 Rama Soundararajan
Rama Soundararajan Ana M. Aparicio2
Ana M. Aparicio2 Sankar N. Maity
Sankar N. Maity- 1Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Genitourinary Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Combined loss of tumor suppressors (TSPs), PTEN, TP53, and RB1, is highly associated with small cell carcinoma of prostate phenotype. Recent genomic studies of human tumors as well as analyses in mouse genetic models have revealed a unique role for these TSPs in dictating epithelial lineage plasticity—a phenomenon that plays a critical role in the development of aggressive variant prostate cancer (PCa) and associated androgen therapy resistance. Here, we summarize recently published key observations on this topic and hypothesize a possible mechanism by which concurrent loss of TSPs could potentially regulate the PCa disease phenotype.
Small-cell prostate carcinoma (SCPC) is a common lethal variant of prostate cancer (PCa) that is androgen receptor (AR) negative and thus represents a mechanism for escape from the potent antiandrogen treatments. The presence of SCPC morphologic characteristics predicts for a distinct clinical course with a dismal prognosis, despite a heightened sensitivity to chemotherapy (1–4). Although often unrecognized, SCPC is frequently found on repeat biopsies of previously diagnosed adenocarcinoma of the prostate during castration-resistant PCa (CRPC) (5, 6) and is present in 10–20% of autopsies of patients who die of the disease (7–10). SCPC also phenotypically overlaps with neuroendocrine prostate cancer (NEPC), and clinicopathologic characteristics of SCPC/NEPC are generally associated with worse progression-free survival (3, 11–14).
Molecular characterization of human CRPC and SCPC/NEPC tumors showed that concurrent alteration of three tumor suppressors (TSPs), PTEN, TP53, and RB1, in which loss of copy numbers of both PTEN and RB1, and loss of copy number as well as missense mutations of TP53 genes are highly enriched in SCPC/NEPC tumors (Table 1) (15–24). Conditional deletion of the TSP genes in mouse luminal epithelial cells of prostate demonstrated new insights into the functional role of these TSPs in therapy resistance (25–28). Studies in mouse models as well as in human PCa cells showed that deletion of both RB1 and TP53 resulted in development of aggressive prostate tumors that are resistant to anti-androgen treatment (27, 28). Deletion of all three TSPs—RB1, TP53, and PTEN—resulted in development of lethal PCa and significantly increased cancer-related death and metastasis in mice (27). Expression analysis of mouse tumors showed that loss of TSPs, induced epithelial lineage plasticity with induction of neuroendocrine markers but loss of AR and epithelial makers. The study further showed that loss of RB1 plays a deterministic role in inducing lineage plasticity when combined with the loss of PTEN and TP53. In contrast, deletion of RB1 by itself showed no impact in prostate tumor development (Table 1).
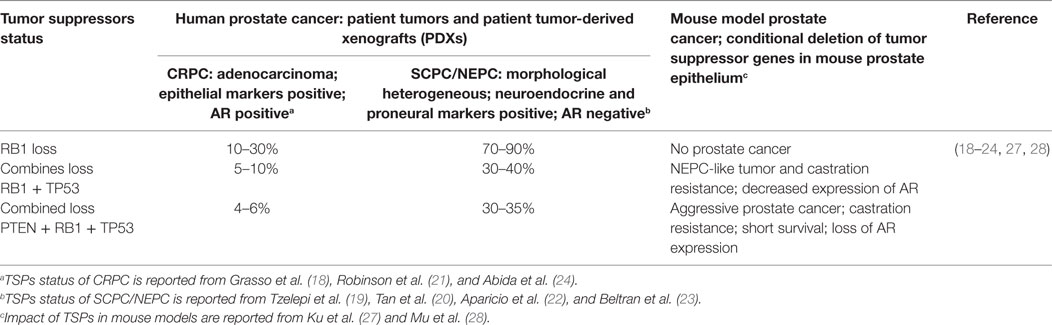
Table 1. Impact of combined loss of three tumor suppressors—PTEN, TP53, and RB1—in human and mouse models of prostate cancer.
The SCPC phenotype can be morphologically heterogeneous and is often described as mixed tumors harboring both typical adenocarcinoma and NEPC, suggesting that the SCPC/NEPC tumors constitute multiple cell types or cells in multiple distinct stages of differentiation. Studies in patient tumor-derived xenografts as well as in human tumors showed that the SCPC phenotype is associated with induction of the pro-neural developmental program, including expression of master transcription factors such as ASCL1, NEUROD1, and BRN2 that determine proneural differentiation (22, 29, 30). Several investigations reported expression of stem and basal cell markers in SCPC/NEPC tumors, suggesting that these tumors might originate from basal cells of the prostate (31, 32). However, analysis of the mouse tumors with conditional deletion of TSPs in luminal epithelial cells strongly indicated that combined loss of TSPs induces lineage plasticity resulting in loss of AR expression but gain of stemness master transcription factor such as SOX2, along with upregulation of NEPC markers (27, 28). Importantly, lineage-tracing experiments demonstrated that anti-androgen therapy, by itself, could induce trans-differentiation of luminal epithelial cells into prostate NEPC-like cells lacking functional PTEN and TP53 TSPs (33). Of note, previous studies of compound deletions of PTEN and TP53 TSPs in mice, wherein gene inactivation was done by a constitutive prostate-specific Cre driver, showed that combined TSP deletions led to development of invasive CRPC phenotype (34, 35). A more recent publication, however, used regulated inducible Cre driver in adult mice that specifically deleted PTEN and TP53 genes in prostate luminal epithelial cells, which allowed for a more detailed study about the role of PTEN and TP53 TSPs in therapy-induced NEPC transdifferentiation (33). Taken together, these studies suggest that cellular reprogramming or trans-differentiation could be a central mechanism potentiating resistance to anti-androgen therapy, in which loss of functional TSPs facilitates alterations in the epithelial lineage. This plasticity is observed in the switch to pro-neural and pluripotent-like cancer cells that are no longer dependent on AR activity.
Role of TSPs in cellular reprogramming or lineage plasticity is a nascent concept. Although the individual function of each TSP has been studied extensively for many years, the impact of combined loss of TSPs in specific cellular pathways that can be linked to therapy-resistance remains largely unclear. Importantly, TP53 loss or RB1 loss events in prostate tumors are not currently druggable. The proteins p53 and Rb1 encoded by TP53 and RB1 genes, respectively, are known to function as nuclear transcriptional activator and repressor, respectively. Loss of p53 function inhibits expression of downstream genes that are direct targets of p53 protein (36), whereas loss of Rb1 results in activation of E2F transcription factors regulating cell cycle genes, the pluripotency (transcription) factor SOX2, as well as heterochromatin regulators (27, 37–39, 40, 41). The SCPC/NEPC tumors also express mutant p53 proteins, which although cannot bind to promoters of p53 target genes, can however indeed regulate transcription via activation of distinct transcription factors or chromatin regulators driving oncogenic pathways (42, 43).
Induction of lineage plasticity due to loss of TSPs suggests that these TSPs likely play a role in maintaining the fundamental state of epithelial differentiation in the prostate. We propose that loss of TSPs in luminal epithelial cells induces epigenetic and transcriptional reprogramming resulting in differential expression of master transcription factors allowing lineage plasticity and thereby, the opportunity for trans-differentiation (Figure 1). Once the stage is set for this cellular fluidity, further molecular “hits” such as epithelial-to-mesenchymal transition (EMT), which can be induced by anti-androgen therapy (44), could then propel transition to SCPC/NEPC. The term EMT is used to describe profound cell biological transitions that convert “epithelial” tissue-resident cells into morphologically and functionally distinct “mesenchymal or mesenchymal-like” cells harboring increased migratory and invasive properties facilitating disease recurrence and progression. The genes expression analyses of both human and mouse prostate tumors demonstrate increased expression of master EMT transcription factors (FOXC2, ZEB1, SNAIL) (45, 46). These factors are known to play a critical role in the inhibition of the epithelial-specific transcriptional program, including inhibiting expression of AR, and instead inducing expression of mesenchymal markers.
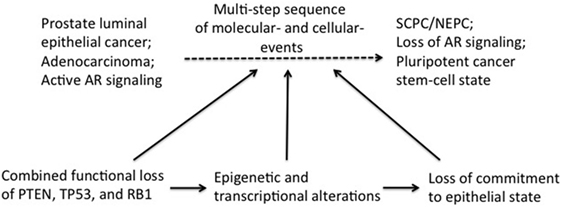
Figure 1. Induction of transdifferentiation of epithelial prostate cancer cells may be brought about in multiple steps involving sequential loss of tumor suppressors (TSPs) function and pluripotency/plasticity events. Combined functional loss of TSPs (PTEN, RB, and P53), epigenetic and transcriptional modifiers, as well as pluripotency and stemness events have each been linked to the altered cellular differentiation process during prostate tumor progression. It is conceivable that these events have a preferential order of occurrence in the history of the tumor development, with each event contributing partially to tumor progression and also setting the stage for the next subsequent event. Combined TSP loss is perhaps an early event in this context, facilitating ensuing complex changes in the epigenome/transcriptome of the early “primed” tumor cell. Together with powerful cell fate modifiers [such as epithelial-to-mesenchymal transition (EMT) and EMT-induced stemness], the changing tumor cell would then be equipped with pluripotency traits needed to fuel self-sustenance.
Interestingly, loss of TP53 and RB1 TSPs is also highly associated with small-cell lung cancer (SCLC), a histological subtype representing nearly 15% of all lung cancers (47). SCLC expresses various neuroendocrine markers including the proneural master transcription factors, ASCL1 and NEUROD1 (48). The non-small-cell lung cancer (NSCLC) subtype, accounting for the majority of the cases, includes adenocarcinoma and is often treated with tyrosine kinase inhibitors targeting epidermal growth factor receptor (EGFR)-activating mutations. SCLC can develop as part of a resistance mechanism to targeted EGFR therapy. Analysis of SCLC tumors utilizing patient tumors and mouse models suggest that the SCLC phenotype can be developed due to transformation or trans-differentiation of NSCLC adenocarcinoma, as a result of RB1 inactivation and/or loss of EGFR expression, as recently reviewed extensively (49, 50). In this regard, it is conceivable that development of both SCPC/NEPC and SCLC can be driven by similar cellular mechanisms involving cell fate changes (47, 50).
Resistance to antiandrogen therapy can also emerge as an AR-independent mechanism without development of the SCPC/NEPC phenotype, as recently revealed in two publications (51, 52). In this case, activation of the fibroblast growth factor and mitogen-activated protein kinase pathways can drive CRPC tumor growth in the absence of both AR and SCPC/NEPC markers (termed as “double negative”) (51). Another mechanism includes activation of the gastrointestinal (GI) lineage transcriptional program in CRPC, in which the resistant tumor cells utilize two hepatocyte nuclear factors (HNF1A and HNF4G), that drive an alternate lineage-specific program due to suppression of AR signaling (52). Since AR plays a key role in driving/sustaining the prostate luminal epithelial lineage program, loss of AR or activation of GI lineage transcription events would alter the luminal epithelial cell fate, in turn resulting in the onset of the AR-independent CRPC phenotype. The specific epigenetic mechanism/s and/or genomic deletion that might be initiating the AR-independent tumors without SCPC/NEPC phenotype are yet to be identified.
In summary, concurrent loss of the TSPs—PTEN, RB1, and TP53—permits powerful cell-fate adaptations (via altered epigenetic and transcriptional rewiring) that together allow the tumor cell a new capacity to transition to a distinct cell fate. Following this necessary and initial event, cell biological processes like EMT determine the ultimate phenotype of these altered tumor cells along with rapid development of therapy resistance, much like in other solid tumors such as breast or lung cancers. We speculate that continued expression of EMT and/or stem-cell factors would sustain the SCPC/NEPC tumors in a pluripotent yet reversible state. The challenge lies in identifying unique and significant modifiers of epithelial lineage plasticity. Understanding the contribution of these “second” hit events such as epigenetic and transcriptional alterations may offer workable therapeutic opportunities overcoming resistance to antiandrogen therapy.
Author Contributions
SM and RS developed conceptual framework and wrote the manuscript. AA, CL, and SAM developed conceptual framework.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the UT MD Anderson Prostate Cancer SPORE-CEP grant (RS), NCI-R01CA200970 (SAM), National Science Foundation-15-597-1605817 (SAM), the Prostate Cancer Foundation, and The UTMDACC Prostate Cancer Moon Shot Program.
References
1. Tetu B, Ro JY, Ayala AG, Johnson DE, Logothetis CJ, Ordonez NG. Small cell carcinoma of the prostate. Part I. A clinicopathologic study of 20 cases. Cancer (1987) 59(10):1803–9. doi:10.1002/1097-0142(19870515)59:10<1803::AID-CNCR2820591019>3.0.CO;2-X
2. Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov (2011) 1(6):466–8. doi:10.1158/2159-8290.CD-11-0259
3. Aparicio A, Tzelepi V, Araujo JC, Guo CC, Liang S, Troncoso P, et al. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient’s tumor: morphological, immunohistochemical, and gene expression profiles. Prostate (2011) 71(8):846–56. doi:10.1002/pros.21301
4. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov (2011) 1(6):487–95. doi:10.1158/2159-8290.CD-11-0130
5. Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang X, Reyes A, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol (2002) 20(14):3072–80. doi:10.1200/JCO.2002.12.065
6. Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol (2008) 32(1):65–71. doi:10.1097/PAS.0b013e318058a96b
7. Turbat-Herrera EA, Herrera GA, Gore I, Lott RL, Grizzle WE, Bonnin JM. Neuroendocrine differentiation in prostatic carcinomas. A retrospective autopsy study. Arch Pathol Lab Med (1988) 112(11):1100–5.
8. Mucci NR, Akdas G, Manely S, Rubin MA. Neuroendocrine expression in metastatic prostate cancer: evaluation of high throughput tissue microarrays to detect heterogeneous protein expression. Hum Pathol (2000) 31(4):406–14. doi:10.1053/hp.2000.7295
9. Tanaka M, Suzuki Y, Takaoka K, Suzuki N, Murakami S, Matsuzaki O, et al. Progression of prostate cancer to neuroendocrine cell tumor. Int J Urol (2001) 8(8):431–6. doi:10.1046/j.1442-2042.2001.00347.x
10. Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res (2004) 64(24):9209–16. doi:10.1158/0008-5472.CAN-04-2442
11. Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res (2013) 19(13):3621–30. doi:10.1158/1078-0432.CCR-12-3791
12. Logothetis CJ, Gallick GE, Maity SN, Kim J, Aparicio A, Efstathiou E, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov (2013) 3(8):849–61. doi:10.1158/2159-8290.CD-12-0460
13. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res (2014) 20(11):2846–50. doi:10.1158/1078-0432.CCR-13-3309
14. Beltran H. Update on the biology and management of neuroendocrine prostate cancer. Clin Adv Hematol Oncol (2016) 14(7):513–5.
15. Heidenberg HB, Sesterhenn IA, Gaddipati JP, Weghorst CM, Buzard GS, Moul JW, et al. Alteration of the tumor suppressor gene p53 in a high fraction of hormone refractory prostate cancer. J Urol (1995) 154(2 Pt 1):414–21. doi:10.1016/S0022-5347(01)67065-4
16. Cronauer MV, Schulz WA, Burchardt T, Ackermann R, Burchardt M. Inhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell lines. Oncogene (2004) 23(20):3541–9. doi:10.1038/sj.onc.1207346
17. Wang X, Deng H, Basu I, Zhu L. Induction of androgen receptor-dependent apoptosis in prostate cancer cells by the retinoblastoma protein. Cancer Res (2004) 64(4):1377–85. doi:10.1158/0008-5472.CAN-03-2428
18. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi:10.1038/nature11125
19. Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res (2012) 18(3):666–77. doi:10.1158/1078-0432.CCR-11-1867
20. Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res (2014) 20(4):890–903. doi:10.1158/1078-0432.CCR-13-1982
21. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161(5):1215–28. doi:10.1016/j.cell.2015.05.001
22. Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res (2016) 22(6):1520–30. doi:10.1158/1078-0432.CCR-15-1259
23. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22(3):298–305. doi:10.1038/nm.4045
24. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol (2017). doi:10.1200/PO.17.00029
25. Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res (2006) 66(16):7889–98. doi:10.1158/0008-5472.CAN-06-0486
26. Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res (2007) 67(12):5683–90. doi:10.1158/0008-5472.CAN-07-0768
27. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (2017) 355(6320):78–83. doi:10.1126/science.aah4199
28. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (2017) 355(6320):84–8. doi:10.1126/science.aah4307
29. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell (2016) 30(4):563–77. doi:10.1016/j.ccell.2016.09.005
30. Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov (2017) 7(1):54–71. doi:10.1158/2159-8290.CD-15-1263
31. Nikitin AY, Matoso A, Roy-Burman P. Prostate stem cells and cancer. Histol Histopathol (2007) 22(9):1043–9. doi:10.14670/HH-22.1043
32. Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, et al. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci U S A (2015) 112(47):E6544–52. doi:10.1073/pnas.1518007112
33. Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov (2017) 7(7):736–49. doi:10.1158/2159-8290.CD-16-1174
34. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature (2005) 436(7051):725–30. doi:10.1038/nature03918
35. Lunardi A, Ala U, Epping MT, Salmena L, Clohessy JG, Webster KA, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet (2013) 45(7):747–55. doi:10.1038/ng.2650
36. Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, et al. Characterization of the p53 cistrome – DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet (2013) 9(8):e1003726. doi:10.1371/journal.pgen.1003726
37. Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol (2005) 7(4):420–8. doi:10.1038/ncb1235
38. Siddiqui H, Fox SR, Gunawardena RW, Knudsen ES. Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol (2007) 211(1):131–7. doi:10.1002/jcp.20913
39. Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest (2010) 120(12):4478–92. doi:10.1172/JCI44239
40. Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell (2015) 16(1):39–50. doi:10.1016/j.stem.2014.10.019
41. Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev (2016) 30(13):1492–502. doi:10.1101/gad.282145.116
42. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature (2015) 525(7568):206–11. doi:10.1038/nature15251
43. Pourebrahim R, Zhang Y, Liu B, Gao R, Xiong S, Lin PP, et al. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev (2017) 31(18):1847–57. doi:10.1101/gad.304972.117
44. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res (2012) 72(2):527–36. doi:10.1158/0008-5472.CAN-11-3004
45. Paranjape AN, Soundararajan R, Werden SJ, Joseph R, Taube JH, Liu H, et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene (2016) 35(46):5963–76. doi:10.1038/onc.2015.498
46. Miao L, Yang L, Li R, Rodrigues DN, Crespo M, Hsieh JT, et al. Disrupting androgen receptor signaling induces snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res (2017) 77(11):3101–12. doi:10.1158/0008-5472.CAN-16-2169
47. Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer (2017) 17(12):725–37. doi:10.1038/nrc.2017.87
48. Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, et al. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep (2016) 16(5):1259–72. doi:10.1016/j.celrep.2016.06.081
49. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun (2015) 6:6377. doi:10.1038/ncomms7377
50. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol (2015) 16(4):e165–72. doi:10.1016/S1470-2045(14)71180-5
51. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell (2017) 32(4):474–489.e476. doi:10.1016/j.ccell.2017.09.003
Keywords: tumor suppressors, neuroendocrine prostate cancer, lineage plasticity, therapy resistance, epithelial-to-mesenchymal transition
Citation: Soundararajan R, Aparicio AM, Logothetis CJ, Mani SA and Maity SN (2018) Function of Tumor Suppressors in Resistance to Antiandrogen Therapy and Luminal Epithelial Plasticity of Aggressive Variant Neuroendocrine Prostate Cancers. Front. Oncol. 8:69. doi: 10.3389/fonc.2018.00069
Received: 13 December 2017; Accepted: 01 March 2018;
Published: 15 March 2018
Edited by:
Stéphane Terry, Institut Gustave Roussy, FranceReviewed by:
Tamara Lotan, Johns Hopkins University, United StatesDavid W. Goodrich, Roswell Park Cancer Institute, United States
Copyright: © 2018 Soundararajan, Aparicio, Logothetis, Mani and Maity. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sankar N. Maity, c21haXR5QG1kYW5kZXJzb24ub3Jn