Abstract
The plasminogen activator (PA) system is an extracellular proteolytic enzyme system associated with various physiological and pathophysiological processes. A large body of evidence support that among the various components of the PA system, urokinase-type plasminogen activator (uPA), its receptor (uPAR), and plasminogen activator inhibitor-1 and -2 (PAI-1 and PAI-2) play a major role in tumor progression and metastasis. The binding of uPA with uPAR is instrumental for the activation of plasminogen to plasmin, which in turn initiates a series of proteolytic cascade to degrade the components of the extracellular matrix, and thereby, cause tumor cell migration from the primary site of origin to a distant secondary organ. The components of the PA system show altered expression patterns in several common malignancies, which have identified them as ideal diagnostic, prognostic, and therapeutic targets to reduce cancer-associated morbidity and mortality. This review summarizes the various components of the PA system and focuses on the role of uPA–uPAR in different biological processes especially in the context of malignancy. We also discuss the current state of knowledge of uPA–uPAR-targeted diagnostic and therapeutic strategies for various malignancies.
Introduction
Tumor metastasis is a multistep process initiated when cancer cells acquire the ability to invade the surrounding matrix and migrate to seed distant organs via hematogenous or lymphatic routes (1). Almost 90% of the cancer-related deaths in human are caused due to the metastatic spread of the tumor cells (2, 3). Even though therapeutic strategies targeting the primary tumors have been improved markedly over the years, targeting tumor metastasis has only seen a minimal to modest success. Since the pathogenesis of metastasis involves a series of sequential events regulated by different molecular determinants, it stands to reason that therapeutic modalities targeting the key molecules and signaling pathways involved in the metastatic cascade may serve as an effective therapeutic strategy to block cancer progression.
One of the major events that underlie metastasis is the proteolytic degradation of the extracellular matrix (ECM) to promote tumor cell invasion, migration, and homing to distant organs (4). Even though several protease systems are implicated in this process, a large body of evidence identified the uPA–urokinase-type plasminogen activator receptor (uPAR) system as a central player in mediating proteolysis during cancer invasion and metastasis (5, 6). Further studies have indicated that the functionality of the uPA–uPAR system is not only limited to proteolysis. In fact, the present consensus suggests that the uPA–uPAR system plays a broader role in multiple stages of cancer starting from tumorigenesis to metastasis (6, 7). Elevated expression of the components of the uPA–uPAR system has been shown to be related to adverse patient outcomes in different types of cancer (8–12). As such, the components of the uPA–uPAR system have been identified as excellent candidates for anticancer therapies (13, 14). This review is aimed to summarize our current knowledge on the role of the uPA–uPAR system in cancer.
The Plasminogen Activator (PA) System
The PA system was initially thought to play a role in the dissolution of clots formed by the fibrins (15). However, later studies have demonstrated that the PA system has additional functions in other biological processes such as embryogenesis, angiogenesis, cell migration, wound healing, inflammatory response, as well as apoptotic cell death (15). In cancer, the PA system plays a dominant role in tumor growth, angiogenesis, tumor cell invasion, migration, and metastasis.
The major physiological function of the PA system is to convert the inactive plasminogen to plasmin, which can be mediated by two types of PAs: the tissue type plasminogen activator (tPA) and uPA. Different inhibitory proteins have also been identified, which primarily regulate the plasminogen activation by both tPA and uPA. Among these, the two well-characterized endogenous inhibitors of tPA and uPA are plasminogen activator inhibitor-1 (PAI-1) and PAI-2. On the other hand, plasmin activation is regulated by inhibitors such as α2-antiplasmin and α2-macroglobulin (Figure 1).
Figure 1
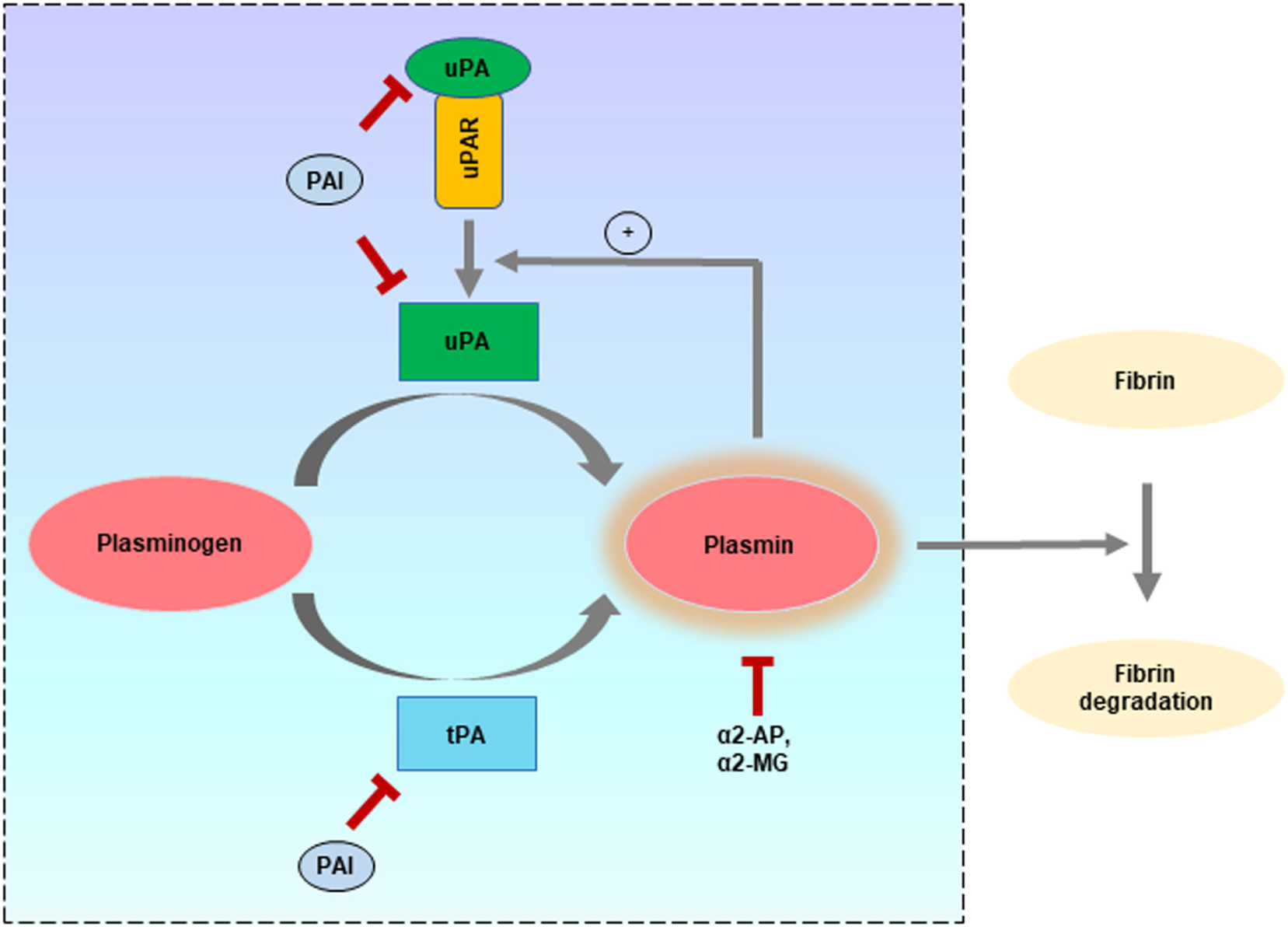
Different components of the plasminogen activator (PA) system and role in fibrinolysis. Schematic representation of the ability of type plasminogen activator (tPA) and uPA to independently activate “plasminogen” to form the active proteolytic enzyme “plasmin” which can mediate fibrinolysis to keep the blood free from clotting. In addition to their fibrinolytic effects, tPA and uPA are implicated in many other physiological and pathophysiological processes. Both tPA and uPA can be inhibited by plasminogen activator inhibitors (PAI) such as plasminogen activator inhibitor-1 and PAI-2, while plasmin can be inhibited by α2-antiplasmin (α2-AP) and α2-macroglobulin (α2-MG). The different components belonging to the PA system are enclosed within the square region.
The activation of plasminogen by tPA and uPA is under temporal and spatial regulation (16). tPA is mainly synthesized by the endothelial cells and functions in clot lysis (17). uPA can also function to protect from the deposition of fibrin and has been used as fibrinolytic/thrombolytic agent (18). Even though both tPA and uPA are present in tumor cells, uPA is more commonly associated with cancer progression (19). For this distinct role, much attention has been given to understand the functionality of the uPA–uPAR system in cancer, which has led to its identification, characterization, and validation as a prognostic, diagnostic, and therapeutic target in several common cancers (18, 20–22). In the following section, the structure and functional significance of uPA, uPAR, and the two inhibitors (PAI-1 and PAI-2) are briefly discussed.
Urokinase-Type Plasminogen Activator (uPA)
The “Urokinase-type Plasminogen Activator” or simply “urokinase” (uPA) is a key serine protease involved in the conversion of inactive plasminogen into active plasmin, which in turn functions in a range of events of the metastatic cascade (23). It was first identified in the urine in 1947 by MacFarlane and Pilling who reported on the fibrinolytic activity of a novel “unnamed” protein (24). Half a decade later, Sobel and colleagues named this “unnamed” protein as “urokinase” (25). Further studies have reported the presence of urokinase in plasma, seminal fluid, and the ECM of many tissues (23, 26).
uPA is synthesized and released as a single polypeptide chain glycosylated zymogen called pro-uPA (411 amino acids) which consists of three domains: a growth factor domain (GFD) that shares homology with the epidermal growth factor (EGF), a kringle domain (KD), and a serine protease domain (27, 28). The GFD (spanning from 1 to 49 amino acids) and KD (50–131 amino acids) reside at the N-terminus while the catalytic serine protease domain (159–411 amino acids) resides at the “C-terminus.” In between the N-terminal and C-terminal region, there is a linker region (132–158 amino acids) (29). Once the pro-uPA is secreted, it undergoes cleavage of the peptide bond between Lys158 and IIe159 located at the linker region to produce a two-chain form of uPA linked via a disulfide bond (30) (Figure 2). Petersen et al. demonstrated that the single-chain pro-uPA is 250-fold less potent to generate active plasmin than the two-chain uPA (31). Different proteases have been reported to function in mediating the cleavage of pro-uPA. Plasmin most effectively converts the pro-uPA into active uPA (30). Among the other proteases that can activate pro-uPA includes cathepsin B and L, nerve growth factor-g, trypsin, kallikrein, thermolysin, and mast cell tryptase (30, 32–34). In addition, proteases such as elastase and thrombin can cause cleavage at different sites of pro-uPA to produce high-molecular weight uPA (30).
Figure 2
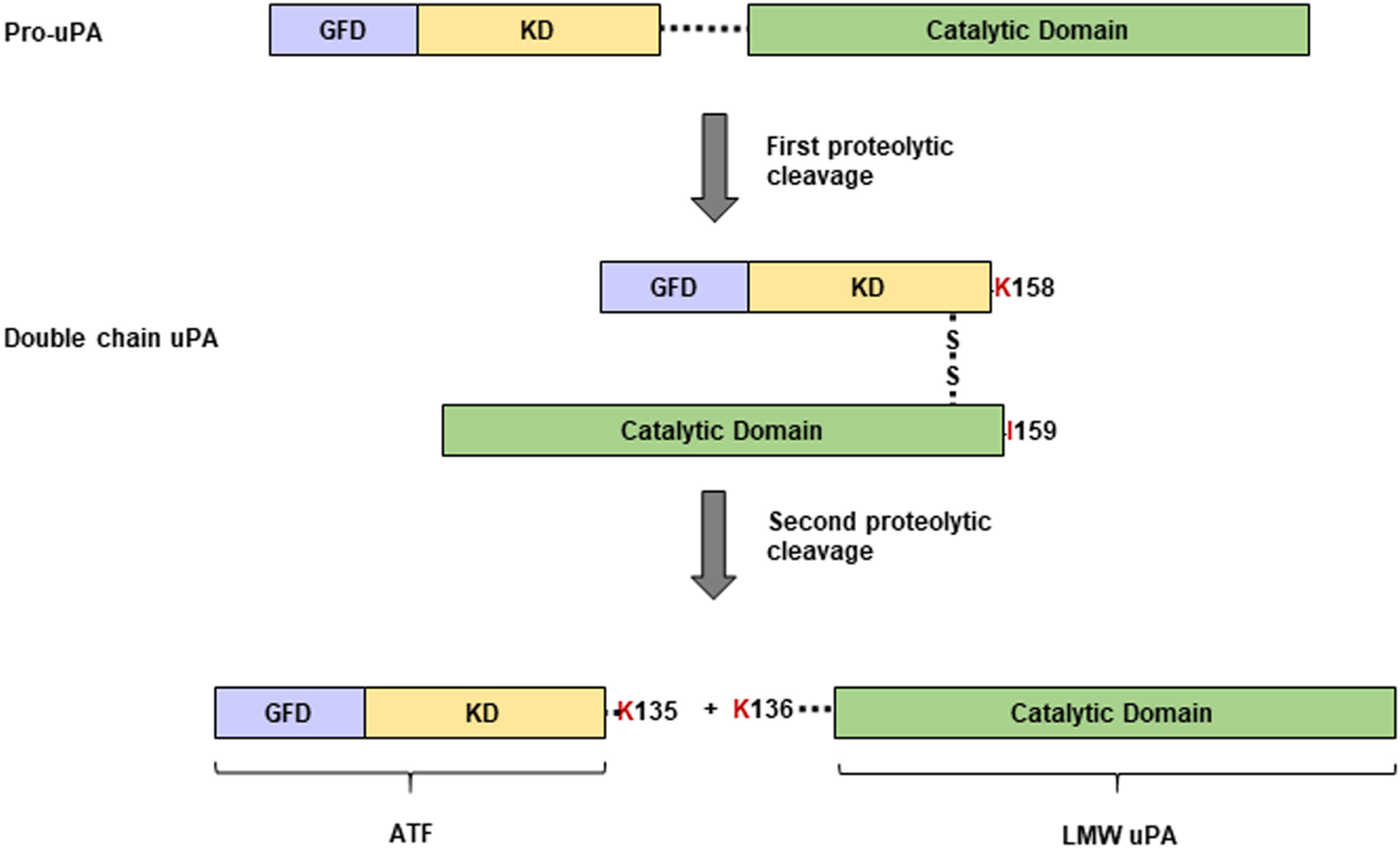
Structure of pro-uPA and uPA. The pro-uPA containing a growth factor domain (GFD), a kringle domain (KD), and a catalytic serine protease domain is secreted as a single-chain precursor that undergoes proteolytic cleavage between the Lys158 and Ile159 peptide bond to generate the two-chain form of uPA. By action of a second proteolytic cleavage, the two-chain form of uPA can be further cut between Lys135 and Lys136 resulting in the formation of an inactive amino-terminal fragment (ATF) as well as a catalytically active low-molecular weight form of uPA (LMW uPA).
Following another round of proteolysis at the peptide bond between Lys135 and Lys136, the two-chain form of uPA can be further cleaved into two parts: (1) a catalytically active low-molecular weight form of uPA having the serine protease domain and (2) an inactive amino-terminal fragment (ATF) that consists of GFD and the KD (28). We have previously shown that the ATF can function as a mitogen (35–38). Due to the presence of GFD, the ATF, the two-chain form of uPA as well as the zymogen pro-uPA can all bind to uPAR at almost similar affinity (28). Binding of uPA to uPAR magnifies its ability to convert plasminogen to plasmin (39). Interestingly, the binding of the catalytically inactive pro-uPA to uPAR can also induce plasmin activation (40). It is speculated that binding to uPAR causes a conformational change in pro-uPA that renders it the ability to activate plasmin even after not being converted into active uPA (40).
uPA has a very high degree of substrate specificity, and plasminogen is its major substrate (41). However, it has been demonstrated that the scatter factor and pro-hepatocyte growth factors and PAI-1 can also be activated by the action of uPA in a manner independent of plasminogen (42, 43). The activity of uPA is regulated by the PAIs and endocytosis (43).
The expression of the gene (PLAU) encoding uPA is minimal in normal cells whereas in tumor cells the expression increases several folds. So, what triggers the expression of uPA in cancer cells? The answer lies in the fact that the PLAU gene expression can be induced by different types of growth factors, hormones, cytokines, as well as by morphological changes of the cells (43). The proximal minimal promoter and the enhancer element located upstream of the transcription start site of the PLAU gene are the two regions that regulate its basal and inducible expression, respectively (44). The proximal minimal promoter, associated with the basal expression of the PLAU gene, consists of a GC/GA-rich region that resides upstream of the TATA box sequence. This region is recognized by specificity protein 1 (Sp1) and Sp3 transcription factors that aid in the transcription of the basal PLAU gene (7). On the other hand, transcription factors Ets1 and Ets2 that binds to the enhancer region located about 2 kb upstream is primarily responsible for the induction of the PLAU gene (44). The basal or activated transcription of the PLAU gene is regulated by the Jun kinase and mitogen-activated protein kinase signaling pathways (45, 46). Interestingly, the signaling pathways regulating PLAU transcription are activated by different types of extracellular stimuli (growth factors, cytokines, etc.) that are frequently elevated during cancer (44). This provides an answer for the elevated expression of the PLAU gene seen in cancer. Other cis-regulatory sites for nuclear factor (NF)-κB, β-catenin, and T-cell factor (TCF) binding have also been demonstrated to be relevant for PLAU gene expression (44).
In addition, DNA methylation-mediated epigenetic mechanisms have been found to be regulating PLAU gene expression (47–49) Using surgical biopsy samples from breast cancer patients, our lab has shown that increased DNA hypomethylation at the promoter of the PLAU gene correlates with its increased expression in the more aggressive form of the disease (50).
Urokinase-Type Plasminogen Activator Receptor
The uPAR (CD87) has the ability to localize both pro-uPA and active uPA to the cell surface (39) (Figure 3). It is a single polypeptide chain (313 amino acids) cysteine-rich glycoprotein having three distinct domains (D1, D2, and D3) that are linked via two flexible linker sequences (51). The presence of uPAR was first reported in human monocytes and U937 monocytoid cell line (52, 53). This protein is devoid of any transmembrane and intracellular domains, and its C-terminal end is covalently connected to the cell membrane by a glysocylphosphatidylinositol (GPI) anchor (54). Llinas et al. deduced the crystal structure of uPAR and found that the receptor-binding module of uPA binds to the central cavity of uPAR leaving the external receptor surface available for binding with other proteins (55). This implies that the function of uPAR is not only limited to uPA binding and its subsequent activation. Having additional binding site other than uPA helps uPAR to interact with integrins, vitronectin, and different types of transmembrane receptors to facilitate downstream intracellular signaling mediated by well-known effector molecules such as the focal adhesion kinase (FAK), src, and Akt (56) (Figure 3). A number of these effector molecules are widely known to be involved in cancer progression. Different proteases such as matrix metalloproteases (MMPs), plasmin, chymotrypsin, and uPA can cause cleavage of the uPAR protein within the D1/D2 linker region resulting in the formation of a truncated uPAR (19, 54, 57). While all three domains (D1, D2, and D3) of uPAR can bind to uPA, only the D2 and D3 domains can bind to other proteins such as integrins, G protein-coupled receptors (GPCR), and different receptor tyrosine kinases (58–61). In addition, cleavage at the GPI anchor produces a soluble form of uPAR (suPAR) that is present at a very low level in the blood normally (62), but its circulatory level is elevated in cancer (63). The suPAR can activate the G protein-coupled chemotactic receptor FPRL1/LXA4R by functioning as an endogenous chemotactic agonist (64). The uPAR protein can be internalized by the cation-independent, mannose 6-phosphate/insulin-like growth factor II receptor (CIMPR), which facilitates the redistribution of the unoccupied uPAR (65).
Figure 3
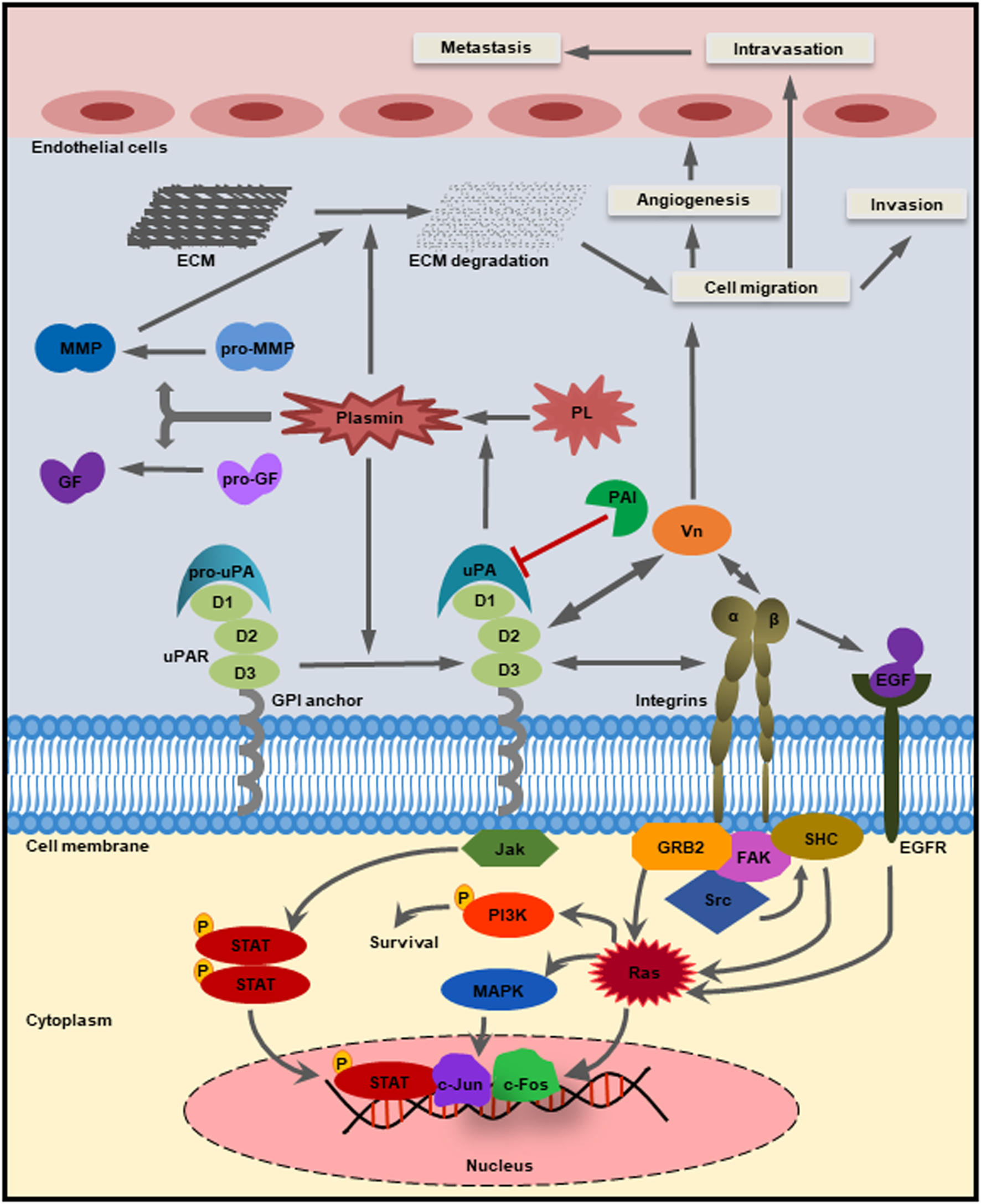
Schematic diagram of the uPA–urokinase-type plasminogen activator receptor (uPAR)-mediated pathways. The glysocylphosphatidylinositol (GPI)-anchored receptor uPAR consisting of three domains (D1, D2, and D3) has the ability to bind the zymogen pro-uPA as well as the active uPA through the growth factor domain. The catalytically active form of uPA then converts inactive plasminogen into plasmin, which in turn can cleave and activate GFs, matrix metalloproteases (MMPs), as well as the extracellular matrix (ECM). The activated MMPs can directly cause the degradation of ECM and thereby release various growth factors. Plasminogen activator inhibitor-1 can inhibit the catalytic activity of both uPA and plasmin. Apart from uPA, uPAR also binds to integrins and other cell surface receptors to activate different intracellular signaling pathways [example: Jak–STAT, PI3K, focal adhesion kinase (FAK), and Rac] and regulates cellular processes such as cell proliferation, survival, migration, invasion, angiogenesis, and metastasis.
The expression of uPAR is elevated in the tumor tissues but not in the surrounding normal tissues, which makes it an attractive therapeutic target (66). More recent evidence indicates that signaling pathways activated by uPAR helps the cancer cells to escape and reduce the cytotoxic effect of anticancer drugs (67). The elevated expression of the gene encoding uPAR (PLAUR) in cancer can be regulated by different mechanisms. Transcription factors such as Sp1, NF-κB, TCF, and hypoxia-inducible factor 1α, that are frequently activated by different types of cancer-related signaling pathways, binds to the cis-acting elements located upstream of the PLAUR gene to trigger its elevated expression in cancer (7). Another important mechanism by which the expression of the PLAUR gene gets amplified is through the cooperativity between human epidermal growth factor receptor 2 (HER2) and PLAUR (68). Using a Real-time quantitative PCR based assay, Pierga et al. first reported on a correlative pattern in the expression of HER2 and PLAUR genes in the disseminated tumor cells from breast cancer patients (69). Later on, Ming et al. showed co-amplification of HER2 and PLAUR when blood and tissue of patients with advanced recurrent breast cancer were analyzed (70). They found that higher the expression of HER2 in the tumor cells, the higher is the possibility of co-amplification of PLAUR in the primary tumors and circulating tumor cells. On the other hand, the HER2 non-amplified tumors had a significantly lower rate of PLAUR amplification. However, the exact mechanism of such co-amplification is yet to be elucidated. Nevertheless, such cooperativity of HER2 and PLAUR suggests the importance of simultaneous targeting of both Her2 and uPAR in breast cancer patients.
Plasminogen Activator Inhibitors (PAI)
Plasminogen activator inhibitor-1 and PAI-2 are serine protease inhibitors belonging to the serpin family that can cause the neutralization of uPA (71). PAI-1 is more potent than PAI-2 to cause the inhibition of uPA (72). While PAI-1 is predominantly found in the extracellular region, PAI-2 is mainly localized in the cytoplasm for reasons still unknown (73).
Plasminogen activator inhibitor-1, as an inhibitor of PA system, is expected to be associated with anticancer functions by inhibiting the activity of uPA–uPAR complex. Intriguingly, the overexpression of PAI-1 shows a completely opposite effect in cancer patients. Higher expression of PAI-1 promotes tumor growth and as such, it is associated with poor prognosis (73–75). This indicates the fact that PAI-1 has ligands other than the PA system, which takes part in promoting tumor growth (76). In its active conformation, the PAI-1 protein can interact with the integrin binding site on vitronectin and thereby inhibit cellular migration (77). However, PAI-1 interaction binding with uPA (uPA–PAI-1 complex) diminishes its vitronectin binding affinity (77). Both PAI-1 and PAI-2 expressions are altered in cancer. Unlike PAI-1, elevated levels of PAI-2 are associated with the decrease in tumor growth and metastasis (73).
The genes encoding PAI-1 (SERPINE1) and PAI-2 (SERPINB2) are regulated by different types of growth factors [transforming growth factor beta (TGF-β) and insulin-like growth factor 1], hormones (insulin), and cytokines (tumor necrosis factor α) that typically shows aberrant expression in cancer (7).
Physiological Role of the uPA–uPAR System
As discussed earlier, the uPA system plays an important role in activating the plasmin from its inactive plasminogen form by proteolytic cleavage. Once activated the plasmin system causes degradation of fibrin, several blood clotting factors, and ECM (78). This, in turn, acts as a homeostatic mechanism in the normal physiologic wound healing. In addition, the components of the uPA–uPAR system are involved in the proteolytic activation of a number of growth factors and cytokines (for example, basic fibroblast growth factor; TGF-β; and interleukin-1 beta) that are involved in myelopoiesis (79, 80).
Degryse et al. demonstrated that the ATF of uPA alone could induce chemotaxis in rat smooth muscle cells through its binding with uPAR (81). Later studies by Mukhina et al. have further streamlined the crucial role of the KD of the ATF in mediating chemotaxis (82). These results suggest that the proteolytic domain of uPA is not required for chemotaxis. Furthermore, antibody-mediated inhibition of uPAR blocked chemotaxis whereas rescue with exogenous uPAR reversed these effects (83). The distribution of uPAR is also different between actively migrating and non-migrating cells. While uPAR is more preferentially distributed on the apical side or the focal contacts on the surface of non-migrating cells (84, 85), however, during migration, it redistributes at the leading edge of the migrating cells (86). In this way, uPAR regulates the concentration and activity of uPA at the required sites on the surface of migrating cells.
It has been shown that the components of the uPA system are expressed in various types of hemopoietic cells (78). More importantly, their levels are altered during the course of infections suggesting the role of the uPA–uPAR system in mediating different types of immune response (87–89). In response to bacterial infection, proinflammatory cytokines such as TNF-α and IL-1β are released, which in turn increases uPA expression and secretion by different types of monocytes, neutrophils, epithelial and endothelial cells (87). Upon its local release, uPA causes the activation of neutrophils, primes them to produce superoxide and potentiates their migration by a pathway that can be either dependent or independent of uPAR (90–93). When uPA is knocked out, it hampered the ability of the mice immune system to recruit neutrophils and macrophages in response to the exposure to Cryptococcus neoformans (strain 52D) bacteria, ultimately leading to uncontrolled infection and death of the animals (94). Like uPA, its receptor uPAR also plays an important role in innate immune response through the regulation of cell adhesion, migration in a manner dependent or independent of uPA (88). The uPAR deficient mice also failed to recruit neutrophils and macrophages at the site of bacterial infection (95, 96).
The uPA system has also been implicated in adaptive immunity. The expressions of both uPA and its receptor uPAR are augmented during T-cell activation compared with their levels in resting or naive T cells (97). In uPA-deficient mice, the T helper 1 and 2 (Th1 and Th2) effector lymphocytes lose the ability to respond to pathogen attack (98, 99). On the other hand, blockade of uPAR restricts the migration of leukocytes in vitro (100) and also hampers the recruitment of T-cells in vivo (101). Collectively, all these studies make it evident that the uPA–uPAR system is at the fringe between fibrinolysis, immune response mechanisms, and pathology (88). In addition, the uPA system is also involved in the male reproductive system where it can enhance sperm mobility, stimulate acrosome reaction, and promote fertilization (102). These district effects of uPA led Qin et al. to demonstrate that targeting uPA may serve as a novel strategy for male contraception (103). These and other physiological functions of the uPA–uPAR system are summarized in Figure 4.
Figure 4
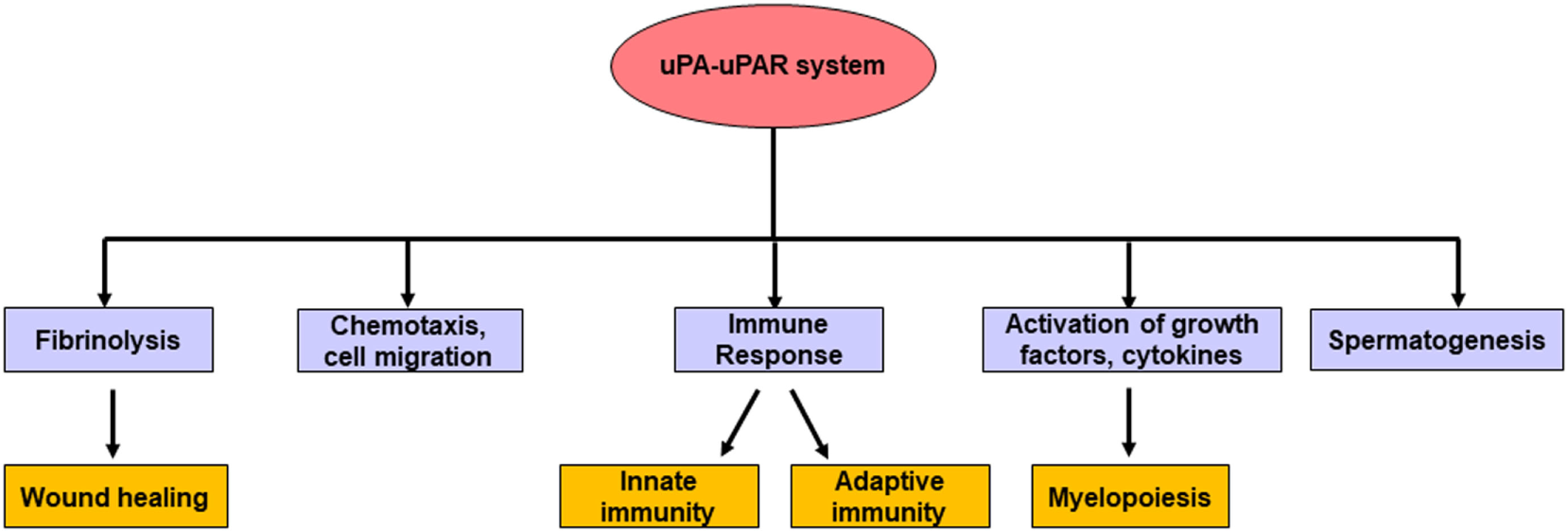
The major physiological roles of the uPA–urokinase-type plasminogen activator receptor (uPAR) system. In normal physiologic conditions, apart from fibrinolysis, the uPA–uPAR system takes part in different biological processes such as the generation of matured myeloid cells, spermatogenesis, chemotaxis, cell migration, wound healing, as well as mediating different types of immune response.
Function of the uPA–uPAR System in Cancer Progression
The following section summarizes the functional role of the uPA system in different steps of cancer progression.
Tumorigenesis and Suppression of Apoptosis
The key mechanism that underlies tumorigenesis includes a combination of enhanced cell proliferation and suppression of apoptotic cell death (6). It has been shown that the components of the uPA system can increase cell proliferation through the proteolytic activation of different types of growth factors such as vascular endothelial growth factor (VEGF), EGF, fibroblast growth factor-2 (FGF-2), and TGF-β as well as adhesion molecules such as the α5β1 integrins (6, 104, 105). Several studies have demonstrated the involvement of the uPA–uPAR system during the early stages of tumor formation. For example, the progression of melanoma was impaired in uPA-deficient mice (106). However, inhibition of uPA did not impair the progression of pancreatic cancer in the well-characterized RIP-Tag2 transgenic mice (107), suggesting that the effect of the uPA system in mediating early tumorigenesis is also dependent on cancer types.
Furthermore, the uPA–uPAR system has been implicated in the inhibition of apoptosis (108, 109). Subramanian et al. have shown that RNAi-mediated inhibition of both uPA and uPAR simultaneously triggers apoptosis in breast cancer cells through the upregulation of different caspase proteins (110). Later studies, using targeted antibodies against uPAR (ATN-658) showed increased apoptosis in ovarian cancer cells both in vitro and in vivo (111). PAI-1, however, has been shown to play a dual role in both augmenting and inhibiting apoptosis. In prostate cancer, PAI-1 expression showed association with increased apoptosis of the endothelial cells of the tumor vasculature (112). In other studies, Chen et al. showed that active PAI-1 causes inhibition of caspase-3 and thereby decreases apoptosis (108). Therefore, PAI-1 is hailed as a “double-edged sword” during apoptosis (113).
Regulation of the Switch between Dormancy and Tumorigenicity
Cancer dormancy is a stage in cancer progression where the disease remains in an asymptomatic state (114). During tumor dormancy, the cells stop dividing but continue to survive in a quiescent state. Cancer cells may remain at this dormant stage even for decades (114). Once favorable conditions for growth are available, the cells restart proliferation. The switch from a dormant state to a tumorigenic state is one of the underlying causes of disease relapse in clinical settings, which has led to its classification into two broad types, tumor mass dormancy and cellular dormancy. In tumor mass dormancy, the cells continue to divide, but the expansion in tumor size is limited by reduced blood supply or the presence of an active immune system. On the other hand, during cellular dormancy, the tumor growth is halted at the G0/G1 phase of cell cycle. Several mechanisms can regulate the switch from a dormant stage. One such mechanism is mediated by the cross talk between the cancer cells and the surrounding microenvironment through the interaction between uPAR and the integrins (104, 115, 116). In cancer cells, the uPAR–integrin interaction causes the recruitment of FAK and EGFR and thereby induces the mitogenic Raf–MEK–ERK signaling pathway (117, 118). It has been shown that inhibition of uPAR, integrin β1, FAK or EGFR alone, or in combination induces dormancy and thereby results in tumor suppression (115, 118, 119).
Degradation of the ECM
Extracellular matrix acts as a barrier to confine the tumor cells within its primary site of origin, and proteolytic degradation of the ECM marks a key event during tumor growth, invasion, and metastasis. The proteases that can cause the degradation of the ECM include uPA, plasmin, cathepsins, and different types of MMPs (120). During metastasis, tumor cells cause ECM degradation at various occasions to escape from the primary site of origin to migrate at a distant tissue through the route of the bloodstream. The components of the uPA–uPAR system can breakdown the ECM through the activation of plasmin or the MMPs (6) (Figure 3). Degradation of ECM causes the release of different types of growth factors, which acts as a feedback loop to enhance the expression of different components of the uPA–uPAR system as well as the regulation of various steps of metastasis (7).
Angiogenesis
Angiogenesis is a biological process by which new blood vessels are developed from the already existing vessels. In tumor microenvironment, the newly formed blood vessels provide oxygen and other nutrients to aid tumor cell growth, invasion, and metastasis (121). The uPA-mediated degradation of the ECM is crucial for the initiation of angiogenesis (7). uPA induces the release of different types of proangiogenic growth factors such as VEGF, FGF-2 that plays a key role in endothelial cell proliferation and invasion (122). The binding of uPAR with vitronectin promotes cell adhesion and migration (123) (Figure 3). uPAR also represses the expression of a key angiogenesis regulator called phosphatase and tensin homolog (PTEN) and thereby promotes angiogenesis (124). Loss of function assays using shRNAs against both uPA and uPAR showed inhibition of angiogenesis signaling by both granulocyte-macrophage colony-stimulating factor (GM-CSF) and VEGF (125, 126). In other studies, inhibition of uPA/uPAR alone or in combination has been shown to repress the expression of Notch-1 (127). This impairs the cross talk of Notch-1 signaling to NF-κB and the PI3K/AKT/mTOR pathways and thereby inhibits invasion and angiogenesis. Whether PAI-1 is proangiogenic or antiangiogenic in cancer is still not clear because of contradictory evidence by different groups (128, 129).
Cell Adhesion and Migration
Tumor cells need to migrate from the primary site of origin to a distant organ to cause metastasis. This makes cell migration a crucial step during metastasis. Cell migration is dependent on different components of the adhesome, which regulates the attachment and detachment of the cells from the ECM (6). The components of the uPA systems are known to increase cell adhesion and migration during metastatic spread of the tumor cells (130). The role of uPAR has been studied quite extensively in this regard. It is now established that uPAR connects the uPA system to GPCR signaling as well as some other proteins such as cytokeratin 8, α-enolase to regulate cell adhesion, and migration (64, 131, 132). PAI-1 can decrease cell migration by repressing the interaction between vitronectin and αvβ3 integrin (77).
Cell Invasion and Metastasis
Epithelial–mesenchymal transition (EMT) is a biological process by which polarized epithelial cells are transformed into highly invasive mesenchymal cells with greater migratory capabilities, increased resistance to apoptotic cell deaths as well as enhanced capabilities to produce ECM proteins (133, 134). Elevated uPAR expression during hypoxic conditions activates downstream Akt and Rac1 signaling pathways leading to the promotion of EMT as well as cellular invasion (135). In addition, blocking the expression of uPAR repressed EMT induced by hypoxia. This suggested that uPAR plays a role in EMT. Other studies have shown that antibody-based targeting uPAR also inhibits tumor invasion and metastasis both in vitro and in vivo (136).
Emerging evidence suggests that tumor cells secrete different types of extracellular vesicles that have pro-metastatic effects (137, 138). Higher levels of uPA and PAI-1 have been detected in the extracellular vesicles of different tumor cells suggesting a possible involvement of the uPA axis in exosome-mediated tumorigenesis and metastasis (139–141). However, detailed studies are warranted in future to verify the phenomena.
Prognostic Role of the uPA–uPAR System in Cancer
Aberrant expression of the components of the uPA–uPAR system has been detected in a wide variety of cancer (142). This opened up newer avenues to develop therapeutic strategies to attain better clinical outcomes in cancer patients (20, 21). In the following section and Table 1, the role of different components of the uPA–uPAR system in the prognosis of different types of cancer is discussed.
Table 1
| Type of cancer | Component | Effect on prognosis | Reference |
|---|---|---|---|
| Breast | uPA and plasminogen activator inhibitor-1 (PAI-1) | Independent prognostic markers for poor relapse-free and overall survival | (145) |
| uPAR | Poor prognosis and metastasis during the advanced stages of breast cancer | (69) | |
|
|
|||
| Prostate | uPA and uPAR | Increased aggressiveness, postoperative progression, and metastasis | (154) |
| PAI-1 | Relation to pathological stage, surgical margin status only | (156) | |
| Soluble form of uPAR (suPAR) | Poor overall survival in patients with prostate cancer | (155) | |
|
|
|||
| Ovarian | uPA and PAI-1 | Predicts overall survival of advanced staged patients; however, the effect is not consistent between different studies | (161, 162) |
| suPAR | Associated with poor survival in preoperative patients; shows stronger prognostic value for assessing the effectiveness of chemotherapy | (157, 160) | |
|
|
|||
| Cervical | uPA and PAI-1 | Elevated in invasive cervical carcinoma; predicts the risk of lymph node metastasis | (163, 164) |
| PAI-2 | Elevated in invasive cervical carcinoma | (163) | |
|
|
|||
| Endometrial | uPA and suPAR | Elevated in the plasma of endometrial cancer patients | (165, 167) |
| uPAR | Correlated with advanced stage endometrial cancer | (166) | |
| PAI-1 | Associated with shorter relapse-free and overall survival | (168) | |
|
|
|||
| Soft-tissue sarcoma | uPA | Associated with increased metastasis and recurrence | (169) |
|
|
|||
| Melanoma | uPA and PAI-1 | Higher levels in patients is likely to have prognostic significance | (171) |
|
|
|||
| Colorectal | uPA | Prognostic marker for survival and metastasis | (173, 175) |
| uPAR, PAI-1, and PAI-2 | Higher expression is associated with poor response to therapy | (176) | |
| suPAR | Higher preoperative level is associated with poor survival | (172) | |
|
|
|||
| Lung | uPA, uPAR, PAI-1, and PAI-2 | Expression is increased in the tumor tissues of non-small cell lung cancer | (183) |
| suPAR | Prognostic marker for lung cancer patients | (181) | |
|
|
|||
| Pancreatic | uPA | Increased gene expression is associated with poor survival in pancreatic ductal adenocarcinoma (PDAC) | (185) |
| uPAR | Marker to differentiate PDAC and chronic pancreatitis | (186) | |
| suPAR | Increased urinary level is associated with poor patient outcome in PDAC | (187) | |
| PAI-2 | Gene encoding PAI-2 protein is frequently deleted in PDAC and thereby increase metastasis | (185) | |
|
|
|||
| Gastric | uPA | Associated with poor patient outcome.Can be used as a prognostic marker (this is contested by some studies) | (188, 190) |
| uPAR | Associated with poor patient outcome | (188) | |
| PAI-1 | Has role as a prognostic marker, which is contested by some studies | (188, 190) | |
|
|
|||
| Oral | uPA, uPAR, PAI-1, and PAI-2 | Elevated in oral squamous cell carcinoma tumors | (191) |
| uPA and PAI-1 | Prognostic factor for relapse-free survival | (192) | |
|
|
|||
| Esophageal | uPA and PAI-1 | uPA/PAI-1 ratio is correlated with invasion | (192) |
| uPA | Associated with poor overall survival | (193) | |
| PAI-2 | Protects from local invasion | (193) | |
|
|
|||
| Liver | uPA, uPAR, and PAI-1 | Higher expression in tumor tissues likely contributes to increased invasion and metastasis | (194) |
| uPAR and PAI-1 | Correlation with poor prognosis | (194) | |
|
|
|||
| Laryngeal | uPA and PAI-2 | Independent prognostic factors for survival | (195) |
|
|
|||
| Head and neck | uPA and PAI-1 | Expression is increased in the tumors, which is likely to provide prognostic value | (197) |
| suPAR | Elevated in the plasma | (198) | |
|
|
|||
| Kidney | PAI-1 | Influence cause-specific survival | (199) |
|
|
|||
| Bladder | uPA, PAI-1, and PAI-2 | Elevated in the tumor samples | (200) |
|
|
|||
| Acute myeloid leukemia | uPAR | Higher expression correlated with aggressiveness of the disease | (201) |
| suPAR | Higher level is correlated to chemotherapy resistance | (202) | |
Selected roles of the uPA–urokinase-type plasminogen activator receptor (uPAR) system in the prognosis of different types of cancers.
More than three decades ago, O’Grady et al. demonstrated an elevation of uPA activity in the malignant breast tumors compared with the benign ones (143). Later on, Duffy et al. showed an association between uPA activity in primary breast tumors with tumor size and metastasis (144). Since then a great deal of effort has been made to discover he potential role of uPA and all the components of the uPA system in cancer. It is now evident that the various compartments of the uPA oncogenic pathways are involved in mediating breast cancer progression and metastatic spread. A surprisingly high level of PAI-1 has been implicated in the adverse outcome in breast cancer patients. Using a cohort comprising 2,780 patients with breast cancer, Foekens et al. have demonstrated that the levels of uPA and PAI-1 can be used as independent prognostic markers for poor relapse-free survival as well as of overall survival (145). Both uPA and PAI-1 are categorized as the best available biomarkers after estrogen receptor and HER2 and are among the first to attain level-of-evidence 1 in breast cancer (146, 147). Therefore, the American Society of Clinical Oncology (ASCO) recommended an enzyme-linked immunosorbent assay (ELISA)-based assay to determine uPA/PAI-1 role as a predictive marker for benefit from chemotherapy and as a poor prognostic marker for disease recurrence and survival (148). A follow-up study spanning over 7 years further confirmed the association between invasiveness and increased uPA and PAI-1 levels in patients with breast cancer (149). In recent years, mRNA-based assays such as Oncotype DX (150) and MammaPrint (151) have also shown promising results for predicting outcome in breast cancer. Compared with these assays, the ELISA-based measurement to evaluate uPA and PAI-1 protein is less cumbersome and inexpensive (152). Therefore, it is more practical to use this system for predicting patient outcome in breast cancer. More recently, a prospective randomized multicenter trial called “Chemo-N0” that spanned over a period of 10 years validated the long-term prognostic and predictive impact of uPA and PAI-1 ELISA for the risk assessment as well as treatment decision in patients with node-negative breast cancer (74). Higher level of PAI-2 demonstrated a weak association with favorable outcome in breast cancer (153). Elevated uPAR expression has been linked with poor prognosis and metastasis during the advanced stages of breast cancer (69).
In prostate cancer, high levels of uPA and its receptor uPAR in the plasma correlated with increased aggressiveness, postoperative progression, and metastasis (154). Furthermore, increased suPAR in the circulation showed association with a decrease in overall survival in patients with prostate cancer (155). Using radical prostatectomy specimens from 153 patients, Kumano et al. showed that the expression of uPA and uPAR has a strong correlation with prostate cancer prognosis (156). Furthermore, the expression of PAI-1 expression only showed relation to surgical margin and pathological stage whereas PAI-2 expression did not show any association with prostate cancer prognosis (156).
In ovarian cancer, elevated level of suPAR in the serum from preoperative ovarian cancer patients but not postoperative patients showed association with poor survival (157). The same group has demonstrated a higher level of urinary suPAR in ovarian cancer patients (158). Later studies have found that a portion of the urinary suPAR is present in a cleaved form which is similar to the one in ovarian cancer tissue extracts (159). In another study by Ljuca et al., suPAR, showed a stronger prognostic value for assessing the effectiveness of chemotherapy than carcinoembryonic antigen (CEA) and uPA in FIGO II and III ovarian cancer patients (160). Using an ELISA-based assay from the tumor tissue extracts, Kuhn et al. demonstrated that both uPA and PAI-1 have prognostic significance in predicting overall survival of the patients who have advanced ovarian cancer stage FIGO IIIc (161). However, this observation is not consistent with other studies where uPA and PAI-1 increase did not show any prognostic significance in ovarian cancer patients (162). Further studies are required to demonstrate the exact prognostic role of uPA and PAI-1 in ovarian cancer.
Daneri-Navarro et al. reported an elevated level of uPA, PAI-1 and PAI-2 in the tissue extracts of invasive cervical carcinoma compared with normal tissues (163). Other studies have shown that assessment of uPA and PAI-1 localization in the cervical tissues can predict the risk of lymph node metastasis (164). An increase in the plasma uPA level was demonstrated in patients with cervical as well as endometrial cancer compared with the control group (165). They also reported that the plasma PAI-1 and tPA levels remained unaltered (166). Furthermore, higher levels of plasma suPAR have been identified in endometrial cancer patients than the controls (167). In another study, Memarzadeh et al. found that elevated expression of uPAR in the surgically excised endometrial tissue is positively correlated with advanced stage endometrial cancer (166). Higher levels of PAI-1 protein showed shorter relapse free and overall survival in stage IB and II endometrial cancer patients (168).
Using frozen tumor tissues from 69 patients, Choong et al. detected an association between higher levels of uPA with increased metastasis and recurrence of soft-tissue sarcoma (169). This study indicated a prognostic role of uPA for patients with soft-tissue sarcoma.
In melanoma, a correlation between uPA expression and metastasis was observed in an earlier study using 45 freshly frozen tumors (170). The same study also confirmed the presence of uPAR in one-third of the melanoma tumors (170). Later on, Stabuc et al. reported significantly higher levels of uPA and PAI-1 in melanoma patients compared with normal individuals and further suggested that uPA and PAI-1 might provide prognostic significance in patients with melanoma (171).
In colorectal cancer patients, an elevated preoperative level of suPAR in the plasma demonstrated an association with poor survival (172). In addition, uPA and PAI-1 levels are increased in blood and tissue of the colorectal cancer patients (173, 174). Furthermore, it has been demonstrated that circulating uPA and PAI-1 can serve as better prognostic markers than the commonly used colorectal cancer markers CEA and the gastrointestinal cancer-associated carbohydrate antigen (19–9) (173). Yang et al. suggested that uPA and uPAR can be used as independent prognostic factors for colorectal cancer patient survival, metastasis, as well as therapeutic targets (175). Halamkova et al. showed a statistically significant correlation between the expression of uPAR, PAI-2 protein, and the grade of the colorectal tumor (176). Analysis of the colorectal cancer metastasis in the liver revealed an increase of PAI-1 in the liver metastases compared with primary colorectal carcinomas (177). A number of studies have demonstrated increased levels of uPA, uPAR, and PAI-1 in tissues collected from patients with hepatocellular carcinoma (HCC) as compared with non-cancerous control tissues from the liver (178, 179). In addition, higher expression of these molecules has been linked to HCC metastasis.
Chen et al. demonstrated significantly higher levels of uPA and uPAR in the circulation of lung cancer patients compared with the controls (180). In addition, suPAR has been demonstrated as a prognostic marker for lung cancer patients (181). In small cell lung cancer, the most aggressive form of lung cancer, an association between elevated levels of domain 1 of uPAR (uPAR-DI) in the blood circulation and short overall survival has been demonstrated (182). In non-small cell lung cancer (NSCLC), elevated expression of uPA, uPAR, PAI-1, and PAI-2 proteins were observed in the cancerous tissue compared with the normal/adjacent tissue (183). However, their elevated expression did not show any significant correlation with patient survival.
In pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, the components of the uPA system are frequently altered (184). Recent studies by Harris et al. have demonstrated that the gene encoding PAI-2 (SERPINB2) is frequently deleted in PDAC (185). Such deletion may cause an increase in metastasis since PAI-2 is the inhibitor of pro-metastatic uPA. Indeed, when a large cohort of patients with resected PDAC was analyzed, it was found that the expression of the gene encoding uPA (PLAU) is significantly increased in these patients. Elevated expression in PLAU showed significant association with poorer survival following pancreatectomy. In another study, Chen et al. showed that PLAUR can discriminate between PDAC and chronic pancreatitis (CP) with highest accuracy (186). Further studies have demonstrated an elevation of urinary suPAR in PDAC patients compared with the patients with CP (187). Such increase in urinary suPAR in PDAC also correlated with poor patient outcome.
In gastric cancer, uPA and uPAR expression show association with poor patient outcome (188). Both uPA and PAI-1 show prognostic significance in gastric cancer, and their levels are elevated during the advanced stage of the disease (188). Moreover, lower expression of uPA, uPAR, and PAI-1 showed correlation with improved patient survival (189). Using an immunohistochemical method, Luebke et al. could not verify the prognostic significance of uPA and PAI-1 in their study conducted on 105 gastric cancer patients (190). Therefore, further studies are needed to assess the prognostic significance of the uPA system in gastric cancer.
Elevated levels of uPA, uPAR, PAI-1, and PAI-2 are observed in oral squamous cell carcinoma (OSCC) tumors (191). In addition, there is a strong correlation between uPA, uPAR, and PAI-1 expression and aggressiveness of the tumor (191). Hundsdorfer et al. showed that uPA and PAI-1 could be used to assess relapse-free survival in OSCC patients (192).
In esophageal cancer, uPA/PAI-1 ratio shows association with invasive properties of the tumor (192). Shiomi et al. have demonstrated that patients with uPA-positive tumor showed poorer overall survival compared with those with uPA-negative tumor (193). In addition, they have found that less local invasion of esophageal cancer cells in patients with PAI-2 positive fibroblasts suggesting its protective role in tumor invasion.
Zheng et al. showed that the expression of uPA, uPAR, and PAI-1 is significantly higher in HCC cells compared with normal liver tissues and this may contribute increased metastasis (194). Furthermore, uPAR and PAI-1 levels are associated with poor HCC prognosis.
In laryngeal cancer, uPA and PAI-2 have been shown to be strong prognostic factors to determine survival while PAI-1 did not show significant correlation in prognosis (195). It has been shown that the expression of both uPA and uPAR are significantly elevated in laryngeal squamous cell carcinoma compared with the normal peripheral tissues around cancer, which is likely to contribute to increased invasion and metastasis (196).
Strojan et al. found elevated expression of both uPA and PAI-1 in the tumors from the patients with head and neck cancer, which may provide prognostic value to determine invasiveness and metastasis (197). Moreover, significantly elevated levels of suPAR were detected in the plasma of the patients with head and neck cancer, which may provide valuable information for disease prognosis (198).
Ohba et al. reported that PAI-1 has the ability to influence the survival of renal cell carcinoma (RCC) patients (199). Span et al. found that the levels of PAI-1 and PAI-2 but not uPA were significantly elevated in RCC samples compared with normal control tissue (200). On the other hand, the expression of uPA, PAI-1, and PAI-2 were all increased in bladder tumors when compared with normal tissues (200). In acute myeloid leukemia patients, the higher expression of uPAR along with some other morphological characteristics correlated with aggressiveness of the disease (201). Mustjoki et al. found that elevated expression of suPAR showed association with chemotherapy resistance (202). Collectively, these studies have demonstrated a dominant role of the uPA–uPAR in a large number of common cancers, which supports its proposed function as a major player in tumor progression.
Diagnostic Role of the uPA–uPAR System
Since the uPA–uPAR system plays a crucial role in the progression of cancer, a plenty of efforts have been made to develop diagnostic modalities targeting this axis (203–205). Figure 5 contains a brief summary of some of the promising use of the uPA–uPAR system in cancer diagnosis.
Figure 5
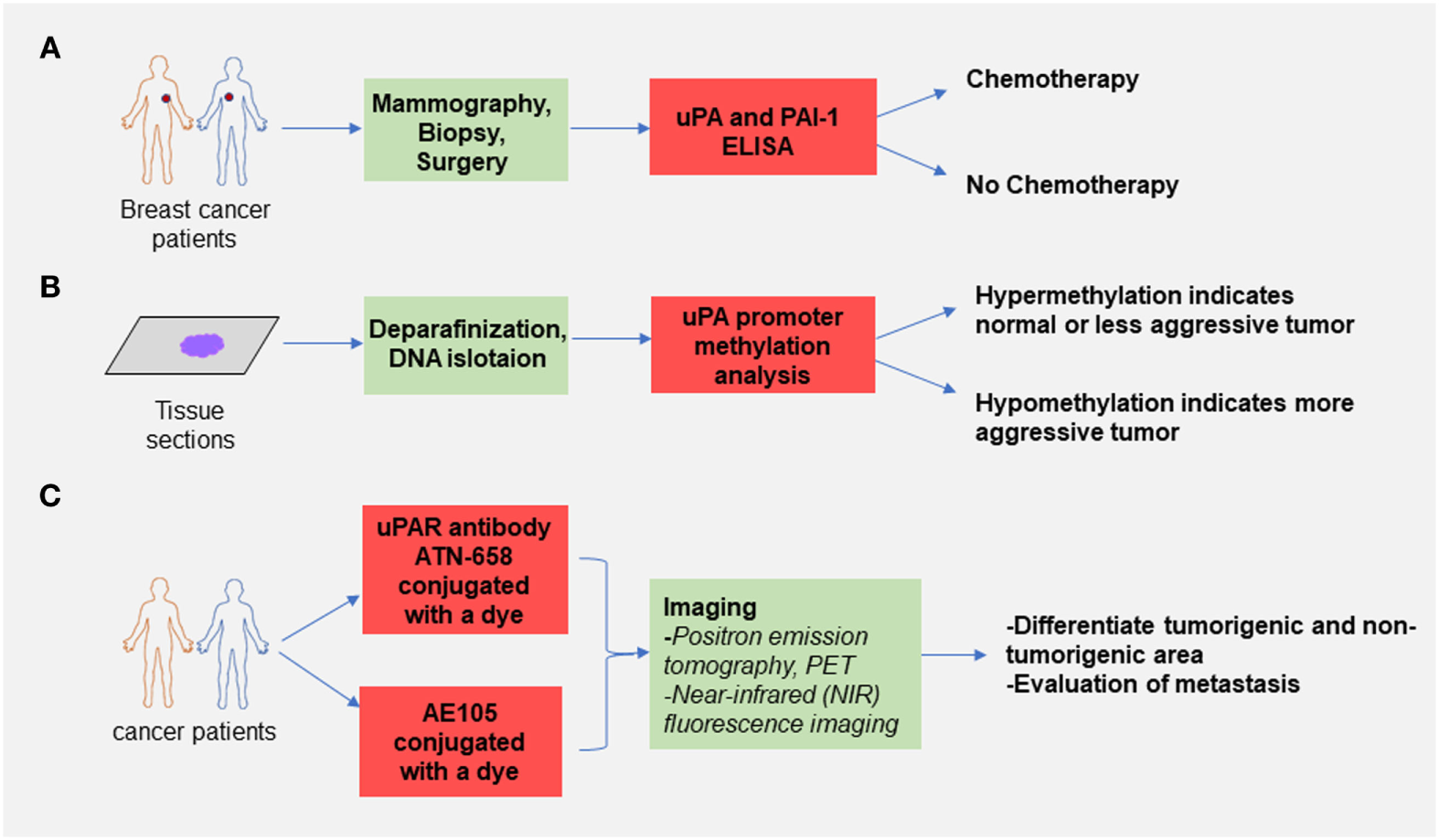
Selected use of the uPA–urokinase-type plasminogen activator receptor (uPAR) system in cancer diagnosis. (A) uPA and plasminogen activator inhibitor-1 (PAI-1) enzyme-linked immunosorbent assay can assess the risk of breast cancer recurrence and based on the results obtained from the assay clinicians decide if chemotherapy is needed after surgery. This method is already used by many oncologists. (B) Assessment of promoter methylation status of the gene encoding uPA can be used to predict the aggressiveness of the primary tumor. It has been shown that the CpG sites on the uPA promoter are hypomethylated in high-grade tumors compared with the control and less aggressive tumors. This method can be used to determine the invasiveness of cancer. (C) Conjugation of a labeled dye with antibodies (ATN-658 for uPAR) or peptide targeting uPA–uPAR interaction (AE105) can distinguish between the tumorigenic and non-tumorigenic area, and this method showed great diagnostic potential in several known cancers.
uPA and PAI As Diagnostic Biomarker
Using ELISA, Jänicke et al. were the first to determine the amount of uPA protein in breast cancer tissue (206). They found that the uPA antigen is elevated in the primary breast tissues, which correlated with poor breast cancer prognosis of the patients. A similar association was also reported for PAI-1 by the same group in 1991 (207). Later on, the ASCO recommended uPA and PAI-1 biomarker testing for breast cancer risk assessment, and also to decide on the appropriate adjuvant chemotherapies to be given to the patients (148).
Enzyme-linked immunosorbent assay-based methods are still considered as the clinically relevant system for assessment of uPA and PAI-1 in the context of breast cancer outcomes. As such commercial ELISA kits for uPA/PAI-1 have been developed. FEMTELLE® is one such kit that is used to assess the risk of recurrence in primary breast cancer and helps to predict if the chemotherapy is beneficial after surgery (208). However, the main problem with all the ELISA-based assays is the need for fresh or fresh-frozen tissue samples (209). Hence, the possibilities of using some other methods including histological analysis that do not require fresh or fresh-frozen tissue samples have also been investigated.
Several groups have assessed uPA and PAI-1 mRNA expression to diagnose cancer (210, 211). The main advantage of using mRNA is that it can be extracted from the formalin-fixed tissues unlike the ELISA-based assays. However, there are discrepancies between studies on the effect of uPA and PAI-1 mRNA expression in cancer diagnosis (210–212). More research is needed to come up with a concrete conclusion.
Another interesting avenue that employs the assessment of uPA to diagnose cancer is through the analysis of alterations of the methylation status of the promoter DNA. The major advantage of this method compared with the other techniques is the fact that DNA is more stable and can be isolated from the formalin-fixed, paraffin-embedded samples (209). Aberrant DNA methylation is seen in almost all cancer. Both hypermethylation-mediated inactivation of tumor suppressor genes and hypomethylation-mediated activation of pro-metastatic genes are key characteristics of the cancer cells, which makes DNA methylation as a suitable diagnostic and therapeutic target (213). More than a decade ago, our lab demonstrated that there is an inverse correlation between uPA expression and promoter methylation for different grades of breast cancer (50). We demonstrated that the percentage of CpG methylation at the uPA promoter is decreased with the progression of breast cancer (50). As such uPA promoter methylation status can be used as an early detection marker. Gao et al. have demonstrated a similar inverse correlation between PAI-1 promoter methylation and gene expression in cancer (214).
uPAR As an Imaging Agent in Malignancy
An important avenue where much attention has been deployed is the development of non-invasive imaging/diagnostic agents targeting the uPA system. In particular, imaging strategies targeting uPAR has been used in different types of cancer (204, 205). Using rodent models of breast and prostate cancer, we have shown that uPAR is a viable imaging target for cancer diagnosis (215). Briefly, an 125I-labeled anti-rat uPAR antibody was injected into animals bearing prostate and breast cancer and the uptake of radiolabel in primary tumors as well as some of the common sites of metastasis including the liver, lung, spleen, and lymph nodes was determined. On the other hand, control animals with no tumors or the tumor-bearing animals injected with 125I-labeled control IgG antibody showed a very minimum radioactivity levels suggesting that uPAR as a diagnostic/imaging target for cancer progression and metastasis. Since then a plethora of studies have been done to validate and improve the diagnostic potential of uPAR in different types of cancer. Among these, well-known examples are peptide antagonist targeting uPA–uPAR interaction (AE105) and monoclonal antibodies against uPAR (ATN-658). Both these molecules have been used for multimodal imaging for cancer diagnosis. The main idea behind the multimodal imaging is to conjugate a single target to either a radionuclide or a near-infrared (NIR) fluorescent dye that can distinguish the tumor tissue from the surrounding non-tumorigenic area before [positron emission tomography (PET) or single-photon emission computed tomography] or during (NIR fluorescence imaging) surgery (216).
AE105 is a smart peptide composed of nine amino acids and is used for intraoperative guidance at the time of surgery as well for the evaluation of metastasis (204, 217). On the other hand, different classes of antibodies such as ATN-658 (targeting uPAR), ATN-291 (targeting uPA) for cancer imaging has been reported (205, 218). The ATN-658 is a humanized antibody that is currently being used by us and others.
While both AE105 and ATN-658 have antitumor activity, however, they exhibit different imaging timeframes. Since it is a small molecule of 1 kD, the imaging timeframes of AE105 are usually within several hours due to short half-life (216, 217). On the other hand, ATN-658 has much longer half-life in the serum, which enables the imaging timeframes to go up to days (64, 216). AE105 cannot target uPAR when it is already attached to uPA and therefore the imaging intensity of the tumors targeted with AE105 peptide-based agents may not always provide information on the actual levels of uPAR. On the other hand, ATN-658 binding to uPAR is independent of the uPA–uPAR interaction, which helps to provide more accurate diagnostic information. Recently, a phase 1 clinical trial using AE105 has been completed in patients with breast, prostate, and bladder cancers (217). In these studies, AE105 was conjugated to the organic macrocyclic chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (also called DOTA) and then labeled with 64Cu for PET imaging and found that administration of the agent is safe in cancer patients with a favorable biodistribution and stability. The clinical trial for ATN-658 imaging is expected to start soon by us and others. Taken together, these studies have shown great promise for the use of uPAR as a diagnostic/imaging target in malignancy.
Therapeutic Targeting of the uPA–uPAR System
Many studies spanning more than two decades have made attempts to target the uPA system to block cancer (219, 220). Some of the most notable ones are listed below.
Inhibition of Proteolytic Activity of uPA
The earliest attempts to block uPA activation focused on the development of agents to inhibit its catalytic activity. Ossowski et al. used an antibody-based approach to block the enzymatic activity of uPA that was able to block local invasion but failed to inhibit distant metastasis in vivo (221). Later studies using small molecule inhibitors proved to be more effective in achieving the inhibition of the enzymatic activity of uPA. By modifying the chemical structure of amiloride, Towle et al. were able to develop a novel class of inhibitors of uPA known as the 4-substituted benzo(b)thiophene-2-carboxamidines (222). Two compounds belonging to this family known as B-428 and B-623 were able to inhibit uPA activity with median inhibition concentration (IC50) values of 0.32 and 0.07 µM and inhibitory constant (Ki) values of 0.53 and 0.16 mM (222). We have previously shown that use of a small molecule inhibitor called B-428 can reduce prostate cancer growth and metastasis in vivo (223). Further work in our lab has shown that B-428 has an additive effect in repressing breast cancer growth and metastasis when used in combination with tamoxifen (224). Other inhibitors of uPA activity have also been used and some of them have shown very promising results in the clinical trials. For example, MESUPRON® (also known as WX-671) and its pro-drug WX-UK1 have shown promising results in the clinical trials for the treatment of different types of solid tumors (225–227).
Inhibitors of the uPA–uPAR Interaction
More than 20 years ago, Crowley et al. showed that competitive removal of uPA from its receptor by the action of a catalytically inactive analog (Ser 356 → Ala) inhibits prostate cancer metastasis in vivo (228). Since then many inhibitors have been developed to cause an interruption in the uPA–uPAR binding (229–231).
Another approach to inhibit the uPA–uPAR interaction functions by blocking the linker peptide of uPA. It has been shown that phosphorylation at Ser138 residue of uPA, which is located in the linker region, causes inhibition of its uPAR-dependent myelomonocytic adherence and motility (232). Our group was the first to demonstrate that an 8-mer (acetyl-KPSSPPEE-amino) non-competitive capped peptide antagonist of the uPA–uPAR interaction called Å6 can inhibit tumor growth and metastasis in vivo (22). In addition, we showed that Å6 enhances the anticancer effects of hormone therapy when used in combination with Tamoxifen in a rat syngeneic model of breast cancer (233). Boyd et al. showed that Å6 treatment prolonged survival of mice bearing prostate cancer cells and also reduced lymph node metastases in vivo (234). In a xenograft model of glioblastoma, the combination of Å6 with standard mode of therapy, i.e., cisplatin showed enhanced antitumor and antiangiogenesis effect than either cisplatin or Å6 alone (235). Piotrowicz et al. have showed that Å6 can also inhibit B16-F10 melanoma cell migration and metastasis in vivo (236). Taken together, the results from these studies accelerated the clinical evaluation of using Å6 for different types of cancer. Phase 1 and 2 clinical trials using Å6 have shown a promising efficacy as well as safety profile (237). However, it has shown a modest anticancer effect for the treatment of gynecologic cancers in Phase 2 clinical trials (238).
Targeting uPAR in Malignancy
In this age of targeted therapies and precision medicine, the use of antibodies directed against a specific metastatic or oncoprotein (for example, HER2 in breast cancer) has shown a great promise. We and others have also used a number of antibodies targeting uPAR to block its interaction with uPA or the integrins (41, 136, 215, 220). More than a decade ago, our group showed that a polyclonal antibody raised against rat uPAR can cause inhibition of breast cancer growth and metastasis in vivo (215). Later on, by using a monoclonal antibody (ATN-658) raised against human uPAR protein, our group showed the ability of ATN-658 to block prostate cancer cell proliferation, invasion, and metastasis (136). The main advantage of using ATN-658 is that it can bind to uPA-occupied uPAR unlike many other predecessor antibodies that targeted uPAR (41). Moreover, it has shown anticancer activity in a wide range of cancers (111, 136, 239). Taken together, the preclinical results showed great promise for the use of ATN-658 in clinical trials for further evaluation of its safety and efficacy in a human population.
Transcriptional Repression of the Components of the uPA System
Various techniques aiming to repress the expression of the uPA system have been evaluated that includes the use of antisense oligonucleotides or RNA interference (RNAi), ribozymes, etc. Antisense oligonucleotides targeted against uPAR have shown to cause a reduction in cell proliferation, invasion, and metastasis in different types of cancer (240–242). RNAi-mediated inhibition of both uPA and uPAR in human glioma cells increased apoptosis as well repressed the PI3K/AKT pathway (243). A specific ribozyme targeting uPAR mRNA has been used to disrupt uPAR translation in human osteosarcoma cells in vitro (244).
We have shown a DNA hypermethylation-mediated inhibition of uPA in different types of cancers both in vitro and in vivo (245, 246). Pakneshan et al. have shown that the uPA promoter is hypomethylated in patients with aggressive breast cancer (50). By using the universal methyl group donor S-adenosylmethionine (SAM), we could reverse the hypomethylated state of uPA and thereby cause a decrease in the expression of uPA (245). SAM treatment decreases tumor cell proliferation, invasion, and metastasis (247). It has also been shown that SAM causes transcriptional repression of a number of pro-metastatic genes apart from uPA. This is interesting because SAM is an approved dietary supplement that can function as a chemopreventive agent, and SAM-mediated repression of uPA would help the patients to take fewer amounts of the agents targeted against the uPA system and thus reduce the probability of any potential side effects. Apart from SAM, different types of growth factors, hormones, and cytokines have been shown to repress uPA and uPAR expression (248, 249).
Targeting PAI-1 Using RNA Aptamers and Peptide Inhibitors
RNA aptamers are oligonucleotides that are capable of folding into complex structures and then bind to various macromolecules with high affinity and selectivity (250). Two RNA aptamers WT-15 and SM-20 have been shown to disrupt the PAI-1 and vitronectin interaction without causing any effect on the ability of PAI-1 to inhibit uPA (251). It has been shown that the vitronectin and PAI-1 interaction promotes metastasis by causing the detachment of tumor cells from the ECM (252). Therefore, by repressing this interaction, the RNA aptamers WT-15 and SM-20 show an antimetastatic effect in cancer. Another way to inhibit PAI-1 is using peptides such as paionin-4-D1D2 that can stimulate PAI-1 conversion into its latent form (253).
Use of Toxins and Nanobins Combine with uPA Agents
Conjugation of cytotoxic drugs with agents directed against various components of the uPA system has been evaluated as a therapeutic strategy. For example, conjugation of DOTA and 213-Bi with the GFD of uPA has shown to cause a cytotoxic effect on ovarian cancer cells expressing uPAR (254). Several groups have targeted the ATF region of uPA by conjugating with a toxin. For example, conjugation of the ATF with the catalytic component of the diphtheria toxin (DTAT) reduced glioblastoma tumor growth (255, 256). Huang et al. demonstrated the cytotoxic effect of DTAT in a mouse model of human metastatic NSCLC to the brain (257). They have shown that treatment with DTAT significantly prolonged survival in the treated animals compared with the controls. In another study, a truncated form of Pseudomonas exotoxin (PE) conjugated with ATF showed cytotoxic effects in cell lines from different types of cancer (258).
In the recent years, some researchers have tried to combine the agents targeting the components of uPA system with the nanobin technology. Nanobins are liposomal nanoparticle drug encapsulation and formulation system by which more precise targeting of the tumor tissue can be done (259). The major advantages of the nanobins include increased circulation half-life, lesser off-target effects, and more specific delivery of the therapeutic payloads into the tumor cells (260). Nanobins can be easily conjugated with a target-specific antibody to potentiate a highly specific antibody-directed cellular cytotoxicity (260, 261). The uPA antibody ATN-291 has been conjugated with nanobins, which enhanced its internalization by the uPA or uPAR expressing tumor cells compared with the cells that do not express uPA or uPAR (58, 260). In addition, nanoparticles conjugated with ATF to target uPAR have been demonstrated by several groups (58). For example, Yang et al. conjugated iron oxide nanoparticles to ATF (ATF-IO) for delivery to breast cancer cells expressing uPAR and further suggested that ATF-IO nanoparticles can be potentially used as molecularly targeted, dual-modality imaging agents (262). In another study, Abdalla et al. have conjugated IO and noscapine to ATF and demonstrated that these conjugates have cytotoxic effects on prostate cancer cells (263). In a recent study, Carriero et al. used a Retro-Inverso (RI) approach to block the interaction between uPAR and G protein-coupled formyl peptide receptors and observed a significant reduction in tumor growth and decreased the number of circulating tumor cells and pulmonary metastases in immunocompromised mice injected with sarcoma cells (264). Further studies are warranted in this attractive area of uPAR-targeted therapeutic agents in different types of cancer.
Conclusion and Future Perspectives
Research over the past 30 years has demonstrated that the uPA–uPAR axis has a pleiotropic effect in different stages of cancer. Abnormalities in the levels of the components of the uPA–uPAR are frequently observed in a number of malignancies. Further mechanistic studies identified it as an oncogenic pathway for tumor growth proliferation and metastatic spread. Therefore, it has become a target for cancer prognosis, diagnosis, and therapeutic interventions. The preclinical data by us and others have demonstrated that targeting this axis shows association with tumor suppression and reduction of metastasis. In the next few years, the therapeutic and diagnostic potential of the uPA–uPAR system will continue to be evaluated for different types of cancer in clinical settings in combination with chemotherapeutic agents, targeted therapies and novels agents that alter tumor cell growth, signaling, and immune mechanism. Based on the strong preclinical data, these studies are expected to provide better diagnostic and therapeutic options for the patients to improve cancer-associated morbidity and mortality.
Statements
Author contributions
All authors listed have made a substantial intellectual contribution and approved it for publication.
Funding
This work was supported by a grant MOP 130410 from the Canadian Institutes for Health Research (CIHR) to SR. NM is the recipient of a studentship award from Research Institute, McGill University Health Centre.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Barkan D Green JE Chambers AF . Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer (2010) 46(7):1181–8.10.1016/j.ejca.2010.02.027
2
Mehlen P Puisieux A . Metastasis: a question of life or death. Nat Rev Cancer (2006) 6(6):449–58.10.1038/nrc1886
3
Weigelt B Peterse JL Van’t Veer LJ . Breast cancer metastasis: markers and models. Nat Rev Cancer (2005) 5(8):591.10.1038/nrc1670
4
Rabbani SA Mazar AP . Evaluating distant metastases in breast cancer: from biology to outcomes. Cancer Metastasis Rev (2007) 26(3–4):663–74.10.1007/s10555-007-9085-8
5
Pillay V Dass CR Choong PF . The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol (2007) 25(1):33–9.10.1016/j.tibtech.2006.10.011
6
Duffy MJ . The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des (2004) 10(1):39–49.10.2174/1381612043453559
7
Su S-C Lin C-W Yang W-E Fan W-L Yang S-F . The urokinase-type plasminogen activator (uPA) system as a biomarker and therapeutic target in human malignancies. Expert Opin Ther Targets (2016) 20(5):551–66.10.1517/14728222.2016.1113260
8
Duffy M Duggan C . The urokinase plasminogen activator system: a rich source of tumour markers for the individualised management of patients with cancer. Clin Biochem (2004) 37(7):541–8.10.1016/j.clinbiochem.2004.05.013
9
Duffy MJ . Urokinase plasminogen activator and its inhibitor, PAI-1, as prognostic markers in breast cancer: from pilot to level 1 evidence studies. Clin Chem(2002) 48(8):1194–7.
10
Shin SJ Kim KO Kim MK Lee KH Hyun MS Kim KJ et al Expression of E-cadherin and uPA and their association with the prognosis of pancreatic cancer. Jpn J Clin Oncol (2005) 35(6):342–8.10.1093/jjco/hyi154
11
Taubert H Würl P Greither T Kappler M Bache M Lautenschläger C et al Co-detection of members of the urokinase plasminogen activator system in tumour tissue and serum correlates with a poor prognosis for soft-tissue sarcoma patients. Br J Cancer (2010) 102(4):731.10.1038/sj.bjc.6605520
12
Pappot H Pedersen AN Brünner N Christensen IJ . The complex between urokinase (uPA) and its type-1 inhibitor (PAI-1) in pulmonary adenocarcinoma: relation to prognosis. Lung Cancer (2006) 51(2):193–200.10.1016/j.lungcan.2005.10.002
13
Sperl S Mueller MM Wilhelm OG Schmitt M Magdolen V Moroder L . The uPA/uPA receptor system as a target for tumor therapy. Drug News Perspect (2001) 14(7):401–11.10.1358/dnp.2001.14.7.858423
14
Muehlenweg B Sperl S Magdolen V Schmitt M Harbeck N . Interference with the urokinase plasminogen activator system: a promising therapy concept for solid tumours. Expert Opin Biol Ther (2001) 1(4):683–91.10.1517/14712598.1.4.683
15
McMahon BJ Kwaan HC . Components of the plasminogen-plasmin system as biologic markers for cancer. Adv Exp Med Biol (2015) 867:145–56.10.1007/978-94-017-7215-0_10
16
Stoppelli MP . The Plasminogen Activation System in Cell Invasion. Austin, TX: Landes Bioscience (2013).
17
O’Rourke J Jiang X Hao Z Cone RE Hand AR . Distribution of sympathetic tissue plasminogen activator (tPA) to a distant microvasculature. J Neurosci Res (2005) 79(6):727–33.10.1002/jnr.20366
18
Leurer C Rabbani SA . Chapter 6: Plasminogen activator system—diagnostic, prognostic and therapeutic implications in breast cancer. In: GunduzM, editor. A Concise Review of Molecular Pathology of Breast Cancer. Rijeka: InTech (2015).10.5772/59429
19
Mekkawy AH Pourgholami MH Morris DL . Involvement of urokinase-type plasminogen activator system in cancer: an overview. Med Res Rev (2014) 34(5):918–56.10.1002/med.21308
20
Mazar AP Ahn RW O’Halloran TV . Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr Pharm Des (2011) 17(19):1970–8.10.2174/138161211796718152
21
Cianfrocca M Kimmel K Gallo J Cardoso T Brown M Hudes G et al Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2), a beta integrin antagonist, in patients with solid tumours. Br J Cancer (2006) 94(11):1621.10.1038/sj.bjc.6603171
22
Guo Y Higazi AA-R Arakelian A Sachais BS Cines D Goldfarb RH et al A peptide derived from the nonreceptor binding region of urokinase plasminogen activator (uPA) inhibits tumor progression and angiogenesis and induces tumor cell death in vivo. FASEB J (2000) 14(10):1400–10.10.1096/fj.14.10.1400
23
Choong PF Nadesapillai AP . Urokinase plasminogen activator system: a multifunctional role in tumor progression and metastasis. Clin Orthop Relat Res (2003) 415:S46–58.10.1097/01.blo0000093845.72468.bd
24
MacFarlane R Pilling J . Fibrinolytic activity of normal urine. Nature (1947) 159(4049):779.10.1038/159779a0
25
Sobel G Mohler S Jones N Dowdy A Guest M , editors. Urokinase-an activator of plasma profibrinolysin extracted from urine. Am J Physiol (1952) 171(3):768–9.
26
Danø K Andreasen P Grøndahl-Hansen J Kristensen P Nielsen L Skriver L . Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res (1985) 44:139–266.10.1016/S0065-230X(08)60028-7
27
Wang Y Dang J Johnson LK Selhamer JJ Doe WF . Structure of the human urokinase receptor gene and its similarity to CD59 and the Ly-6 family. FEBS J (1995) 227(1–2):116–22.
28
Appella E Robinson E Ullrich S Stoppelli M Corti A Cassani G et al The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem (1987) 262(10):4437–40.
29
Kristensen P Larsson L-I Nielsen LS Grøndahl-Hansen J Andreasen PA Danø K . Human endothelial cells contain one type of plasminogen activator. FEBS Lett (1984) 168(1):33–7.10.1016/0014-5793(84)80201-X
30
Schmitt M Goretzki L Jänicke F Calvete J Eulitz M Kobayashi H et al Biological and clinical relevance of the urokinase-type plasminogen activator (uPA) in breast cancer. Biomed Biochim Acta (1990) 50(4–6):731–41.
31
Petersen L Lund L Nielsen L Danø K Skriver L . One-chain urokinase-type plasminogen activator from human sarcoma cells is a proenzyme with little or no intrinsic activity. J Biol Chem (1988) 263(23):11189–95.
32
Stack MS Johnson DA . Human mast cell tryptase activates single-chain urinary-type plasminogen activator (pro-urokinase). J Biol Chem (1994) 269(13):9416–9.
33
Van Veldhuizen PJ Sadasivan R Cherian R Wyatt A . Urokinase-type plasminogen activator expression in human prostate carcinomas. Am J Med Sci (1996) 312(1):8–11.10.1016/S0002-9629(15)41739-2
34
Goretzki L Schmitt M Mann K Calvete J Chucholowski N Kramer M et al Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett (1992) 297(1–2):112–8.10.1016/0014-5793(92)80339-I
35
Koutsilieris M Rabbani SA Bennett H Goltzman D . Characteristics of prostate-derived growth factors for cells of the osteoblast phenotype. J Clin Invest (1987) 80(4):941.10.1172/JCI113186
36
Koutsilieris M Rabbani S Goltzman D . Effects of human prostatic mitogens on rat bone cells and fibroblasts. J Endocrinol (1987) 115(3):447–N.10.1677/joe.0.1150447
37
Rabbani SA Desjardins J Bell AW Banville D Mazar A Henkin J et al An amino-terminal fragment of urokinase isolated from a prostate cancer cell line (PC-3) is mitogenic for osteoblast-like cells. Biochem Biophys Res Commun (1990) 173(3):1058–64.10.1016/S0006-291X(05)80893-9
38
Rabbani SA Mazar A Bernier S Haq M Bolivar I Henkin J et al Structural requirements for the growth factor activity of the amino-terminal domain of urokinase. J Biol Chem (1992) 267(20):14151–6.
39
Ellis V Behrendt N Danø K . Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem (1991) 266(19):12752–8.
40
Higazi AA-R Cohen RL Henkin J Kniss D Schwartz BS Cines DB . Enhancement of the enzymatic activity of single-chain urokinase plasminogen activator by soluble urokinase receptor. J Biol Chem (1995) 270(29):17375–80.10.1074/jbc.270.29.17375
41
Xu X Cai Y Wei Y Donate F Juarez J Parry G et al Identification of a new epitope in uPAR as a target for the cancer therapeutic monoclonal antibody ATN-658, a structural homolog of the uPAR binding integrin CD11b (αM). PLoS One (2014) 9(1):e85349.10.1371/journal.pone.0085349
42
Mars WM Zarnegar R Michalopoulos GK . Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol (1993) 143(3):949.
43
Irigoyen J Munoz-Canoves P Montero L Koziczak M Nagamine Y . The plasminogen activator system: biology and regulation. Cell Mol Life Sci (1999) 56(1):104–32.10.1007/PL00000615
44
Nagamine Y Medcalf RL Muñoz-Cánoves P . Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb Haemost (2005) 93(4):661–75.
45
Benasciutti E Pages G Kenzior O Folk W Blasi F Crippa MP . MAPK and JNK transduction pathways can phosphorylate Sp1 to activate the uPA minimal promoter element and endogenous gene transcription. Blood (2004) 104(1):256–62.10.1182/blood-2003-08-2661
46
D’Orazio D Besser D Marksitzer R Kunz C Hume DA Kiefer B et al Cooperation of two PEA3/AP1 sites in uPA gene induction by TPA and FGF-2. Gene (1997) 201(1):179–87.10.1016/S0378-1119(97)00445-9
47
Pakneshan P Xing RH Rabbani SA . Methylation status of uPA promoter as a molecular mechanism regulating prostate cancer invasion and growth in vitro and in vivo. FASEB J (2003) 17(9):1081–8.10.1096/fj.02-0973com
48
Guo Y Pakneshan P Gladu J Slack A Szyf M Rabbani SA . Regulation of DNA methylation in human breast cancer effect on the urokinase-type plasminogen activator gene production and tumor invasion. J Biol Chem (2002) 277(44):41571–9.10.1074/jbc.M201864200
49
Xing RH Rabbani SA . Transcriptional regulation of urokinase (uPA) gene expression in breast cancer cells: role of DNA methylation. Int J Cancer (1999) 81(3):443–50.10.1002/(SICI)1097-0215(19990505)81:3<443::AID-IJC19>3.0.CO;2-T
50
Pakneshan P Têtu B Rabbani SA . Demethylation of urokinase promoter as a prognostic marker in patients with breast carcinoma. Clin Cancer Res (2004) 10(9):3035–41.10.1158/1078-0432.CCR-03-0545
51
Fowler B Mackman N Parmer RJ Miles LA . Binding of human single chain urokinase to Chinese hamster ovary cells and cloning of hamster u-PAR. Thromb Haemost (1998) 80(1):148–54.
52
Stoppelli MP Corti A Soffientini A Cassani G Blasi F Assoian RK . Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc Natl Acad Sci U S A (1985) 82(15):4939–43.10.1073/pnas.82.15.4939
53
Vassalli J-D Baccino D Belin D . A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J Cell Biol (1985) 100(1):86–92.10.1083/jcb.100.1.86
54
Ploug M Rønne E Behrendt N Jensen AL Blasi F Danø K . Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem (1991) 266(3):1926–33.
55
Llinas P Le Du MH Gårdsvoll H Danø K Ploug M Gilquin B et al Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J (2005) 24(9):1655–63.10.1038/sj.emboj.7600635
56
Degryse B . The urokinase receptor and integrins constitute a cell migration signalosome. Cancer Degradome (2008):451–74.10.1007/978-0-387-69057-5_23
57
Montuori N Visconte V Rossi G Ragno P . Soluble and cleaved forms of the urokinase-receptor: degradation products or active molecules. Thromb Haemost (2005) 93(2):192–8.
58
O’Halloran TV Ahn R Hankins P Swindell E Mazar AP . The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics (2013) 3(7):496.10.7150/thno.4953
59
Kugler MC Wei Y Chapman HA . Urokinase receptor and integrin interactions. Curr Pharm Des (2003) 9(19):1565–74.10.2174/1381612033454658
60
Hu J Jo M Cavenee WK Furnari F VandenBerg SR Gonias SL . Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc Natl Acad Sci U S A (2011) 108(38):15984–9.10.1073/pnas.1113416108
61
Webb DJ Nguyen D Gonias SL . Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and matrigel invasion. J Cell Sci (2000) 113(1):123–34.
62
Rønne E Pappot H Grøndahl-Hansen J Høyer-Hansen G Plesner T Hansen NE et al The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol (1995) 89(3):576–81.10.1111/j.1365-2141.1995.tb08366.x
63
Lomholt A Høyer-Hansen G Nielsen H Christensen I . Intact and cleaved forms of the urokinase receptor enhance discrimination of cancer from non-malignant conditions in patients presenting with symptoms related to colorectal cancer. Br J Cancer (2009) 101(6):992.10.1038/sj.bjc.6605228
64
Resnati M Pallavicini I Wang J Oppenheim J Serhan C Romano M et al The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci U S A (2002) 99(3):1359–64.10.1073/pnas.022652999
65
Nykjaer A Christensen EI Vorum H Hager H Petersen CM Røigaard H et al Mannose 6-phosphate/insulin-like growth factor-II receptor targets the urokinase receptor to lysosomes via a novel binding interaction. J Cell Biol (1998) 141(3):815–28.10.1083/jcb.141.3.815
66
Mazar AP . Urokinase plasminogen activator receptor choreographs multiple ligand interactions: implications for tumor progression and therapy. Clin Cancer Res (2008) 14(18):5649–55.10.1158/1078-0432.CCR-07-4863
67
Gonias SL Hu J . Urokinase receptor and resistance to targeted anticancer agents. Front Pharmacol (2015) 6:1–6.10.3389/fphar.2015.00154
68
Chandran VI Eppenberger-Castori S Venkatesh T Vine KL Ranson M . HER2 and uPAR cooperativity contribute to metastatic phenotype of HER2-positive breast cancer. Oncoscience (2015) 2(3):207.10.18632/oncoscience.146
69
Pierga JY Bonneton C Magdelénat H Vincent-Salomon A Nos C Boudou E et al Real-time quantitative PCR determination of urokinase-type plasminogen activator receptor (uPAR) expression of isolated micrometastatic cells from bone marrow of breast cancer patients. Int J Cancer (2005) 114(2):291–8.10.1002/ijc.20698
70
Meng S Tripathy D Shete S Ashfaq R Saboorian H Haley B et al uPAR and HER-2 gene status in individual breast cancer cells from blood and tissues. Proc Natl Acad Sci U S A (2006) 103(46):17361–5.10.1073/pnas.0608113103
71
Andreasen PA Georg B Lund LR Riccio A Stacey SN . Plasminogen activator inhibitors: hormonally regulated serpins. Mol Cell Endocrinol (1990) 68(1):1–19.10.1016/0303-7207(90)90164-4
72
Thorsen S Philips M Selmer J Lecander I Åstedt B . Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. FEBS J (1988) 175(1):33–9.
73
Croucher DR Saunders DN Lobov S Ranson M . Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat Rev Cancer (2008) 8(7):535.10.1038/nrc2400
74
Harbeck N Schmitt M Meisner C Friedel C Untch M Schmidt M et al Ten-year analysis of the prospective multicentre Chemo-N0 trial validates American Society of Clinical Oncology (ASCO)-recommended biomarkers uPA and PAI-1 for therapy decision making in node-negative breast cancer patients. Eur J Cancer (2013) 49(8):1825–35.10.1016/j.ejca.2013.01.007
75
Halamkova J Kiss I Pavlovsky Z Jarkovsky J Tomasek J Tucek S et al Clinical relevance of uPA, uPAR, PAI 1 and PAI 2 tissue expression and plasma PAI 1 level in colorectal carcinoma patients. Hepatogastroenterology (2011) 58(112):1918–25.10.5754/hge10232
76
Lee C-C Huang T-S . Plasminogen activator inhibitor-1: the expression, biological functions, and effects on tumorigenesis and tumor cell adhesion and migration. J Cancer Mol (2005) 1(1):25–36.
77
Stefansson S Lawrence DA . The serpin PAI-1 inhibits cell migration by blocking integrin alphavbeta3 binding to vitronectin. Nature (1996) 383(6599):441.10.1038/383441a0
78
Plesner T Behrendt N Ploug M . Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem Cells (1997) 15(6):398–408.10.1002/stem.150398
79
Naldini L Tamagnone L Vigna E Sachs M Hartmann G Birchmeier W et al Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J (1992) 11(13):4825.
80
Hannocks M-J Oliver L Gabrilove JL Wilson EL . Regulation of proteolytic activity in human bone marrow stromal cells by basic fibroblast growth factor, interleukin-1, and transforming growth factor beta. Blood (1992) 79(5):1178–84.
81
Degryse B Resnati M Rabbani SA Villa A Fazioli F Blasi F . Src-dependence and pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. Blood (1999) 94(2):649–62.
82
Mukhina S Stepanova V Traktouev D Poliakov A Beabealashvilly R Gursky Y et al The chemotactic action of urokinase on smooth muscle cells is dependent on its kringle domain characterization of interactions and contribution to chemotaxis. J Biol Chem (2000) 275(22):16450–8.10.1074/jbc.M909080199
83
Resnati M Guttinger M Valcamonica S Sidenius N Blasi F Fazioli F . Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO J (1996) 15(7):1572.
84
Myöhänen H Stephens R Hedman K Tapiovaara H Rønne E Høyer-Hansen G et al Distribution and lateral mobility of the urokinase-receptor complex at the cell surface. J Histochem Cytochem (1993) 41(9):1291–301.10.1177/41.9.8394852
85
Limongi P Resnati M Hernandez-Marrero L Cremona O Blasi F Fazioli F . Biosynthesis and apical localization of the urokinase receptor in polarized MDCK epithelial cells. FEBS Lett (1995) 369(2–3):207–11.10.1016/0014-5793(95)00742-R
86
Estreicher A Mühlhauser J Carpentier J-L Orci L Vassalli J-D . The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J Cell Biol (1990) 111(2):783–92.10.1083/jcb.111.2.783
87
Blasi F . Urokinase and urokinase receptor: a paracrine/autocrine system regulating cell migration and invasiveness. Bioessays (1993) 15(2):105–11.10.1002/bies.950150206
88
Mondino A Blasi F . uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol (2004) 25(8):450–5.10.1016/j.it.2004.06.004
89
Sanderson-Smith ML Zhang Y Ly D Donahue D Hollands A Nizet V et al A key role for the urokinase plasminogen activator (uPA) in invasive Group A streptococcal infection. PLoS Pathog (2013) 9(7):e1003469.10.1371/journal.ppat.1003469
90
Abraham E Gyetko MR Kuhn K Arcaroli J Strassheim D Park JS et al Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol (2003) 170(11):5644–51.10.4049/jimmunol.170.11.5644
91
Cao D Mizukami IF Garni-Wagner BA Kindzelskii A Todd R Boxer LA et al Human urokinase-type plasminogen activator primes neutrophils for superoxide anion release. Possible roles of complement receptor type 3 and calcium. J Immunol (1995) 154(4):1817–29.
92
May AE Kanse SM Lund LR Gisler RH Imhof BA Preissner KT . Urokinase receptor (CD87) regulates leukocyte recruitment via β2 integrins in vivo. J Exp Med (1998) 188(6):1029–37.10.1084/jem.188.6.1029
93
Gyetko MR Sitrin RG Fuller J Todd RF Petty H Standiford TJ . Function of the urokinase receptor (CD87) in neutrophil chemotaxis. J Leukoc Biol (1995) 58(5):533–8.10.1002/jlb.58.5.533
94
Gyetko MR Chen GH McDonald RA Goodman R Huffnagle GB Wilkinson CC et al Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. A murine transgenic model. J Clin Invest (1996) 97(8):1818.10.1172/JCI118611
95
Rijneveld AW Levi M Florquin S Speelman P Carmeliet P van Der Poll T . Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. J Immunol (2002) 168(7):3507–11.10.4049/jimmunol.168.7.3507
96
Gyetko MR Sud S Kendall T Fuller JA Newstead MW Standiford TJ . Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J Immunol (2000) 165(3):1513–9.10.4049/jimmunol.165.3.1513
97
Nykjaer A Møller B Todd R Christensen T Andreasen PA Gliemann J et al Urokinase receptor. An activation antigen in human T lymphocytes. J Immunol (1994) 152(2):505–16.
98
Gyetko MR Sud S Chensue SW . Urokinase-deficient mice fail to generate a type 2 immune response following schistosomal antigen challenge. Infect Immun (2004) 72(1):461–7.10.1128/IAI.72.1.461-467.2004
99
Gyetko MR Sud S Chen G-H Fuller JA Chensue SW Toews GB . Urokinase-type plasminogen activator is required for the generation of a type 1 immune response to pulmonary Cryptococcus neoformans infection. J Immunol (2002) 168(2):801–9.10.4049/jimmunol.168.2.801
100
Gyetko MR Todd R III Wilkinson CC Sitrin RG . The urokinase receptor is required for human monocyte chemotaxis in vitro. J Clin Invest (1994) 93(4):1380.10.1172/JCI117114
101
Gyetko MR Sud S Sonstein J Polak T Sud A Curtis JL . Cutting edge: antigen-driven lymphocyte recruitment to the lung is diminished in the absence of urokinase-type plasminogen activator (uPA) receptor, but is independent of uPA. J Immunol (2001) 167(10):5539–42.10.4049/jimmunol.167.10.5539
102
Zheng P Zou R Liu Y . Source of plasminogen activator in rhesus monkey semen and its possible role in sperm capacitation. Sheng Li Xue Bao (2001) 53(1):45–50.
103
Qin Y Han Y Xiong C-L Li H-G Hu L Zhang L . Urokinase-type plasminogen activator: a new target for male contraception?Asian J Androl (2015) 17(2):269.10.4103/1008-682X.143316
104
Aguirre-Ghiso JA Liu D Mignatti A Kovalski K Ossowski L . Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell (2001) 12(4):863–79.10.1091/mbc.12.4.863
105
Ulisse S Baldini E Sorrenti S D’Armiento M . The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets (2009) 9(1):32–71.10.2174/156800909787314002
106
Shapiro RL Duquette JG Roses DF Nunes I Harris MN Kamino H et al Induction of primary cutaneous melanocytic neoplasms in urokinase-type plasminogen activator (uPA)-deficient and wild-type mice: cellular blue nevi invade but do not progress to malignant melanoma in uPA-deficient animals. Cancer Res (1996) 56(15):3597–604.
107
Bergers G Brekken R McMahon G Vu TH Itoh T Tamaki K et al Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2(10):737–44.10.1038/35036374
108
Chen Y Kelm RJ Budd RC Sobel BE Schneider DJ . Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem (2004) 92(1):178–88.10.1002/jcb.20058
109
Soeda S Oda M Ochiai T Shimeno H . Deficient release of plasminogen activator inhibitor-1 from astrocytes triggers apoptosis in neuronal cells. Brain Res Mol Brain Res (2001) 91(1):96–103.10.1016/S0169-328X(01)00133-4
110
Subramanian R Gondi CS Lakka SS Jutla A Rao JS . siRNA-mediated simultaneous downregulation of uPA and its receptor inhibits angiogenesis and invasiveness triggering apoptosis in breast cancer cells. Int J Oncol (2006) 28(4):831–9.10.3892/ijo.28.4.831
111
Kenny HA Leonhardt P Ladanyi A Yamada SD Montag A Im HK et al Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res (2011) 17(3):459–71.10.1158/1078-0432.CCR-10-2258
112
Chen S-C Henry DO Reczek PR Wong MK . Plasminogen activator inhibitor-1 inhibits prostate tumor growth through endothelial apoptosis. Mol Cancer Ther (2008) 7(5):1227–36.10.1158/1535-7163.MCT-08-0051
113
Balsara RD Ploplis VA . Plasminogen activator inhibitor-1: the double-edged sword in apoptosis. Thromb Haemost (2008) 100(6):1029–36.10.1160/TH08-07-0427
114
Aguirre-Ghiso JA . Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7(11):834–46.10.1038/nrc2256
115
Ghiso JAA Kovalski K Ossowski L . Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol (1999) 147(1):89–104.10.1083/jcb.147.1.89
116
Montuori N Pesapane A Rossi FW Giudice V De Paulis A Selleri C et al Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl Med UniSa (2016) 15:15–21.
117
Liu D Ghiso JAA Estrada Y Ossowski L . EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell (2002) 1(5):445–57.10.1016/S1535-6108(02)00072-7
118
Aguirre-Ghiso JA Estrada Y Liu D Ossowski L . ERKMAPK activity as a determinant of tumor growth and dormancy; regulation by p38SAPK. Cancer Res (2003) 63(7):1684–95.
119
Ghiso JA . Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene (2002) 21:2513–24.10.1038/sj.onc.1205342
120
Duffy M . The role of proteolytic enzymes in cancer invasion and metastasis. Clin Exp Metastasis (1992) 10(3):145–55.10.1007/BF00132746
121
Nyberg P Salo T Kalluri R . Tumor microenvironment and angiogenesis. Front Biosci (2008) 13(7):6537–53.10.2741/3173
122
Gerwins P Sköldenberg E Claesson-Welsh L . Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Crit Rev Oncol Hematol (2000) 34(3):185–94.10.1016/S1040-8428(00)00062-7
123
Preissner KT Reuning U , editors. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin Thromb Hemost (2011) 37(4):408–24.10.1055/s-0031-1276590
124
Unseld M Chilla A Pausz C Mawas R Breuss J Zielinski C et al PTEN expression in endothelial cells is down-regulated by uPAR to promote angiogenesis. Thromb Haemost (2015) 114(2):379–89.10.1160/TH15-01-0016
125
Raghu H Nalla AK Gondi CS Gujrati M Dinh DH Rao JS . uPA and uPAR shRNA inhibit angiogenesis via enhanced secretion of SVEGFR1 independent of GM-CSF but dependent on TIMP-1 in endothelial and glioblastoma cells. Mol Oncol (2012) 6(1):33–47.10.1016/j.molonc.2011.11.008
126
Gondi CS Lakka SS Dinh DH Olivero WC Gujrati M Rao JS . Intraperitoneal injection of a hairpin RNA–expressing plasmid targeting urokinase-type plasminogen activator (uPA) receptor and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin Cancer Res (2007) 13(14):4051–60.10.1158/1078-0432.CCR-06-3032
127
Raghu H Gondi CS Dinh DH Gujrati M Rao JS . Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch-1 receptor. Mol Cancer (2011) 10(1):130.10.1186/1476-4598-10-130
128
Stefansson S Petitclerc E Wong MK McMahon GA Brooks PC Lawrence DA . Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J Biol Chem (2001) 276(11):8135–41.10.1074/jbc.M007609200
129
Bajou K Noël A Gerard R Masson V Brunner N Holst-Hansen C et al Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med (1998) 4(8):923–8.10.1038/nm0898-923
130
Andreasen PA Kjøller L Christensen L Duffy MJ . The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer (1997) 72(1):1–22.10.1002/(SICI)1097-0215(19970703)72:1<1::AID-IJC1>3.0.CO;2-Z
131
Hsiao K-C Shih N-Y Fang H-L Huang T-S Kuo C-C Chu P-Y et al Surface α-enolase promotes extracellular matrix degradation and tumor metastasis and represents a new therapeutic target. PLoS One (2013) 8(7):e69354.10.1371/journal.pone.0069354
132
Obermajer N Doljak B Kos J . Cytokeratin 8 ectoplasmic domain binds urokinase-type plasminogen activator to breast tumor cells and modulates their adhesion, growth and invasiveness. Mol Cancer (2009) 8(1):88.10.1186/1476-4598-8-88
133
Kalluri R Weinberg RA . The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420.10.1172/JCI39104
134
Kalluri R Neilson EG . Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest (2003) 112(12):1776.10.1172/JCI200320530
135
Lester RD Jo M Montel V Takimoto S Gonias SL . uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J Cell Biol (2007) 178(3):425–36.10.1083/jcb.200701092
136
Rabbani SA Ateeq B Arakelian A Valentino ML Shaw DE Dauffenbach LM et al An anti-urokinase plasminogen activator receptor antibody (ATN-658) blocks prostate cancer invasion, migration, growth, and experimental skeletal metastasis in vitro and in vivo. Neoplasia (2010) 12(10):778–88.10.1593/neo.10296
137
Zhang Y Wang X-F . A niche role for cancer exosomes in metastasis. Nat Cell Biol (2015) 17(6):709–11.10.1038/ncb3181
138
Alderton GK . Metastasis: exosomes drive premetastatic niche formation. Nat Rev Cancer (2012) 12(7):447.10.1038/nrc3304
139
Min L Shen J Tu C Hornicek F Duan Z . The roles and implications of exosomes in sarcoma. Cancer Metastasis Rev (2016) 35(3):377–90.10.1007/s10555-016-9630-4
140
Endo-Munoz L Cai N Cumming A Macklin R de Long LM Topkas E et al Progression of osteosarcoma from a non-metastatic to a metastatic phenotype is causally associated with activation of an autocrine and paracrine uPA Axis. PLoS One (2015) 10(8):e0133592.10.1371/journal.pone.0133592
141
Harris DA Patel SH Gucek M Hendrix A Westbroek W Taraska JW . Exosomes released from breast cancer carcinomas stimulate cell movement. PLoS One (2015) 10(3):e0117495.10.1371/journal.pone.0117495
142
Dass K Ahmad A Azmi AS Sarkar SH Sarkar FH . Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev (2008) 34(2):122–36.10.1016/j.ctrv.2007.10.005
143
O’Grady P Lijnen H Duffy M . Multiple forms of plasminogen activator in human breast tumors. Cancer Res (1985) 45:6216–8.
144
Duffy MJ O’Siorain L O’Grady P Devaney D Fennelly JJ Lijnen HJ . Urokinase-plasminogen activator, a marker for aggressive breast carcinomas. Preliminary report. Cancer (1988) 62(3):531–3.
145
Foekens JA Peters HA Look MP Portengen H Schmitt M Kramer MD et al The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res (2000) 60(3):636–43.
146
Jänicke F Prechtl A Thomssen C Harbeck N Meisner C Untch M et al Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst (2001) 93(12):913–20.10.1093/jnci/93.12.913
147
Look MP van Putten W Duffy M . Pooled analysis of prognostic impact of tumor biological factors uPA and PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst (2002) 94:116–28.10.1093/jnci/94.2.116
148
Harris L Fritsche H Mennel R Norton L Ravdin P Taube S et al American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol (2007) 25(33):5287–312.10.1200/JCO.2007.14.2364
149
Jelisavac-Cosic S Sirotkovic-Skerlev M Kulic A Jakic-Razumovic J Kovac Z Vrbanec D . Prognostic significance of urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor (PAI-1) in patients with primary invasive ductal breast carcinoma-a 7.5-year follow-up study. Tumori (2011) 97(4):532–9.
150
Paik S Shak S Tang G Kim C Baker J Cronin M et al A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med (2004) 351(27):2817–26.10.1056/NEJMoa041588
151
Knauer M Mook S Rutgers EJ Bender RA Hauptmann M Van de Vijver MJ et al The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat (2010) 120(3):655–61.10.1007/s10549-010-0814-2
152
Nicolini A Ferrari P Duffy MJ . Prognostic and predictive biomarkers in breast cancer: past, present and future. Semin Cancer Biol (2017).10.1016/j.semcancer.2017.08.010
153
Foekens JA Buessecker F Peters HA Krainick U van Putten WL Look MP et al Plasminogen activator inhibitor-2: prognostic relevance in 1012 patients with primary breast cancer. Cancer Res (1995) 55(7):1423–7.
154
Shariat SF Roehrborn CG McConnell JD Park S Alam N Wheeler TM et al Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J Clin Oncol (2007) 25(4):349–55.10.1200/JCO.2006.05.6853
155
Kjellman A Akre O Gustafsson O Høyer-Hansen G Lilja H Norming U et al Soluble urokinase plasminogen activator receptor as a prognostic marker in men participating in prostate cancer screening. J Intern Med (2011) 269(3):299–305.10.1111/j.1365-2796.2010.02284.x
156
Kumano M Miyake H Muramaki M Furukawa J Takenaka A Fujisawa M , editors. Expression of urokinase-type plasminogen activator system in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy. Urol Oncol (2009) 27(2):180–6.10.1016/j.urolonc.2008.01.012
157
Sier CF Stephens R Bizik J Mariani A Bassan M Pedersen N et al The level of urokinase-type plasminogen activator receptor is increased in serum of ovarian cancer patients. Cancer Res (1998) 58(9):1843–9.
158
Sier CF Nicoletti I Santovito ML Frandsen T Aletti G Ferrari A et al Metabolism of tumour-derived urokinase receptor and receptor fragments in cancer patients and xenografted mice. Thromb Haemost (2004) 91(2):403–11.
159
Sier C Sidenius N Mariani A Aletti G Agape V Ferrari A et al Presence of urokinase-type plasminogen activator receptor in urine of cancer patients and its possible clinical relevance. Lab Invest (1999) 79(6):717–22.
160
Ljuca D Fatusić Z Iljazović E Ahmetović B . Monitoring of chemotherapy successfulness of platina/taxol chemotherapy protocol by using determination of serum urokinase plasminogen activator (uPA) and soluble urokinase plasminogen activator receptor (suPAR) in patients with ovarian carcinoma FIGO II and III stage. Bosn J Basic Med Sci (2007) 7(2):113–8.
161
Kuhn W Schmalfeldt B Reuning U Pache L Berger U Ulm K et al Prognostic significance of urokinase (uPA) and its inhibitor PAI-1 for survival in advanced ovarian carcinoma stage FIGO IIIc. Br J Cancer (1999) 79(11–12):1746.10.1038/sj.bjc.6690278
162
Hoffmann G Pollow K Weikel W Strittmatter H-J Bach J Schaffrath M et al Urokinase and plasminogen activator-inhibitor (PAI-1) status in primary ovarian carcinomas and ovarian metastases compared to benign ovarian tumors as a function of histopathological parameters. Clin Chem Lab Med (1999) 37(1):47–54.10.1515/CCLM.1999.007
163
Daneri-Navarro A Macias-Lopez G Oceguera-Villanueva A Del Toro-Arreola S Bravo-Cuellar A Perez-Montfort R et al Urokinase-type plasminogen activator and plasminogen activator inhibitors (PAI-1 and PAI-2) in extracts of invasive cervical carcinoma and precursor lesions. Eur J Cancer (1998) 34(4):566–9.10.1016/S0959-8049(97)10038-7
164
Kobayashi H Fujishiro S Terao T . Impact of urokinase-type plasminogen activator and its inhibitor type 1 on prognosis in cervical cancer of the uterus. Cancer Res (1994) 54(24):6539–48.
165
Koelbl H Kirchheimer JC Tatra G Christ G Binder BR . Increased plasma levels of urokinase-type plasminogen activator with endometrial and cervical cancer. Obstet Gynecol (1988) 72(2):252–6.
166
Memarzadeh S Kozak KR Chang L Natarajan S Shintaku P Reddy ST et al Urokinase plasminogen activator receptor: prognostic biomarker for endometrial cancer. Proc Natl Acad Sci U S A (2002) 99(16):10647–52.10.1073/pnas.152127499
167
Riisbro R Stephens R Brünner N Christensen I Nielsen H Heilmann L et al Soluble urokinase plasminogen activator receptor in preoperatively obtained plasma from patients with gynecological cancer or benign gynecological diseases. Gynecol Oncol (2001) 82(3):523–31.10.1006/gyno.2001.6324
168
Dariusz S Agnieszka M Elzbieta R Danuta O-N Maciej Z Piotr D et al A potency of plasminogen activation system in long-term prognosis of endometrial cancer: a pilot study. Eur J Obstet Gynecol Reprod Biol (2012) 163(2):193–9.10.1016/j.ejogrb.2012.03.031
169
Choong PF Ferno M Akerman M Willen H Langstrom E Gustafson P et al Urokinase plasminogen activator levels and prognosis in 69 soft tissue sarcomas. J Bone Joint Surg Br Vol (1997) 79(4S):411–2.
170
de Vries TJ Mooy CM van Balken MR Luyten GP Quax PH Verspaget HW et al Components of the plasminogen activation system in uveal melanoma—a clinico-pathological study. J Pathol (1995) 175(1):59–67.10.1002/path.1711750110
171
Stabuc B Markovic J Bartenjev I Vrhovec I Medved U Kocijancic B . Urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 and type 2 in stage I malignant melanoma. Oncol Rep (2003) 10(3):635–9.10.3892/or.10.3.635
172
Riisbro R Christensen IJ Nielsen HJ Brünner N Nilbert M Fernebro E . Preoperative plasma soluble urokinase plasminogen activator receptor as a prognostic marker in rectal cancer patients. An EORTC-Receptor and Biomarker Group collaboration. Int J Biol Markers (2005) 20(2):93–102.10.1177/172460080502000203
173
Herszényi L Farinati F Cardin R István G Molnár LD Hritz I et al Tumor marker utility and prognostic relevance of cathepsin B, cathepsin L, urokinase-type plasminogen activator, plasminogen activator inhibitor type-1, CEA and CA 19-9 in colorectal cancer. BMC Cancer (2008) 8(1):194.10.1186/1471-2407-8-194
174
Papadopoulou S Scorilas A Yotis J Arnogianaki N Plataniotis G Agnanti N et al Significance of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 (PAI-1) expression in human colorectal carcinomas. Tumor Biol (2002) 23(3):170–8.10.1159/000064033
175
Yang JL Seetoo DQ Wang Y Ranson M Berney CR Ham JM et al Urokinase-type plasminogen activator and its receptor in colorectal cancer: independent prognostic factors of metastasis and cancer-specific survival and potential therapeutic targets. Int J Cancer (2000) 89(5):431–9.10.1002/1097-0215(20000920)89:5<431::AID-IJC6>3.0.CO;2-V
176
Halamkova J Kiss I Pavlovsky Z Tomasek J Jarkovsky J Cech Z et al Clinical significance of the plasminogen activator system in relation to grade of tumor and treatment response in colorectal carcinoma patients. Neoplasma (2011) 58(5):377–85.10.4149/neo_2011_05_377
177
Sier CF Vloedgraven HJ Ganesh S Griffioen G Quax PH Verheijen JH et al Inactive urokinase and increased levels of its inhibitor type 1 in colorectal cancer liver metastasis. Gastroenterology (1994) 107(5):1449–56.10.1016/0016-5085(94)90549-5
178
Dubuisson L Monvoisin A Nielsen B Le Bail B Bioulac-Sage P Rosenbaum J . Expression and cellular localization of the urokinase-type plasminogen activator and its receptor in human hepatocellular carcinoma. J Pathol (2000) 190(2):190–5.10.1002/(SICI)1096-9896(200002)190:2<190::AID-PATH511>3.0.CO;2-H
179
Zheng Q Tang Z Wu Z Shi D Song H . Inhibitor of plasminogen activator 1 (PAI-1) in hepatocellular carcinoma. Zhonghua Wai Ke Za Zhi (1998) 36(8):474–6.
180
Chen Q Fei J Wu L Jiang Z Wu Y Zheng Y et al Detection of cathepsin B, cathepsin L, cystatin C, urokinase plasminogen activator and urokinase plasminogen activator receptor in the sera of lung cancer patients. Oncol Lett (2011) 2(4):693–9.10.3892/ol.2011.302
181
Langkilde A Hansen TW Ladelund S Linneberg A Andersen O Haugaard SB et al Increased plasma soluble uPAR level is a risk marker of respiratory cancer in initially cancer-free individuals. Cancer Epidemiol Biomarkers Prev (2011) 20:609–18.10.1158/1055-9965.EPI-10-1009
182
Almasi CE Drivsholm L Pappot H Høyer-Hansen G Christensen IJ . The liberated domain I of urokinase plasminogen activator receptor – a new tumour marker in small cell lung cancer. APMIS (2013) 121(3):189–96.10.1111/j.1600-0463.2012.02955.x
183
Salden M Splinter T Peters H Look M Timmermans M van Meerbeeck J et al The urokinase-type plasminogen activator system in resected non-small-cell lung cancer. Ann Oncol (2000) 11(3):327–32.10.1023/A:1008312801800
184
Witkiewicz AK McMillan EA Balaji U Baek G Lin W-C Mansour J et al Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun (2015) 6:6744.10.1038/ncomms7744
185
Harris N Vennin C Conway J Vine KL Pinese M Cowley M et al SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene (2017) 36(30):4288–98.10.1038/onc.2017.63
186
Chen Y Zheng B Robbins DH Lewin DN Mikhitarian K Graham A et al Accurate discrimination of pancreatic ductal adenocarcinoma and chronic pancreatitis using multimarker expression data and samples obtained by minimally invasive fine needle aspiration. Int J Cancer (2007) 120(7):1511–7.10.1002/ijc.22487
187
Sorio C Mafficini A Furlan F Barbi S Bonora A Brocco G et al Elevated urinary levels of urokinase-type plasminogen activator receptor (uPAR) in pancreatic ductal adenocarcinoma identify a clinically high-risk group. BMC Cancer (2011) 11(1):448.10.1186/1471-2407-11-448
188
Beyer B Heiss MM Simon EH Gruetzner KU Babic R Jauch KW et al Urokinase system expression in gastric carcinoma. Cancer (2006) 106(5):1026–35.10.1002/cncr.21682
189
Plebani M Herszènyi L Carraro P De Paoli M Roveroni G Cardin R et al Urokinase-type plasminogen activator receptor in gastric cancer: tissue expression and prognostic role. Clin Exp Metastasis (1997) 15(4):418–26.10.1023/A:1018454305889
190
Luebke T Baldus S Spieker D Grass G Bollschweiler E Schneider P et al Is the urokinase-type plasminogen activator system a reliable prognostic factor in gastric cancer? Int J Biol Markers (2006) 21(3):162–9.10.5301/JBM.2008.3029
191
Baker E Leaper D Hayter J Dickenson A . Plasminogen activator system in oral squamous cell carcinoma. Br J Oral Maxillofac Surg (2007) 45(8):623–7.10.1016/j.bjoms.2007.04.021
192
Hundsdorfer B Zeilhofer H-F Bock KP Dettmar P Schmitt M Kolk A et al Tumour-associated urokinase-type plasminogen activator (uPA) and its inhibitor PAI-1 in normal and neoplastic tissues of patients with squamous cell cancer of the oral cavity—clinical relevance and prognostic value. J Craniomaxillofac Surg (2005) 33(3):191–6.10.1016/j.jcms.2004.12.005
193
Shiomi H Eguchi Y Tani T Kodama M Hattori T . Cellular distribution and clinical value of urokinase-type plasminogen activator, its receptor, and plasminogen activator inhibitor-2 in esophageal squamous cell carcinoma. Am J Pathol (2000) 156(2):567–75.10.1016/S0002-9440(10)64761-X
194
Zheng Q Tang Z Wu Z . Urokinase-type plasminogen activator (uPA), uPA receptor (uPA-R) and inhibitors (PA I-1) expression in hepatocellular carcinoma in relation to cancer invasion/metastasis and prognosis. Zhonghua Zhong Liu Za Zhi (1998) 20(1):57–9.
195
Zhao E Han D Yu Z Fan E Li Y Zhou Z . Prognostic value of the urokinase-type plasminogen activator and its inhibitors in squamous cell carcinoma of human larynx. J Clin Otolaryngol (2002) 16(11):599–602.
196
Wang D Wang T . Expressions and clinical significance of urokinase-type activator (uPA) and uPA receptor (uPAR) in laryngeal squamous cell carcinoma. J Clin Otolaryngol (2005) 19(12):529–31.
197
Strojan P Budihna M Šmid L Vrhovec I Škrk J . Urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor type 1 (PAI-1) in tissue and serum of head and neck squamous cell carcinoma patients. Eur J Cancer (1998) 34(8):1193–7.10.1016/S0959-8049(98)00029-X
198
Schmidt M Hoppe F . Increased levels of urokinase receptor in plasma of head and neck squamous cell carcinoma patients. Acta Otolaryngol (1999) 119(8):949–53.10.1080/00016489950180342
199
Ohba K Miyata Y Kanda S Koga S Hayashi T Kanetake H . Expression of urokinase-type plasminogen activator, urokinase-type plasminogen activator receptor and plasminogen activator inhibitors in patients with renal cell carcinoma: correlation with tumor associated macrophage and prognosis. J Urol (2005) 174(2):461–5.10.1097/01.ju.0000165150.46006.92
200
Span PN Witjes JA Grebenchtchikov N Geurts-Moespot A Moonen PM Aalders TW et al Components of the plasminogen activator system and their complexes in renal cell and bladder cancer: comparison between normal and matched cancerous tissues. BJU Int (2008) 102(2):177–82.10.1111/j.1464-410X.2008.07568.x
201
Lanza F Castoldi G Castagnari B Todd R Moretti S Spisani S et al Expression and functional role of urokinase-type plasminogen activator receptor in normal and acute leukaemic cells. Br J Haematol (1998) 103:110–23.10.1046/j.1365-2141.1998.00932.x
202
Mustjoki S Alitalo R Stephens RW Vaheri A . Blast cell-surface and plasma soluble urokinase receptor in acute leukemia patients: relationship to classification and response to therapy. Thromb Haemost (1999) 81:705–10.
203
Rasch MG Lund IK Almasi CE Hoyer-Hansen G . Intact and cleaved uPAR forms: diagnostic and prognostic value in cancer. Front Biosci (2008) 13:6752–62.10.2741/3186
204
Li D Liu S Shan H Conti P Li Z . Urokinase plasminogen activator receptor (uPAR) targeted nuclear imaging and radionuclide therapy. Theranostics (2013) 3(7):507.10.7150/thno.5557
205
Yang D Severin GW Dougherty CA Lombardi R Chen D Van Dort ME et al Antibody-based PET of uPA/uPAR signaling with broad applicability for cancer imaging. Oncotarget (2016) 7(45):73912.10.18632/oncotarget.12528
206
Jänicke F Schmitt M Ulm K Gössner W Graeff H . Urokinase-type plasminogen activator antigen and early relapse in breast cancer. Lancet (1989) 334(8670):1049.10.1016/S0140-6736(89)91070-2
207
Jänicke F Schmitt M Graeff H , editors. Clinical relevance of the urokinase-type and tissue-type plasminogen activators and of their type 1 inhibitor in breast cancer. Semin Thromb Hemost (1991) 17(3):303–12.10.1055/s-2007-1002624
208
Mengele K Napieralski R Magdolen V Reuning U Gkazepis A Sweep F et al Characteristics of the level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn (2010) 10(7):947–62.10.1586/erm.10.73
209
Schmitt M Mengele K Gkazepis A Napieralski R Magdolen V Reuning U et al Assessment of urokinase-type plasminogen activator and its inhibitor PAI-1 in breast cancer tissue: historical aspects and future prospects. Breast Care (2008) 3(Suppl 2):3–10.10.1159/000151737
210
Spyratos F Bouchet C Tozlu S Labroquere M Vignaud S Becette V et al Prognostic value of uPA, PAI-1 and PAI-2 mRNA expression in primary breast cancer. Anticancer Res (2002) 22(5):2997–3003.
211
Lamy P-J Verjat T Servanton A-C Paye M Leissner P Mougin B . Urokinase-type plasminogen activator and plasminogen activator inhibitor type-1 mRNA assessment in breast cancer by means of NASBA: correlation with protein expression. Am J Clin Pathol (2007) 128(3):404–13.10.1309/K4JAF2NMD5EJU67Y
212
Al-Janabi O Taubert H Lohse-Fischer A Fröhner M Wach S Stöhr R et al Association of tissue mRNA and serum antigen levels of members of the urokinase-type plasminogen activator system with clinical and prognostic parameters in prostate cancer. Biomed Res Int (2014) 2014:972587.10.1155/2014/972587
213
Mahmood N Rabbani SA . DNA methylation and breast cancer: mechanistic and therapeutic applications. Trends Cancer Res (2017) 12:1–18.
214
Gao S Skeldal S Krogdahll A Sørensen JA Andreasen P . CpG methylation of the PAI-1 gene 5’ flanking region is inversely correlated with PAI-1 mRNA levels in human cell lines. Thromb Haemostas (2005) 94:651–60.
215
Rabbani SA Gladu J . Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res (2002) 62(8):2390–7.
216
Baart VM Boonstra MC Sier CF . uPAR directed-imaging of head-and-neck cancer. Oncotarget (2017) 8(13):20519.10.18632/oncotarget.16240
217
Persson M Skovgaard D Brandt-Larsen M Christensen C Madsen J Nielsen CH et al First-in-human uPAR PET: imaging of cancer aggressiveness. Theranostics (2015) 5(12):1303.10.7150/thno.12956
218
Boonstra MC van Driel PB van Willigen DM Stammes MA Prevoo HA Tummers QR et al uPAR-targeted multimodal tracer for pre-and intraoperative imaging in cancer surgery. Oncotarget (2015) 6(16):14260.10.18632/oncotarget.3680
219
Sidenius N Blasi F . The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev (2003) 22(2–3):205–22.10.1023/A:1023099415940
220
Lund IK Illemann M Thurison T Christensen IJ Høyer-Hansen G . uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr Drug Targets (2011) 12(12):1744–60.10.2174/138945011797635902
221
Ossowski L Russo-Payne H Wilson EL . Inhibition of urokinase-type plasminogen activator by antibodies: the effect on dissemination of a human tumor in the nude mouse. Cancer Res (1991) 51(1):274–81.
222
Towle MJ Lee A Maduakor EC Schwartz CE Bridges AJ Littlefield BA . Inhibition of urokinase by 4-substituted benzo [b] thiophene-2-carboxamidines: an important new class of selective synthetic urokinase inhibitor. Cancer Res (1993) 53(11):2553–9.
223
Rabbani S Harakidas P Davidson DJ Henkin J Mazar AP . Prevention of prostate-cancer metastasis in vivo by a novel synthetic inhibitor of urokinase-type plasminogen activator (uPA). Int J Cancer (1995) 63(6):840–5.10.1002/ijc.2910630615
224
Xing RH Mazar A Henkin J Rabbani SA . Prevention of breast cancer growth, invasion, and metastasis by antiestrogen tamoxifen alone or in combination with urokinase inhibitor B-428. Cancer Res (1997) 57(16):3585–93.
225
Meyer JE Brocks C Graefe H Mala C Thäns N Bürgle M et al The oral serine protease inhibitor WX-671–first experience in patients with advanced head and neck carcinoma. Breast Care (2008) 3(Suppl 2):20–4.10.1159/000151736
226
Goldstein LJ . Experience in phase I trials and an upcoming phase II study with uPA inhibitors in metastatic breast cancer. Breast Care (2008) 3(Suppl 2):25–8.10.1159/000151733
227
Schmitt M Harbeck N Brünner N Jänicke F Meisner C Mühlenweg B et al Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn (2011) 11(6):617–34.10.1586/erm.11.47
228
Crowley CW Cohen RL Lucas BK Liu G Shuman MA Levinson AD . Prevention of metastasis by inhibition of the urokinase receptor. Proc Natl Acad Sci U S A (1993) 90(11):5021–5.10.1073/pnas.90.11.5021
229
Goodson RJ Doyle MV Kaufman SE Rosenberg S . High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc Natl Acad Sci U S A (1994) 91(15):7129–33.10.1073/pnas.91.15.7129
230
Ploug M Østergaard S Gårdsvoll H Kovalski K Holst-Hansen C Holm A et al Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry (2001) 40(40):12157–68.10.1021/bi010662g
231
Bürgle M Koppitz M Riemer C Kessler H König B Weidle UH et al Inhibition of the interaction of urokinase-type plasminogen activator (uPA) with its receptor (uPAR) by synthetic peptides. Biol Chem (1997) 378(3–4):231–8.
232
Franco P Iaccarino C Chiaradonna F Brandazza A Iavarone C Mastronicola MR et al Phosphorylation of human pro-urokinase on Ser138/303 impairs its receptor-dependent ability to promote myelomonocytic adherence and motility. J Cell Biol (1997) 137(3):779–91.10.1083/jcb.137.3.779
233
Guo Y Mazar AP Lebrun J-J Rabbani SA . An antiangiogenic urokinase-derived peptide combined with tamoxifen decreases tumor growth and metastasis in a syngeneic model of breast cancer. Cancer Res (2002) 62(16):4678–84.
234
Boyd DD Kim S-J Wang H Jones TR Gallick GE . A urokinase-derived peptide (Å6) increases survival of mice bearing orthotopically grown prostate cancer and reduces lymph node metastasis. Am J Pathol (2003) 162(2):619–26.10.1016/S0002-9440(10)63855-2
235
Mishima K Mazar AP Gown A Skelly M Ji X-D Wang X-D et al A peptide derived from the non-receptor-binding region of urokinase plasminogen activator inhibits glioblastoma growth and angiogenesis in vivo in combination with cisplatin. Proc Natl Acad Sci U S A (2000) 97(15):8484–9.10.1073/pnas.150239497
236
Piotrowicz RS Damaj BB Hachicha M Incardona F Howell SB Finlayson M . A6 peptide activates CD44 adhesive activity, induces FAK and MEK phosphorylation, and inhibits the migration and metastasis of CD44-expressing cells. Mol Cancer Ther (2011) 10(11):2072–82.10.1158/1535-7163.MCT-11-0351
237
Finlayson M . Modulation of CD44 activity by A6-peptide. Front Immunol (2015) 6:135.10.3389/fimmu.2015.00135
238
Gold MA Brady WE Lankes HA Rose PG Kelley JL De Geest K et al A phase II study of a urokinase-derived peptide (A6) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol (2012) 125(3):635–9.10.1016/j.ygyno.2012.03.023
239
Van Buren G Gray MJ Dallas NA Xia L Lim SJ Fan F et al Targeting the urokinase plasminogen activator receptor with a monoclonal antibody impairs the growth of human colorectal cancer in the liver. Cancer (2009) 115(14):3360–8.10.1002/cncr.24371
240
D’Alessio S Margheri F Pucci M Del Rosso A Monia BP Bologna M et al Antisense oligodeoxynucleotides for urokinase-plasminogen activator receptor have anti-invasive and anti-proliferative effects in vitro and inhibit spontaneous metastases of human melanoma in mice. Int J Cancer (2004) 110(1):125–33.10.1002/ijc.20077
241
Mohan PM Chintala SK Mohanam S Gladson CL Kim ES Gokaslan ZL et al Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Res (1999) 59(14):3369–73.
242
Margheri F D’Alessio S Serrati S Pucci M Annunziato F Cosmi L et al Effects of blocking urokinase receptor signaling by antisense oligonucleotides in a mouse model of experimental prostate cancer bone metastases. Gene Ther (2005) 12(8):702.10.1038/sj.gt.3302456
243
Gondi CS Kandhukuri N Dinh DH Gujrati M Rao JS . Down-regulation of uPAR and uPA activates caspase-mediated apoptosis and inhibits the PI3K/AKT pathway. Int J Oncol (2007) 31(1):19–27.10.3892/ijo.31.1.19
244
Karikó K Megyeri K Xiao Q Barnathan ES . Lipofectin-aided cell delivery of ribozyme targeted to human urokinase receptor mRNA. FEBS Lett (1994) 352(1):41–4.10.1016/0014-5793(94)00914-7
245
Pakneshan P Szyf M Farias-Eisner R Rabbani SA . Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem (2004) 279(30):31735–44.10.1074/jbc.M401669200
246
Shukeir N Pakneshan P Chen G Szyf M Rabbani SA . Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res (2006) 66(18):9202–10.10.1158/0008-5472.CAN-06-1954
247
Mahmood N Cheishvili D Arakelian A Tanvir I Khan HA Pépin AS et al Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget (2018) 9(4):5169–83.10.18632/oncotarget.23704
248
Koutsilieris M Reyes-Moreno C Sourla A Dimitriadou V Choki I . Growth factors mediate glucocorticoid receptor function and dexamethasone-induced regression of osteoblastic lesions in hormone refractory prostate cancer. Anticancer Res (1997) 17(3A):1461–5.
249
Mustjoki S Tapiovaara H Siren V Vaheri A . Interferons and retinoids enhance and dexamethasone suppresses urokinase-mediated plasminogen activation in promyelocytic leukemia cells. Leukemia (1998) 12(2):164–74.10.1038/sj.leu.2400918
250
Sundaram P Kurniawan H Byrne ME Wower J . Therapeutic RNA aptamers in clinical trials. Eur J Pharm Sci (2013) 48(1):259–71.10.1016/j.ejps.2012.10.014
251
Blake CM Sullenger BA Lawrence DA Fortenberry YM . Antimetastatic potential of PAI-1 – specific RNA aptamers. Oligonucleotides (2009) 19(2):117–28.10.1089/oli.2008.0177
252
Dupont DM Madsen JB Kristensen T Bodker JS Blouse GE Wind T et al Biochemical properties of plasminogen activator inhibitor-1. Front Biosci (2009) 14:1337–61.10.2741/3312
253
Mathiasen L Dupont DM Christensen A Blouse GE Jensen JK Gils A et al A peptide accelerating the conversion of plasminogen activator inhibitor-1 to an inactive latent state. Mol Pharmacol (2008) 74(3):641–53.10.1124/mol.108.046417
254
Knör S Sato S Huber T Morgenstern A Bruchertseifer F Schmitt M et al Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in α-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging (2008) 35(1):53–64.10.1007/s00259-007-0582-3
255
Vallera DA Li C Jin N Panoskaltsis-Mortari A Hall WA . Targeting urokinase-type plasminogen activator receptor on human glioblastoma tumors with diphtheria toxin fusion protein DTAT. J Natl Cancer Inst (2002) 94(8):597–606.10.1093/jnci/94.8.597
256
Hall WA Vallera DA . Efficacy of antiangiogenic targeted toxins against glioblastoma multiforme. Neurosurg Focus (2006) 20(4):E23.10.3171/foc.2006.20.4.15
257
Huang J Li YM Massague J Sicheneder A Vallera DA Hall WA . Intracerebral infusion of the bispecific targeted toxin DTATEGF in a mouse xenograft model of a human metastatic non-small cell lung cancer. J Neurooncol (2012) 109(2):229–38.10.1007/s11060-012-0904-6
258
Rajagopal V Kreitman RJ . Recombinant toxins that bind to the urokinase receptor are cytotoxic without requiring binding to the α2-macroglobulin receptor. J Biol Chem (2000) 275(11):7566–73.10.1074/jbc.275.11.7566
259
Adair JH Parette MP Altınoglu EI Kester M . Nanoparticulate alternatives for drug delivery. ACS Nano (2010) 4(9):4967–70.10.1021/nn102324e
260
Zhang Y Kenny HA Swindell EP Mitra AK Hankins PL Ahn RW et al Urokinase plasminogen activator system-targeted delivery of nanobins as a novel ovarian cancer therapy. Mol Cancer Ther (2013) 12(12):2628–39.10.1158/1535-7163.MCT-13-0204
261
Kirpotin DB Noble CO Hayes ME Huang Z Kornaga T Zhou Y et al 7 building and characterizing antibody-targeted lipidic nanotherapeutics. Methods Enzymol (2012) 502:139.10.1016/B978-0-12-416039-2.00007-0
262
Yang L Peng X-H Wang YA Wang X Cao Z Ni C et al Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res (2009) 15(14):4722–32.10.1158/1078-0432.CCR-08-3289
263
Abdalla MO Karna P Sajja HK Mao H Yates C Turner T et al Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release (2011) 149(3):314–22.10.1016/j.jconrel.2010.10.030
264
Carriero MV Bifulco K Ingangi V Costantini S Botti G Ragone C et al Retro-inverso urokinase receptor antagonists for the treatment of metastatic sarcomas. Sci Rep (2017) 7:1312.10.1038/s41598-017-01425-9
Summary
Keywords
uPA, urokinase-type plasminogen activator receptor, plasminogen activator system, plasminogen activator inhibitor-1, PAI-2, ATN-658, metastasis, cancer imaging
Citation
Mahmood N, Mihalcioiu C and Rabbani SA (2018) Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 8:24. doi: 10.3389/fonc.2018.00024
Received
24 October 2017
Accepted
24 January 2018
Published
12 February 2018
Volume
8 - 2018
Edited by
Francois X. Claret, University of Texas MD Anderson Cancer Center, United States
Reviewed by
Kara Lea Vine, University of Wollongong, Australia; Harikumar K. B., Rajiv Gandhi Centre for Biotechnology, India
Updates
Copyright
© 2018 Mahmood, Mihalcioiu and Rabbani.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shafaat A. Rabbani, shafaat.rabbani@mcgill.ca
Specialty section: This article was submitted to Cancer Molecular Targets and Therapeutics, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.