 Stuart L. Cramer1*†
Stuart L. Cramer1*† Aubrey L. Miller2
Aubrey L. Miller2 Joseph G. Pressey1†Tracy L. Gamblin2
Joseph G. Pressey1†Tracy L. Gamblin2 Elizabeth A. Beierle3Brian D. Kulbersh3
Elizabeth A. Beierle3Brian D. Kulbersh3 Patrick L. Garcia2Leona N. Council4,5Rupa Radhakrishnan6Skyler V. Hendrix2,7
Patrick L. Garcia2Leona N. Council4,5Rupa Radhakrishnan6Skyler V. Hendrix2,7 David R. Kelly4,8
David R. Kelly4,8 Raymond G. Watts1†
Raymond G. Watts1† Karina J. Yoon2*†
Karina J. Yoon2*†
- 1Department of Pediatrics, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Department of Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Department of Surgery, University of Alabama at Birmingham, Birmingham, AL, United States
- 4Department of Pathology, University of Alabama at Birmingham, Birmingham, AL, United States
- 5The Birmingham Veterans Administration Medical Center, Birmingham, AL, United States
- 6Department of Radiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 7Biomedical Science Program, UAB Honors College, University of Alabama at Birmingham, Birmingham, AL, United States
- 8Department of Pathology and Laboratory Medicine, Children’s of Alabama, Birmingham, AL, United States
Therapy for rhabdomyosarcoma (RMS) has generally been limited to combinations of conventional cytotoxic agents similar to regimens originally developed in the late 1960s. Recently, identification of molecular alterations through next-generation sequencing of individual tumor specimens has facilitated the use of more targeted therapeutic approaches for various malignancies. Such targeted therapies have revolutionized treatment for some cancer types. However, malignancies common in children, thus far, have been less amenable to such targeted therapies. This report describes the clinical course of an 8-year-old female with embryonal RMS having anaplastic features. This patient experienced multiple relapses after receiving various established and experimental therapies. Genomic testing of this RMS subtype revealed mutations in BCOR, ARID1A, and SETD2 genes, each of which contributes to epigenetic regulation and interacts with or modifies the activity of histone deacetylases (HDAC). Based on these findings, the patient was treated with the HDAC inhibitor vorinostat as a single agent. The tumor responded transiently followed by subsequent disease progression. We also examined the efficacy of vorinostat in a patient-derived xenograft (PDX) model developed using tumor tissue obtained from the patient’s most recent tumor resection. The antitumor activity of vorinostat observed with the PDX model reflected clinical observations in that obvious areas of tumor necrosis were evident following exposure to vorinostat. Histologic sections of tumors harvested from PDX tumor-bearing mice treated with vorinostat demonstrated induction of necrosis by this agent. We propose that the evaluation of clinical efficacy in this type of preclinical model merits further evaluation to determine if PDX models predict tumor sensitivity to specific agents and/or combination therapies.
Introduction
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood. This tumor type is characterized by myoblastic differentiation and expression of skeletal muscle markers such as desmin, myogenin, and/or MYOD1. Embryonal RMS (eRMS), the most common subtype, usually occurs before 10 years of age. A secondary subtype, anaplastic eRMS, is characterized by enlarged hyperchromatic nuclei and TP53 mutations, and is associated with poor outcome (1, 2). Chemotherapy, surgery, and/or radiation comprise standard therapy for patients with RMS. Chemotherapeutic regimens frequently include vincristine, actinomycin D, cyclophosphamide, and inhibitors of type-1 topoisomerase or mammalian target of rapamycin (mTOR) (3, 4).
Here, we report the case of a patient who presented with an anaplastic eRMS of the left parapharyngeal pterygopalatine fossa. At diagnosis, the tumor mass measured 3.9 cm × 3.1 cm × 3.6 cm. The patient’s treatment history included multiple chemotherapeutic regimens, palliative radiation (XRT), and debulking procedures (Table 1). Responses were transient, and several localized relapses were documented during the 4 years of treatment. Tissue from final surgical resection on 4/22/2014 was used to establish the patient-derived xenograft (PDX) model described in this study and to submit for sequencing (Foundation Medicine, Cambridge, MA, USA). Sequence analysis identified mutations in genes encoding BCOR, ARID1A, and SETD2 (Table 2). These mutations would be predicted to increase histone deacetylases (HDAC) activity or confer gain of function or constitutive activation of HDAC. Therefore, the patient was treated with the HDAC inhibitor vorinostat for 6 months.

Table 1. Summary of relevant clinical history.

Table 2. Genetic alterations identified by sequencing.
Treatment with vorinostat was initiated 4 months after surgical resection in April 2014. Tumor specimens obtained prior to administration of vorinostat provided the opportunity to establish a PDX model from this patient’s tumor, and to use this model to determine if preclinical data characterizing the efficacy of vorinostat reflected the efficacy of this agent in the clinic. A goal of the study was to evaluate whether this type of model might be used to predict efficacy in the clinic, as an approach toward personalized medicine.
Background
There are no published studies describing the utility of vorinostat in treating RMS, but several preclinical studies have been published. Keshelava et al. demonstrated that vorinostat had IC50 values of 0.88–9.77 µM in four RMS cell lines (RD, Rh41, Rh18, and Rh30) in vitro, but had little or no effect in five RMS xenograft models (Rh30, Rh30R, Rh41, Rh18, and Rh36) (5). A second study by Vleeshouwer-Neumann et al. reported that vorinostat suppressed the growth of RD, 381 T, and SMS–CTR eRMS cell lines and also inhibited the migration of these cells in vitro (6). Furthermore, these investigators observed that vorinostat had antiproliferative effects in a zebrafish transgenic eRMS model.
Tumor DNA Sequencing
Sequencing of the 400 genes in the Pediatric Cancer Prone Gene Panel was performed by Foundation One (CLIA Certified Sequencing Foundation, Cambridge, MA, USA) using DNA extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissue. Mutations were identified in genes encoding BCOR, ARID1A, and SETD2, which are involved in DNA methylation and chromatin remodeling and each of which affects HDAC-associated cell processes (7–9).
BCOR
BCOR (BCL6 corepressor; Polycomb group repressive complex-1 variant) mediates BCL6 function in diffuse large B-cell lymphoma (10, 11). BCOR inhibits histone methylation (H3 Lys-4:H3K4me3 and Lys-36:H3K36me2), an activity regulated by HDAC (7, 12). Studies with mesenchymal stem cells obtained from patients with oculofacialcardiodental syndrome (OFDC) indicate that mutations of BCOR enhance mesenchymal stem cell proliferation (7). Mutations in this gene are also associated with myelodysplastic syndromes, pediatric acute myeloid leukemia, and medulloblastoma (13–15).
ARID1A
ARID1A (AT-rich interaction domain 1A) is a member of the SWI/SNF family and has helicase and ATPase activities (16). ARID1A regulates transcription by altering the structure of chromatin (8). ARID1A also contributes to regulation of cell-cycle progression and is involved in DNA damage repair (17, 18). Mutations in ARID1A have been identified in ovarian, endometrial, and uterine tumors (19–22). Inactivating mutations in ARID1A suggest that wild-type ARID1A may act as a tumor suppressor (23). In vitro studies indicate that ovarian cancer cells harboring ARID1A mutations are relatively sensitive to the histone methyltransferase EZH2 inhibitor and to nutlin, which inhibits MDM2–p53 interaction (24, 25).
SETD2
SETD2 (SET domain containing 2) is a histone methyl transferase that methylates Lys36 of histone H3 (H3K36Me3) (9, 26). Loss-of-function mutations of this gene promote renal cancer progression and decrease expression of H3K36Me3 in clear cell renal cell carcinoma (27). SETD2–H3K36Me3 pathway alterations are associated with development of leukemia (28).
HDAC
Histone deacetylases regulate the ratio of acetylated and deacetylated histones, and are associated with gene silencing by modification of chromatin structure (29, 30). Alterations in HDAC genes and/or HDAC expression are related to multiple human pathologies including cancer (31–35). For example, increased HDAC1 and HDAC2 expression is reported in colon cancer cells compared with non-oncogenic adenoma cells, and decreased expression of HDAC1 arrests the growth of this tumor cell type (36, 37).
Each of the epigenetic alterations described above has been reported to contribute to tumor phenotypes (9, 13, 15, 27, 28, 38, 39), and each of the three proteins encoded by genes identified as harboring mutations contributes directly or indirectly to HDAC-associated functions. The HDAC inhibitor vorinostat (suberoylanilide hydroxamic acid, SAHA) is approved for refractory T-cell lymphoma and is being evaluated in multiple clinical trials for other types of cancers (40). Based on the patient’s tumor characteristics, the FDA-approved status of vorinostat, the known maximum tolerated dose in pediatric patients, and preclinical information in the literature, the patient was treated with vorinostat.
Vorinostat (Suberoylanilide Hydroxamic Acid, an HDAC Inhibitor)
Vorinostat was approved in the USA in 2006 for the treatment of cutaneous T-cell lymphoma (CTCL) in patients with progressive or recurrent disease (41). A Phase-I COG clinical trial demonstrated that vorinostat was well tolerated at 230 mg/m2/day in children with recurrent solid tumors (42). Vorinostat inhibits the activity of HDACs 1 and 2, thereby increasing the ratio of deactylated/acetylated histones and suppressing tumor cell proliferation in vitro and in vivo. Current literature suggests that the likely mechanism of this suppression is that HDAC deacetylation selectively activates transcription of genes that induce cell differentiation and/or apoptosis (43, 44).
Discussion
Ethics Statement
Protocols involving human subjects were approved by the Institutional Review Board (IRB) of the University of Alabama at Birmingham (Birmingham, AL, USA). Written informed assent and consent were obtained from the patient and her family to use tumor specimens for research purposes. Written informed consent was also obtained from the patient’s parent agreeing to publication of the report.
Clinical Course Associated with Vorinostat Treatment
Treatment consisted of 28-day cycles at 200 mg/day (42). Approximately 4 weeks after the initiation of treatment, magnetic resonance imaging (MRI) of brain, orbit, and neck revealed a reduction of the tumor mass centered at the left infratemporal fossa from 8.9 cm × 4.9 cm × 6.5 cm to 8.2 cm × 4.7 × 6.0 cm (Figure 1). The dose of vorinostat was increased to 300 mg/day 3 days/week and 200 mg/day 4 days/week. Within 2 weeks of dose escalation, the patient developed severe thrombocytopenia associated with nasopharyngeal hemorrhage requiring packed red blood cell resuscitation. The dose was then de-escalated to 200 mg/day. Over the next 16 weeks, MRIs documented stabilization of the infratemporal fossa tumor mass, with sustained necrosis. While receiving vorinostat, the patient showed symptomatic improvement and was weaned from patient-controlled analgesia. Following this transient response, the patient developed neck swelling due to tumor progression and tumor rupture traversing the skin. Two months after the 6-month regimen of single-agent vorinostat had been discontinued, the patient succumbed to her disease.
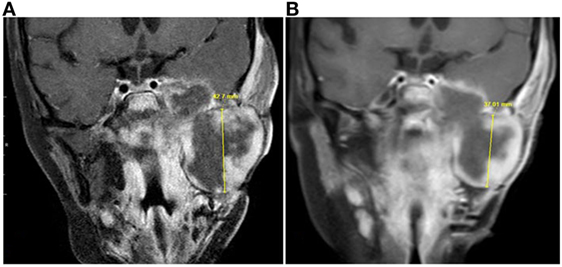
Figure 1. Magnetic resonance imaging (MRI) taken prior to and during vorinostat treatment. Coronal contrast-enhanced T1-weighted MRI images from (A) July 2014 and (B) September 2014 showed decrease in size of the infratemporal component of the tumor. Craniocaudal measurement showed the greatest change, although this component of the tumor had decreased in size in other dimensions as well.
Response to Vorinostat of Mice Bearing Patient-Derived Xenografts
Ethics Statement
Protocols involving animal use were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham (Birmingham, AL, USA).
A tumor specimen harvested 4 months prior to initiating vorinostat therapy was implanted subcutaneously into immunocompromised mice (SCID) within 1 h of tumor resection. When the tumor volume of the donor mouse reached ~800–1,000 mm3, the tumor was harvested, divided, and transplanted into a cohort of mice for evaluation of vorinostat efficacy. When tumor volume reached ~300 mm3, tumor-bearing mice were randomized into two groups (N = 10/group) and received 50 mg/kg vorinostat or vehicle (vehicle control) intraperitoneally daily for 21 days. This dose is equivalent to a clinical dose of ~200 mg (45). Tumors were measured with Vernier calipers (Fowler/Slyvac, Newtown, MA, USA) twice weekly, and tumor volumes calculated using the equation v = (π/6)d3. Twenty-four hours after completion of treatment, mice were euthanized, and tumor tissue was harvested and archived as both formalin fixed paraffin embedded (FFPE) and snap frozen in liquid nitrogen. Tumor volumes were compared by two-way analysis of variance (ANOVA) followed by Bonferroni posttest (GraphPad Prism 5.0). Values presented equal mean ± SEM.
As shown in Figure 2A, the 21-day treatment of vorinostat did not inhibit anaplastic eRMS tumor growth in the PDX model. Immunostaining for the proliferation marker Ki67 (Figure 2B) showed no difference between drug- and vehicle-treated groups. However, interestingly, vorinostat-treated tumors had obvious necrotic regions compared with vehicle controls, as determined by histopathologic analysis (LNC; Figure 2C). Although there were no differences in tumor volumes between vorinostat-treated and vehicle control-treated groups by the end of the 21-day treatment study, we observed that vorinostat-treated mice had “softened” tumors starting on day 7, which we regarded as consequent to drug treatment (Figure 2C) and which may correspond with the necrosis observed by MRI imaging of the primary tumor in situ (Figure 1).
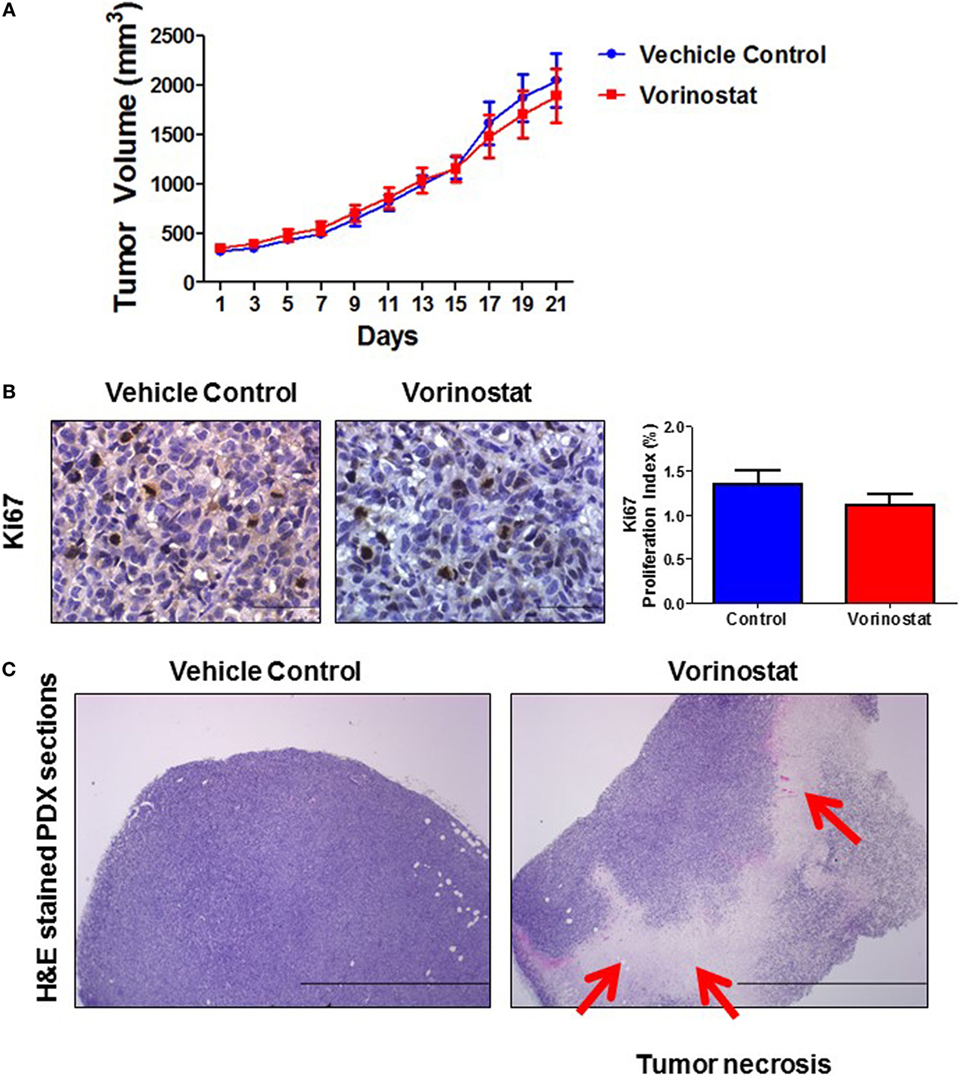
Figure 2. Monotherapy of vorinostat which did not suppress tumor growth in a patient-derived xenograft model (COA/UAB-15), but did induce tumor necrosis. (A) When tumor volumes reached ~300 mm3, 50 mg/kg vorinostat or vehicle was administered once a day to tumor-bearing mice (N = 10/group) for 21 days. (B) Immunohistological analysis for the proliferation marker Ki67 showed no difference in growth fraction between vorinostat- and vehicle-treated mice group. (C) H&E staining of formalin-fixed paraffin-embedded tissue prepared from tumor harvested from vehicle-treated or vorinostat-treated mice 24 h after the last day of treatment. Red arrows indicate examples of necrotic areas.
Concluding Remarks
In this study, we report evaluation of the efficacy of vorinostat, a non-standard treatment, for a patient with anaplastic eRMS and in a parallel preclinical study using a PDX model developed from resected tumor tissue. The patient was treated with the HDAC inhibitor vorinostat, based on the mutational status of her recurrent chemorefractory tumor. Clinically, vorinostat treatment induced a transient tumor regression, followed by tumor progression (increase in tumor volume at primary site). Necrotic areas of the tumor following vorinostat treatment were documented by MRI (Figure 1). The preclinical evaluation of vorinostat efficacy using the PDX model reflected clinical observations with respect to induction of tumor necrosis. Notably, this is the first report demonstrating that vorinostat induces necrosis in vivo (Figure 2C). We propose that more successful treatment for solid tumors lies in understanding the molecular and genetic characteristics that confer specific malignant phenotypes, and in the use of well-characterized preclinical models for evaluating novel agents with potential efficacy.
Ethics Statement
Ethics statement: Protocols involving human subjects were approved by the Institutional Review Board (IRB) of the University of Alabama at Birmingham (Birmingham, AL, USA). Written informed assent and consent were obtained from the patient and her family to use tumor specimens for research purposes. Written informed consent was also obtained from the patient’s parent agreeing to publication of the report. Protocols involving animal use were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham (Birmingham, AL, USA).
Author Contributions
SC, JP, and KY contributed to the conception and design of the study. AM, TG, PG, and SH contributed to the acquisition of data (coordinate specimen acquisition and performed experiments). SC, JP, AM, LC, RR, and DK contributed to the analysis and interpretation of data. SC, JP, and KY contributed to the writing of the manuscript. BK and EB contributed to the clinical resection. RW contributed to the administrative or material support. SC and KY contributed to the study supervision. All authors contributed to review and revision of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to the patient and the patient’s parents who agreed to participate in this study. This work was supported in part by the Pediatric Tumor Bank and Tumorgraft Initiative from the Department of Pediatrics, University of Alabama at Birmingham, and Ribbons of Hope Foundation Alabama for pediatric cancer research.
References
1. Qualman S, Lynch J, Bridge J, Parham D, Teot L, Meyer W, et al. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer (2008) 113(11):3242–7. doi:10.1002/cncr.23929
2. Hettmer S, Archer NM, Somers GR, Novokmet A, Wagers AJ, Diller L, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer (2014) 120(7):1068–75. doi:10.1002/cncr.28507
3. Hawkins DS, Anderson JR, Mascarenhas L, McCowage GB, Rodeberg DA, Wolden SL, et al. Vincristine, dactinomycin, cyclophosphamide (VAC) versus VAC/V plus irinotecan (VI) for intermediate-risk rhabdomyosarcoma (IRRMS): a report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. J Clin Oncol (2014) 32:15s; Abstract retrieved from 2014 ASCO Annual Meeting. (No. 10004). doi:10.1200/jco.2014.32.15_suppl.10004
4. Mascarenhas L, Meyer W, Lyden E, Rodeberg D, Indelicato D, Linardic C, et al. Randomized phase II trial of bevacizumab and temsirolimus in combination with vinorelbine (V) and cyclophosphamide (C) for first relapse/disease progression of rhabdomyosarcoma (RMS): a report from the Children’s Oncology Group (COG). J Clin Oncol (2014) 32:15s; Abstract retrieved from 2014 ASCO Annual Meeting. (No. 10003). doi:10.1200/jco.2014.32.15_suppl.10003
5. Keshelava N, Houghton PJ, Morton CL, Lock RB, Carol H, Keir ST, et al. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr Blood Cancer (2009) 53(3):505–8. doi:10.1002/pbc.21988
6. Vleeshouwer-Neumann T, Phelps M, Bammler TK, MacDonald JW, Jenkins I, Chen EY. Histone deacetylase inhibitors antagonize distinct pathways to suppress tumorigenesis of embryonal rhabdomyosarcoma. PLoS One (2015) 10(12):e0144320. doi:10.1371/journal.pone.0144320
7. Fan Z, Yamaza T, Lee JS, Yu J, Wang S, Fan G, et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol (2009) 11(8):1002–9. doi:10.1038/ncb1913
8. Guan B, Wang TL, Shih IeM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res (2011) 71(21):6718–27. doi:10.1158/0008-5472.CAN-11-1562
9. Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun (2014) 5:3469. doi:10.1038/ncomms4469
10. Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev (2000) 14(14):1810–23. doi:10.1101/gad.14.14.1810
11. Cao Q, Gearhart MD, Gery S, Shojaee S, Yang H, Sun H, et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia (2016) 30(5):1155–65. doi:10.1038/leu.2016.2
12. Choi WI, Jeon BN, Yoon JH, Koh DI, Kim MH, Yu MY, et al. The proto-oncoprotein FBI-1 interacts with MBD3 to recruit the Mi-2/NuRD-HDAC complex and BCoR and to silence p21WAF/CDKN1A by DNA methylation. Nucleic Acids Res (2013) 41(13):6403–20. doi:10.1093/nar/gkt359
13. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature (2012) 488(7409):106–10. doi:10.1038/nature11329
14. Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood (2013) 122(18):3169–77. doi:10.1182/blood-2012-11-469619
15. de Rooij JD, van den Heuvel-Eibrink MM, Hermkens MC, Verboon LJ, Arentsen-Peters ST, Fornerod M, et al. BCOR and BCORL1 mutations in pediatric acute myeloid leukemia. Haematologica (2015) 100(5):e194–5. doi:10.3324/haematol.2014.117796
16. Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol (2010) 102(2–3):122–8. doi:10.1016/j.pbiomolbio.2010.05.001
17. Wu RC, Wang TL, Shih IeM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther (2014) 15(6):655–64. doi:10.4161/cbt.28411
18. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov (2015) 5(7):752–67. doi:10.1158/2159-8290.CD-14-0849
19. Jones S, Wang TL, Shih IeM, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science (2010) 330(6001):228–31. doi:10.1126/science.1196333
20. Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol (2011) 35(5):625–32. doi:10.1097/PAS.0b013e318212782a
21. Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol (2011) 224(3):328–33. doi:10.1002/path.2911
22. Takeda T, Banno K, Okawa R, Yanokura M, Iijima M, Irie-Kunitomi H, et al. ARID1A gene mutation in ovarian and endometrial cancers (review). Oncol Rep (2016) 35(2):607–13. doi:10.3892/or.2015.4421
23. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov (2013) 3(1):35–43. doi:10.1158/2159-8290.CD-12-0361
24. Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med (2015) 21(3):231–8. doi:10.1038/nm.3799
25. Bitler BG, Fatkhutdinov N, Zhang R. Potential therapeutic targets in ARID1A-mutated cancers. Expert Opin Ther Targets (2015) 19(11):1419–22. doi:10.1517/14728222.2015.1062879
26. Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J (2008) 27(2):406–20. doi:10.1038/sj.emboj.7601967
27. Kanu N, Gronroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene (2015) 34(46):5699–708. doi:10.1038/onc.2015.24
28. Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet (2014) 46(3):287–93. doi:10.1038/ng.2894
29. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer (2001) 1(3):194–202. doi:10.1038/35106079
30. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi:10.1038/nrd4360
31. Krusche CA, Wulfing P, Kersting C, Vloet A, Bocker W, Kiesel L, et al. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat (2005) 90(1):15–23. doi:10.1007/s10549-004-1668-2
32. Weichert W, Roske A, Gekeler V, Beckers T, Ebert MP, Pross M, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol (2008) 9(2):139–48. doi:10.1016/S1470-2045(08)70004-4
33. Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer (2008) 98(3):604–10. doi:10.1038/sj.bjc.6604199
34. Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res (2008) 14(6):1669–77. doi:10.1158/1078-0432.CCR-07-0990
35. Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, et al. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer (2011) 74(2):300–4. doi:10.1016/j.lungcan.2011.02.019
36. Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, et al. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol (2007) 60(11):1205–10. doi:10.1136/jcp.2005.029165
37. Yang H, Salz T, Zajac-Kaye M, Liao D, Huang S, Qiu Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J (2014) 28(10):4265–79. doi:10.1096/fj.14-250654
38. Cornen S, Adelaide J, Bertucci F, Finetti P, Guille A, Birnbaum DJ, et al. Mutations and deletions of ARID1A in breast tumors. Oncogene (2012) 31(38):4255–6. doi:10.1038/onc.2011.598
39. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood (2014) 123(10):1487–98. doi:10.1182/blood-2013-05-500264
40. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest (2014) 124(1):30–9. doi:10.1172/JCI69738
41. Grant S, Easley C, Kirkpatrick P. Vorinostat. Nat Rev Drug Discov (2007) 6(1):21–2. doi:10.1038/nrd2227
42. Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol (2010) 28(22):3623–9. doi:10.1200/JCO.2009.25.9119
43. Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, et al. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin Cancer Res (2011) 17(12):3956–68. doi:10.1158/1078-0432.CCR-10-3412
44. Bellarosa D, Bressan A, Bigioni M, Parlani M, Maggi CA, Binaschi M. SAHA/Vorinostat induces the expression of the CD137 receptor/ligand system and enhances apoptosis mediated by soluble CD137 receptor in a human breast cancer cell line. Int J Oncol (2012) 41(4):1486–94. doi:10.3892/ijo.2012.1551
Keywords: anaplastic embryonal rhabdomyosarcoma, patient-derived xenograft, case study, vorinostat, targeted therapy
Citation: Cramer SL, Miller AL, Pressey JG, Gamblin TL, Beierle EA, Kulbersh BD, Garcia PL, Council LN, Radhakrishnan R, Hendrix SV, Kelly DR, Watts RG and Yoon KJ (2018) Pediatric Anaplastic Embryonal Rhabdomyosarcoma: Targeted Therapy Guided by Genetic Analysis and a Patient-Derived Xenograft Study. Front. Oncol. 7:327. doi: 10.3389/fonc.2017.00327
Received: 01 May 2017; Accepted: 18 December 2017;
Published: 11 January 2018
Edited by:
Giuseppe Giaccone, Georgetown University, United StatesReviewed by:
Victor C. Kok, Asia University, TaiwanFrederic Barr, National Cancer Institute (NIH), United States
Copyright: © 2018 Cramer, Miller, Pressey, Gamblin, Beierle, Kulbersh, Garcia, Council, Radhakrishnan, Hendrix, Kelly, Watts and Yoon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart L. Cramer, c3R1YXJ0LmNyYW1lckBwYWxtZXR0b2hlYWx0aC5vcmc=;
Karina J. Yoon, a3lvb25AdWFiLmVkdQ==
†Present address: Stuart L. Cramer, Palmetto Health Children’s Hospital, Columbia, SC, United States;
Joseph G. Pressey, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States;
Raymond G. Watts, Department of Pediatrics, LSUHSC School of Medicine, New Orleans, LA, United States