 Célia Cintas
Célia Cintas Julie Guillermet-Guibert
Julie Guillermet-Guibert- 1INSERM U1037, CRCT, Université Toulouse III Paul Sabatier, Toulouse, France
- 2Laboratoire d’Excellence TouCAN, Toulouse, France
Introduction
One of the most common events in human cancer is hyperactivation of the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway, generally described as a consequence of genetic alterations of pathway members. The core components of the pathway are depicted in Figure 1A. There are four members of the class I PI3Ks, which act upstream of this pathway, performing the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3). This lipid produced in the inner leaflet of the plasma membrane controls a range of cellular actions including cell growth, migration, metabolism, survival and proliferation. Class I PI3K activity in vertebrates regulates both physiological and pathological processes (1).
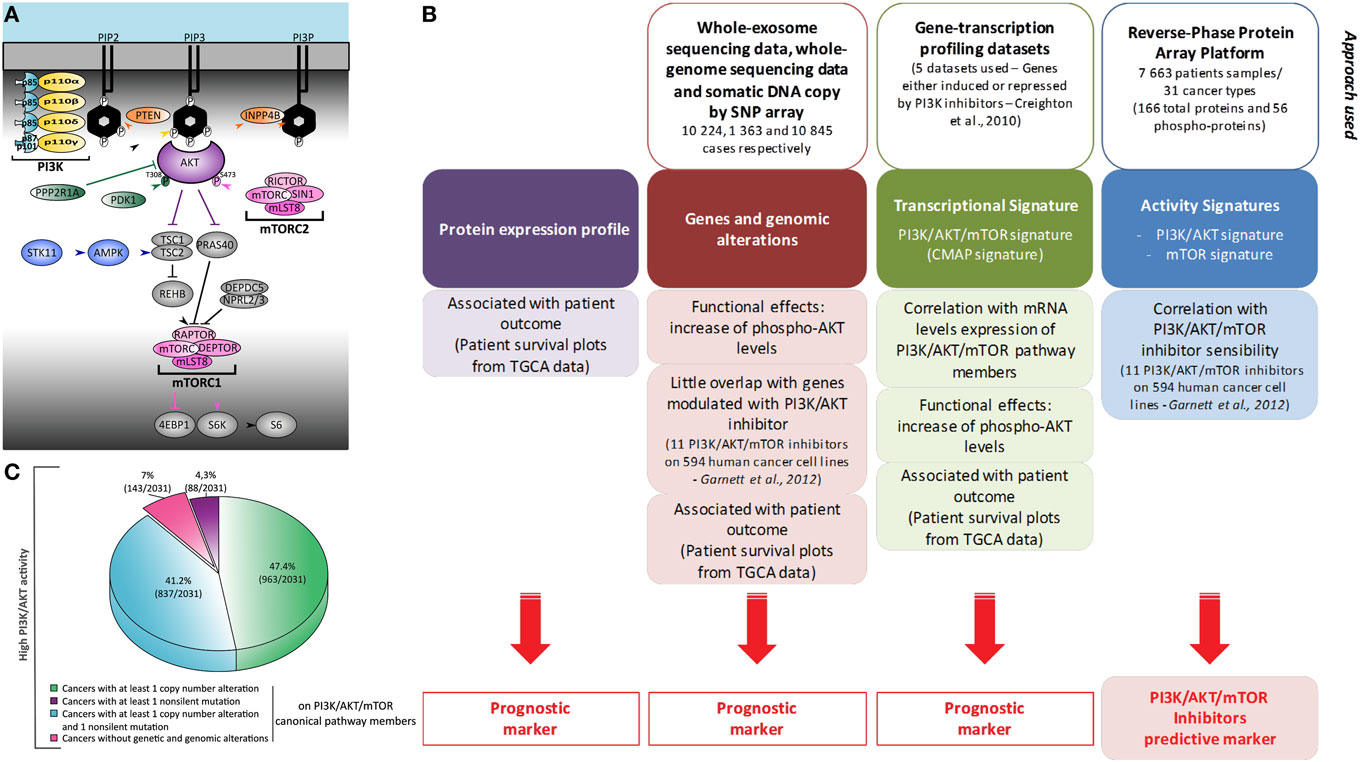
Figure 1. Heterogeneity of phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) activation in cancer: global and comprehensive mapping by a multiscale integrated approach. (A) Representation of PI3K/AKT/mTOR canonic members. Most omics results use data obtained under treatment with pan-PI3K inhibitors which still display relative isoform specificity, or with PI3Kα-selective inhibitors associated with genetic alterations. Usually, only the expression of PIK3CA and PIK3CB is studied. Production of PIP3 at the plasma membrane is, however, performed by four enzymes: PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ. They are composed of a regulatory subunit (p85 or p101/p87) and a catalytic subunit (p110α, p110β, p110δ, or p110γ). (B) This schematic summarizes the bioinformatic meta-analysis performed in the study by Zhang et al. (2) using two PI3K/AKT/mTOR transcriptional signatures of Creighton et al. and Garnett et al. (3, 4). (C) Representation of the molecular alterations found in cancer patients with high PI3K/AKT score. Molecular alterations in patients with reverse-phase protein array (RPPA) score values ≥0.5 are shown. These RPPA scores were normalized to SDs from the median across all cancers.
Phosphatidylinositol-3-kinase activity has been implicated in a variety of different cancers, hence this class of enzymes is a prime drug target for anticancer therapies (5). However, initial phase I/II clinical trials of small molecule PI3K inhibitors show that the predictive markers of efficiency of these drugs need to be improved. The presence of PIK3CA mutation in the primary tumor alone is not a sufficient predictive marker of efficiency (6, 7). Signal-targeted therapy would benefit from the identification of patients more likely to respond.
A Recent Multiscale Omics Approach Mapped PI3K/AKT/mTOR Activation in Cancer
Zhang et al. analyzed in an unbiased fashion both known molecular mechanisms by which the PI3K/AKT/mTOR pathway is upregulated in human cancers, as well as other possibly unrelated genetic alterations (2). They examined The Cancer Genome Atlas open access omics data (including genomic mutations by whole-genome sequencing, gene copy number by single-nucleotide polymorphism array, or RNA expression by whole-exome sequencing) across 11,219 human cancers representing 32 distinct major types. The authors also used reverse-phase protein array (RPPA) analysis to assess the level of expression of 166 total proteins and 56 phosphorylated proteins (Figure 1B). Phosphoproteome-based PI3K/AKT and mTOR activity signatures (p-AKTS473/T308, p-GSK3S9, p-PRAS40T246, p-TSC2T1462 and p-mTORS2448, p-RICTORT1135, p-4EBP1S65/T34/T46/T70, p-S6KT389, p-S6S235/S236/S240/S244, respectively) by RPPA analysis were found to be correlated. Transcriptomics analysis found that the levels of expression of a selected list of members of the PI3K/AKT pathways were not correlated with the activation of PI3K or mTOR signaling nodes by RPPA. However, the authors confirmed a correlation between the transcriptional expression and alterations in the copy number of the so-called core genes. All mutated residues of members of the PI3K/AKT/mTOR pathway resulted in the expected increase in activity of the PI3K/AKT/mTOR pathway. Therefore, modifications to DNA, mRNA expression, and phosphoprotein levels were found to be functionally relevant in the hyperactivation of the PI3K pathway associated with cancers.
Not all Cases of Increased PI3K/AKT/mTOR Pathway Activity Can be Explained by the Canonic Genetic Alterations Associated with PI3K Signaling
In most scenarios, increased AKT activity can be explained by genetic or genomic alterations to members of the PI3K/AKT/mTOR pathway; however, this is not the case for all instances of AKT hyperactivation (Figure 1C). In 764 of 7,099 tumors, including mostly lower grade glioma, pheochromocytoma and paraganglioma, prostate adenocarcinoma, and kidney renal clear cell carcinoma, the level of phospho-AKT was increased without any of the genetic or genomic alterations described as being functionally coupled to this pathway. In addition, upregulation of mTOR pathway activity is also associated with non-canonical alterations, such as IDH1 (Isocitrate dehydrogenase 1) or VHL (Von Hippel-Lindau syndrome) mutations, miRNA modulation, and ERK, SRC, and NDRG1 activation. Further work is required to understand the complex mechanisms involved in upregulation of the PI3K/AKT/mTOR pathway by non-canonical alterations.
Cellular Activity of the PI3K/AKT/mTOR Pathway is a Predictive Marker of Sensitivity to PI3K/mTOR Inhibitors
Analysis of knock-down (shRNAs) data for pathway effectors (3, 4) and a correlation matrix of PI3K/AKT/mTOR, MYC and active KRAS gene signatures, showed a convergence of MYC and KRAS oncogenic signaling pathways with the PI3K/AKT/mTOR transcriptomic signature. However, the PI3K/AKT/mTOR transcriptomic signature was not a predictive marker of PI3K/mTOR inhibitor efficacy, but was associated with patient outcome/survival. Instead, a list of 146 genes significantly associated with tumor cell sensitivity to pathway inhibitors correlated with the PI3K/AKT phosphoproteomic signature. Therefore, functional assessment of the PI3K pathway was linked with the sensitivity to its inhibitors, while its final transcriptomic targets were only indicative of the impact of this pathway in the malignancy of each cancer (Figure 1B).
Both Pan-PI3K and Isoform-Selective Compounds are Progressing in the Clinic
There are four members of the class I PI3Ks: PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ, and these have different functions in physiopathology (1). PI3Kα and PI3Kβ are ubiquitously expressed, whereas PI3Kγ and PI3Kδ are preferentially expressed in leukocytes or the vascular system, but are also overexpressed in some primary tumors. This led to the concept that isoform-specific functions of PI3K enzymes in non-transformed cells become redundant in transformed cells due to a possible dysregulation of upstream signaling. However, several research teams, including us, show that certain cancers are solely dependent on one isoform of PI3K. For example, a prostate cancer model induced by inactivation of the tumor suppressor gene PTEN (a phosphatase that negatively regulates the PI3K/AKT signaling pathway—see Figure 1A) depends only on the PI3K activity of PI3Kβ. In addition, thyroid tumors induced by inactivation of PTEN depend only on PI3Kα activity, suggesting a tissue-context phenomenon (8, 9). Selective engagement by receptor tyrosine kinases (RTKs) or G-protein-coupled receptors (GPCRs) to specific PI3K isoforms when all the RTK-linked (α, β, and δ) PI3Ks or all the GPCR-linked PI3Ks (β and γ) are expressed in a given cell type remains to be demonstrated (Figure 1A). Indeed, the two GPCR-activated class I PI3Ks were found to be redundant in various cell systems (10). In the context of tumor initiation, evidence clearly shows isoform specificity (11, 12). In neoplastic cells harboring high levels of genomic instability, isoform selectivity could be transient, because, once specific isoforms are inhibited, other class I enzymes can become activated by other mechanisms (13, 14). For example, inhibition of PI3Kβ relieves feedback inhibition of RTKs, thus reactivating PI3Kα (14). Conversely, inhibition of PI3Kα (in tumors harboring RTK activation or an activating oncogenic mutation in PIK3CA) leads to the activation of PI3Kβ by inactivation of the tumor suppressor PTEN (15). With limited success in patients receiving dose-limiting toxicity amounts of pan-PI3K inhibitors, literature suggests that a selective inhibition of one isoform could lead to a better efficacy/toxicity ratio. Novel, isoform-selective inhibitors are currently progressing quickly in the clinic (16, 17). Moreover, isoform-sparing compounds have been successfully developed to target the oncogenic driving PI3K and the immune restricted PI3Ks, in particular in the highly inflammatory triple-negative breast cancer setting (18). These isoform-selective or isoform-sparing inhibitors still have serious toxicity issues, as seen with the FDA-approved idelalisib. The main resistance mechanism of pan-class I PI3K inhibitors is via reactivation of the MAPK pathway. However, most patients treated with the pan-PI3K inhibitor buparlisib (BKM120) and the MEK1/2 inhibitor trametinib (GSK1120212) experienced severe grade 3/4 adverse events, and 31% of them arrested their treatment (19). On-target toxicity is also major issue for PI3K inhibitors, hence more balanced pan-PI3K, pan-PI3K/mTOR, or dual isoform PI3K inhibitors are being developed.
Many questions need to be answered regarding clinically relevant PI3K inhibition in cancer. This inhibition can be either isoform-specific or pan-PI3K, the later possibly with a selective inhibition efficiency for each isoform. Some of the more pertinent questions are outlined below:
i- While it was obvious to hit PI3Kδ in hematological malignancies due to the overexpression of this isoform in this cell type, how can we predict the sensitivity to isoform-specific drugs for the two ubiquitously expressed yet critical for cancerogenesis PI3Ks, PI3Kα, and PI3Kβ?
ii- Should we take into account the activity of stromal PI3Kγ and PI3Kδ in solid cancers?
iii- Are inter-isoform compensatory mechanisms similar for all types of cancers?
iv- How can we predict which other isoforms are involved in resistance mechanisms?
It thus remains critical to delineate for each type of cancer and ultimately each individual patient which PI3K isoform(s) to target, and the level of inhibition at which each of these isoforms needs to be targeted so as to reach an optimal cost benefit ratio.
Conclusion/Perspectives
Zhang et al. assessed PI3K/AKT and mTOR activity signatures, the PI3K/AKT/mTOR transcriptional signature and genetic and genomic alterations of canonical members of the pathways, to strongly demonstrate the major contribution of PI3K/AKT/mTOR pathway deregulation to poor survival in different types of cancer. Direct mutation of genes of the pathway is not the only common mechanism of activation of this pathway in cancer cells. It is also necessary to consider other non-canonical members of the PI3K/AKT/mTOR pathway to evaluate its activity, as well as other apparently unrelated genetic or genomic alterations. Such examples of meta-analysis largely pave the way to a better understanding of PI3K oncogenic signaling regulation and interconnection with its transcriptional targets in cancers. Finally, the first specific PI3K inhibitor to be approved for clinical use is a PI3Kδ-selective inhibitor, which has shown high efficacy in phase III clinical trials for B cell lymphoma (20). Evidence for PI3K isoform selectivity in tumors should not be neglected in these large-scale omics studies. An analysis of PI3K signaling taking into account its entire complexity (isoform selectivity) is important in order to establish optimal antitumoral therapies and select patients likely to benefit from PI3K-targeted therapies.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JG-G is a member of COST action EU-Pancreas BM1204.
Funding
JG-G’s laboratory belongs to Toucan, Laboratoire d’Excellence, ANR, an integrated research program on Signal-targeted Drug Resistance. CC’s salary was funded by French Charity Ligue Nationale Contre le Cancer (GB/MA/VSP-10443). JG-G’s laboratory for this topic was/is funded by Europe EU-ERG FP7 (270696 PaCa/PI3K), Arc PJA20141201744, Toucan, and MSCA-ITN/ETN PhD-PI3K (Project ID: 675392).
References
1. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol (2010) 11:329–41. doi:10.1038/nrm2882
2. Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell (2017) 31:820–32.e3. doi:10.1016/j.ccell.2017.04.013
3. Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res (2010) 12:R40. doi:10.1186/bcr2594
4. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature (2012) 483:570–5. doi:10.1038/nature11005
5. Pons-Tostivint E, Thibault B, Guillermet-Guibert J. Targeting PI3K signaling in combination cancer therapy. Trends Cancer (2017) 3:454–69. doi:10.1016/j.trecan.2017.04.002
6. Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther (2014) 13:1117–29. doi:10.1158/1535-7163.MCT-13-0865
7. Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol (2015) 16:1324–34. doi:10.1016/S1470-2045(15)00188-6
8. Berenjeno IM, Guillermet-Guibert J, Pearce W, Gray A, Fleming S, Vanhaesebroeck B. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J (2012) 442:151–9. doi:10.1042/BJ20111741
9. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer (2015) 15:7–24. doi:10.1038/nrc3860
10. Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A (2008) 105:8292–7. doi:10.1073/pnas.0707761105
11. Baer R, Cintas C, Dufresne M, Cassant-Sourdy S, Schonhuber N, Planque L, et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev (2014) 28:2621–35. doi:10.1101/gad.249409.114
12. Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, et al. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev (2012) 26:1573–86. doi:10.1101/gad.191973.112
13. Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, et al. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer Cell (2015) 27:97–108. doi:10.1016/j.ccell.2014.11.007
14. Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell (2015) 27:109–22. doi:10.1016/j.ccell.2014.11.008
15. Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature (2015) 518:240–4. doi:10.1038/nature13948
16. Juric D, de Bono JS, LoRusso PM, Nemunaitis J, Heath EI, Kwak EL, et al. A first-in-human, phase I, dose-escalation study of TAK-117, a selective PI3Kalpha isoform inhibitor, in patients with advanced solid malignancies. Clin Cancer Res (2017) 23(17):5015–23. doi:10.1158/1078-0432.CCR-16-2888
17. Mateo J, Ganji G, Lemech C, Burris HA, Han SW, Swales K, et al. A first-time-in-human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin Cancer Res (2017) 23:5981–92. doi:10.1158/1078-0432.CCR-17-0725
18. Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res (2014) 16:406. doi:10.1186/s13058-014-0406-x
19. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res (2015) 21:730–8. doi:10.1158/1078-0432.CCR-14-1814
Keywords: phosphatidylinositol-3-kinase pathway, proteomics, transcriptomics, genomics, data integration, precision medicine, predictor of therapeutic response, isoform specificity
Citation: Cintas C and Guillermet-Guibert J (2018) Heterogeneity of Phosphatidylinositol-3-Kinase (PI3K)/AKT/Mammalian Target of Rapamycin Activation in Cancer: Is PI3K Isoform Specificity Important? Front. Oncol. 7:330. doi: 10.3389/fonc.2017.00330
Received: 31 October 2017; Accepted: 26 December 2017;
Published: 22 January 2018
Edited by:
Ruggero De Maria, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Emilio Hirsch, Università degli Studi di Torino, ItalyCopyright: © 2018 Cintas and Guillermet-Guibert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie Guillermet-Guibert, anVsaWUuZ3VpbGxlcm1ldEBpbnNlcm0uZnI=