 Younguk Sun
Younguk Sun Bo-Rui Chen
Bo-Rui Chen Aniruddha Deshpande
Aniruddha Deshpande- Tumor Initiation and Maintenance Program, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, United States
The importance of epigenetic dysregulation to acute myeloid leukemia (AML) pathophysiology has become increasingly apparent in recent years. Epigenetic regulators, including readers, writers, and erasers, are recurrently dysregulated by way of chromosomal translocations, somatic mutations, or genomic amplification in AML and many of these alterations are directly implicated in AML pathogenesis. Mutations in epigenetic regulators are often discovered in founder clones and persist after therapy, indicating that they may contribute to a premalignant state poised for the acquisition of cooperating mutations and frank malignancy. Apart from the proto-oncogenic impact of these mutations, the AML epigenome is also shaped by other epigenetic factors that are not mutated but co-opted by AML oncogenes, presenting with actionable vulnerabilities in this disease. Targeting the AML epigenome might also be important for eradicating AML leukemia stem cells, which can be critical for disease maintenance and resistance to therapy. In this review, we describe the importance of epigenetic regulators in AML. We also summarize evidence implicating specific epigenetic regulators in AML pathobiology and discuss emerging epigenome-based therapies for the treatment of AML in the clinic.
Introduction
Acute myeloid leukemia (AML) is a clonal malignancy resulting from the transformation of hematopoietic stem and progenitor cells. AML is marked by enhanced proliferation and impaired differentiation of immature myeloid progenitors. Over the past few decades, strategies for treating AML have remained largely unchanged, although survival outcomes have improved, especially in younger patients (1). Despite these improvements, approximately 60% of young patients with AML eventually succumb to disease even after treatment with intensive therapies (2). In patients over 60 years of age, a population that has an increased frequency of AML, survival outcomes are much more dismal; less than 5% of patients are alive 5 years after diagnosis (3). There are several reasons why AML cure rates have plateaued. First, therapeutic approaches that have shown success in younger patients are often extremely aggressive and are, therefore, not tolerated well by elderly patients with frailty and other comorbidities. Treatment-related toxicity also results from the fact that standard therapies do not discriminate between normal and leukemic cells, resulting in severe toxicities. Second, although patient selection based on morphologic and cytogenetic features is routinely used for guiding treatment strategies and risk stratification, current therapeutic approaches do not adequately address the inherent molecular heterogeneity of AML. Last, current treatments that target the leukemic bulk may spare leukemia stem cells (LSCs) that provide a reservoir of premalignant or malignant clones that can regenerate the tumor. This is of great significance for AML therapy. Most patients who go into remission after treatment will relapse within the first few years, which diminishes their rate of survival substantially. Therefore, safer and more effective therapies are urgently required for the majority of AML patients with severely limited effective treatment options. A better understanding of the molecular landscape of AML and the biology of LSCs may, therefore, aid the design of much more targeted therapies for AML. We will discuss advances in our understanding of these processes in more detail in the following section with a focus the contribution of epigenetic regulators to AML heterogeneity and for the emergence and sustenance of LSCs.
Epigenetic Regulators and the AML Mutational Landscape
Acute myeloid leukemia is highly heterogeneous in terms of its underlying genetics, pathobiology, and clinical manifestation. Even though the morphological and cytogenetic heterogeneity of AML has been recognized for several years, the marked molecular heterogeneity has only come to the fore recently. Emerging evidence from genome-scale studies propelled by advances in next-generation sequencing (NGS) has substantially broadened our knowledge of the spectrum and frequency of mutations in AML. Characterization of the genomic AML landscape has led to the identification of recurrent mutations in a number of previously uncharacterized genes in AML. The classes of genes mutated in AML include transcription factors, kinases, cell cycle regulators, spliceosomal genes, and epigenetic regulators. The observation that genes encoding epigenetic regulators are among the most commonly occurring mutated factors in AML, strongly points to a role of epigenome dysregulation in AML pathogenesis. These mutations in epigenetic regulators encompass a broad spectrum of epigenetic writer, eraser, and reader proteins which will be the focus of this review. The epigenome is dynamically regulated through chemical modification of DNA and RNA as well as the histone proteins around which DNA is packaged. Our genomes harbor a number of enzymes that deposit these chemical marks (writers), or remove them (erasers), dedicated to specific modifications of DNA or chromatin. Proteins with specialized domains that can selectively bind to specific DNA, RNA, or histone modifications (readers) also abound, indicating a well-orchestrated mechanism for relaying epigenetic marks to downstream effectors. The coordinated action of epigenetic reader, writer, and eraser proteins is important for regulation of various cellular processes, including transcription, DNA replication, cell cycle control, and the DNA damage response. Recurrent genomic alterations in epigenetic writer, reader, and eraser proteins, such as DNA methyltransferase 3A (DNMT3A), TET1/2, IDH1/IDH2, EZH2, mixed-lineage leukemia (MLL), NSD1/3, AF10, ENL, and other epigenetic regulators have been cataloged in AML, inspiring a wave of preclinical studies aimed at uncovering causal links between epigenome dysregulation and leukemogenesis (see Figure 1 and Table 1). These studies are yielding important actionable information that can be rationally applied to the development of epigenome-based therapies for AML patients.
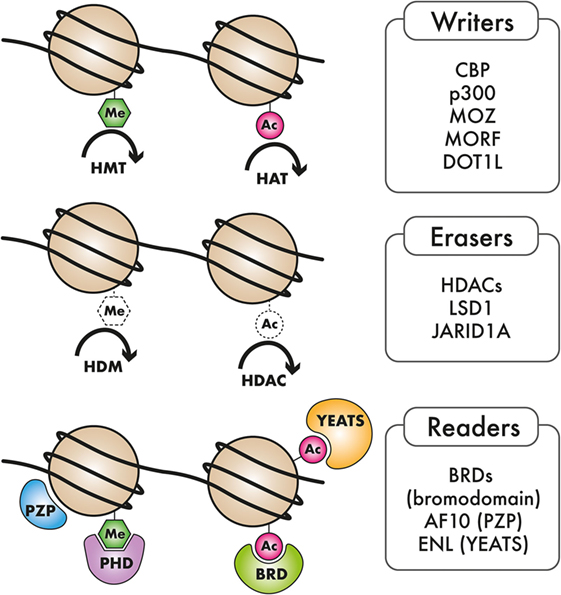
Figure 1. Types of epigenetic regulators mutated in AML: Epigenetic “writers,” such as DNA methyltransferases, histone methyltransferase (HMT), and histone acetyltransferases (HAT), deposit methylation and/or acetylation on DNA or on histones. These epigenetic marks may be removed by epigenetic “erasers,” including histone demethylase (HDM) and histone deacetylase complexes (HDACs). Epigenetic “readers” are highly specialized proteins that specifically bind to distinct epigenetic marks to convey this information to downstream effectors.
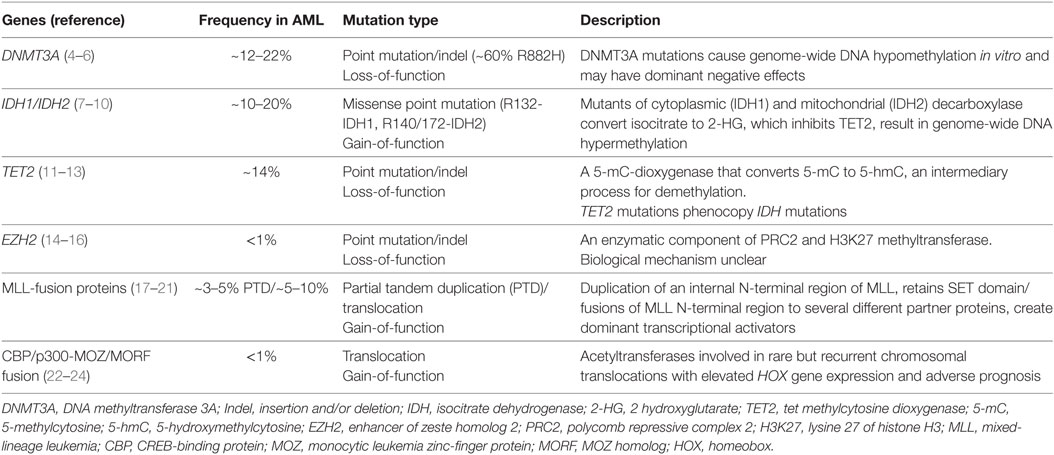
Table 1. Frequency and role of recurrently mutated epigenetic regulators in acute myeloid leukemia (AML).
LSCs and the Epigenome
The failure of “debulking” strategies in AML can now at least partly be attributed to AML–LSCs. Several lines of evidence demonstrate that AML emerges from a subset of cells with stem-cell-like properties [reviewed in Ref. (25, 26)]. It is now well documented that long-lived normal hematopoietic stem cells (HSCs) can accumulate mutations bearing the potential to trigger myeloid transformation in later life (27–30). These mutant HSC clones can eventually transform into LSCs, a population of cells with stem-cell properties that have the ability to sustain and propagate the tumor. Alternatively, certain AML-specific mutations in downstream hematopoietic progenitors can also initiate a transcriptional program reminiscent of HSCs, converting them to self-renewing LSCs (31, 32). It is now clear that stemness attributes in cancer are much more fluid than previously imagined, especially in constantly evolving neoplastic cells that display an enormous amount of genetic and epigenetic instability. Therefore, instead of the presence of a fixed, immutable population of cancer stem cells, there is evidence suggesting that cancer cells can switch between stem-like and non-stem-like states within the tumor, making the cancer stem cell a “moving target.” Such extraordinary plasticity of tumor cells requires rapid adaptations to changing micro-environmental cues as well as to the selective pressures mounted by aggressive therapeutic interventions typically used in cancer patients. This exceptional plasticity is likely to be provided by rapid and reversible epigenetic, rather than genetic changes in cancer cells. This is especially likely since epigenetic changes govern key steps in the transition of stem cells to their differentiated progeny in the process of normal hematopoiesis (33). Consistent with this notion, it is no surprise that almost all of the epigenetic regulators with recurrent AML-associated mutations have important roles in HSC self-renewal, survival, or differentiation. Importantly, studies have shown that mutations in epigenetic modifiers, including DNMT3A and IDH1/IDH2, occur in early pre-leukemic HSCs (29, 34, 35), while signaling pathway mutations in genes that confer proliferative advantage, such as NPM1 (nucleophosmin 1), FLT3-ITD (internal tandem duplication of the FLT3 gene), and KRAS/NRAS, are acquired later during the development of AML (36). Strikingly, there is increasing evidence that mutations that lead to clonal expansion of HSCs are acquired during normal aging, a process that is termed “clonal hematopoiesis.” Individuals with clonal hematopoiesis have an increased risk of progression to myeloid neoplasia and lower overall survival. Interestingly, a large proportion of the mutations observed in normal elderly individuals with clonal hematopoiesis are in epigenetic regulators (27–30). These striking observations indicate that mutations in epigenetic regulators may establish a leukemia-predisposing epigenetic state in premalignant HSC clones. These HSC clones may then be poised to transform into fully leukemic LSCs upon acquisition of secondary mutations with complementary oncogenic activities. Taken together, therapeutic targeting of the epigenome may turn out to be an attractive strategy for targeting AML–LSCs and may provide lasting curative benefit, especially in combination with traditional “debulking” strategies.
Epigenetic Regulators in AML Pathogenesis
Ever since chromosomal translocations and fusion oncogenes were discovered in AML several years ago, it was apparent that chromatin modulators such as the “writers” MLL1/KMT2A, CBP/p300, and NSD1/KMT3B might have causative roles in AML pathogenesis. MLL1, CBP, and NSD1/3 are involved in recurrent chromosomal translocations in a fraction of AML patients. These translocations were discovered early because they could be observed using methods, such as karyotyping and fluorescence in situ hybridization, that enabled identification of gross genetic aberrations in AML cells. However, these chromatin modifier mutations only accounted for a minor fraction of AML patients. There was little evidence for the direct genomic alteration of epigenetic regulators in the vast majority of AML. This scenario changed dramatically with the recent explosion in NGS, whereby mutations in several novel genes not previously implicated in AML pathogenesis were identified. Recent NGS-based discovery efforts in AML have demonstrated that epigenetic regulators comprise one of the most frequently mutated classes of genes in AML, accentuating the role of the epigenome in AML pathogenesis. Recurrent mutations in DNA methyltransferases (DNMTs), isocitrate dehydrogenases (IDH1/IDH2), methylcytosine dioxygenases of the ten-eleven-translocated (TET) family, and human homologs of the Drosophila polycomb complex such as Enhancer of Zeste 2 (EZH2) and additional sex-combs like genes (ASXL1/2) have been discovered in AML and myelodysplastic syndromes and myeloproliferative neoplasms (MDS/MPN), and many of these mutations have been causally linked to myeloid transformation in murine models. The role of these epigenetic modifiers in AML pathobiology and studies exploring these proteins as druggable targets will be described in detail below. Apart from genes mentioned above, there are a number of examples of epigenome modulators that are not directly mutated but nevertheless implicated in AML pathogenesis. Several chromatin modifiers have been discovered as selective dependencies in specific AML subtypes as discussed in the Section “DNMT Mutations.”
DNMT Mutations
DNA methylation is an important process in development that involves the addition of a methyl group to the carbon-5 position of cytosine in CpG dinucleotides, leading to the formation of 5-methylcytosine (5-mC). The DNMT family, including DNMT1, DNMT3A, and DNMT3B encode methyltransferases that catalyze this reaction. DNMT3A and DNMT3B are largely de novo DNMTs, whereas DNMT1 predominantly plays a role in the maintenance of DNA methylation (37). CpG clusters are enriched in regions upstream of genes (CpG islands) and increased methylation of CpG islands leads to transcriptional silencing of the downstream gene. Recurrent mutations in DNMT3A are observed in 12–22% of AML and always present as heterozygous mutations. DNMT3A mutations are associated with poor prognosis and decreased overall survival (4). A majority of these mutations lead to premature truncation of DNMT3A protein through nonsense or frame-shift mutations in the protein-coding region. Approximately 60% of DNMT3A-mutated AML patients harbor a missense mutation in the arginine 822 residue that diminishes its methyltransferase activity while reducing its binding affinity to DNA, which has been proposed to have a dominant negative function over the wild-type DNMT3A protein (5). DNMT3A mutations have been observed in non-leukemic T-cells from AML patients as well as in normal elderly individuals with no signs of leukemia, suggesting their provenance from an early, premalignant multipotent cell (27, 35). The mechanisms of leukemogenesis by DNMT3A are not entirely clear; however, studies have shown that heterozygous Dnmt3a ablation in mice leads to an expansion of the HSC pool (38), myeloid skewing and a predisposition to myeloid malignancies that may require additional genetic alterations. These studies reinforce the notion that the DNMT3A mutation, perhaps like mutations in other epigenetic regulators, do not lead to frank leukemic transformation on their own, but rather create a premalignant state that lays the ground for malignancy. Recently, it was also reported that mutant DNMT3A (R882H) interacts with the Polycomb repressive complex 1 (PRC1) to silence genes, suggesting that PRC1 activity could be an attractive target in DNMT3A-mutant tumors (39).
Isocitrate Dehydrogenase (IDH) Mutations
Isocitrate dehydrogenases are key components of the tricarboxylic acid cycle responsible for oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). The IDH1 and IDH2 proteins are nicotinamide adenine dinucleotide phosphate (NADP+)-dependent enzymes that mediate a number of important cellular processes including lipid metabolism, glucose sensing, and oxidative phosphorylation (7). IDH1 and IDH2 mutations are found at a frequency of 10–20%, and these mutations are more common in the cytogenetically normal sub-group of AML. IDH1 and IDH2 mutations are mutually exclusive and result in a gain of neomorphic activity (8). Specifically, gain-of-function IDH mutations convert the metabolite α-KG to the structurally similar I-2-hydroxyglutarate (2-HG). 2-HG acts as an “oncometabolite” since its accumulation in leukemic cells interferes with the enzymatic functions of several chromatin modifiers that use α-KG as a cofactor. Mechanistic investigations into the model of action of IDH mutations have shown that hematopoietic specific IDH1 (R132H) mutation using a conditional knock-in strategy expands HSC and myeloid progenitor compartments but fail to show signs of overt AML (40). Similar results were demonstrated by Heuser and colleagues using a retroviral bone marrow transplantation model which showed that mutant IDH overexpression was not sufficient to cause AML, but could do so in the presence of the Hoxa9 oncogene (9). These results suggest that similar to DNMT3A, IDH mutations may also need secondary mutations for initiation of frank malignancy in AML. Strikingly, the same group also demonstrated that in vivo injection of the oncometabolite 2-HG, could recapitulate most, but not all of the oncogenic effects of the IDH1 mutation (10). These interesting observations reinforced the role of 2-HG as an oncometabolite but also suggested that IDH1 may have additional oncogenic functions beyond its role in 2-HG accumulation. The exact role of chromatin modifying enzymes and epigenomic modifications in relaying the consequences of IDH mutation to oncogenic transcription remains to be determined.
TET Family Mutations
One of the most important classes of enzymes affected by IDH mutations is the TET family of methylcytosine dioxygenases. Normally, TET2, with the cofactor molecule α-ketoglutarate (α-KG), converts 5-mC to 5-hydroxymethylcytosine (5-hmC), which can then be demethylated back to cytosine via a series of intermediate steps (11) This TET-enzyme catalyzed CpG demethylation is an important step in the dynamic regulation of DNA methylation associated regulation of cellular processes. Inactivating mutations in TET enzymes lead to decreased hydroxylation of methyl-CpG sites (12, 41) resulting in aberrant CpG hypermethylation, decreased expression of key differentiating enzymes, and inhibition of normal cellular differentiation (42). Several studies have examined the function of TET2 inactivation in mice, Tet2 deletion leads to hematopoietic defects including enhanced HSC self-renewal and myeloid expansion, correlating with global loss of 5-hmC in primitive hematopoietic populations (43–45). It was recently described that restoration of TET function using an inducible shRNA model of TET-induced AML or through the administration of Vitamin C, which is a cofactor for α-KG dependent dioxygenases reverses leukemogenicity induced by the mutant TET protein (13). These exciting results imply that metabolic control of TET activity could be harnessed for therapeutic benefit in patients with TET mutations. Notably, cytosine methylation signatures of TET2-mutated AML show significant overlaps with those found in IDH1/IDH2 mutated patients and IDH1/IDH2 and TET2 mutations are mutually exclusive in AML (8), signaling a common mechanism of leukemogenesis based on aberrant DNA methylation. Recently another addition to this sub-group was made due to the discovery that mutations in the Wilms tumor gene WT1, which are found in approximately 10% of AML, are also mutually exclusive with TET and IDH mutations and display global cytosine hydroxymethylation profiles reminiscent of IDH and TET mutated AML. Levine and colleagues, who reported these observations, went on to demonstrate that WT1 physically interacts with TET proteins, TET2 and TET3, and compromises TET functions. It was concluded in this study that IDH1/IDH2, TET2, and WT1 mutations define a common AML subtype with overlapping disordered DNA 5-hmC profiles (46). Taken together, these results imply that dysregulated DNA methylation, achieved either through mutations in DNMT3A, IDH1/IDH2, TET2, or WT1, play an important role in the pathogenesis of a large proportion of AML patients. This information may help identify common targeted therapies for patients with mutations in these functionally related genes.
MLL/KMT2A Tandem Duplications
The MLL/KMT2A gene was one of the first epigenetic regulators known to be involved in leukemia pathogenesis. MLL is a chromatin writer, a SET-domain containing lysine methyltransferase belonging to the Drosophila Trithorax family of proteins. Approximately 3–5% of de novo AML present with in-frame partial tandem duplications of MLL exons 3–9 or 3–11 (17). This mutation is associated with a poor prognosis (18, 19). The MLL partial tandem duplication (MLL-PTD) duplicated the N-terminal AT-hook region of MLL, in addition to a domain that preferentially binds to unmethylated CpG sites and a transcriptional repression domain (20, 21). Mice carrying the MLL-PTD mutation show developmental abnormalities and dysregulated Hox gene expression similar to AML patients with the MLL-PTD mutation (47), but require additional leukemogenic driver mutations such as the Flt3-internal tandem duplication (48) for overt leukemogenesis. Intriguingly, a recent study from Koeffler and colleagues aimed at capturing the mutational landscape of MLL-PTD AML demonstrated that MLL-PTD mutations co-occur with several other mutations, including FLT3-ITD, DNMT3A, IDH1, TET2, cohesion genes, and splicing factors, but not NPM1 which is the most commonly mutated gene in AML (49). These studies suggest that MLL-PTD and NPM1 mutations may act through overlapping mechanisms. Furthermore, ordering of mutations in this study suggested that the MLL-PTD mutation was a secondary mutation that was undetected in remission in contrast to persistent mutations in epigenetic regulators, such as IDH2/DNMT3A and TET2.
MLL/KMT2A Translocations
In addition to tandem duplications of MLL that are observed in AML, the chromosomal band 11q23 is also involved in chromosomal translocations that fuse MLL to a partner gene on another chromosome. MLL fuses to several different partner genes; more than 80 different MLL-fusion partners have been discovered to date (50, 51). MLL-fusions are observed in 5–10% of adult AML and approximately 15–20% of AML in infants (50). In infant ALL, the frequency of MLL-rearrangements is as high as 70% (50), highlighting the role of these fusions in leukemogenesis. The binding of MLL-fusions to their target promoters is contingent upon the interaction of the N-terminal part of MLL with the LEDGF protein, an interaction that is bridged by the protein Menin (MEN1). The MLL–Menin interaction, therefore, is an attractive target for therapy and small-molecule compounds targeting this interaction have been developed (52–55). MLL-fusion protein expression activates a cascade of downstream transcriptional programs, one of the most important of which is the clustered homeobox (HOX) genes and their cofactor MEIS1. These HOX/MEIS genes are crucial for perpetuating the highly self-renewing state that is triggered by MLL-fusion protein expression in transformed hematopoietic progenitors. Indeed, several recent studies have shown that oncogenesis by MLL-fusion proteins requires the coordinate action of a number of chromatin factors that are essential and rate limiting for the transcriptional activation of HOX/MEIS genes. A prime example of this is the histone methyltransferase (HMT) DOT1L. The DOT1L protein biochemically interacts with several of the most common MLL-fusion partners, including AF4, AF9, ENL, AF10, and AF17 (56–59). All of these fusion partners retain the DOT1L interacting motif in their respective MLL-fusion events, and this interaction has been shown to be necessary and sufficient for oncogenic transcriptional activation functions by MLL-fusion proteins. Based on structure–function assays, genetic studies, and small-molecule inhibitor investigations, DOT1L has emerged as a clear therapeutic target in MLL-rearranged AML and clinical trials are currently ongoing (60) as described later in the review. Interestingly, DOT1L seems to be generally involved in HOX/MEIS regulation and other models of AML where HOX/MEIS activation is observed are sensitive to genetic and/or pharmacological DOT1L inhibition. These include AML driven by MLL-fusion proteins that do not recruit DOT1L, MLL-tandem duplications, nucleoporin 98 (NUP98)–NSD1 fusions, NPM1 mutations or mutations in the DNMT3A gene (59, 61–63). Strikingly, MLL–Menin inhibitors also seem to show broad activity against diverse HOX-activating AML oncogenes, suggesting that both these proteins are involved in an epigenetic network that is broadly essential for sustaining HOX gene expression (61). MLL-fusion transformed cells have also been shown to be sensitive to the depletion of several other chromatin factors, including PRC1 and polycomb repressive complex 2 (PRC2) complex proteins (64–69), the histone acetyltransferases (HATs) MOF, the arginine methyltransferase PRMT1 (70), and the MLL methyltransferase paralog MLL2 (71). Another interesting aspect of MLL-rearrangements is the involvement of chromatin readers. Many of the common fusion partners of MLL have chromatin-reading domains that recognize specific histone modifications and these reader–histone interactions and their transcriptional consequences are only recently being uncovered. AF9 and its paralog ENL harbor YEATS domains in their N-terminal region that bind to specific acetylated or crotonylated histone residues (72, 73). AF10 and AF17 on the other hand, have N-terminal PHD-zinc finger-PHD (PZP) domains that specifically recognize unmethylated H3K27 (74). Even though the chromatin reader modules of these MLL-fusion partners are excluded from MLL-fusion proteins themselves, chromatin reading by some of the wild-type, non-rearranged MLL-fusion partners, such as AF10 and ENL, have been shown to be important for MLL-leukemogenesis (73–75). Intriguingly, MLL-rearranged AML cells, which were dependent on AF10 or ENL for their proliferation, were found to be insensitive to the inactivation of their closely related paralogs AF17 or AF9, respectively. Even though this mystery of differential sensitivity is still unresolved, the fact that chromatin reading by specific PZP and YEATS domains are critical for MLL-leukemogenesis opens up the exciting possibility of targeting MLL-rearranged leukemias using selective small-molecule inhibitors of these chromatin-reading modules that are likely to be developed in the near future.
PRC Dysregulation in AML
Polycomb group (PcG) proteins are transcriptional repressors that regulate key fundamental processes, including cellular identity, differentiation, and stem cell plasticity (76). PcG proteins have highly conserved roles throughout evolution in the silencing of transcription through specific histone modifications. PcG proteins are constituents of two major multi-subunit complexes, PRC1 and 2, which have distinct effects on chromatin, gene expression, and developmental regulation. The PRC2 complex consists of four core constituents: The Drosophila enhancer of zeste homolog (EZH2), embryonic ectoderm development, suppressor of zeste homolog, and RbAp46/48, also known as RBBP4. PRC1 composition is more variable with only two core components RING1A and RING1B which complex together with the proteins BMI1, MEL18, or NSPC1 (76). The PRC2 complex is involved in histone 3 lysine 27 mono, di, and trimethylation, a function that shows high evolutionary conservation as a major facilitator of gene silencing. EZH2, the enzymatic component of PRC2 is mutated in myeloid malignancies, most commonly in MDS, chronic myelomonocytic leukemia (CMML), and primary myelofibrosis and rarely in AML (14–16). These mutations are missense or frame-shift mutations, which are predicted to lead to EZH2 loss of function. Interestingly, in diffuse large B-cell lymphoma (DLBCL), approximately 20% of patients bear activating EZH2 mutations (77), suggesting that PRC2 may have contrasting context-dependent roles in oncogenesis. Wild-type Ezh2 depletion in murine hematopoietic progenitors leads to myeloproliferative effects (78), whereas depletion of non-enzymatic PRC2 components such as Eed leads to severe lethal myelo- and lympho-proliferative disorders (79). These results indicate that further investigations are required to clarify the roles of EZH2 and PRC2 activity in leukemogenesis.
Of the PRC1 components, the BMI1 oncogene is implicated in the self-renewal of normal as well as leukemic stem cells in AML (80). Despite the apparent importance of BMI1 in normal and leukemic stem cells, mutations in this PRC1 component or any other members of the PRC1 complex have not been identified in AML.
Demethylase Mutations
Mutations in the histone 3 lysine 27 demethylase UTX are found in a variety of human cancers, including multiple myeloma, esophageal squamous cell carcinomas, and renal cell carcinoma (81). In myeloid malignancies, UTX mutations are found in 8% of patients with CMML and approximately 10% of patients with CMML-derived secondary AML. Most of these mutations were adjacent to the Jumonji C domain of UTX, which is required for the demethylase activity of UTX, suggesting that UTX loss of function may contribute to leukemogenesis. The JARID1A (KDM5A) H3K4 demethylase is fused to NUP98 in approximately 10% of pediatric acute megakaryoblastic leukemia resulting in the cytogenetically cryptic NUP98–JARID1A translocation. These fusions are believed to compromise normal functions both of NUP98 as well as JARID1A, leading to leukemogenesis (82). Exact consequences of demethylase mutations in these rare AML subtypes and their role in leukemogenesis remain to be discovered.
NSD Gene Fusions
Nuclear receptor-binding SET domain protein 1 is a HMT that is involved in recurrent chromosomal translocations with the NUP98 gene that are usually cryptic. NUP98–NSD1 fusions are found at a significantly increased frequency in pediatric as compared to adult patients (approximately 5 vs 1.4% of AML, respectively) (83, 84). In both adult and pediatric AML, NUP98–NSD1 translocations confer a poor prognosis and are enriched in the cytogenetically normal AML cohort. Mechanistically, NUP98–NSD1 fusions drive abnormal expression of HOX/MEIS oncogenes and this activation is dependent on the H3K36 methyltransferase activity of NSD1. NSD1-driven H3K36 methylation repels PRC2 complex proteins from the HOX/MEIS and other NUP98–NSD1 target genes, leading to sustained transcriptional activation and oncogenesis. NUP98-fusions with NSD3, a close homolog of NSD1 have also been reported in AML (85), further highlighting the role of this family of proteins in AML pathogenesis.
CBP/p300 and MOZ–MORF Fusions
The monocytic leukemia zinc-finger MOZ (MYST3) protein and its paralog MORF (MYST4) are HATs involved in recurring chromosomal rearrangements in AML. The balanced chromosomal translocation t(8;16)(p11;p13), which is found in <1% of AML patients, leads to in-frame fusions of MOZ with the HAT CBP (22). Another common partner of MOZ is TIF2, a member of the p160 family of nuclear receptor co-activators (86). MOZ-TIF2 expression in murine hematopoietic progenitor cells leads to aberrant Hoxa gene activation, increased self-renewal, and transformation in in vitro and in vivo assays. Notably, TIF2 interacts with CBP, indicating a common thread that links MOZ-fusions is the enlisting of CBP/p300 HAT activity. Consistent with this notion, MORF–CBP fusions, as well as fusions of either MOZ or MORF to the CBP homolog p300 have also been observed in AML, signifying common mechanisms linking these paralogous pairs of HATs to leukemogenesis. HOX gene activation is also observed in AML cells bearing MOZ–CBP fusions, similar to MLL and NUP98-fusion proteins (23). Even though patterns of HOX gene activation vary depending on which HOX-activating fusion protein is present in AML cells, HOX gene activation seems to be causally linked to transformation in all these AML subtypes based on preclinical studies.
Hijacking of Chromatin Modulators by AML Oncogenes
Apart from the epigenetic regulator mutations mentioned above, there are a number of examples of epigenome modulators that are not directly mutated but nevertheless implicated in AML pathogenesis. In recent years, several chromatin modifiers have been discovered as selective dependencies in specific AML subtypes as discussed briefly in the Section “The Advantage of Epigenetic Therapies.” Some of the most striking examples of epigenetic regulator hijacking for AML pathogenesis are observed in studies with oncogenic fusion proteins. Co-option of histone methyl and acetyltransferases, such as DOT1L by MLL-fusion proteins has been discussed in detail in the Section “CBP/p300 and MOZ–MORF Fusions.” In addition, a number of AML fusion proteins interfere with functioning of the PRC1 and PRC2 complexes. The promyelocytic leukemia–retinoic acid receptor (PML–RAR) fusion, which is seen in approximately 95% of the cases of acute promyelocytic leukemia (APL) (87, 88) can participate in biochemical interactions with several PRC2 complex proteins, recruiting repressive epigenetic modifications on target loci, while the other PML fusion oncoprotein PLZF–RARA binds to PRC1 complex members (89, 90). In separate studies, the PML–RARA fusion protein has also been shown to enlist the gene silencing activity of DNMT3A and HDAC3 complexes through biochemical interactions with the fusion protein (91–93). Similarly, the AML1/ETO fusion protein, a product of the recurrent t(8;21)(q22;q22) translocation, one of the most common cytogenetic abnormalities in AML, participates in biochemical interactions with chromatin modulatory proteins. AML1–ETO interacts with the protein arginine methyltransferase PRMT1. PRMT1 knockdown reduces the transcription of AML1–ETO target genes, implicating PRMT1 activity in AML1–ETO pathogenesis (94). AML1–ETO also acts as a transcriptional repressor and the repressive mechanisms of AML1–ETO have been shown to be facilitated by biochemical interactions with repressive complexes, such as N-CoR, mSin3A, SMRT, and HDAC1 (95–101).
The Advantage of Epigenetic Therapies
The last few years have seen a wave of unprecedented activity in the development of novel therapeutic agents and treatment strategies for AML. These include novel monoclonal antibody-based therapies, potent small-molecule inhibitors of signaling pathway mutations, tyrosine kinases, nuclear export, and immunotherapy. Most of these approaches are guided by specific mutations found recurrently in AML patients, which may herald a new era of precision medicine in AML. This strategy has been used with great success for more than a decade in the treatment of chronic myeloid leukemia and APL, but has largely failed in AML due to the absence of a single defining mutation event or hitherto intractable molecular targets. The recurrent prevalence of epigenetic regulator mutations in subsets of AML as well as broad epigenomic reprogramming across AML subtypes has ignited vigorous efforts to therapeutically target the AML epigenome. One of the biggest advantages of exploiting the epigenome as a therapeutic target is that, in contrast to the genomic alterations observed in AML cells that are difficult to reverse, epigenetic abnormalities can be reverted using pharmacological agents. Many epigenetic regulators such as DNA and histone modifying proteins have enzymatic activity, which is considered more amenable to therapeutic targeting using small-molecule inhibitors than other classes of proteins such as transcription factors. Another consideration is that since mutations in chromatin modulators are often observed in founding AML clones, targeting mutated epigenetic regulators may also eliminate LSCs, thereby striking at the root of AML and prevent relapse. For all of these reasons, the AML epigenome has emerged as one of the most exciting frontiers for drug discovery in recent years. Recent advances in preclinical and clinical development of epigenome-based therapies in AML will be discussed in the Section “Emerging Epigenome-Based Therapies in AML.”
Emerging Epigenome-Based Therapies in AML
Some of the early epigenome-based strategies have focused on broad-based epigenomic reprogramming aimed at restoring the altered epigenomic configurations in AML cells. This kind of broad epigenomic reprogrammig—for example, with the use of DNMT or histone deacetylase complex (HDAC) inhibitors—has been shown to reverse the commonly observed silencing of tumor suppressor genes (TSG) and restore normal differentiation. Since these epigenetic processes are involved in both silencing as well as activation of transcription dependent on the epigenetic mark and the chromatin context, it may be very difficult to identify which subset of AML may benefit most from broad-based epigenomic reprogramming therapies. More targeted therapies require the identification of specific silenced TSG or activated oncogenes for targeted therapeutics (Figure 2). Nevertheless, broad-based inhibition of DNA methylation and histone deacetylation using DNMT and HDAC inhibitors has been explored extensively as a therapeutic strategy in AML. The DNMT inhibitors azacitidine (AZA) and decitabine (DAC) are extensively used in MDS and also in patients with AML, where they show benefit, especially in elderly AML patients (102). Drugs, such as valproic acid (VPA), panobinostat and vorinostat, are some of the HDAC inhibitors approved for clinical use. More recently, after the identification of epigenetic regulator mutations, efforts have intensified to precisely target the oncogenic activity of those mutant proteins. This approach is particularly promising, as it may finally lead to precisely targeted therapies in patients with non-APL AML. Finally, as mentioned previously, there is compelling evidence that some AML-activated oncogenic transcriptional programs are specifically dependent on chromatin regulatory proteins, marking these chromatin regulators as attractive candidates for therapy. Prominent examples are the HMT DOT1L that regulates HOX gene expression and the bromodomain-containing protein BRD4, which regulates the expression of super-enhancer linked genes in AML and other cancers. These newly discovered dependencies present hitherto unexplored epigenetic vulnerabilities for therapeutic intervention (see Table 2). In the Section “Broad Epigenomic Reprogramming As a Therapeutic Strategy in AML,” pharmacological strategies that employ broad epigenetic reprogramming, specific targeting of mutated epigenetic regulators, or selective inhibition of cancer-specific epigenetic vulnerabilities will be discussed.
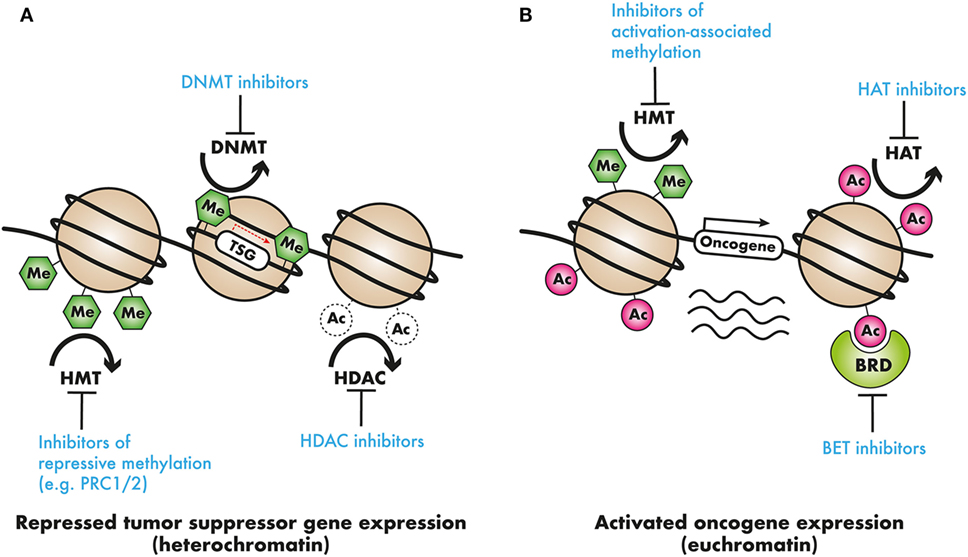
Figure 2. Epigenetic modifiers in cancer as clinical targets: (A) tumor suppressor genes (TSG) may be silenced by chromatin compaction resulting from DNA methylation or histone deacetylation, or repressive histone methylation. DNA methyltransferase (DNMT) inhibitors, demethylating agents, histone deacetylase (HDAC) inhibitors, or inhibitors of repressive histone modifying complexes such as PRC2 may restore the expression of these TSGs. (B) In contrast, oncogene activation by means of activation-associated histone hypermethylation, or histone hyperacetylation could be countered with the use of selective histone methyltransferase (HMT) or acetyltransferase histone acetyltransferase (HAT) inhibitors. Additionally, readers that recruit these activation-associated marks, such as the AF10 PZP domain, the AF9 or ENL YEATS domain, and the BRD4 bromodomains, and recruit transcriptional complexes present targets for pharmacological intervention.
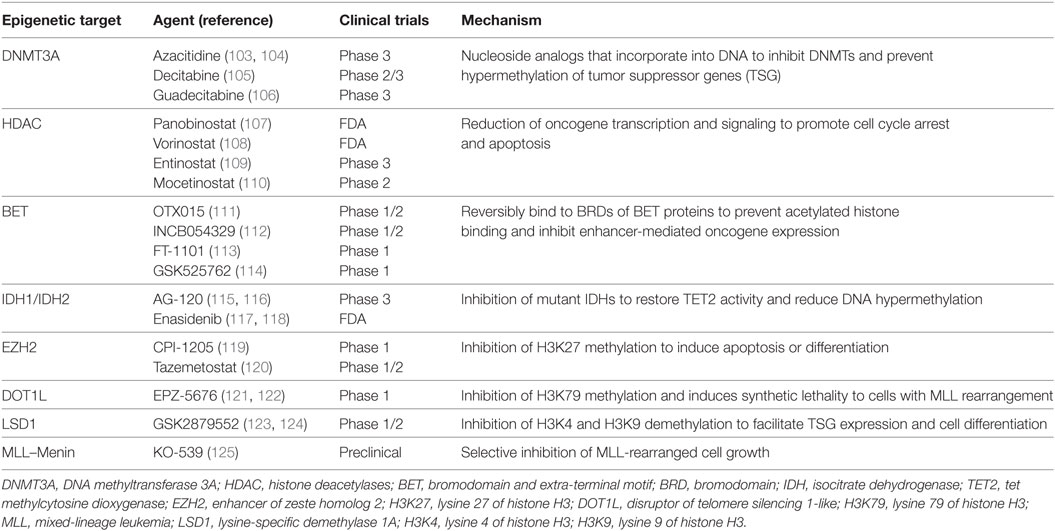
Table 2. Current status of select pharmacological agents targeting epigenetic regulators.
Broad Epigenomic Reprogramming As a Therapeutic Strategy in AML
DNMT Inhibitors
DNA methylation is dysregulated in most cancers including leukemia and has been the preferred target for cancer therapy since the development of hypomethylating agents (HMA). The HMAs AZA and DAC are nucleoside analogs and inhibitors of the DNMT enzymes DMNT1 and DMNT3. Investigational treatment with AZA and DAC in AML started more than 40 years ago [reviewed in Ref (102).]. AZA and DAC are now established as standard options for the treatment of older patients who do not tolerate standard intensive therapy. HMAs are thought to reactivate epigenetically silenced TSG through hypomethylation. Interestingly, HMAs seem to act indirectly through epigenetic reprogramming, rather than through direct cytotoxicity, as indicated by the delayed and prolonged responses (126, 127). Yet, a few caveats exist to the first-generation HMAs. Primary and secondary resistance to HMAs has been commonly reported (128, 129) and both AZA and DAC are degraded in plasma by the enzyme cytidine deaminase. This has promoted the development of second-generation HMAs with enhanced pharmacology and pharmacodynamic properties like guadecitabine, which has shown encouraging results in early clinical trials (130). Even though HMAs have provided much-needed options for older patients, their efficacy as single agent is limited. A number of studies have reported successful early findings from combination trials with HMAs with other agents used in AML such as tyrosine kinase inhibitors (102).
HDAC Inhibitors
Histone deacetylase complex inhibitors were initially identified in screens aimed at identifying factors that induce differentiation in leukemia cells (131). Histone acetylation is a major epigenetic mechanism that is carefully maintained by the interplay of HDACs and HATs (132). HDACs enzymatically remove the acetyl group from histones to serve as critical regulators of gene expression. Besides histones, many non-histone proteins that can be reversibly acetylated have been identified and are reported to be involved in a wide range of cellular processes, including gene expression, translation, DNA repair, metabolism, and cell structure (133). Many of these acetylated proteins are known to play roles in tumorigenesis, tumor progression, and metastasis (134). Along with HMAs, histone deacetylase inhibitors (HDACi) were the first epigenetically targeted inhibitors to be FDA approved for the treatment of cancer in the United States. HDAC inhibitors were historically identified based on their ability to induce tumor cell differentiation (135). Inhibition of class I HDACs targets expression of genes involved in cell cycle protein expression, cell cycle arrest in the G2/M phase, and apoptosis. HDAC inhibitors may help reactivate epigenetically silenced TSG including p21 and TP53. VPA was investigated in AML as the inhibitor of class I histone deacetylases. Unfortunately, the response rates of VPA for monotherapy in AML have been relatively low. Several other HDACi have been also tested as monotherapy in myeloid cancers, including romidepsin/depsipeptide (136, 137), entinostat (138), and mocetinostat (139). Likewise, these were found to be insufficient to further develop as a single agent in AML with the overall response rate ranging from 0 to 16%, with transient blast clearance and hematological improvement. Instead, when used in combination with agents with known antileukemia activity, including DNMTi (e.g., AZA, DAC) and chemotherapies, HDACi have shown a decreased time to response and an increase of overall response (107, 108, 140–150). Combination therapy based on the second-generation HDACi vorinostat or entinostat yielded an increased complete remission rate as compared to historical controls (151). The second-generation pan-HDACi panobinostat modulates gene expression by inducing hyperacetylation of core histone proteins, H3 and H4, and was shown to exhibit antitumor activity against several hematologic tumors, both in vitro and in vivo (107, 152). Even though potent and orally bioavailable, panobinostat yields modest result as a single agent in elderly patients with AML. Adding non-selective HDACi to combination schedules often results in increased toxicities which can lead to dose reduction and early treatment discontinuation (144, 153–157). Therefore, isozyme-selective HDACi with improved safety profiles may overcome this hurdle and provide additional clinical benefit to patients.
Bromodomain and Extra-Terminal Motif Protein (BET) Inhibitors
The bromodomain and extra-terminal (BET) protein family serve as transcriptional adapter molecules that facilitate transcription (158–160). They comprise bromodomain-containing protein (BRD) 2, BRD3, and BRD4, which are universally expressed, while BRDT expression is limited to the testes (160, 161). Diverse functions of BET proteins include histone modification to chromatin remodeling and ultimately lead to transcriptional activation (162) and are essential for cellular homeostasis (160, 163–166). The most well-characterized function of BET proteins is their binding to acetylated lysine restudies through tandem N-terminal bromodomains, These bromodomains are specialized epigenetic reader modules that are essential for high-level expression of oncogenes such as Myc by promoting enhancer activity (167, 168). Recently, they have also been implicated in transcriptional dysregulation in many cancer types, with BRD4 identified as a key player in AML (167, 169–172). BET inhibitors (BETi) reversibly bind the bromodomains of BET proteins. In a variety of human AML cell lines, suppression of BRD4 was shown to suppress MYC effectively suggesting a potential target for cancer treatment (111, 172). OTX015, a thienotriazolodiazepine, is a small-molecule oral inhibitor of BRD 2/3/4 demonstrated to induce apoptosis in a variety of leukemia cell lines and human AML samples (173). BETi have raised great interest as a novel treatment approach, and ongoing phase 1 trials are investigating their single-agent activities along with combination therapies with other novel agents.
Lysine-Specific Demethylase 1 (LSD1) Inhibitors
Lysine-specific demethylase 1 has emerged as a promising therapeutic target in multiple cancers, notably in AML (174–179). Its main role is demethylation of H3K4me1/2 and H3K9me1/2 and LSD1 has been shown to dynamically affect a wide range of transcriptional programs in a context-specific manner, acting either as a transcriptional repressor or as an activator (180–183). Pharmacologic inhibition or genetic knockdown of LSD1 in human leukemia cells induces differentiation (123). GSK2879552, an oral LSD1 inhibitor, is currently being investigated as a monotherapy in a phase 1 study for patients with relapsed/refractory AML (NCT02177812). In leukemia cell lines, there appears to be synergism between HDAC and LSD1 inhibitors which supports a clinical trial for further exploration (124). To date, the only HDACi to be evaluated preclinically in combination with an LSD1 inhibitor (SP2509) in AML is the pan-HDACi panobinostat. Treatment with SP2509 and panobinostat resulted in synergistic in vitro cytotoxic effects and significantly improved the survival of mice engrafted with AML cells without overt toxicity (178).
EZH2 Inhibitors
As mentioned previously, the exact role of EZH2 in AML is not entirely clear. Studies using an MLL-AF9 leukemia model have shown that PRC2 activity is required for MLL-rearranged AML. Inactivation of Eed, the critical component of PRC2 prolonged survival and reduced tumor burden in leukemic mice (22). These results were recapitulated with the use of UNC1999, a small-molecule inhibitor of both EZH1 and 2 which upregulated PRC2 target genes such as p16 and p19 in MLL-rearranged leukemia cells and strongly suppressed transformation (64, 184). A number of potent and selective EZH2 and PRC2 inhibitors are being tested in clinical trials in other malignancies where PRC2 activity has demonstrated proto-oncogenic roles such as DLBCL and synovial sarcoma, and it remains to be studied which subsets of AML may benefit from PRC2 antagonist therapies.
Targeting of Mutated Epigenetic Regulators
IDH Inhibitors
Given the high prevalence of IDH mutations in AML as well as in low-grade glioma, intensive efforts are on to develop clinical-grade IDH inhibitors. AGI-6780, a potent and selective allosteric inhibitor of the IDH2-R140Q mutations was recently reported to significantly induce differentiation in primary AML cells bearing IDH2-R140Q in ex vivo cultures. More recently another potent small-molecule inhibitor AG-221 (enasidenib) was developed that was shown to confer significant survival benefits in a mouse model of IDH mutant leukemia and also in a xenografts model of primary human AML (185). These exciting studies catapulted IDH inhibitors into clinical trials with very encouraging results, leading to the FDA approval of AG-221 for the treatment of patients with relapsed or refractory AML with IDH mutations. Considering that the IDH mutations were only first discovered less than 10 years ago (186), the fact that IDH inhibitors have already been approved for use is an astonishing success story for precision medicine in AML, although the long-term benefits of IDH inhibitors for AML patients remain to be seen.
Targeting Epigenetic Dependencies
DOT1L Inhibitors
An S-adenosyl-methionine competitive inhibitor of DOT1L (EPZ-4777) was developed by Epizyme Inc. as a potent and selective inhibitor of the methyltransferase activity of DOT1L (187). Using this compound as a tool, several studies preclinical studies were performed to show that MLL-rearranged AML was highly sensitive to pharmacological DOT1L inhibition (59, 187–190). Subsequently, using structure-guided design and optimization of a series of aminonucleoside compounds, the small-molecule EPZ-5676 was developed as a more potent DOT1L with better pharmacological properties than EPZ-4777 (92). Preliminary studies demonstrated potent single-agent antitumor effects of EPZ-5676 in preclinical models of MLL-rearranged AML, and synergistic effects with other standard chemotherapeutic drugs (63, 191). EPZ-5676 is being evaluated in clinical trials for adult and pediatric patients with relapsed or refractory AML with MLL-rearrangements (122, 187). EPZ-5676 was well-tolerated in initial studies and showed efficacy in a few patients, but several other patients showed moderate to no response, possibly due to pharmacokinetic limitations of the drug. Continued investigation of EPZ-5676 in patients with MLL gene rearrangements is warranted and results from the Phase I/II trials are awaited. Next-generation DOT1L inhibitors with improved pharmacological properties are being developed and are likely to show more pronounced efficacy in the clinic.
MLL–Menin Inhibitors
The MLL–Menin interaction is retained in all MLL-fusion proteins (192–195). Preclinical studies have demonstrated a critical role for Menin in leukemic transformations mediated by numerous MLL-fusion proteins. Genetic disruption of the MLL–Menin fusion protein interaction abrogates oncogenic properties of MLL-fusion proteins and blocks the development of acute leukemia in vivo (195). Recently, small-molecule inhibitors of the MLL–Menin interaction MI-463 and MI-503 were developed, and they were used to demonstrate that pharmacologic inhibition of the MLL–Menin interaction blocks progression of MLL leukemia in vivo without impairing normal hematopoiesis (54). These studies have prompted the development of more potent clinical-grade MLL–Menin inhibitors.
Concluding Remarks
These are still early days for targeted epigenetic therapies, but the prospects are very exciting. There are several challenges ahead that warrant consideration before epigenetic therapies become the mainstay of AML treatment strategies. First, a lot more needs to be done in terms of preclinical and basic research in order to define exact consequences of epigenetic regulator mutations that have been discovered in AML. This will require the development of faithful genetically engineered mouse models that recapitulate AML mutations, combined with detailed studies on normal and leukemic hematopoiesis. Characterization of the impact of these mutations on normal physiological processes in general and hematopoiesis, in particular will be helpful in predicting potential toxicities. Second, barring few exceptions, it is not entirely clear which subsets of AML may benefit from a particular epigenome-based therapy. Matching patients appropriately to epigenetic therapies will require detailed characterization and sensitivity studies including in vitro and in vivo inhibitor or genetic screens or epigenomic studies aimed at identifying specific “epigenetic lesions” and their respective drivers. Finally, there is an urgent need for the development of more potent and more selective small-molecules targeting epigenetic regulators. This is a rapidly developing field and selectively small-molecule inhibitors of class-specific HDACs, HATs, as well as DNA and HMT are being developed by several academic investigators and pharmaceutical companies. The next decade will see unprecedented activity in preclinical and clinical investigation of epigenome-based therapies.
Author Contributions
AD, YS, and BC conceived and drafted the manuscript. YS prepared the illustrations.
Conflict of Interest Statement
AD is a consultant for A2A Pharmaceuticals (New Jersey) and Salgomed Inc. (San Diego). Y-CS and B-RC declare no conflicts of interest.
Acknowledgments
The authors would like to apologize to their colleagues whose work could not be cited due to space limitations. The authors would like to acknowledge members of the Deshpande laboratory, particularly Dr. Anagha Deshpande for critical reading of the review and insightful comments and Laura Nelson for administrative support. AD is an American Society of Hematology and V-Foundation Scholar and is also supported by the following grants: NIH R00 CA154880, NIH/NCI P30 CA030199 Cancer Center Sponsored Grant, and the San Diego NCI Cancer Centers (C3) #PTC2017.
References
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373(12):1136–52. doi:10.1056/NEJMra1406184
2. Alibhai SM, Leach M, Minden MD, Brandwein J. Outcomes and quality of care in acute myeloid leukemia over 40 years. Cancer (2009) 115(13):2903–11. doi:10.1002/cncr.24373
3. Menzin J, Lang K, Earle CC, Kerney D, Mallick R. The outcomes and costs of acute myeloid leukemia among the elderly. Arch Intern Med (2002) 162(14):1597–603. doi:10.1001/archinte.162.14.1597
4. Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood (2012) 119(24):5824–31. doi:10.1182/blood-2011-07-367961
5. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell (2014) 25(4):442–54. doi:10.1016/j.ccr.2014.02.010
6. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet (2011) 43(4):309–15. doi:10.1038/ng.788
7. Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A (2011) 108(8):3270–5. doi:10.1073/pnas.1019393108
8. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18(6):553–67. doi:10.1016/j.ccr.2010.11.015
9. Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood (2013) 122(16):2877–87. doi:10.1182/blood-2013-03-491571
10. Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Goparaju R, Schwarzer A, et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia (2016) 30(8):1708–15. doi:10.1038/leu.2016.71
11. Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol (2013) 14(6):341–56. doi:10.1038/nrm3589
12. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333(6047):1303–7. doi:10.1126/science.1210944
13. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell (2017) 170(6):1079–95.e20. doi:10.1016/j.cell.2017.07.032
14. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet (2010) 42(8):722–6. doi:10.1038/ng.621
15. Ernst T, Pflug A, Rinke J, Ernst J, Bierbach U, Beck JF, et al. A somatic EZH2 mutation in childhood acute myeloid leukemia. Leukemia (2012) 26(7):1701–3. doi:10.1038/leu.2012.16
16. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet (2010) 42(8):665–7. doi:10.1038/ng.620
17. Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, et al. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res (1998) 58(1):55–9.
18. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med (2012) 366(12):1079–89. doi:10.1056/NEJMoa1112304
19. Dohner K, Tobis K, Ulrich R, Frohling S, Benner A, Schlenk RF, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol (2002) 20(15):3254–61. doi:10.1200/JCO.2002.09.088
20. Schichman SA, Caligiuri MA, Gu Y, Strout MP, Canaani E, Bloomfield CD, et al. ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci U S A (1994) 91(13):6236–9. doi:10.1073/pnas.91.13.6236
21. Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res (2002) 30(4):958–65. doi:10.1093/nar/30.4.958
22. Rozman M, Camos M, Colomer D, Villamor N, Esteve J, Costa D, et al. Type I MOZ/CBP (MYST3/CREBBP) is the most common chimeric transcript in acute myeloid leukemia with t(8;16)(p11;p13) translocation. Genes Chromosomes Cancer (2004) 40(2):140–5. doi:10.1002/gcc.20022
23. Camos M, Esteve J, Jares P, Colomer D, Rozman M, Villamor N, et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res (2006) 66(14):6947–54. doi:10.1158/0008-5472.CAN-05-4601
24. Schmidt HH. MYST3/CREBBP (MOZ/CBP) and CREBBP/MYST3 (CBP/MOZ) transcripts in AML with t(8;16)(p11;p13). Genes Chromosomes Cancer (2005) 42(2):207–8, author reply 9. doi:10.1002/gcc.20112
25. Reinisch A, Chan SM, Thomas D, Majeti R. Biology and clinical relevance of acute myeloid leukemia stem cells. Semin Hematol (2015) 52(3):150–64. doi:10.1053/j.seminhematol.2015.03.008
26. Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood (2017) 129(12):1627–35. doi:10.1182/blood-2016-10-696039
27. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi:10.1056/NEJMoa1408617
28. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126(1):9–16. doi:10.1182/blood-2015-03-631747
29. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20(12):1472–8. doi:10.1038/nm.3733
30. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature (2017) 547(7661):104–8. doi:10.1038/nature22993
31. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature (2006) 442(7104):818–22. doi:10.1038/nature04980
32. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell (2004) 6(6):587–96. doi:10.1016/j.ccr.2004.10.015
33. Langstein J, Milsom MD, Lipka DB. Impact of DNA methylation programming on normal and pre-leukemic hematopoiesis. Semin Cancer Biol (2017). doi:10.1016/j.semcancer.2017.09.008
34. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell (2012) 150(2):264–78. doi:10.1016/j.cell.2012.06.023
35. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature (2014) 506(7488):328–33. doi:10.1038/nature13038
36. Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A (2014) 111(7):2548–53. doi:10.1073/pnas.1324297111
37. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet (2013) 14(3):204–20. doi:10.1038/nrg3354
38. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet (2011) 44(1):23–31. doi:10.1038/ng.1009
39. Koya J, Kataoka K, Sato T, Bando M, Kato Y, Tsuruta-Kishino T, et al. DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nat Commun (2016) 7:10924. doi:10.1038/ncomms10924
40. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature (2012) 488(7413):656–9. doi:10.1038/nature11323
41. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (2011) 333(6047):1300–3. doi:10.1126/science.1210597
42. Fathi AT, Wander SA, Faramand R, Emadi A. Biochemical, epigenetic, and metabolic approaches to target IDH mutations in acute myeloid leukemia. Semin Hematol (2015) 52(3):165–71. doi:10.1053/j.seminhematol.2015.03.002
43. Guillamot M, Cimmino L, Aifantis I. The impact of DNA methylation in hematopoietic malignancies. Trends Cancer (2016) 2(2):70–83. doi:10.1016/j.trecan.2015.12.006
44. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell (2011) 20(1):11–24. doi:10.1016/j.ccr.2011.06.001
45. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20(1):25–38. doi:10.1016/j.ccr.2011.06.003
46. Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep (2014) 9(5):1841–55. doi:10.1016/j.celrep.2014.11.004
47. Dorrance AM, Liu S, Chong A, Pulley B, Nemer D, Guimond M, et al. The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood (2008) 112(6):2508–11. doi:10.1182/blood-2008-01-134338
48. Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood (2012) 120(5):1130–6. doi:10.1182/blood-2012-03-415067
49. Sun QY, Ding LW, Tan KT, Chien W, Mayakonda A, Lin DC, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia (2017) 31(1):1–10. doi:10.1038/leu.2016.160
50. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer (2007) 7(11):823–33. doi:10.1038/nrc2253
51. Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol (2012) 7:283–301. doi:10.1146/annurev-pathol-011811-132434
52. He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem (2014) 57(4):1543–56. doi:10.1021/jm401868d
53. Manka J, Daniels RN, Dawson E, Daniels JS, Southall N, Jadhav A, et al. Inhibitors of the menin-mixed lineage leukemia (MLL) interaction. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD (2010).
54. Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell (2015) 27(4):589–602. doi:10.1016/j.ccell.2015.02.016
55. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol (2012) 8(3):277–84. doi:10.1038/nchembio.773
56. Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell (2005) 121(2):167–78. doi:10.1016/j.cell.2005.02.020
57. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell (2010) 17(2):198–212. doi:10.1016/j.ccr.2009.12.040
58. Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev (2010) 24(6):574–89. doi:10.1101/gad.1898410
59. Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell (2014) 26(6):896–908. doi:10.1016/j.ccell.2014.10.009
60. Stein EM, Tallman MS. Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L. Curr Opin Hematol (2015) 22(2):92–6. doi:10.1097/MOH.0000000000000123
61. Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov (2016) 6(10):1166–81. doi:10.1158/2159-8290.CD-16-0237
62. Kuhn MW, Hadler MJ, Daigle SR, Koche RP, Krivtsov AV, Olhava EJ, et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica (2015) 100(5):e190–3. doi:10.3324/haematol.2014.115337
63. Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood (2016) 128(7):971–81. doi:10.1182/blood-2015-11-684225
64. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A (2012) 109(13):5028–33. doi:10.1073/pnas.1202258109
65. Danis E, Yamauchi T, Echanique K, Haladyna J, Kalkur R, Riedel S, et al. Inactivation of Eed impedes MLL-AF9-mediated leukemogenesis through Cdkn2a-dependent and Cdkn2a-independent mechanisms in a murine model. Exp Hematol (2015) 43(11):930–5.e6. doi:10.1016/j.exphem.2015.06.005
66. Fujita S, Honma D, Adachi N, Araki K, Takamatsu E, Katsumoto T, et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia (2017). doi:10.1038/leu.2017.300
67. Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood (2012) 120(5):1107–17. doi:10.1182/blood-2011-11-394932
68. Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia (2011) 25(8):1335–43. doi:10.1038/leu.2011.85
69. Smith LL, Yeung J, Zeisig BB, Popov N, Huijbers I, Barnes J, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell (2011) 8(6):649–62. doi:10.1016/j.stem.2011.05.004
70. Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol (2007) 9(10):1208–15. doi:10.1038/ncb1642
71. Chen Y, Anastassiadis K, Kranz A, Stewart AF, Arndt K, Waskow C, et al. MLL2, Not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell (2017) 31(6):755–70.e6. doi:10.1016/j.ccell.2017.05.002
72. Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell (2014) 159(3):558–71. doi:10.1016/j.cell.2014.09.049
73. Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature (2017) 543(7644):265–9. doi:10.1038/nature21687
74. Chen S, Yang Z, Wilkinson AW, Deshpande AJ, Sidoli S, Krajewski K, et al. The PZP domain of AF10 senses unmodified H3K27 to regulate DOT1L-mediated methylation of H3K79. Mol Cell (2015) 60(2):319–27. doi:10.1016/j.molcel.2015.08.019
75. Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature (2017) 543(7644):270–4. doi:10.1038/nature21688
76. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature (2011) 469(7330):343–9. doi:10.1038/nature09784
77. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi:10.1038/ng.518
78. Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol (2003) 4(2):124–31. doi:10.1038/ni876
79. Lessard J, Schumacher A, Thorsteinsdottir U, van Lohuizen M, Magnuson T, Sauvageau G. Functional antagonism of the Polycomb-Group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev (1999) 13(20):2691–703. doi:10.1101/gad.13.20.2691
80. Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature (2003) 423(6937):255–60. doi:10.1038/nature01572
81. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet (2009) 41(5):521–3. doi:10.1038/ng.349
82. de Rooij JD, Hollink IH, Arentsen-Peters ST, van Galen JF, Berna Beverloo H, Baruchel A, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia (2013) 27(12):2280–8. doi:10.1038/leu.2013.87
83. Thol F, Kolking B, Hollink IH, Damm F, van den Heuvel-Eibrink MM, Michel Zwaan C, et al. Analysis of NUP98/NSD1 translocations in adult AML and MDS patients. Leukemia (2013) 27(3):750–4. doi:10.1038/leu.2012.249
84. Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood (2011) 118(13):3645–56. doi:10.1182/blood-2011-04-346643
85. Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. NUP98-NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet (2009) 190(2):108–12. doi:10.1016/j.cancergencyto.2008.12.008
86. Carapeti M, Aguiar RC, Watmore AE, Goldman JM, Cross NC. Consistent fusion of MOZ and TIF2 in AML with inv(8)(p11q13). Cancer Genet Cytogenet (1999) 113(1):70–2. doi:10.1016/S0165-4608(99)00007-2
87. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood (1999) 93(10):3167–215.
88. Martens JHA, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, et al. PML-RAR alpha/RXR alters the epigenetic landscape in acute PROMYELOCYTIC leukemia. Cancer Cell (2010) 17(2):173–85. doi:10.1016/j.ccr.2009.12.042
89. Villa R, Pasini D, Gutierrez A, Morey L, Occhionorelli M, Vire E, et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell (2007) 11(6):513–25. doi:10.1016/j.ccr.2007.04.009
90. Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, et al. The PRC1 polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev (2009) 23(10):1195–206. doi:10.1101/gad.512009
91. Villa R, Morey L, Raker VA, Buschbeck M, Gutierrez A, De Santis F, et al. The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc Natl Acad Sci U S A (2006) 103(5):1400–5. doi:10.1073/pnas.0509343103
92. Subramanyam D, Belair CD, Barry-Holson KQ, Lin H, Kogan SC, Passegue E, et al. PML-RAR{alpha} and Dnmt3a1 cooperate in vivo to promote acute promyelocytic leukemia. Cancer Res (2010) 70(21):8792–801. doi:10.1158/0008-5472.CAN-08-4481
93. Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science (2002) 295(5557):1079–82. doi:10.1126/science.1065173
94. Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood (2012) 119(21):4953–62. doi:10.1182/blood-2011-04-347476
95. Hug BA, Lee SY, Kinsler EL, Zhang J, Lazar MA. Cooperative function of Aml1-ETO corepressor recruitment domains in the expansion of primary bone marrow cells. Cancer Res (2002) 62(10):2906–12.
96. Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A (1998) 95(18):10860–5. doi:10.1073/pnas.95.18.10860
97. Nimer SD, Moore MAS. Effects of the leukemia-associated AML1-ETO protein on hematopoietic stem and progenitor cells. Oncogene (2004) 23(24):4249–54. doi:10.1038/sj.onc.1207673
98. Wildonger J, Mann RS. The t(8;21) translocation converts AML1 into a constitutive transcriptional repressor. Development (2005) 132(10):2263–72. doi:10.1242/dev.01824
99. Minucci S, Nervi C, Lo Coco F, Pelicci PG. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene (2001) 20(24):3110–5. doi:10.1038/sj.onc.1204336
100. Lin RJ, Nagy L, Inoue S, Shao WL, Miller WH, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature (1998) 391(6669):811–4. doi:10.1038/35895
101. Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature (1998) 391(6669):815–8. doi:10.1038/35901
102. Gardin C, Dombret H. Hypomethylating agents as a therapy for AML. Curr Hematol Malig Rep (2017) 12(1):1–10. doi:10.1007/s11899-017-0363-4
103. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell (1980) 20(1):85–93. doi:10.1016/0092-8674(80)90237-8
104. Qin T, Youssef EM, Jelinek J, Chen R, Yang AS, Garcia-Manero G, et al. Effect of cytarabine and decitabine in combination in human leukemic cell lines. Clin Cancer Res (2007) 13(14):4225–32. doi:10.1158/1078-0432.CCR-06-2762
105. Pastore F, Levine RL. Epigenetic regulators and their impact on therapy in acute myeloid leukemia. Haematologica (2016) 101(3):269–78. doi:10.3324/haematol.2015.140822
106. Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood (2016) 127(1):71–8. doi:10.1182/blood-2015-07-604538
107. Tan P, Wei A, Mithraprabhu S, Cummings N, Liu HB, Perugini M, et al. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J (2014) 4:e170. doi:10.1038/bcj.2013.68
108. Kirschbaum M, Gojo I, Goldberg SL, Bredeson C, Kujawski LA, Yang A, et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br J Haematol (2014) 167(2):185–93. doi:10.1111/bjh.13016
109. Prebet T, Sun Z, Figueroa ME, Ketterling R, Melnick A, Greenberg PL, et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US Leukemia Intergroup trial E1905. J Clin Oncol (2014) 32(12):1242–8. doi:10.1200/JCO.2013.50.3102
110. Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol (2017) 178(3):434–41. doi:10.1111/bjh.14698
111. Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget (2015) 6(19):17698–712. doi:10.18632/oncotarget.4131
112. Koblish HK, Hansbury M, Hall L, Wang LC, Zhang Y, Covington M, et al. The BET inhibitor INCB054329 enhances the activity of checkpoint modulation in syngeneic tumor models. Cancer Res (2016) 76:4904. doi:10.1158/1538-7445.Am2016-4904
113. Millan DS, Morales MAA, Barr KJ, Cardillo D, Collis A, Dinsmore CJ, et al. FT-1101: a structurally distinct pan-BET bromodomain inhibitor with activity in preclinical models of hematologic malignancies. Blood (2015) 126(23):1367.
114. Borthakur G, Dawson MA, Stein EM, Karadimitris A, Huntly BJP, Dickinson MJ, et al. A Phase I/II open-label, dose escalation study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK525762 in subjects with relapsed, refractory hematologic malignancies. Blood (2016) 128(22):5223.
115. de Botton S, Pollyea DA, Stein EM, DiNardo C, Fathi AT, Roboz GJ, et al. Clinical safety and activity of Ag-120, a first-in-class, potent inhibitor of the Idh1 mutant protein, in a phase 1 study of patients with advanced Idh1-mutant hematologic malignancies. Haematologica (2015) 100:214–5.
116. Pollyea DA, de Botton S, Fathi AT, Stein EM, Tallman MS, Agresta S, et al. Clinical safety and activity in a phase I trial of AG-120, a first in class, selective, potent inhibitor of the IDH1-mutant protein, in patients with IDH1 mutant positive advanced hematologic malignancies. Eur J Cancer (2014) 50:195. doi:10.1016/S0959-8049(14)70722-1
117. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood (2017) 130(6):722–31. doi:10.1182/blood-2017-04-779405
118. Stein EM. IDH2 inhibition in AML: finally progress? Best Pract Res Clin Haematol (2015) 28(2–3):112–5. doi:10.1016/j.beha.2015.10.016
119. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492(7427):108. doi:10.1038/nature11606
120. Kurmasheva RT, Sammons M, Favours E, Wu J, Kurmashev D, Cosmopoulos K, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2017) 64(3). doi:10.1002/pbc.26218
121. Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Jongen-Lavrencic M, et al. A Phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in adults with relapsed or refractory leukemia: safety, clinical activity, exposure and target inhibition. Blood (2015) 126(23):2547.
122. Stein EM, Garcia-Manero G, Rizzieri DA, Savona M, Tibes R, Altman JK, et al. The DOT1L Inhibitor EPZ-5676: safety and activity in relapsed/refractory patients with MLL-rearranged leukemia. Blood (2014) 124(21):387.
123. Harris WJ, Huang X, Lynch JT, Somervaille T. Pharmacological inhibition or genetic knockdown of Kdm1a (Lsd1 or Aof2) induces differentiation of MLL acute myeloid leukaemia stem cells. Blood (2011) 118(21):58.
124. Ramirez L, Singh M, Chandra J. HDAC and LSD1 inhibitors synergize to induce cell death in acute leukemia cells. Blood (2011) 118(21):1427.
125. Yue LY, Du JJ, Ye F, Chen ZF, Li LC, Lian FL, et al. Identification of novel small-molecule inhibitors targeting menin-MLL interaction, repurposing the antidiarrheal loperamide. Org Biomol Chem (2016) 14(36):8503–19. doi:10.1039/c6ob01248e
126. Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JP. Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood (2008) 111(4):2382–4. doi:10.1182/blood-2007-07-103960
127. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell (2012) 21(3):430–46. doi:10.1016/j.ccr.2011.12.029
128. Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS One (2011) 6(8):e23372. doi:10.1371/journal.pone.0023372
129. Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol (2011) 29(24):3322–7. doi:10.1200/JCO.2011.35.8135
130. Issa JJ, Roboz G, Rizzieri D, Jabbour E, Stock W, O’Connell C, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol (2015) 16(9):1099–110. doi:10.1016/S1470-2045(15)00038-8
131. Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A (1998) 95(6):3003–7. doi:10.1073/pnas.95.6.3003
132. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5(9):769–84. doi:10.1038/nrd2133
133. Peng L, Seto E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb Exp Pharmacol (2011) 206:39–56. doi:10.1007/978-3-642-21631-2_3
134. Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther (2010) 10(6):935–54. doi:10.1586/era.10.62
135. Leder A, Leder P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell (1975) 5(3):319–22. doi:10.1016/0092-8674(75)90107-5
136. Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood (2005) 105(3):959–67. doi:10.1182/blood-2004-05-1693
137. Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res (2008) 14(3):826–32. doi:10.1158/1078-0432.CCR-07-0318
138. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood (2007) 109(7):2781–90. doi:10.1182/blood-2006-05-021873
139. DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T, et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia (2013) 27(8):1628–36. doi:10.1038/leu.2013.38
140. Bose P, Grant S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leuk Res (2014) 38(8):862–5. doi:10.1016/j.leukres.2014.06.007
141. Gojo I, Tan M, Fang HB, Sadowska M, Lapidus R, Baer MR, et al. Translational phase I trial of vorinostat (suberoylanilide hydroxamic acid) combined with cytarabine and etoposide in patients with relapsed, refractory, or high-risk acute myeloid leukemia. Clin Cancer Res (2013) 19(7):1838–51. doi:10.1158/1078-0432.CCR-12-3165
142. Garcia-Manero G, Tambaro FP, Bekele NB, Yang H, Ravandi F, Jabbour E, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J Clin Oncol (2012) 30(18):2204–10. doi:10.1200/JCO.2011.38.3265
143. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood (2006) 108(10):3271–9. doi:10.1182/blood-2006-03-009142
144. Lubbert M, Kuendgen A. Combining DNA methyltransferase and histone deacetylase inhibition to treat acute myeloid leukemia/myelodysplastic syndrome: achievements and challenges. Cancer (2015) 121(4):498–501. doi:10.1002/cncr.29083
145. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood (2016) 127(1):42–52. doi:10.1182/blood-2015-07-604512
146. Voso MT, Lo-Coco F, Fianchi L. Epigenetic therapy of myelodysplastic syndromes and acute myeloid leukemia. Curr Opin Oncol (2015) 27(6):532–9. doi:10.1097/CCO.0000000000000231
147. Issa JP, Garcia-Manero G, Huang X, Cortes J, Ravandi F, Jabbour E, et al. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer (2015) 121(4):556–61. doi:10.1002/cncr.29085
148. Fredly H, Ersvaer E, Kittang AO, Tsykunova G, Gjertsen BT, Bruserud O. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin Epigenetics (2013) 5(1):13. doi:10.1186/1868-7083-5-13
149. Raffoux E, Cras A, Recher C, Boelle PY, de Labarthe A, Turlure P, et al. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget (2010) 1(1):34–42. doi:10.18632/oncotarget.100518
150. Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood (2007) 110(7):2302–8. doi:10.1182/blood-2007-03-078576
151. Silverman LR, Verma A, Odchimar-Reissig R, Cozza A, Najfeld V, Licht JD, et al. A phase I/II study of vorinostat, an oral histone deacetylase inhibitor, in combination with azacitidine in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Initial results of the phase I trial: A New York Cancer Consortium. J Clin Oncol (2008) 26(15):7000. doi:10.1200/jco.2008.26.15_suppl.7000
152. Wahaib K, Beggs AE, Campbell H, Kodali L, Ford PD. Panobinostat: a histone deacetylase inhibitor for the treatment of relapsed or refractory multiple myeloma. Am J Health Syst Pharm (2016) 73(7):441–50. doi:10.2146/ajhp150487
153. Dasari A, Gore L, Messersmith WA, Diab S, Jimeno A, Weekes CD, et al. A phase I study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Invest New Drugs (2013) 31(1):115–25. doi:10.1007/s10637-012-9812-z
154. Jones SF, Bendell JC, Infante JR, Spigel DR, Thompson DS, Yardley DA, et al. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin Adv Hematol Oncol (2011) 9(3):225–30.
155. Matulonis U, Berlin S, Lee H, Whalen C, Obermayer E, Penson R, et al. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother Pharmacol (2015) 76(2):417–23. doi:10.1007/s00280-015-2813-9
156. San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol (2014) 15(11):1195–206. doi:10.1016/S1470-2045(14)70440-1
157. Tarhini AA, Zahoor H, McLaughlin B, Gooding WE, Schmitz JC, Siegfried JM, et al. Phase I trial of carboplatin and etoposide in combination with panobinostat in patients with lung cancer. Anticancer Res (2013) 33(10):4475–81.
158. Florence B, Faller DV. You bet-cha: a novel family of transcriptional regulators. Front Biosci (2001) 6:D1008–18. doi:10.2741/A663
159. Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett (2002) 513(1):124–8. doi:10.1016/S0014-5793(01)03309-9
160. Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem (2007) 282(18):13141–5. doi:10.1074/jbc.R700001200
161. Pivot-Pajot C, Caron C, Govin J, Vion A, Rousseaux S, Khochbin S. Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol Cell Biol (2003) 23(15):5354–65. doi:10.1128/MCB.23.15.5354-5365.2003
162. Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol (2007) 14(11):1025–40. doi:10.1038/nsmb1338
163. Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol (2002) 22(11):3794–802. doi:10.1128/MCB.22.11.3794-3802.2002
164. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A (2003) 100(15):8758–63. doi:10.1073/pnas.1433065100
165. Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell (2004) 13(1):33–43. doi:10.1016/S1097-2765(03)00482-9
166. Shang E, Wang X, Wen D, Greenberg DA, Wolgemuth DJ. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev Dyn (2009) 238(4):908–17. doi:10.1002/dvdy.21911
167. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell (2011) 146(6):904–17. doi:10.1016/j.cell.2011.08.017
168. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153(2):320–34. doi:10.1016/j.cell.2013.03.036
169. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature (2011) 478(7370):529–33. doi:10.1038/nature10509
170. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia (2014) 28(2):311–20. doi:10.1038/leu.2013.338
171. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A (2011) 108(40):16669–74. doi:10.1073/pnas.1108190108
172. Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature (2015) 525(7570):543–7. doi:10.1038/nature14898
173. Coude MM, Berrou J, Bertrand S, Riveiro E, Herait P, Baruchel A, et al. Preclinical study of the bromodomain inhibitor OTX015 in acute myeloid (AML) and lymphoid (ALL) leukemias. Blood (2013) 122(21):4218.
174. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell (2005) 19(6):857–64. doi:10.1016/j.molcel.2005.08.027
175. Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric human protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics (2008) 7(10):2019–27. doi:10.1074/mcp.R800013-MCP200
176. Lokken AA, Zeleznik-Le NJ. Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML. Cancer Cell (2012) 21(4):451–3. doi:10.1016/j.ccr.2012.03.027
177. Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med (2012) 18(4):605–11. doi:10.1038/nm.2661
178. Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia (2014) 28(11):2155–64. doi:10.1038/leu.2014.119
179. Niebel D, Kirfel J, Janzen V, Holler T, Majores M, Gutgemann I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood (2014) 124(1):151–2. doi:10.1182/blood-2014-04-569525
180. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature (2005) 437(7057):436–9. doi:10.1038/nature04020
181. Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet (2009) 41(1):125–9. doi:10.1038/ng.268
182. Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature (2007) 446(7138):882–7. doi:10.1038/nature05671
183. Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell (2011) 20(4):457–71. doi:10.1016/j.ccr.2011.09.001
184. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood (2015) 125(2):346–57. doi:10.1182/blood-2014-06-581082
185. Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov (2017) 7(5):478–93. doi:10.1158/2159-8290.CD-16-1034
186. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med (2009) 361(11):1058–66. doi:10.1056/NEJMoa0903840
187. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell (2011) 20(1):53–65. doi:10.1016/j.ccr.2011.06.009
188. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell (2011) 20(1):66–78. doi:10.1016/j.ccr.2011.06.010
189. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood (2013) 121(13):2533–41. doi:10.1182/blood-2012-11-465120
190. Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia (2013) 27(4):813–22. doi:10.1038/leu.2012.327
191. Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther (2014) 350(3):646–56. doi:10.1124/jpet.114.214577
192. Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res (2007) 67(15):7275–83. doi:10.1158/0008-5472.CAN-06-2369
193. Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J Biol Chem (2010) 285(52):40690–8. doi:10.1074/jbc.M110.172783
194. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell (2008) 14(1):36–46. doi:10.1016/j.ccr.2008.05.003
Keywords: acute myeloid leukemia, epigenetic therapy, leukemia stem cell, epigenome, chromatin modification
Citation: Sun Y, Chen B and Deshpande A (2018) Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 8:41. doi: 10.3389/fonc.2018.00041
Received: 16 October 2017; Accepted: 07 February 2018;
Published: 23 February 2018
Edited by:
Keisuke Ito, Albert Einstein College of Medicine, United StatesReviewed by:
Takaomi Sanda, National University of Singapore, SingaporeMyriam Alcalay, Istituto Europeo di Oncologia, Italy
Copyright: © 2018 Sun, Chen and Deshpande. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aniruddha Deshpande, YWRlc2hwYW5kZUBzYnBkaXNjb3Zlcnkub3Jn