 Sun-Hee Kim
Sun-Hee Kim Jason Roszik
Jason Roszik Elizabeth A. Grimm
Elizabeth A. Grimm Suhendan Ekmekcioglu
Suhendan Ekmekcioglu- Department of Melanoma Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
The progression from neoplastic initiation to malignancy happens in part because of the failure of immune surveillance. Cancer cells successfully escape immune recognition and elimination and create an immune-suppressive microenvironment. A suppressive metabolic microenvironment may also contribute to ineffective T-cell function. Tumor progression is characterized by a complex network of interactions among different cell types that cooperatively exploit metabolic reprogramming. As we start to recognize that cancer cells use different metabolism processes than normal cells do, a better understanding of the functional mechanisms of the regulation and reprogramming of the metabolic landscape in cancer cells is crucial to successful immunotherapy strategies. However, the exact role of metabolism in T cells and in the tumor microenvironment is not known. One pathway that plays an important role in the regulation of immune cell reactivity is arginine metabolism, which has complex cellular functions. l-arginine and its downstream metabolites (e.g., ornithine and citrulline) could be essential to T-cell activation and thus modulate innate and adaptive immunity to further promote tumor survival and growth. Identifying metabolic targets that mediate immunosuppression and are fundamental to sustaining tumor growth is key to increasing the efficacy of immunotherapies.
Introduction
Metabolic pathways and their intermediates have been shown to contribute to the formation of the immunosuppressive tumor features that exclude immune cells from the tumor microenvironment or impair their ability to respond. Manipulating these metabolic changes can strengthen antitumor immune responses by recovering the functions of the T cells. Thus, cancer immunotherapy strategies focus on metabolism to promote immune cells’ acquisition of sufficient metabolites to preserve their antitumor responses.
Tumor progression is characterized by a complex network of communication among different cell types and cancer cells that stimulates the metabolic reprogramming of the cancer cells and thus influences their functionality. Our research suggests that deleterious tumor inflammatory processes occur in response to microenvironmental communications and are driven by intrinsic inflammatory processes (1). Much recent interest and new immune system data suggests that cellular bioenergy links metabolism with inflammation and immunity to protect normal cells from immune death (i.e., checkpoint inhibition) and to restore homeostasis with minimal tissue damage (2), and provides a complex balance to make it beneficial for the host. However, cancer cells with these self-protective mechanisms become resistant to immune attack. Clinically evident cancers are those that have evaded the immune response. Inflammation and immune evasion are hallmarks of cancer progression, as evidenced by the fact that cancer patients with fewer immune cells in their biopsied tissues have poorer responses to immunotherapies (3). These cells may include the direct involvement of immune cells, including macrophages, T cells, B cells, natural killer cells, dendritic cells, and myeloid-derived suppressor cells (MDSCs) (4).
Some of the mechanisms the immune system uses to fight infectious diseases also promote immune suppression in the tumor microenvironment. Tumor resistance to the immune response is assisted by the immunoediting of the immune signals induced by the tumors and their metabolites (2), possibly including that of checkpoint inhibitor upregulation, via inflammatory mechanisms mediated by arginine metabolism (5). This review presents the role of arginine metabolism in the major inflammatory processes that we have found to drive the survival of melanoma and other cancers, including those processes underlying these tumors’ resistance to therapy, particularly immunotherapy.
Arginine Metabolism as a Major Requirement for Inflammatory Networks and Products
Human tumors support an immunosuppressive microenvironment that often prevents effective immunotherapy. Because of their effectiveness and the long-term responses they elicit, cancer immunotherapies have changed the landscape of cancer care. Despite the major breakthroughs in cancer immunotherapy of the past decade, the success to date does not meet the promise of the approach. Different physiological mechanisms can prevent durable responses to immunotherapy. Some of these mechanisms have been characterized molecularly and may be triggered by the tumor microenvironment, resulting in impaired immune effector cell function.
Arginine metabolism is one of the mechanisms responsible for tumor progression and it is highly compartmentalized due to expression of enzymes involved in arginine metabolism in various cells. l-arginine is a multipurpose amino acid that also serves as a precursor for multiple metabolites, including polyamines and nitric oxide (NO), which have strong immunomodulatory properties (Figure 1). l-arginine is the substrate for four enzymes, several of which exist as multiple isoforms: NO synthases (NOSs), arginases (ARGs), glycine aminidotransferase, and l-arginine decarboxylase (6). To encounter the enzymes involved in its metabolism, l-arginine must be transported through the plasma membrane via cationic amino acid transporters (CATs) and metabolized by NOS enzymes (7). Using the Cancer Cell Line Encyclopedia database, we found that CAT2B expression varies among human melanoma cells and that a subset of human melanoma cells has a very high level of CAT2B expression, but that CAT1 expression is similar among all the cell lines. CAT1 is constitutively expressed in most tissues, whereas CAT2B is cytokine inducible (8). Thus, we speculate that the diverse expression of CAT2B may be associated with the inflammatory factors expressed in human melanoma cells.
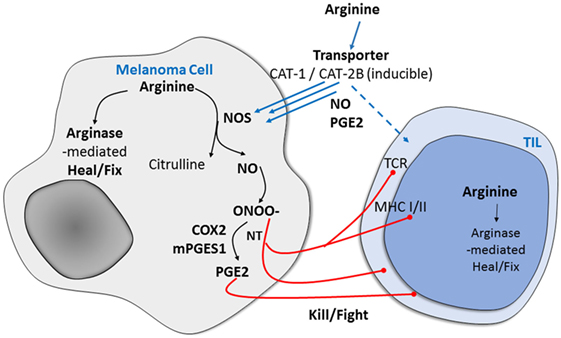
Figure 1. Potential mechanism of arginine metabolism in cancer cells versus immune-infiltrating cells in human melanoma. After arginine enters mammalian cells through the membrane-bound transporters CAT1 and CAT2B, it is metabolized by one of the NO synthase (NOS) enzymes to produce nitric oxide (NO), which is associated with immune suppression and results in the expression of specific markers and functional changes.
After arginine enters mammalian cells through the membrane-bound transporters CAT1 and CAT2B, it is metabolized by one of the NOS enzymes. The enzymatic production of NO is cell-type specific, with cytokine-driven inducible NOS (iNOS) noted initially for the burst of higher levels as part of the pathogen defense system. Neuronal cells use neuronal NOS to produce NO for signaling. The third NOS, endothelial NOS, regulates NO production in endothelia and is responsible for vascular relaxation (9). In previous studies, we found that the selectively increased metabolism of l-arginine in melanoma and other human tumors is associated with immune suppression and neovascularization and that the arginine metabolic product of NO results in the expression of specific markers of tumor progression and in functional changes (10, 11). We and others have also found that increased arginine metabolism in melanoma tumors and their microenvironment mediates a previously unappreciated system that is fundamental to sustaining the growth of many cancers while specifically causing immunosuppression (11–14).
The metabolism of arginine by a family of NOSs generates NO, which can affect molecular pathways in melanoma cells and the melanoma microenvironment by defined posttranslational modifications. NO is a relatively stable intracellular molecule that diffuses through lipid bilayers and persists for at least 7 s (15). NO can react with superoxide to form peroxynitrite, which rapidly forms two posttranslational modifications on proteins, nitrosylation of thiols and nitration of tyrosines (NT) (16, 17). A second arginine metabolism via ARG leads to proline and polyamine production, which may also contribute to tumor progression. Therefore, overall arginine metabolism depends on the activity of the NOS and ARG enzyme families. NOS oxidizes arginine to citrulline and NO, and ARG hydrolyzes arginine into ornithine and urea. We and others have demonstrated that many human tumors, including melanoma, express iNOS (18–23). iNOS expression and NO production have been shown to promote many cancers’ growth, survival, and resistance to therapy. One recent study of the genomic and transcriptomic features of the response of metastatic melanoma to anti-programmed cell death protein 1 (PD-1) therapy revealed that a transcriptional signature is related to innate anti-PD-1 resistance (24). Using the same clinical dataset, we found that iNOS expression in metastatic melanoma is associated with response to anti-PD-1 therapy (unpublished data). Therefore, understanding the regulation of immune cell response and functions of tumor-expressed iNOS will provide significant opportunities for therapeutic intervention. ARG-1 activity is associated with polarized, protumoral M2 tumor-associated macrophages (25). Suppression of T-cell activation, proliferation, and differentiation by macrophage ARG-1 dominated by M2 macrophages and interactions between macrophage metabolism and M1/M2 polarization is an urgent matter to be resolved in cancer. Together, ARG and NOS enzymes metabolize arginine and are critical components of immune suppression pathways, and the metabolic products of these enzymes are imperative mediators of T-cell function in cancer (12).
The importance of arginine metabolism as a novel field of investigation includes that of arginine depletion retarding the growth of some cancers (26), whereas others report that arginine supplementation enhances antitumor effects, probably by enhancing immune function (11, 27). Arginine supplementation has been discovered to stimulate T cell and natural killer cell activity and promotes the production of pro-inflammatory cytokines (28, 29). The roles of arginine in the context of NOS and ARG activity and in tumor-derived NO need to be understood.
Arginine Metabolism and NOS and Cyclooxygenase
Preliminary data from our laboratory and others (24, 25) indicate that NO activates cyclooxygenase-2 (COX-2) and other inflammatory mediators, thereby creating a pro-oxidant microenvironment that supports cancer cell growth and suppresses antitumor immunity. iNOS/NO positively regulates the production of COX-2, microsomal prostaglandin E synthase-1 (mPGES1), and prostaglandin E2 (PGE2). COX-2 and iNOS can be produced concurrently in several inflammatory conditions (24). In our previous study, a tissue microarray analysis of stage III melanoma showed that most mPGES1-positive tissues were also positive for iNOS and had co-localized mPGES1 and iNOS expression. n addition, transient iNOS expression and NO donors dramatically enhanced PGE2 production, suggesting that iNOS and NO are upstream of PGE2 biosynthesis in melanoma cells. We found that mPGES1 was tyrosine-nitrated by NO. Our data suggest that iNOS regulates mPGES1 activity through a posttranslational NT modification, which ultimately enhances PGE2 production; that iNOS activates the mPGES1/PGE2 pathway; and that the cross talk between iNOS and mPGES1 promotes inflammation, which favors melanoma progression (24).
In melanoma and other cancers, PGE2 plays a role in immune responses, acting as both a pro-inflammatory mediator and a potent immune suppressant (26). In melanoma, specifically, tumor-derived COX activity is the key suppressor of type I interferon-mediated tumor elimination and induces an inflammatory signature associated with progression (27). The PGE2-mediated stimulation of immunosuppressive cells, including T regulatory cells (Tregs) and MDSCs, may also be an important mechanism of immune suppression. PGE2 has an important role in redirecting the differentiation of human dendritic cells into MDSCs, and the inhibition of COX-2 or PGE2 receptors abolishes MDSCs’ functions and their CXCR4-CXCL12-mediated attraction to the cancer environment (28). PGE2 stimulates the de novo conversion of Tregs from naïve CD4+ T cells (29), and Tregs expressing PGE2 receptors are preferentially recruited to factors expressed by COX-2-expressing tumor cells (30). Elevated MDSCs and Tregs suppress T-cell infiltration into the tumor site and/or inhibit T-cell activity. In a previous study of stage III melanoma, our tissue microarray analysis demonstrated that high mPGES1 staining intensity was significantly associated with low CD8 levels, and we found that patients whose tumors had a high-mPGES1, low-CD8 expression signature had a significantly increased risk of death (31).
Several recent reports suggest that targeting PGE2 metabolism could help reduce programmed death-ligand 1 (PD-L1)-mediated immune suppression (27, 32, 33). One study showed that COX-2 and PD-L1 were expressed in both primary melanoma lesions and non-matching lymph node metastases and that the inhibition of COX-2 activity by celecoxib downregulated PD-L1 expression in both BRAFV600E A375 and NRASQ61R SK-MEL-2 melanoma cell lines (32). In another study, an analysis of murine bone marrow cells cocultured with murine MBT-2 bladder tumor cells demonstrated that the COX-2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and MDSCs (33). A third study showed that conventional nonsteroidal anti-inflammatory drugs and COX-2 inhibitors combined with an anti-PD-1 monoclonal antibody promoted a much more rapid regression of mouse melanoma tumors than the anti-PD-1 antibody alone did (27). This result suggests that the COX-2/mPGES1/PGE2 pathway contributes to immune evasion and that nonsteroidal anti-inflammatory drugs and COX-2 inhibitors are useful adjuvants to immune-based anticancer therapies.
Our data show not only the NO-mediated modulation of tumor-infiltrating leukocyte (TIL) growth but also the COX-2-mediated production of PGE2 in human melanoma. Other potential sources of NO from arginine metabolism in cancers include MDSCs, which are a heterogeneous population of myeloid cells that often infiltrate cancers and sites of inflammation and infection and have a remarkable ability to suppress T-cell responses (30). We have previously reported that iNOS expression in melanoma is significantly associated with apoptosis resistance and shorter patient survival (18, 31). We recently observed global NT expression in stage IIIc and IV melanomas in humans. NT is a marker of NO’s reaction with reactive oxygen species to form peroxynitrite, which irreversibly nitrates tyrosines. NT expression in tissues is associated with poor TIL growth and poorer response to TIL-based therapy. Protein-associated NT has been identified as a surrogate marker of in situ inflammation, but its role in cellular immune response has not been studied broadly. One intriguing study showed that the oxidation of tyrosine to NT on either T-cell receptor or major histocompatibility complex (MHC) molecules can disturb CD8 T cells’ recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein (32). This study also demonstrated that T cells’ specific recognition of nitrated epitopes is not limited to MHC class II epitopes but that the conversion of tyrosine to nitrotyrosine can also intensely disturb these cells’ recognition of MHC class I-restricted epitopes. If, like CD4 T cells, CD8 T cells can recognize epitopes containing an NT posttranslational modification as antigenic, this would certainly affect TIL activation in melanoma patients.
Conclusion
Following the success of checkpoint inhibitors in many tumor types, immunotherapy has become a major focus of cancer researchers and clinicians. Our mission is to guide oncology research by improving our understanding of the role of tumors’ interactions with their microenvironment and with immune cells functions in particular. Arginine metabolism is one of the mechanisms responsible for immune response to tumor progression. Because different cells express enzymes involved in the process, arginine metabolism is highly dysregulated in cancer. The concept of antitumor immune response, based on usage of arginine via NOS and ARG, is timely topic to focus on to set our knowledge regarding their role in current immunotherapy approaches. The extent to which immune cells and tumor cells in the tumor microenvironment respond to arginine metabolism could predict better immunotherapy outcomes.
Author Contributions
All authors (S-HK, JR, EG, and SE) were involved in building the concept and participated in the writing based on their expertise area. S-HK was involved mainly in writing; JR was involved in concept and analyses, EG was involved in sharing her expertise, reviewing, and editing; and SE was mainly involved in writing, concept design, reviewing, and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by MD Anderson’s Melanoma SPORE (P50 CA093459; S-HK, EG, and SE); the National Institutes of Health through MD Anderson’s Cancer Center Support Grant (P30-CA016672; EG); the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (EAG); MD Anderson’s Melanoma Moon Shot Program (JR and EG); and the Jim Mulva Foundation; and AIM Foundations. We also thank Joseph A. Munch of the MD Anderson Department of Scientific Publications for carefully editing the manuscript and providing valuable comments.
References
1. Ekmekcioglu S, Davies MA, Tanese K, Roszik J, Shin-Sim M, Bassett RL Jr, et al. Inflammatory marker testing identifies CD74 expression in melanoma tumor cells, and its expression associates with favorable survival for stage III melanoma. Clin Cancer Res (2016) 22:3016–24. doi:10.1158/1078-0432.CCR-15-2226
2. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell (2010) 140:771–6. doi:10.1016/j.cell.2010.03.006
3. Reuben A, Spencer CN, Prieto PA, Gopalakrishnan V, Reddy SM, Miller JP, et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom Med (2017) 2:10. doi:10.1038/s41525-017-0013-8
4. Veglia F, Gabrilovich DI. Dendritic cells in cancer: the role revisited. Curr Opin Immunol (2017) 45:43–51. doi:10.1016/j.coi.2017.01.002
5. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
6. Popolo A, Adesso S, Pinto A, Autore G, Marzocco S. l-arginine and its metabolites in kidney and cardiovascular disease. Amino Acids (2014) 46:2271–86. doi:10.1007/s00726-014-1825-9
7. Hatzoglou M, Fernandez J, Yaman I, Closs E, Regulation of cationic amino acid transport: the story of the CAT-1 transporter. Annu Rev Nutr (2004) 24:377–99. doi:10.1146/annurev.nutr.23.011702.073120
8. Simmons WW, Closs EI, Cunningham JM, Smith TW, Kelly RA. Cytokines and insulin induce cationic amino acid transporter (CAT) expression in cardiac myocytes. Regulation of l-arginine transport and no production by CAT-1, CAT-2A, and CAT-2B. J Biol Chem (1996) 271:11694–702. doi:10.1074/jbc.271.20.11694
9. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33:829–37. doi:10.1093/eurheartj/ehr304
10. Wang Y, Hu S, Gabisi AM Jr, Er JA, Pope A, Burstein G, et al. Developing an irreversible inhibitor of human DDAH-1, an enzyme upregulated in melanoma. ChemMedChem (2014) 9:792–7. doi:10.1002/cmdc.201300557
11. Yoon JK, Frankel AE, Feun LG, Ekmekcioglu S, Kim KB. Arginine deprivation therapy for malignant melanoma. Clin Pharmacol (2013) 5:11–9. doi:10.2147/CPAA.S37350
12. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. l-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–842e813. doi:10.1016/j.cell.2016.09.031
13. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res (2015) 75:275–83. doi:10.1158/0008-5472.CAN-14-1491
14. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via l-arginine metabolism. Cancer Res (2009) 69:3086–94. doi:10.1158/0008-5472.CAN-08-2826
15. Habib S, Ali A. Biochemistry of nitric oxide. Indian J Clin Biochem (2011) 26:3–17. doi:10.1007/s12291-011-0108-4
16. Kovacs I, Lindermayr C. Nitric oxide-based protein modification: formation and site-specificity of protein S-nitrosylation. Front Plant Sci (2013) 4:137. doi:10.3389/fpls.2013.00137
17. Yakovlev VA, Mikkelsen RB. Protein tyrosine nitration in cellular signal transduction pathways. J Recept Signal Transduct Res (2010) 30:420–9. doi:10.3109/10799893.2010.513991
18. Ekmekcioglu S, Ellerhorst JA, Prieto VG, Johnson MM, Broemeling LD, Grimm EA. Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer (2006) 119:861–6. doi:10.1002/ijc.21767
19. Johansson CC, Egyhazi S, Masucci G, Harlin H, Mougiakakos D, Poschke I, et al. Prognostic significance of tumor iNOS and COX-2 in stage III malignant cutaneous melanoma. Cancer Immunol Immunother (2009) 58:1085–94. doi:10.1007/s00262-008-0631-1
20. Grimm EA, Sikora AG, Ekmekcioglu S. Molecular pathways: inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin Cancer Res (2013) 19:5557–63. doi:10.1158/1078-0432.CCR-12-1554
21. Heinecke JL, Ridnour LA, Cheng RY, Switzer CH, Lizardo MM, Khanna C, et al. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proc Natl Acad Sci U S A (2014) 111:6323–8. doi:10.1073/pnas.1401799111
22. Tanese K, Grimm EA, Ekmekcioglu S. The role of melanoma tumor-derived nitric oxide in the tumor inflammatory microenvironment: its impact on the chemokine expression profile, including suppression of CXCL10. Int J Cancer (2012) 131:891–901. doi:10.1002/ijc.26451
23. Ekmekcioglu S, Grimm EA, Roszik J. Targeting iNOS to increase efficacy of immunotherapies. Hum Vaccin Immunother (2017) 13:1105–8. doi:10.1080/21645515.2016.1276682
24. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165:35–44. doi:10.1016/j.cell.2016.02.065
25. Tham M, Tan KW, Keeble J, Wang X, Hubert S, Barron L, et al. Melanoma-initiating cells exploit M2 macrophage TGFbeta and arginase pathway for survival and proliferation. Oncotarget (2014) 5:12027–42. doi:10.18632/oncotarget.2482
27. Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des (2008) 14:1049–57. doi:10.2174/138161208784246199
28. Rodriguez PC, Quiceno DG, Ochoa AC. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi:10.1182/blood-2006-06-031856
29. Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, Vasson MP, et al. Altered functions of natural killer cells in response to l-arginine availability. Cell Immunol (2012) 280:182–90. doi:10.1016/j.cellimm.2012.11.018
30. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
31. Tang CH, Grimm EA. Depletion of endogenous nitric oxide enhances cisplatin-induced apoptosis in a p53-dependent manner in melanoma cell lines. J Biol Chem (2004) 279:288–98. doi:10.1074/jbc.M310821200
32. Visigalli R, Bussolati O, Sala R, Barilli A, Rotoli BM, Parolari A, et al. The stimulation of arginine transport by TNFalpha in human endothelial cells depends on NF-kappaB activation. Biochim Biophys Acta (2004) 1664:45–52. doi:10.1016/j.bbamem.2004.04.001
Keywords: arginine, NO synthase, arginase-1, cyclooxygenase-2, microsomal prostaglandin E synthase-1, immunotherapy, metabolism, immune response
Citation: Kim S-H, Roszik J, Grimm EA and Ekmekcioglu S (2018) Impact of l-Arginine Metabolism on Immune Response and Anticancer Immunotherapy. Front. Oncol. 8:67. doi: 10.3389/fonc.2018.00067
Received: 18 December 2017; Accepted: 01 March 2018;
Published: 16 March 2018
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), United StatesReviewed by:
Pin Wu, Zhejiang University, ChinaWilliam K. Decker, Baylor College of Medicine, United States
Copyright: © 2018 Kim, Roszik, Grimm and Ekmekcioglu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suhendan Ekmekcioglu, c2VrbWVrY2lvZ2x1QG1kYW5kZXJzb24ub3Jn