ATP production by the enzyme (MTHD1L) or NADPH generation in the reaction catalyzed by 10-formyTHF dehydrogenase (ALDH1L2). If ATP is generated, 10-formylTHF is converted into a format that is transported into the cytosol and used by trifunctional MTHFD1 enzyme to regenerate 10-formylTHF for purine synthesis, 5,10-methylene-THF for thymidylate synthesis and homocysteine remethylation in the methionine cycle. The unidirectionality of the folate cycle seems to be provided by more oxidative mitochondrial redox state that favors use of NAD(P)+ by mitochondrial MTHD2(L).
Besides production of NADPH, the folate cycle contributes to production of GSH by intersecting with the methionine cycle (Figure 4). Considering the role of methionine and homocysteine in the TSP (cysteine synthesis), as well as that glycine is product of serine metabolism (folate cycle), it is not surprising that serine depletion results in reduced level of glutathione (143), while activation of serine synthesis is now well identified as a bypass of glycolysis flux contributing to GSH synthesis (136, 144).
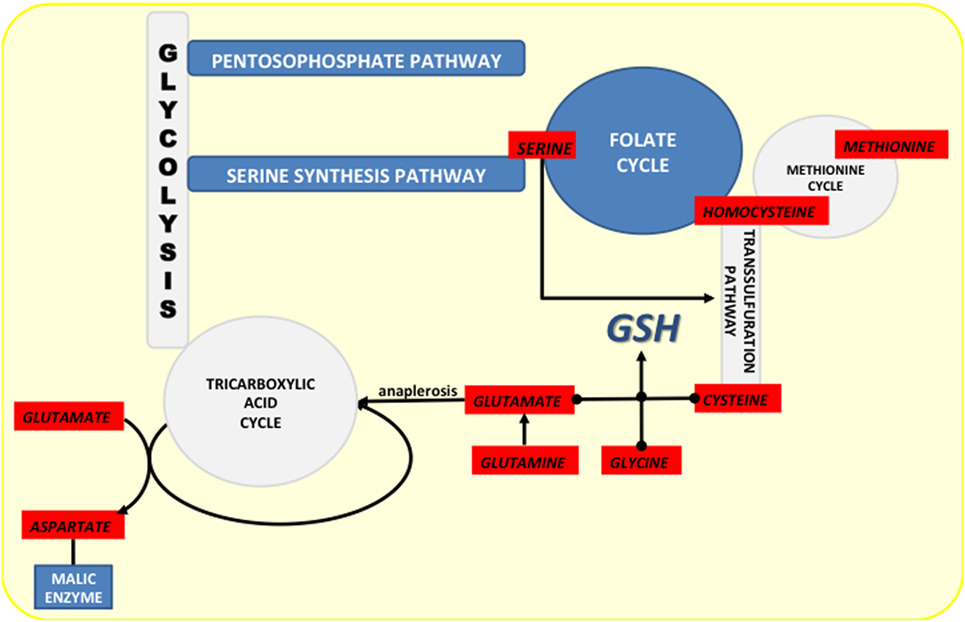
Figure 4. Crossroads of NADPH-producing pathways (marked dark blue) and the pathways from which they diverge or to which they converge (marked light blue). Amino acids involved in these pathways are marked in red.
Serine, just like cysteine, can be transported into the cell by different transporters [such as the sodium-dependent transport system ASC that will be mentioned later in the text, and transporter system A, as well as sodium-independent system asc (145, 146)], or synthesized de novo from glycolytic intermediate 3-phosphoglycerate through the SSP. Highly proliferating cancer cells both in culture conditions and in vivo consume significant amount of exogenous serine (143, 147). Consequently, serine depletion both in vitro and in vivo decreases proliferation and induces metabolic remodeling, commencing with SSP induction, to replenish cellular serine pool (143).
Serine Synthesis Pathway
The importance of serine for cancer physiology came from earlier studies that showed increased flux through the SSP in cancer cells (148). However, this was somewhere neglected until the recent discovery that the first enzyme of SSP, phosphoglycerate dehydrogenase (PHGDH), is genetically amplified in breast cancer and melanoma (149, 150), and overexpression of the SSP components are correlated with poorer prognosis in breast cancer patients (151). Consistently, suppression of PHGDH in cell lines characterized with elevated expression of this enzyme decreases cell proliferation and serine synthesis. What is even more interesting is that in non-tumorigenic breast cancer cells, overexpression of PHGDH alone lead to disruption of the acinar cellular morphology and predisposed them to neoplastic transformation (149, 152), making the PHGDH a bona fide oncogene (153).
Amplification of PHGDH de-sensitizes tumors to exogenous serine levels but also represents a vulnerability point for potential cancer treatment. Namely, PHGDH knockdown strongly decreased proliferation and some of the SSP outputs [such as α-ketoglutarate (α-KG)] only in cells with amplified PHGDH expression (150). Interestingly, PHGDH also prevents conversion of glycine to serine suggesting that the folate cycle relies exclusively on serine synthesis in PHGDH overexpressing tumors (154). This was demonstrated by PHGDH knockdown decreasing cell proliferation even when exogenous serine was present (154).
Several other oncogenes also induce expression of the SSP enzymes, such as c-Myc and HER2 (155, 156). Also, in line with its involvement in maintaining redox balance, the SSP enzyme expression is induced by NRF2 in an ATF4-dependent manner in NSCLC cells (136). Interestingly, Maddocks and coworkers (143) showed that serine can be a vulnerable point of cancer metabolism even in tumors that do not have multiplication of the PHGDH gene, but lack p53. Namely, they showed that the p53–p21 axis is fundamental for metabolic adaptation upon serine deprivation, while loss of p53 in the conditions of serine depletion leads to impaired glycolysis and elevated ROS levels.
Interestingly, pharmacological inhibition of the SSP could also influence flux through the PPP. Namely, inhibition of the SSP would increase intracellular levels of 3-phosphoglycerate, which has been shown to inhibit 6-phosphogluconate dehydrogenase that catalyzes the second step in the oxidative PPP (157).
Glutamate and NADPH Production
In addition to the PPP and folate cycle, MEs are known to regulate NADPH/NADP+ balance, which is seemingly dependent of glutamine metabolism in cancer. One of the main metabolic characteristics of many cancers, besides the Warburg effect (158, 159), is increased consumption of glutamine to the extent where exogenous level of this AA limit tumor cell survival. This “glutamine addiction” has been recognized for more than 50 years (160, 161); however, diverse contributions of glutamine to intermediary metabolism, cell signaling, and gene expression are still not fully understood (162).
The vast majority of glutamine in the cell is converted into glutamate either by cytoplasmic glutaminase (GLS1) or by the mitochondrial isoform of this enzyme (GLS2). Glutamate is then converted to α-KG by the enzyme glutamate dehydrogenase. α-KG can then have one of two fates (Figure 5). (1) Canonically, produced α-KG enters the TCA and replenishes it, or (2) it is carboxylated to isocitrate, pushing the TCA in the opposite direction (163).
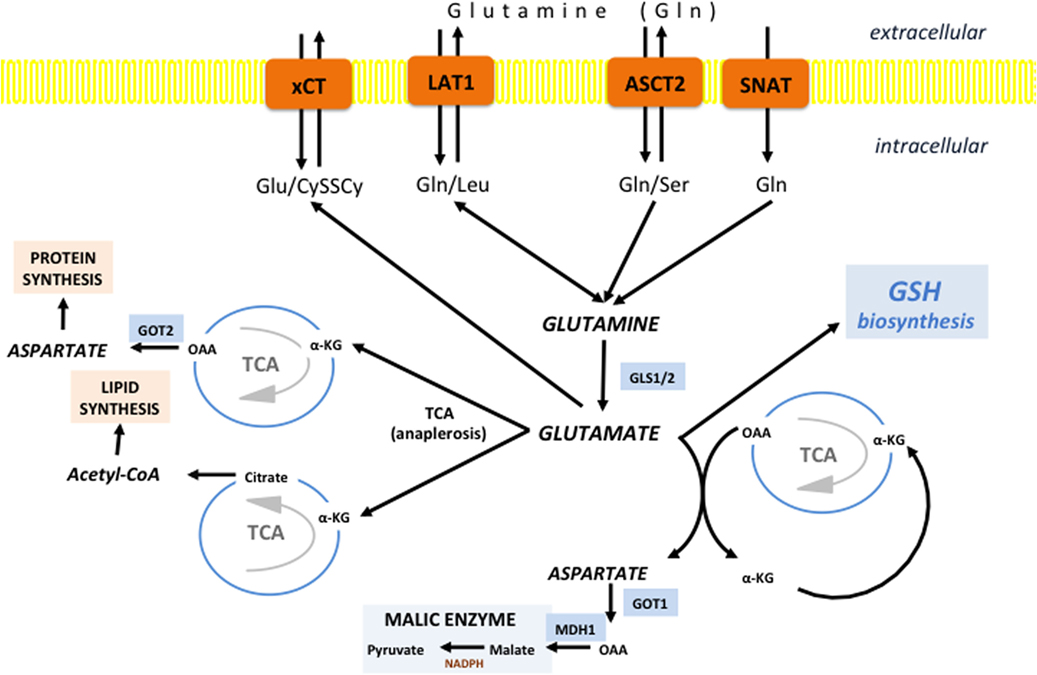
Figure 5. Glutamine/glutamate fates in cancer cells. Different transporters are proposed to fuel the “Glutamine addiction” of cancer cells including alanine-serine-cysteine transporter 2 (ASCT2), SNAT1/2, and L-type amino acid transporter 1 (LAT1). Once inside the cell, Gln can be use for uptake of essential AAs by LAT1. However, the vast majority of Gln is promtly deaminated to glutamate by the action of cytoplasmic or mitochondrial glutaminase (GLS1 and GLS2, respectively). If deaminated in cytosol, Glu is transferred into mitochondria, and there it is further converted into α-ketoglutarate (α-KG) to replenish the tricarboxylic acid (TCA). However, the fate of α-KG can be dual. It can follow normal TCA flow until oxaloacetate (OAA), which is then converted into asparate by aspartate dehydrogenase (GOT2) and translocated into cytoplasm or used for synthesis of asparagine and arginine (protein synthesis). However, if the α-KG is carboxylated to isocitrate and then converted into citrate, citrate is exported into the cytosol where it is used for lipid synthesis in the form of acetyl-CoA. Glutamate-derived aspartate can also be converted into OAA by cytoplasmic GOT1, commonly induced in KRAS-mutated tumors. OAA is then converted first into malate by malate dehydrogenase 1 (MDH1) and then into pyruvate by malic enzyme (ME), generating reducing power in the form of NADPH. Besides involvement in anaplerosis and NADPH production, Glu has an important role as a component of GSH, as well as a substrate for system xc-light chain (xCT) in allowing entrance of cystine into the cell.
When glutamine-derived α-KG follows the canonical pathway, the TCA works normally (clockwise) until oxaloacetate (OAA), which is usually converted into aspartate by aspartate transaminase (GOT2) and exported into the cytosol, or alternatively, it can be converted into asparagine and arginine and fuel protein synthesis. Interestingly, a recent study on KRAS-mutated pancreatic ductal adenocarcinoma (PDAC) showed that GOT2 regulates glutamine flux by producing α-KG and aspartate from glutamate and OAA (164). Aspartate is then shuttled into the cytosol where it is converted back into OAA by cytoplasmic GOT1. The OAA produced is converted first to malate and then to pyruvate and NADPH by the action of cytoplasmic malic enzyme 1 (ME1). Considering that KRAS-mutated PDACs have decreased flux through the PPP (165), glutamine-fueled ME1 in these cells may be seen as a major contributor to the NADPH homeostasis. Indeed, ME1 suppression increased ROS accumulation and decreased tumor cell growth both in vitro and in vivo, while suppressing glutamine utilization and sensitizing cells to oxidative damage (164). Conversely, it remains to be determined if inhibitors of glutamine import or its conversion to glutamate would have the same effects on oxidative status and cell growth.
Oppositely to KRAS, p53 has a negative impact on this NADPH-producing pathway. This was demonstrated by a strong upregulation of MEs (ME1/2) in the absence of functional p53 (166), which were crucial for maintenance of adequate NADPH levels. Here is important to recall the importance of the p53–p21 axis to serine starvation (143) and to anticipate potential resistance mechanisms for serine starvation, in the absence of p53, via upregulation of the ME1/2.
Alanine-Serine-Cysteine Transporter 2 (ASCT2)
Alanine-serine-cysteine transporter 2 (SLC1A5) is a Na+-dependent transporter carrying small neutral AAs such as alanine, serine, cysteine, glutamine, and asparagine (Km ~20 μM) in addition to long-chain AAs such as threonine, valine, and methionine with lower affinity (Km ~300–500 μM). ASCT2 is proposed to play a central role in sustaining cancer cell glutamine homeostasis based on work from Myc-driven cancers, which are particularly addicted to glutamine, and fuel their “glutamine addiction” by promoting high ASCT2 expression (167–169). Also, ASCT2 together with xCT and L-type amino acid transporter 1 (LAT1), comprise the “minimal set” of transporters required for cancer AA homeostasis and the group known to be highly upregulated in cancer (170, 171). Consequently the glutamine import activity of ASCT2 has been proposed to be fundamental for the activity of other AA transporters upregulated in cancer, such as xCT and LAT1 (leucine-for-glutamine exchanger) (171–173). However, recent findings demonstrated that ASCT2 inhibition can be overcame in certain cancer cell types partly by expressing the Na+-dependent glutamine transporters system A amino acid transporter 1–2, questioning the functional redundancy for certain AA transporters in tumor growth (174). Regardless, glutamine import (via ASCT2 or other transporters) is indeed of great importance for normal functioning of LAT1 and xCT. Recent studies showed that cancer cell glutamine addiction might be a direct consequence of xCT activity, which consumes large amounts of glutamate derived from extracellular glutamine thereby restricting nutrient flexibility of the cell (175, 176).
The importance of glutamine in cancer cells often dominates ASCT2 experimental interpretations. However, it is important to remember ASCT2’s ability to transport other AAs such as serine. As mentioned, some cancer cells remain highly dependent on the uptake of exogenous serine (143). Since ASCT2 display a strong affinity for serine, it would be interesting to investigate the role of this transporter in serine metabolism and redox homeostasis in general. Furthermore, the name of ASCT2: alanine–serine–cysteine transporter may be misleading. Namely, ASCT2 is structurally related to the glutamate transporter and neutral AA transporter ASCT1 and when expressed in Xenopus laevis oocyte ASCT2 indeed exhibits Na+-dependent uptake of AA similar to ASCT1 (177). However, the same study of Utsunomiya-Tate and collaborators revealed that ASCT2 exhibits different tissue distribution, as well as substrate selectivity and functional properties when compared to ASCT1. Thus, for example, glutamate uptake by ASCT1 is electrogenic, while in the case of ASCT2 lowering pH enhances uptake, which suggests electroneutral uptake. Also, it seems that cysteine is not a substrate for ASCT2, but an allosteric inhibitor of its activity. In accordance to this are recent findings that mark cysteine as a potent competitive inhibitor of ASCT2 that binds to the site different from the one for substrate and induces efflux of glutamine both in the case of proteoliposomes and in intact cells (178).
Considering that the “minimal set” of transporters required for cancer AA homeostasis comprises ASCT2, while its activity/specificity is still rather debatable, it is of utmost importance to continue research on the biology of this very intriguing AA transporter.
Concluding Remarks
For a long time, the mild pro-oxidative redox state of cancer cells has been recognized as a vulnerable point of these highly metabolically active cells. However, in the context of chemotherapy, we are still struggling to find the adequate approach to the vast majority of ROS-producing therapeutics that encounter cellular resistance and frequent disease relapse. During the past decade, an approach involving suppression of the internal AOD of cancer has attracted more attention. Within highly complex and intertwined AOD system, GSH and NADPH play the most universal and important role in determining the characteristic redox cellular profile. Considering that AA import and metabolism seems to be upstream of these AOD systems, we have emphasized here the specific molecules and pathways that show great, but still insufficiently examined, potential for anticancer therapy from a redox standpoint. In conclusion, the transport and internal synthesis pathways for cysteine, serine, glutamine, and to some extent glycine appear to be the most interesting targets for the development of novel redox-based therapeutics. Targeting AA transport systems (xCT, ASCT2, and SNAT) is promising considering that import of these semi-essential AAs are not required in normal cells, while they are absolutely required for cancer cell survival.
Author Contributions
MV and JP made substantial contributions to conception and design, revised manuscript critically, and gave final approval of the version to be submitted. YC and SP revised manuscript critically and gave final approval of the version to be submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MV was supported by a post-doctoral fellowship from GEMLUC. YC, SP, and JP were funded by the Centre Scientifique de Monaco (CSM) and a Grant from GEMLUC.
Abbreviations
AA(s), amino acid(s); ALDH1L2, 10-formyl-THF dehydrogenase; AOD, antioxidant defense; ARE, antioxidant response element; ASCT2, alanine-serine-cysteine transporter 2; ATM, ataxia telangiectasia mutated gene; eIF2α, eukaryotic initiation factor 2α; ER, endoplasmic reticulum; ETC, electron transport chain; FOXO, forkhead box O; GCL, glutamate–cysteine ligase; GCN2, general control non-derepressable 2; GLS1/2, cytoplasmic/mitochondrial glutaminase; GLUD1, glutamate dehydrogenase; GOT1/2, aspartate transaminase 1/2; GR, glutathione reductase; GS, glutathione synthetase; GSH, glutathione; GSH-Px, glutathione peroxidase; GSTs, glutathione S-transferases; GSSG, glutathione oxidized; [H2O2], hydrogen peroxide; [HO⋅], peroxyl radical; [HO−], hydroxyl anion; KEAP1, Kelch-like ECH-associated protein 1; LAT1, L-type amino acid transporter 1; ME1/2, malic enzyme 1/2; MTHFD, methylene tetrahydrofolate dehydrogenase; mTORC1, mechanistic target of rapamycin complex 1; NAD(P)H, nicotinamide adenine dinucleotide (phosphate); NNT, energy-linked transhydrogenase; [NO⋅], nitric oxide; NOX, NADPH oxidase; NRF2, nuclear factor (erythroid-derived-2)-like 2; [1O2], singlet oxygen; , superoxide anion radical; PDAC, pancreatic ductal adenocarcinoma; PERK, protein kinase RNA-like endoplasmic reticulum kinase; PHGDH, phosphoglycerate dehydrogenase; PPP, pentose phosphate pathway; PRXs, peroxiredoxins; Rb, retinoblastoma; [ROO⋅], peroxyl radicals; [ROOH], organic hydroperoxides; ROS, reactive oxygen species; SHMT2, serine hydroxymethyl transferase 2; SNAT1-2, system A amino acid transporter 1–2; SOD, superoxide dismutase; SSP, serine synthesis pathway; TCA, tricarboxylic acid; THF, tetrahydrofolate; TRXs, thioredoxins; TSC, tuberous sclerosis complex; TSP, trans-sulfuration pathways; CySSCy, cystine (oxidized cysteine); xCT, system xc-light chain.
References
1. Halliwell B. The antioxidant paradox. Lancet (2000) 355(9210):1179–80. doi:10.1016/S0140-6736(00)02075-4
2. Halliwell B. The antioxidant paradox: less paradoxical now? Br J Clin Pharmacol (2013) 75(3):637–44. doi:10.1111/j.1365-2125.2012.04272.x
3. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (Basel) (2014) 6(3):1769–92. doi:10.3390/cancers6031769
4. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature (2009) 458(7239):780–3. doi:10.1038/nature07733
5. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov (2013) 12(12):931–47. doi:10.1038/nrd4002
6. Jones DP, Sies H. The redox code. Antioxid Redox Signal (2015) 23(9):734–46. doi:10.1089/ars.2015.6247
7. Gerschman R, Gilbert DL, Nye SW, Dwyer P, Fenn WO. Oxygen poisoning and x-irradiation: a mechanism in common. Science (1954) 119(3097):623–6. doi:10.1126/science.119.3097.623
8. Gerschman R, Gilbert DL, Nye SW, Fenn WO. Influence of X-irradiation on oxygen poisoning in mice. Proc Soc Exp Biol Med (1954) 86(1):27–9. doi:10.3181/00379727-86-21002
9. Gerschman R, Nadig PW, Snell AC Jr, Nye SW. Effect of high oxygen concentrations on eyes of newborn mice. Am J Physiol (1954) 179(1):115–8.
10. Hayaishi O, Sato Y, Jakoby WB, Stohlman EF. Reversible enzymatic oxidation of bile acids. Arch Biochem Biophys (1955) 56(2):554–5. doi:10.1016/0003-9861(55)90278-2
11. Mason HS, Fowlks WL, Peterson E. Oxygen transfer and electron transport by the phenolase complex1. J Am Chem Soc (1955) 77(10):2914–5. doi:10.1021/ja01615a088
12. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem (1969) 244(22):6049–55.
13. Cadenas E, Sies H. Oxidative stress: excited oxygen species and enzyme activity. Adv Enzyme Regul (1985) 23:217–37. doi:10.1016/0065-2571(85)90049-4
14. D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol (2007) 8(10):813–24. doi:10.1038/nrm2256
15. Egea J, Fabregat I, Frapart YM, Ghezzi P, Gorlach A, Kietzmann T, et al. European contribution to the study of ROS: a summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol (2017) 13:94–162. doi:10.1016/j.redox.2017.05.007
16. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med (2000) 192(7):1001–14. doi:10.1084/jem.192.7.1001
17. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res (1991) 51(3):794–8.
19. Storz P. Reactive oxygen species in tumor progression. Front Biosci (2005) 10:1881–96. doi:10.2741/1667
20. Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J (2007) 401(1):1–11. doi:10.1042/BJ20061131
21. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res (2010) 44(5):479–96. doi:10.3109/10715761003667554
22. Hornsveld M, Dansen TB. The hallmarks of cancer from a redox perspective. Antioxid Redox Signal (2016) 25(6):300–25. doi:10.1089/ars.2015.6580
23. Kong Q, Beel JA, Lillehei KO. A threshold concept for cancer therapy. Med Hypotheses (2000) 55(1):29–35. doi:10.1054/mehy.1999.0982
24. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood (2003) 101(10):4098–104. doi:10.1182/blood-2002-08-2512
25. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat (2004) 7(2):97–110. doi:10.1016/j.drup.2004.01.004
26. Gems D, Partridge L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab (2008) 7(3):200–3. doi:10.1016/j.cmet.2008.01.001
27. Calabrese EJ, Baldwin LA. Defining hormesis. Hum Exp Toxicol (2002) 21(2):91–7. doi:10.1191/0960327102ht217oa
28. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature (2015) 527(7577):186–91. doi:10.1038/nature15726
29. Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med (2001) 7(10):1111–7. doi:10.1038/nm1001-1111
30. Conklin KA. Coenzyme q10 for prevention of anthracycline-induced cardiotoxicity. Integr Cancer Ther (2005) 4(2):110–30. doi:10.1177/1534735405276191
31. Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer (2007) 120(1):175–80. doi:10.1002/ijc.22132
32. Wang S, Zhang H, Cheng L, Evans C, Pan CX. Analysis of the cytotoxic activity of carboplatin and gemcitabine combination. Anticancer Res (2010) 30(11):4573–8.
33. Suzuki S, Okada M, Shibuya K, Seino M, Sato A, Takeda H, et al. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget (2015) 6(1):458–70. doi:10.18632/oncotarget.2693
34. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer (2012) 12(8):564–71. doi:10.1038/nrc3278
35. Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med (2016) 22(7):578–93. doi:10.1016/j.molmed.2016.05.002
36. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol (2010) 220(4):446–51. doi:10.1002/path.2653
37. Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun (2007) 362(4):816–21. doi:10.1016/j.bbrc.2007.08.051
38. Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res (2008) 68(5):1303–9. doi:10.1158/0008-5472.CAN-07-5003
39. Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology (2008) 135(4):1358–68, 1368.e1–4. doi:10.1053/j.gastro.2008.06.082
40. Konstantinopoulos PA, Spentzos D, Fountzilas E, Francoeur N, Sanisetty S, Grammatikos AP, et al. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res (2011) 71(15):5081–9. doi:10.1158/0008-5472.CAN-10-4668
41. Yoo NJ, Kim HR, Kim YR, An CH, Lee SH. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology (2012) 60(6):943–52. doi:10.1111/j.1365-2559.2012.04178.x
42. Akerboom TP, Bilzer M, Sies H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J Biol Chem (1982) 257(8):4248–52.
43. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med (2009) 30(1–2):42–59. doi:10.1016/j.mam.2008.05.005
44. Zhang K, Mack P, Wong KP. Glutathione-related mechanisms in cellular resistance to anticancer drugs. Int J Oncol (1998) 12(4):871–82.
45. Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med (1999) 27(3–4):322–8. doi:10.1016/S0891-5849(99)00051-9
46. Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct (2004) 22(6):343–52. doi:10.1002/cbf.1149
47. Midander J, Deschavanne PJ, Malaise EP, Revesz L. Survival curves of irradiated glutathione-deficient human fibroblasts: indication of a reduced enhancement of radiosensitivity by oxygen and misonidazole. Int J Radiat Oncol Biol Phys (1982) 8(3–4):443–6. doi:10.1016/0360-3016(82)90657-5
48. Ishikawa T. The ATP-dependent glutathione S-conjugate export pump. Trends Biochem Sci (1992) 17(11):463–8. doi:10.1016/0968-0004(92)90489-V
49. Suzuki KT, Tomita T, Ogra Y, Ohmichi M. Glutathione-conjugated arsenics in the potential hepato-enteric circulation in rats. Chem Res Toxicol (2001) 14(12):1604–11. doi:10.1021/tx0155496
50. Vukovic V, Nicklee T, Hedley DW. Differential effects of buthionine sulphoximine in hypoxic and non-hypoxic regions of human cervical carcinoma xenografts. Radiother Oncol (2001) 60(1):69–73. doi:10.1016/S0167-8140(01)00331-0
51. Hoek JB, Rydstrom J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J (1988) 254(1):1–10. doi:10.1042/bj2540001
52. Bucher T, Brauser B, Conze A, Klein F, Langguth O, Sies H. State of oxidation-reduction and state of binding in the cytosolic NADH-system as disclosed by equilibration with extracellular lactate-pyruvate in hemoglobin-free perfused rat liver. Eur J Biochem (1972) 27(2):301–17. doi:10.1111/j.1432-1033.1972.tb01840.x
53. Sies H. Oxidative Stress [Online]. London/Orlando: Academic Press (1985). Available from: http://search.ebscohost.com/login.aspx?direct=true&scope=site&db=nlebk&db=nlabk&AN=893380
54. Puigserver P, Ribot J, Serra F, Gianotti M, Bonet ML, Nadal-Ginard B, et al. Involvement of the retinoblastoma protein in brown and white adipocyte cell differentiation: functional and physical association with the adipogenic transcription factor C/EBPalpha. Eur J Cell Biol (1998) 77(2):117–23. doi:10.1016/S0171-9335(98)80079-4
55. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481(7381):380–4. doi:10.1038/nature10602
56. Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep (2014) 7(5):1679–90. doi:10.1016/j.celrep.2014.04.037
57. Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem (2012) 287(18):14615–20. doi:10.1074/jbc.C112.353946
58. Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem (2013) 288(18):12967–77. doi:10.1074/jbc.M112.396796
59. Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol (2014) 24(7):400–6. doi:10.1016/j.tcb.2014.03.003
60. Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature (2015) 517(7534):302–10. doi:10.1038/nature14190
61. Broer S, Broer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J (2017) 474(12):1935–63. doi:10.1042/BCJ20160822
62. Rebsamen M, Pochini L, Stasyk T, de Araújo MEG, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature (2015) 519:477. doi:10.1038/nature14107
63. Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science (2015) 347(6218):188–94. doi:10.1126/science.1257132
64. Parrott LA, Templeton DJ. Osmotic stress inhibits p70/85 S6 kinase through activation of a protein phosphatase. J Biol Chem (1999) 274(35):24731–6. doi:10.1074/jbc.274.35.24731
65. Huang C, Li J, Ke Q, Leonard SS, Jiang BH, Zhong XS, et al. Ultraviolet-induced phosphorylation of p70(S6K) at Thr(389) and Thr(421)/Ser(424) involves hydrogen peroxide and mammalian target of rapamycin but not Akt and atypical protein kinase C. Cancer Res (2002) 62(20):5689–97.
66. Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem (2005) 280(47):39505–9. doi:10.1074/jbc.M506096200
67. Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, et al. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem (2011) 286(37):32651–60. doi:10.1074/jbc.M111.238014
68. Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol (2015) 17(10):1259–69. doi:10.1038/ncb3230
69. Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A (2010) 107(9):4153–8. doi:10.1073/pnas.0913860107
70. Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy (2010) 6(5):672–3. doi:10.4161/auto.6.5.12509
71. Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep (2016) 6:30033. doi:10.1038/srep30033
72. Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol (2000) 17(8):1232–9. doi:10.1093/oxfordjournals.molbev.a026406
73. Kawahara T, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol (2007) 7:109. doi:10.1186/1471-2148-7-109
74. Rytkonen KT, Storz JF. Evolutionary origins of oxygen sensing in animals. EMBO Rep (2011) 12(1):3–4. doi:10.1038/embor.2010.192
75. Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, et al. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol (2002) 12(15):1279–86. doi:10.1016/S0960-9822(02)01037-0
76. Zhan K, Narasimhan J, Wek RC. Differential activation of eIF2 kinases in response to cellular stresses in Schizosaccharomyces pombe. Genetics (2004) 168(4):1867–75. doi:10.1534/genetics.104.031443
77. Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell (2005) 16(12):5493–501. doi:10.1091/mbc.E05-03-0268
78. Hayes JD, McMahon M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett (2001) 174(2):103–13. doi:10.1016/S0304-3835(01)00695-4
79. He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem (2001) 276(24):20858–65. doi:10.1074/jbc.M101198200
80. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem (1999) 274(37):26071–8. doi:10.1074/jbc.274.37.26071
81. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell (2003) 11(3):619–33. doi:10.1016/S1097-2765(03)00105-9
82. Rajesh K, Krishnamoorthy J, Kazimierczak U, Tenkerian C, Papadakis AI, Wang S, et al. Phosphorylation of the translation initiation factor eIF2alpha at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis (2015) 6:e1591. doi:10.1038/cddis.2014.554
83. Chaveroux C, Lambert-Langlais S, Parry L, Carraro V, Jousse C, Maurin AC, et al. Identification of GCN2 as new redox regulator for oxidative stress prevention in vivo. Biochem Biophys Res Commun (2011) 415(1):120–4. doi:10.1016/j.bbrc.2011.10.027
84. Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep (2017) 19(6):1083–90. doi:10.1016/j.celrep.2017.04.042
85. Meister A, Anderson ME. Glutathione. Annu Rev Biochem (1983) 52(1):711–60. doi:10.1146/annurev.bi.52.070183.003431
86. Yang H, Zeng Y, Lee TD, Yang Y, Ou X, Chen L, et al. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase bytert-butylhydroquinone. J Biol Chem (2002) 277(38):35232–9. doi:10.1074/jbc.M203812200
87. Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem (2005) 280(40):33766–74. doi:10.1074/jbc.M504604200
88. Tu Z, Anders MW. Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem J (1998) 336(Pt 3):675–80. doi:10.1042/bj3360675
89. Fraser JA, Saunders RD, McLellan LI. Drosophila melanogaster glutamate-cysteine ligase activity is regulated by a modifier subunit with a mechanism of action similar to that of the mammalian form. J Biol Chem (2002) 277(2):1158–65. doi:10.1074/jbc.M106683200
90. Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem (2002) 277(51):49446–52. doi:10.1074/jbc.M209372200
91. Banerjee R, Zou CG. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys (2005) 433(1):144–56. doi:10.1016/j.abb.2004.08.037
92. Kim J, Hong SJ, Park JH, Park SY, Kim SW, Cho EY, et al. Expression of cystathionine beta-synthase is downregulated in hepatocellular carcinoma and associated with poor prognosis. Oncol Rep (2009) 21(6):1449–54. doi:10.3892/or_00000373
93. Zhao H, Li Q, Wang J, Su X, Ng KM, Qiu T, et al. Frequent epigenetic silencing of the folate-metabolising gene cystathionine-beta-synthase in gastrointestinal cancer. PLoS One (2012) 7(11):e49683. doi:10.1371/journal.pone.0049683
94. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med (2017) 23(1):120–7. doi:10.1038/nm.4232
95. Bannai S. Transport of cystine and cysteine in mammalian cells. Biochim Biophys Acta (1984) 779(3):289–306. doi:10.1016/0304-4157(84)90014-5
96. Jones DP, Mody VC Jr, Carlson JL, Lynn MJ, Sternberg P Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic Biol Med (2002) 33(9):1290–300. doi:10.1016/S0891-5849(02)01040-7
97. Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev (2008) 88(1):249–86. doi:10.1152/physrev.00018.2006
98. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal (2013) 18(5):522–55. doi:10.1089/ars.2011.4391
99. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem (1986) 261(5):2256–63.
100. Eck HP, Droge W. Influence of the extracellular glutamate concentration on the intracellular cyst(e)ine concentration in macrophages and on the capacity to release cysteine. Biol Chem Hoppe Seyler (1989) 370(2):109–13. doi:10.1515/bchm3.1989.370.1.109
101. Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martin R, et al. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers Arch (2001) 442(2):286–96. doi:10.1007/s004240100537
102. Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, et al. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta (2001) 1512(2):335–44. doi:10.1016/S0005-2736(01)00338-8
103. Rimaniol AC, Mialocq P, Clayette P, Dormont D, Gras G. Role of glutamate transporters in the regulation of glutathione levels in human macrophages. Am J Physiol Cell Physiol (2001) 281(6):C1964–70. doi:10.1152/ajpcell.2001.281.6.C1964
104. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A (2002) 99(3):1491–6. doi:10.1073/pnas.022630299
105. Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J Histochem Cytochem (2006) 54(5):549–57. doi:10.1369/jhc.5A6840.2006
106. Bannai S, Sato H, Ishii T, Sugita Y. Induction of cystine transport activity in human fibroblasts by oxygen. J Biol Chem (1989) 264(31):18480–4.
107. Makowske M, Christensen HN. Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. J Biol Chem (1982) 257(10):5663–70.
108. Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature (1999) 402(6762):685–9. doi:10.1038/45280
109. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia (2001) 15(10):1633–40. doi:10.1038/sj.leu.2402238
110. Narang VS, Pauletti GM, Gout PW, Buckley DJ, Buckley AR. Suppression of cystine uptake by sulfasalazine inhibits proliferation of human mammary carcinoma cells. Anticancer Res (2003) 23(6C):4571–9.
111. Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci (2005) 25(31):7101–10. doi:10.1523/JNEUROSCI.5258-04.2005
112. Doxsee DW, Gout PW, Kurita T, Lo M, Buckley AR, Wang Y, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate (2007) 67(2):162–71. doi:10.1002/pros.20508
113. Lo M, Ling V, Wang YZ, Gout PW. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer (2008) 99(3):464–72. doi:10.1038/sj.bjc.6604485
114. Verschoor ML, Singh G. Ets-1 regulates intracellular glutathione levels: key target for resistant ovarian cancer. Mol Cancer (2013) 12(1):138. doi:10.1186/1476-4598-12-138
115. Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol (2014) 34(18):3421–34. doi:10.1128/MCB.00221-14
116. Habib E, Linher-Melville K, Lin HX, Singh G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol (2015) 5:33–42. doi:10.1016/j.redox.2015.03.003
117. Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun (2004) 325(1):109–16. doi:10.1016/j.bbrc.2004.10.009
118. Bannai S, Tateishi N. Role of membrane transport in metabolism and function of glutathione in mammals. J Membr Biol (1986) 89(1):1–8. doi:10.1007/BF01870891
119. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem (2002) 277(47):44765–71. doi:10.1074/jbc.M208704200
120. Chen RS, Song YM, Zhou ZY, Tong T, Li Y, Fu M, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene (2009) 28(4):599–609. doi:10.1038/onc.2008.414
121. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell (2011) 19(3):387–400. doi:10.1016/j.ccr.2011.01.038
122. Ohmura M, Hishiki T, Yamamoto T, Nakanishi T, Kubo A, Tsuchihashi K, et al. Impacts of CD44 knockdown in cancer cells on tumor and host metabolic systems revealed by quantitative imaging mass spectrometry. Nitric Oxide (2015) 46:102–13. doi:10.1016/j.niox.2014.11.005
123. Huang Y, Dai Z, Barbacioru C, Sadee W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res (2005) 65(16):7446–54. doi:10.1158/0008-5472.CAN-04-4267
124. Huang Y. Pharmacogenetics/genomics of membrane transporters in cancer chemotherapy. Cancer Metastasis Rev (2007) 26(1):183–201. doi:10.1007/s10555-007-9050-6
125. Huang Y, Penchala S, Pham AN, Wang J. Genetic variations and gene expression of transporters in drug disposition and response. Expert Opin Drug Metab Toxicol (2008) 4(3):237–54. doi:10.1517/17425255.4.3.237
126. Liu R, Blower PE, Pham AN, Fang J, Dai Z, Wise C, et al. Cystine-glutamate transporter SLC7A11 mediates resistance to geldanamycin but not to 17-(allylamino)-17-demethoxygeldanamycin. Mol Pharmacol (2007) 72(6):1637–46. doi:10.1124/mol.107.039644
127. Pham AN, Blower PE, Alvarado O, Ravula R, Gout PW, Huang Y. Pharmacogenomic approach reveals a role for the x(c)- cystine/glutamate antiporter in growth and celastrol resistance of glioma cell lines. J Pharmacol Exp Ther (2010) 332(3):949–58. doi:10.1124/jpet.109.162248
128. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi:10.1016/j.cell.2012.03.042
129. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov (2016) 15(5):348–66. doi:10.1038/nrd.2015.6
130. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol (2016) 26(3):165–76. doi:10.1016/j.tcb.2015.10.014
131. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3(3):285–96. doi:10.1016/S1535-6108(03)00050-3
132. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget (2016) 7(24):36021–33. doi:10.18632/oncotarget.8651
133. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ (2016) 23(2):270–8. doi:10.1038/cdd.2015.93
134. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature (2014) 510(7504):298–302. doi:10.1038/nature13236
135. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell (2012) 22(1):66–79. doi:10.1016/j.ccr.2012.05.016
136. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet (2015) 47(12):1475–81. doi:10.1038/ng.3421
137. Krupenko SA, Oleinik NV. 10-Formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ (2002) 13(5):227–36.
138. Krupenko NI, Dubard ME, Strickland KC, Moxley KM, Oleinik NV, Krupenko SA. ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J Biol Chem (2010) 285(30):23056–63. doi:10.1074/jbc.M110.128843
139. Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, Teng X, et al. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab (2016) 24(4):640–1. doi:10.1016/j.cmet.2016.09.011
140. Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov (2014) 4(12):1406–17. doi:10.1158/2159-8290.CD-14-0250
141. Moran DM, Trusk PB, Pry K, Paz K, Sidransky D, Bacus SS. KRAS mutation status is associated with enhanced dependency on folate metabolism pathways in non-small cell lung cancer cells. Mol Cancer Ther (2014) 13(6):1611–24. doi:10.1158/1535-7163.MCT-13-0649
142. Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science (2016) 351(6274):728–33. doi:10.1126/science.aad0489
143. Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature (2013) 493(7433):542–6. doi:10.1038/nature11743
144. Nikkanen J, Forsstrom S, Euro L, Paetau I, Kohnz RA, Wang L, et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab (2016) 23(4):635–48. doi:10.1016/j.cmet.2016.01.019
145. Furuya S, Watanabe M. Novel neuroglial and glioglial relationships mediated by L-serine metabolism. Arch Histol Cytol (2003) 66(2):109–21. doi:10.1679/aohc.66.109
146. Kanai Y, Clemencon B, Simonin A, Leuenberger M, Lochner M, Weisstanner M, et al. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol Aspects Med (2013) 34(2–3):108–20. doi:10.1016/j.mam.2013.01.001
147. Jaeken J, Detheux M, Van Maldergem L, Foulon M, Carchon H, Van Schaftingen E. 3-Phosphoglycerate dehydrogenase deficiency: an inborn error of serine biosynthesis. Arch Dis Child (1996) 74(6):542–5. doi:10.1136/adc.74.6.542
148. Snell K, Weber G. Enzymic imbalance in serine metabolism in rat hepatomas. Biochem J (1986) 233(2):617–20. doi:10.1042/bj2330617
149. Locasale JW, Cantley LC. Genetic selection for enhanced serine metabolism in cancer development. Cell Cycle (2011) 10(22):3812–3. doi:10.4161/cc.10.22.18224
150. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476(7360):346–50. doi:10.1038/nature10350
151. Antonov A, Agostini M, Morello M, Minieri M, Melino G, Amelio I. Bioinformatics analysis of the serine and glycine pathway in cancer cells. Oncotarget (2014) 5(22):11004–13. doi:10.18632/oncotarget.2668
152. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet (2011) 43(9):869–74. doi:10.1038/ng.890
153. Frezza C. Cancer metabolism: addicted to serine. Nat Chem Biol (2016) 12(6):389–90. doi:10.1038/nchembio.2086
154. Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol (2016) 12(6):452–8. doi:10.1038/nchembio.2070
155. Bollig-Fischer A, Dewey TG, Ethier SP. Oncogene activation induces metabolic transformation resulting in insulin-independence in human breast cancer cells. PLoS One (2011) 6(3):e17959. doi:10.1371/journal.pone.0017959
156. Nilsson LM, Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW, et al. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet (2012) 8(3):e1002573. doi:10.1371/journal.pgen.1002573
157. Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell (2012) 22(5):585–600. doi:10.1016/j.ccr.2012.09.020
158. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi:10.1085/jgp.8.6.519
159. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell (2008) 13(6):472–82. doi:10.1016/j.ccr.2008.05.005
160. Eagle H. The specific amino acid requirements of a human carcinoma cell (stain HeLa) in tissue culture. J Exp Med (1955) 102(1):37–48. doi:10.1084/jem.102.1.37
161. Kvamme E, Svenneby G. Effect of anaerobiosis and addition of keto acids on glutamine utilization by Ehrlich ascites-tumor cells. Biochim Biophys Acta (1960) 42:187–8. doi:10.1016/0006-3002(60)90779-4
162. Still ER, Yuneva MO. Hopefully devoted to Q: targeting glutamine addiction in cancer. Br J Cancer (2017) 116(11):1375–81. doi:10.1038/bjc.2017.113
163. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev (2008) 18(1):54–61. doi:10.1016/j.gde.2008.02.003
164. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496(7443):101–5. doi:10.1038/nature12040
165. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149(3):656–70. doi:10.1016/j.cell.2012.01.058
166. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature (2013) 493(7434):689–93. doi:10.1038/nature11776
167. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A (2008) 105(48):18782–7. doi:10.1073/pnas.0810199105
168. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458(7239):762–5. doi:10.1038/nature07823
169. Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol (2015) 235(1):90–100. doi:10.1002/path.4429
170. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature (2012) 483(7391):603–7. doi:10.1038/nature11003
171. Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res (2015) 75(9):1782–8. doi:10.1158/0008-5472.CAN-14-3745
172. Banjac A, Perisic T, Sato H, Seiler A, Bannai S, Weiss N, et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene (2008) 27(11):1618–28. doi:10.1038/sj.onc.1210796
173. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136(3):521–34. doi:10.1016/j.cell.2008.11.044
174. Broer A, Rahimi F, Broer S. Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem (2016) 291(25):13194–205. doi:10.1074/jbc.M115.700534
175. Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife (2017) 6:e27713. doi:10.7554/eLife.27713
176. Shin CS, Mishra P, Watrous JD, Carelli V, D’Aurelio M, Jain M, et al. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun (2017) 8:15074. doi:10.1038/ncomms15074
177. Utsunomiya-Tate N, Endou H, Kanai Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem (1996) 271(25):14883–90. doi:10.1074/jbc.271.25.14883
Keywords: cancer, amino acids, redox homeostasis, glutathione, NADPH/NADP+
Citation: Vučetić M, Cormerais Y, Parks SK and Pouysségur J (2017) The Central Role of Amino Acids in Cancer Redox Homeostasis: Vulnerability Points of the Cancer Redox Code. Front. Oncol. 7:319. doi: 10.3389/fonc.2017.00319
Received: 02 October 2017; Accepted: 08 December 2017;
Published: 21 December 2017
Edited by:
Sergio Giannattasio, Istituto di Biomembrane, Bioenergetica e Biotecnologie Molecolari (IBIOM), ItalyReviewed by:
Cesare Indiveri, University of Calabria, ItalyPaula Ludovico, University of Minho, Portugal
Copyright: © 2017 Vučetić, Cormerais, Parks and Pouysségur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milica Vučetić, bWlsaWNhQGNlbnRyZXNjaWVudGlmaXF1ZS5tYw==;
Jacques Pouysségur, amFjcXVlcy5wb3V5c3NlZ3VyQHVuaWNlLmZy