 Alexandre Morrot1,2*†
Alexandre Morrot1,2*† Leonardo Marques da Fonseca3†
Leonardo Marques da Fonseca3† Eduardo J. Salustiano3†
Eduardo J. Salustiano3† Luciana Boffoni Gentile3
Luciana Boffoni Gentile3 Luciana Conde4
Luciana Conde4 Alessandra Almeida Filardy4
Alessandra Almeida Filardy4 Tatiany Nunes Franklim3Kelli Monteiro da Costa3
Tatiany Nunes Franklim3Kelli Monteiro da Costa3 Celio Geraldo Freire-de-Lima3
Celio Geraldo Freire-de-Lima3 Leonardo Freire-de-Lima3*
Leonardo Freire-de-Lima3*
- 1Faculdade de Medicina, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Laboratório de Imunoparasitologia, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz (Fiocruz), Rio de Janeiro, Brazil
- 3Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 4Instituto de Microbiologia, Departamento de Imunologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
The tumor microenvironment (TME) is composed by cellular and non-cellular components. Examples include the following: (i) bone marrow-derived inflammatory cells, (ii) fibroblasts, (iii) blood vessels, (iv) immune cells, and (v) extracellular matrix components. In most cases, this combination of components may result in an inhospitable environment, in which a significant retrenchment in nutrients and oxygen considerably disturbs cell metabolism. Cancer cells are characterized by an enhanced uptake and utilization of glucose, a phenomenon described by Otto Warburg over 90 years ago. One of the main products of this reprogrammed cell metabolism is lactate. “Lactagenic” or lactate-producing cancer cells are characterized by their immunomodulatory properties, since lactate, the end product of the aerobic glycolysis, besides acting as an inducer of cellular signaling phenomena to influence cellular fate, might also play a role as an immunosuppressive metabolite. Over the last 10 years, it has been well accepted that in the TME, the lactate secreted by transformed cells is able to compromise the function and/or assembly of an effective immune response against tumors. Herein, we will discuss recent advances regarding the deleterious effect of high concentrations of lactate on the tumor-infiltrating immune cells, which might characterize an innovative way of understanding the tumor-immune privilege.
Introduction
Cancer as a Metabolic Disease
Historically, cancer has been considered a product of multiple pathologies. In second century AD, the philosopher and physician Claudius Galenus was the first to employ the Greek word onco (swelling) for all types of tumors, leaving Hippocrates’ term karkinos exclusively for malignant tumors. During his time, Galenus asserted that tumors were the result of “black bile” accumulation. It was only during the nineteenth century that this theory was revisited and cancer begun to be perceived as the result of acquired metabolic abnormalities (1). Nowadays, it is well accepted that cancer development and progression is modulated by the disordered growth of cells featuring self-sufficiency of growth signals, evasion of apoptosis, angiogenesis, invasiveness, and metastasis (2, 3). When cells break free from the restraints on cell division, they start assuming inappropriate proliferation rates and distinct metabolic profiles, becoming abnormal in their own way (4, 5). Cells originating from solid tumors may gain the ability to dissolve the extracellular matrix (ECM), invade nearby tissues, reaching the bloodstream or lymphatic vessels, or remain within the boundaries of the original tissue, being characterized as malignant or benign tumors, respectively. Several genomic changes lead normal cells through malignant transformation. These changes can be anything from point mutations and deletions to chromosome rearrangements, as long as they result in irreversible changes affecting cell cycle (6). Any individual suffers several mutations in various cell types during its lifetime, due to diverse exogenous or endogenous factors. Most of these mutations are promptly corrected or lead to apoptosis. The accumulation of uncorrected mutations leads to the development of benign or malignant tumors (3). Loss of tumor suppressor factors, germ-line mutations, and overexpression of oncogenes are some of the changes that may collaborate for the occurrence of somatic mutations that escape DNA repair processes (7, 8).
The tumor microenvironment (TME) comprises both cellular and non-cellular components (9–11). The acellular components include the ECM, as well as soluble signals secreted by transformed and tumor-associated cancer cells. Several cell types associate with tumors, including fibroblasts, endothelial cells, and immune cells. Together, all components form an organ-like structure capable of interacting with the organism as a whole (12–14). To maintain tumor growth, several adaptations may be driven by neoplastic cells. A well-known mechanism is the formation of immature and abnormal vessels, in a phenomenon named neoangiogenesis (15, 16). In this context, both the platelet-derived growth factor and the vascular endothelial growth factor (VEGF) are recognized as the main proangiogenic signals upregulated by cancer cells during tumor growth (15, 17, 18).
The incredible proliferation rate of tumor cells can make a single mutated cell generate a tumor of ≈1 cm in diameter containing over 109 cells. Such a high proliferation ratio demands effective metabolic pathways, capable of meeting the steep energy requirements while supplying the biosynthetic precursors that maintain cell anabolism and redox balance in the neoplastic cell (19). Reprogramming of cellular metabolism has been observed in several types of cancer and is considered a hallmark of this disease (3, 20). The elucidation of these atypical metabolic activities is a lively field in the study of cancer biology, showing great potential for the development of novel therapeutic approaches. Several studies have shown that inhibition of some metabolic pathways of cancer cells is able to prevent tumor growth and metastasis (21, 22).
Metabolic Symbiosis: A Proposed Concept of Energy Management Between Cancer Cells in the TME
The impact of the acidosis induced by lactate and protons in the TME is a hot topic in cancer research (23–25). It is a well-established fact that a high enough lactate production can overcome the cellular proton buffering capability, resulting in a decrease of the cellular pH. Such condition, besides influencing the dynamics of waste and reuse of energy by cancer cells, modulates the function of distinct tumor-associated cell types as well (26–29). Several papers published over the last 10 years, demonstrated that when cancer cells experience low tension of oxygen, the hypoxia-inducible factor-1α (HIF-1α) transcription factor is stabilized, increasing glucose (Glc) uptake and secretion of substantial levels of lactate and protons out of cytoplasm by the monocarboxylated transporter 4 (MCT4) (Figure 1B), promoting a biochemical event termed lactic acidosis. By contrast, when cancer cells are adjacent to blood vessels and oxygen availability is sufficient, the transformed cells preferably use lactate as energy source (29–33). For this reason, lactate should not be considered a waste metabolite. In fact, it is reused by different cell subpopulations in a tumor (28, 29). Recently, Lee and colleagues (34) showed that an oxygen-regulated protein (NDGR3), which is usually degraded under normoxia via the prolyl hydroxylase 2/Von Hippel–Lindau (PDH2/VHL) system, becomes protected from degradation when bound to lactate. The authors demonstrated that when stable, NDRG3 is able to bind the proto-oncogene c-Raf, a serine/threonine-protein kinase, and induce activation of the Raf–ERK pathway, thus promoting cell growth and angiogenesis. However, inhibition of lactate production compromises NDRG3-mediated hypoxia responses (34).
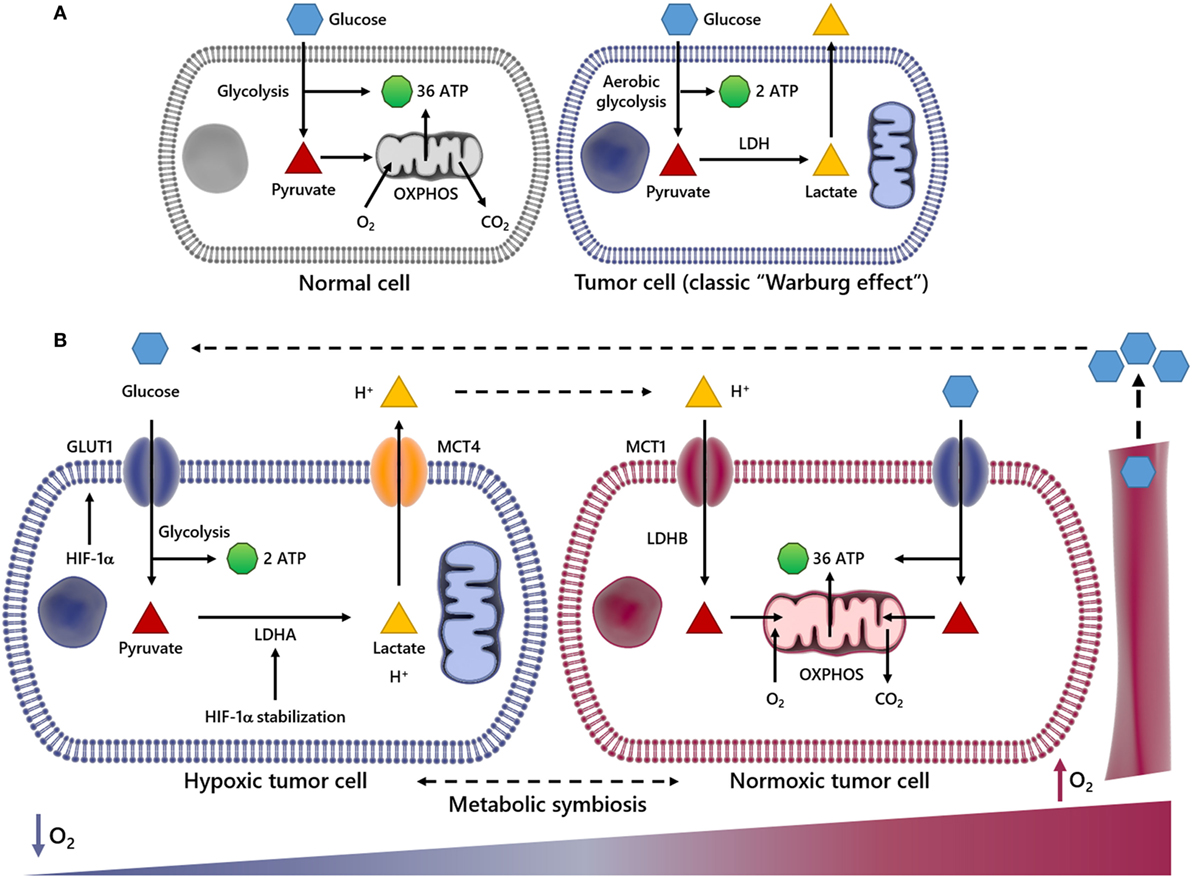
Figure 1. Scheme summarizing metabolic differences between normal and cancer cells and metabolic symbiosis. In normal cells, glucose (Glc) is initially metabolized to pyruvate and further to carbon dioxide (CO2) through tricarboxylic acid cycle and oxidative phosphorylation (OXPHOS) processes in the mitochondria, generating 36 ATP molecules per Glc molecule consumed (A). In this process, O2 is indispensable, since it is used as the final electron acceptor (A). However, in cancer cells undergoing aerobic glycolysis (Warburg effect), Glc is broken down into pyruvate and finally converted into lactate, deviating Glc metabolites from energy production to anabolic process. This event generates two ATP molecules per Glc molecule. The panel (B) illustrates an event named metabolic symbiosis. It has been well documented that when cancer cells are near or distant of blood vessels, they are well or poorly oxygenated, respectively. It is also known that when cancer cells are subject to low oxygen tension (↓O2) hypoxia-inducible factor-1α (HIF-1α) is stabilized, increasing the transcriptional activation of genes encoding glucose transporters (GLUTs), lactate dehydrogenase A (LDHA), as well as the uptake of Glc and secretion of lactate and protons out of cytoplasm through the monocarboxylated transporter 4 (MCT4). However, when transformed cells are close to blood vessels and the availability of O2 is enough, lactate is taken by monocarboxylated transporter 1 (MCT1) and utilized as energy source after conversion into pyruvate by lactate dehydrogenase B (LDHB). In this way, lactate may not be pointed out as a waste metabolite, since it is reused by different cell subpopulations in a tumor.
This metabolic model of lactate reuse in the TME has been described as a metabolic symbiosis, where lactate works as a medium to convey energy from highly glycolytic/hypoxic transformed cells to more oxidative cancer cells (35, 36). In the TME, the uptake of lactate and protons by oxygenated cancer cells occurs in a dynamic way through the monocarboxylated transporter 1 (MCT1) (Figure 1B), which has been previously identified as gatekeeper of this metabolic symbiosis (29). In the same study, the authors demonstrated that cells with inhibited or silenced MCT1 became more sensitive to cell death, which may support lactate management within the TME as a valid therapeutic strategy. Therefore, it would plausible to speculate that the high-lactate concentration at the intercellular space might affect the functionality of diverse tumor-associated cells, including those of the innate and adaptive immune system (see sections below).
The Effect of Lactate in the TME
In normal cell metabolism, the consumed Glc is catabolized into pyruvate, which is then transported to the mitochondria to fuel the tricarboxylic acid cycle in a series of redox reactions. The resulting free electrons go through the electron transport chain (ETC), beginning the oxidative phosphorylation (OXPHOS) and ultimately leading to a high production of ATP (37) (Figure 1A). In the early 1920s, Otto Warburg observed that tumor cells remain in glycolytic state, constitutively absorbing Glc and converting pyruvate to lactate, in the presence of oxygen (Figure 1A). Lactate production is 40-fold greater in tumor cells, so the transport of lactate to the ECM by MCTs (38–41) is essential for the glycolytic switch. This metabolic behavior is named “Warburg Effect,” or aerobic glycolysis, one of the main characteristics studied in cancer metabolism (19) (Figure 1B). Glycolysis produces ATP faster yet less efficiently than OXPHOS, forcing the tumor cell to consume much more Glc than a normal cell to produce enough energy to maintain its high proliferative status. Therefore, the glycolysis is an advantage for the tumor cell only when Glc supply is not limited. The importance of Glc for the metabolism of cancer cells is so evident that low-carbohydrate diet as a therapeutic approach for cancer patients, aiming to starve tumor cells, was described to limit the growth of incurable cancers in a pilot trial with 10 patients (42). In that regard, the uptake of a radioactive Glc analog, [18F]fluorodeoxyGlc, is used as a diagnostic tool for the positron emission tomography (FDG-PET) imaging of highly proliferative tumor regions (43, 44). Currently we know that tumor cells primarily fulfill their energetic needs by the oxidation of Glc, glutamine and other nutrients coupled to the ETC, using oxygen as the final acceptor of electrons (45, 46). In cancer cells, the anaerobic respiration is optional, and there is no mitochondrial defect (47); in fact, tumor cells still retain OXPHOS and mitochondrial activity (39). The reduced mitochondrial activity is a direct result either of oxygen deprivation or activation of HIF-1α (48, 49), which is able to promote the transcriptional activation of genes encoding glucose transporters (GLUTs), as well as glycolytic enzymes, such as lactate dehydrogenase A (LDHA) (50). When the supply of oxygen is low, LDHA is essential to sustain glycolysis and the production of ATP by regenerating NAD+ form NADH. In this way, HIF-1α regulates the production of lactate, the end by-product of this reaction, which consumes two ATP but generates four ATP, generating two net ATP per Glc molecule as seen in (Figures 1A,B) (50). Upstream of HIF-1α and the previously discussed Raf–ERK, the Ras oncogenic pathway seems to be critical for the metabolic reprogramming observed during carcinogenesis. Overexpression of oncogenic H-RasV12 was able to drive immortalized fibroblasts to consume more Glc and to release more lactate (51). Conversely, Ras inhibition in a model of glioblastoma (GBM) effectively shuts down Glc uptake and glycolysis itself, leading to the downregulation of 12 genes from the glycolytic pathway and increased extracellular pH due to reduced lactate efflux (52). The role of PDH2 is prominent in this, since oncogenic H-Ras signaling, as well as hypoxia, triggers oxidative stress, PDH2 dimerization, and inactivation, leading to HIF-1α stabilization and ultimately the OXPHOS to glycolysis shift (53).
An immediate consequence of the Warburg effect is the accumulation of lactate and protons in the TME (23, 54). It has been shown that in patients diagnosed with different stages of cervical cancer, primary tumors exhibiting high-lactate levels often lead to the manifestation of metastatic foci (55). The same group, using human larynx squamous carcinoma cells, showed that increased lactate concentration can augment cell motility and migration, corroborating the data observed in patients (56). In addition, the use of siRNA to inhibit the expression of LDHA, whose expression can be induced by lactate itself, is able to inhibit the migration of glioma cells as well as downregulate active matrix metalloproteinase-2 (57). Increased lactic acid is also responsible for the overexpression of factors related to tumor progression, such as CD44, hyaluronic acid and transforming growth factor-beta (TGF-β) (58) (Figure 2), a pro-carcinogenic cytokine able to activate the epithelial–mesenchymal transition process, an event that permits dissemination of tumor cells from the primary site into the surrounding stroma, setting the stage for metastatic spread (59–61). In addition, due to its antioxidant properties, increased lactate concentrations may offer a degree or resistance against any therapy relying on the production of oxygen reactive species, such as radiotherapy (62). As it stands, further studies on the production of lactate by solid tumors represent an important step toward the understanding of tumor progression and malignancy, as well as for therapy development.
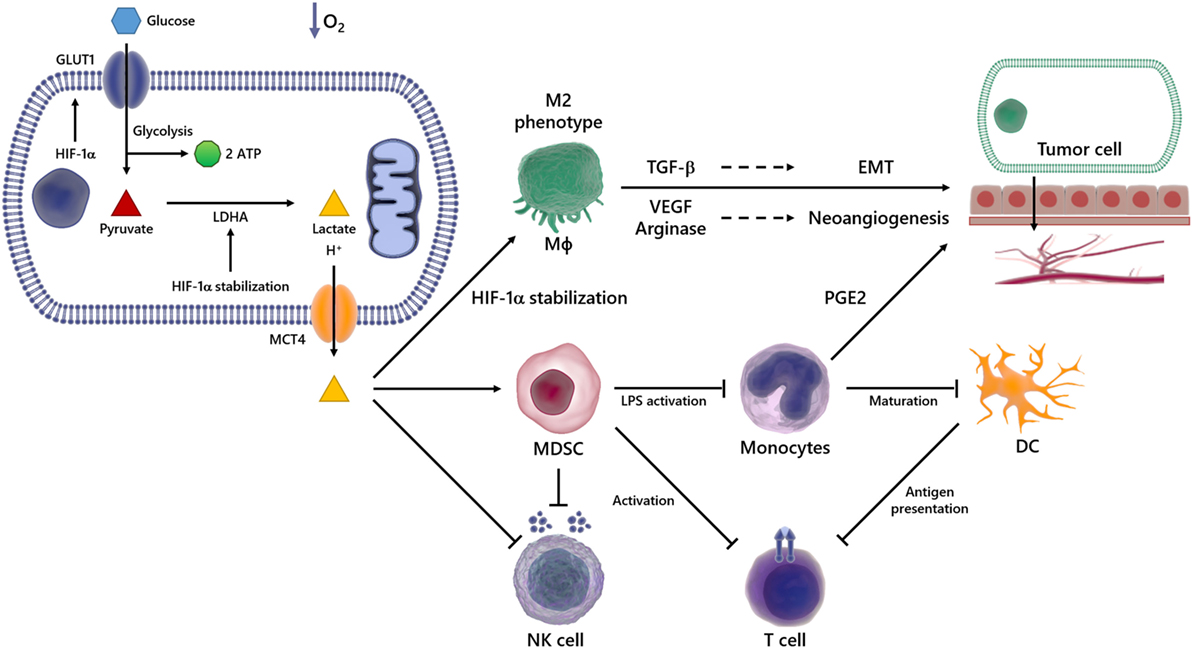
Figure 2. Overview of immunosuppressive effects of lactate in the tumor microenvironment (TME). In a hypoxic environment, Glc enters the cell via glucose transporter (GLUT) 1 and is broken down into pyruvate and then in lactate, which is transported out of the cell via monocarboxylated transporter 4 (MCT4). The lactate produced by transformed cells culminates in an acidified TME. This phenomenon is able to suppress the anticancer immune responses, particularly through impaired T and natural killer (NK) cells activation, reduced antigen presentation, compromised dendritic cell (DC) differentiation and maturation. It also promotes the emergence of the M2 Mϕ, which secretes high levels of pro-carcinogenic cytokines, such as transforming growth factor-beta (TGF-β) and vascular endothelial growth factor (VEGF), involved in processes such as epithelial–mesenchymal transition (EMT) and angiogenesis, events implicated in metastasis and cancer progression.
The TME is characterized by acidity and low oxygen tension (63, 64), events capable of modulating not only the growth and survival of tumor cells but also the recruitment of inflammatory cells that are reeducated in the microenvironment to favor tumor spread and metastasis. In this scenario, various inflammatory cells, such as T lymphocytes, dendritic cells (DCs), natural killer (NK) cells, and macrophages (Mϕ), acquire pro-carcinogenic properties (Figure 2) (65–69). Recruitment and accumulation of those cells in the TME is an essential phenomenon to sustain the tumor growth (70). The immune system’s role in the first phases of tumor establishment is well described, being capable of detecting and destroying cancer cells, halting their growth and spread, in a phenomenon termed immunosurveillance (71, 72), which was initially proposed by Paul Ehrlich and later developed by Sir Frank Macfarlane Burnet and Lewis Thomas (13, 73). Defects in this event might favor tumor progression and, consequently, the acquisition of a malignant phenotype. Any cancer cell that manages to escape death triggered by immune response could still have their proliferation hindered by immune mechanisms, reaching an equilibrium. On the other hand, the immunogenicity is molded through selective pressure exerted by the immune system, in an event termed immunoediting (74–77). Consequently, novel tumor variants emerge, bearing more mutations, making them more likely to evade detection and elimination by immune effector cells like NK and CD8+ T cells (74, 78). The immunoediting stage is the longest phase, and it is characterized by the dynamic coevolution of cancer and immune cells (78–80). Ultimately, cells reach an escape phase, where the accumulation of edited cells drives tumor growth and the manifestation of clinical symptoms (74). The presence of the immune system in the TME undoubtedly compromises tumor growth and, in fact, correlates with favorable prognosis in some cancer types, such as renal, ovarian, colorectal, and breast cancers (74, 81). The expression of molecules able to compromise cell-to-cell interaction (3, 82–84), as well as soluble factors such as VEGF (85), cytokines (86–88), prostaglandin E2 (PGE2) (89), soluble Fas and FasL (90), and soluble MICA (91), all contribute to the appearance of multifaceted local and regional immunosuppressive networks (92–94). For example, the occurrence of the IL-4, TGF-β, IL-13, and IL-10 cytokines in the TME induces the emergence of M2 instead of M1 Mϕ (86, 87). In addition, secretion of nitric oxide, IL-10, arginase-1, IL-6, and VEGF promotes cell death and avoids the antitumor function of immune cells (95–99).
As stated earlier, the TME is rich in lactate (38–41), an immunosuppressive soluble factor that promotes cancer development (54, 74, 100). Particularly, several studies have shown that tumor-derived lactate is capable of inhibiting the activation of immune cells such as monocytes, Mϕ and T lymphocytes (101–103). It has been demonstrated that high LDHA levels are deeply correlated with tumor size, as well as with the clinical stages of the disease (104, 105). Accordingly, the infiltration of immune cells in the TME correlates with high LDHA expression and/or activity (106). Besides lactate accumulation in the primary tumor site, its immunosuppressive properties can outspread to distant sites, thus stimulating invasion and metastasis in a paracrine fashion (107) (Figure 2).
Despite being mainly produced by skeletal muscle, various tissues generate lactate, and its elimination in healthy conditions is handled primarily through the liver and secondarily through the kidneys (108). The citric acid cycle is also a source of lactate, as pyruvate can be diverted to lactate and NAD+ generation through LDH activity (109, 110). The high amounts of lactate in the extracellular microenvironment contribute to lowering the extracellular pH, which can be as low as 6.0–6.5 (111), producing acidosis and inducing angiogenesis and a reduction in efficacy of the immune system (101, 112, 113). Tumor-associated immune cells from myeloid and lymphoid origin can be modulated by hypoxic conditions as well as high levels of lactate, then favoring the acquisition of malignant phenotypes (23, 64, 114–116).
Lactate and Myeloid Cells
Over the last 15 years, several studies demonstrated that tumor-derived lactate modulates the functionality of immune cells, contributing to the establishment of an immunosuppressive microenvironment, which favors the developing of tumors (106, 117–119). Lactate promotes the development of myeloid-derived suppressor cells (MDSCs), the most prominent bone marrow-derived cell population that exerts broadly immunosuppressive functions (106). In the TME, MDSCs potently suppress both innate and adaptive immunity by preventing the maturation of DCs, suppressing NK-cell cytotoxicity, inhibiting T-cell activation, and favoring the differentiation of regulatory T cells (Figure 2) (117).
In addition, lactate suppresses monocytes’ LPS-induced activation by influencing their gene expression. Particularly, the expression of most LPS-induced genes was significantly delayed in the presence of lactate, including TNF, IL-23, CCL2, and CCL17. These effects are mediated by delayed LPS-induced phosphorylation of protein kinase B (AKT) and degradation of IkB, with reduced nuclear accumulation of NFκB (119). Furthermore, lactate stabilizes the transcription factor HIF-1α in monocytes, which ultimately promote the expression of PGE2 and the growth of human colon cancer HCT116 cells (120).
Another suppressive function of lactate is to impair the differentiation of monocytes into Mϕ or DCs in the TME (118, 121–123) and in non-tumor conditions (103, 124) (Figure 2). It was reported that lactate blocks LPS activation of bone marrow-derived Mϕ (BMDMs) (123), and also, in hypoxia or normoxia, lactate drives tumor-associated Mϕ polarization to the “tumor friendly” M2 profile (Figure 2) (125, 126). Mechanistically, lactate stabilizes HIF-1α, which leads to the transcription of a broader set of M2-associated genes, including VEGF, TGF-β and arginase-1, as well as Fizz1, Mgl1, and Mgl2 (Figure 2) (98, 125–128). The role of PDH2 as a regulator of the metabolic reprogramming in Mϕ was observed in both RAW264 cells and in primary BMDM, since transfection with shRNA targeting PDH2 or conditional PDH2 knocking out led to decreased ATP levels along and increased lactate release into the medium (129). M2 Mϕ and their products favor tumor growth and metastasis by suppressing antitumor immune responses, activating and enhancing angiogenesis. Particularly, VEGF triggers the development of neovascularization of the tumor. Similarly, arginase-1 plays an indirect role in angiogenesis through reorganization of the tumor ECM and contributes for the generation of essential metabolites during cell division, such as polyamines, supporting cancer cells growth (130–133). The importance of arginase-1 in tumor development was demonstrated by the use of arginase-1 KO mice, which presented tumors 50% smaller than tumors from wild type mice (125, 134). Finally, distinct studies have shown that when present in high levels, lactate inhibits antigen presentation and IL-12 production by DCs (54, 135) (Figure 2) and enhances IL-10 production as well, generating an immunosuppressive profile in the TME (136, 137).
Lactate and Lymphoid Cells
The immunobiological effects of lactate on immune cells from lymphoid origin have been mainly investigated in NK and T cells. The cytotoxic effect mediated by both cell types is of fundamental importance in immunological surveillance against the emergence and spread of malignant disease. NK cells represent large granular lymphocytes that induce their antitumor responses through the ligation of particular cell-surface receptors (138), such as the natural killer group 2, member D receptor (NKG2D), which induces the release of cytotoxic granules that promote the lysis of cancer cells (139, 140).
An elegant study developed by Husain and colleagues (106) revealed that the low production of lactate in LDHA-depleted tumors was able to improve the cytolytic functions of NK cells. However, when NK cells where pretreated with lactate in vitro, its cytolytic property was compromised and/or abrogated. The molecular mechanism responsible for such effect was investigated, and the authors demonstrated that the decline of NK cytotoxic activity was promoted by the lower expression of granzyme and perforin in lactate-treated cells (106). Furthermore, it was described that lactate works as a potent inhibitor of histone deacetylases, suggesting that lactate might be able to regulate (at transcriptional level) several genes involved not only in cell metabolism but also in immune responses, such as NCR1/NKp46, an activating NK cell receptor (141, 142).
In 2014, Crane and coworkers demonstrated that GBM cells secrete LDH-5, an enzymatically active isoform of LDH (143), that besides catalyzing the conversion of pyruvate to lactate in an efficient way (144), is also capable to upregulate HIF-related pathways (145) and induce the expression of NKG2D ligands on healthy monocytes, thus subverting antitumor immune responses (143). In a previous study realized by the same group, it was demonstrated that TGF-β downregulates NKG2D expression in NK cells in vitro (146), supporting the idea that the elevated production/concentration of TGF-β in acidic TME is one of the main evasion mechanisms adopted by cancer cells (146).
It is well accepted that a robust presence of T cells in the TME is associated with good clinical outcome in distinct types of cancers (147–149). It is important to notice that new progresses in cancer immunotherapy are allied to the use of monoclonal antibodies directed against T cell-immune checkpoints. Examples include CTLA4 (149–151) and PD-1 (152, 153). These outstanding findings undoubtedly confirm the necessity of an effective T cell activation to control tumor growth and spread (147). As with other types of immune cells, cancer cells limit T cell immunity by distinct ways. In this regard, the acidification of the TME is a clear example, and several papers have demonstrated that lactate plays a pivotal role in this process (54, 154–156). As with transformed cells, activated T cells may generate energy through aerobic glycolysis (157–159). The upregulation of glycolytic enzymes intensifies the uptake of Glc and glycolytic rate, favoring the secretion of lactate into the microenvironment (157). It is possible to imagine that when together in the TME, both cancer cells and activated T lymphocytes significantly increase the production/secretion of lactate. As it stands, it has been demonstrated that the very acidic microenvironment suppresses the proliferation and cytokine production by activated T cells (101). A recent study developed by Brand and colleagues revealed that pathophysiological concentrations of lactic acid repeal the upregulation of the nuclear factor of activated T cells in both NK and T cells, which significantly reduced the production of IFN-γ (160). These results corroborate previous findings that lactic acid is able to downmodulate the function of cells from lymphoid origin, then contributing to tumor escape from immune attack.
Concluding Remarks and Perspectives
This review presents a snapshot of metabolic changes in cancer cells, describing how, even in aerobic conditions, transformed cells opt for glycolysis instead of OXPHOS to sustain their energy demand, high proliferation and biosynthesis rates, a process named “Warburg effect.” This metabolic reprogramming culminates in a high-lactate and protons output, which is also exported to the extracellular environment by MCT4, generating acidosis, neoangiogenesis, and immunosuppression, directly modulating the TME. Although several genetic, biochemical, and pathophysiologic mechanisms have been identified as causes of malignancy in high-lactate tumors, it remains unclear why seemingly identical tumors may exhibit extreme differences in their lactate levels. Although it is certainly another challenge for future research in this field, several reports point out that high-lactate amounts help in generating a hostile microenvironment for normal cells, affecting the activation and differentiation of effector immune cells as well as antigen presentation and the production of cytokines. Future studies, particularly in solid tumors characterized by highly acidic environments, are needed to better understand the effect of lactate and other “waste” metabolites on cancer progression. The participation of lactate in TME and its immunosuppressive actions not only make it crucial for tumor survival and growth but also turns it into an interesting and promising candidate to therapeutic target in cancer chemotherapy. Following this reasoning, classic and novel drugs that modulate TME pH might be useful as potential immunomodulatory tools in cancer patients, particularly in combination with immunotherapeutic strategies.
Author Contributions
Wrote the paper: AM, LF, LG, LC, AF, TF, KC, ES, CF-d-L, and LF-d-L. All the authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from Fundação do Câncer, Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq).
References
1. Hajdu SI, Vadmal M, Tang P. A note from history: landmarks in history of cancer, part 7. Cancer (2015) 121(15):2480–513. doi:10.1002/cncr.29365
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
4. Zhang YS, Duchamp M, Oklu R, Ellisen LW, Langer R, Khademhosseini A. Bioprinting the cancer microenvironment. ACS Biomater Sci Eng (2016) 2(10):1710–21. doi:10.1021/acsbiomaterials.6b00246
5. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J (2017) 36(10):1302–15. doi:10.15252/embj.201696151
7. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell (1990) 61(5):759–67. doi:10.1016/0092-8674(90)90186-I
8. Solomon E, Borrow J, Goddard AD. Chromosome aberrations and cancer. Science (1991) 254(5035):1153–60. doi:10.1126/science.1957167
9. Novikova MV, Khromova NV, Kopnin PB. Components of the hepatocellular carcinoma microenvironment and their role in tumor progression. Biochemistry (Mosc) (2017) 82(8):861–73. doi:10.1134/S0006297917080016
10. Timaner M, Beyar-Katz O, Shaked Y. Analysis of the stromal cellular components of the solid tumor microenvironment using flow cytometry. Curr Protoc Cell Biol (2016) 70:1981–82. doi:10.1002/0471143030.cb1918s70
11. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi:10.1038/nm.3394
12. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell (2010) 18(6):884–901. doi:10.1016/j.devcel.2010.05.012
13. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res (1970) 13:1–27. doi:10.1159/000386035
14. Andon FT, Digifico E, Maeda A, Erreni M, Mantovani A, Alonso MJ, et al. Targeting tumor associated macrophages: the new challenge for nanomedicine. Semin Immunol (2017) 34:103–13. doi:10.1016/j.smim.2017.09.004
15. Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res (2006) 66(11):5527–36. doi:10.1158/0008-5472.CAN-05-4128
16. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag (2006) 2(3):213–9. doi:10.2147/vhrm.2006.2.3.213
17. Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev (1987) 6(4):559–93. doi:10.1007/BF00047468
18. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi:10.1158/0008-5472.CAN-06-1278
19. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
20. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi:10.1016/j.cmet.2015.12.006
21. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell (2013) 24(2):213–28. doi:10.1016/j.ccr.2013.06.014
22. Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A (2015) 112(21):6539–44. doi:10.1073/pnas.1507228112
23. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer (2017) 17(10):577–93. doi:10.1038/nrc.2017.77
24. Dang CV, Kim JW. Convergence of cancer metabolism and immunity: an overview. Biomol Ther (Seoul) (2018) 26(1):4–9. doi:10.4062/biomolther.2017.194
25. Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol (2011) 2:49. doi:10.3389/fphar.2011.00049
26. Danhier P, Banski P, Payen VL, Grasso D, Ippolito L, Sonveaux P, et al. Cancer metabolism in space and time: beyond the Warburg effect. Biochim Biophys Acta (2017) 1858(8):556–72. doi:10.1016/j.bbabio.2017.02.001
27. Kobliakov VA. Role of proton pumps in tumorigenesis. Biochemistry (Mosc) (2017) 82(4):401–12. doi:10.1134/S0006297917040010
28. Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest (2008) 118(12):3835–7. doi:10.1172/JCI37373
29. Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118(12):3930–42. doi:10.1172/JCI36843
30. Strickaert A, Saiselet M, Dom G, De Deken X, Dumont JE, Feron O, et al. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene (2017) 36(19):2637–42. doi:10.1038/onc.2016.411
31. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci (2016) 73(7):1333–48. doi:10.1007/s00018-015-2098-5
32. Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, et al. Proton channels and exchangers in cancer. Biochim Biophys Acta (2015) 1848(10 Pt B):2715–26. doi:10.1016/j.bbamem.2014.10.015
33. Perez-Escuredo J, Van Hee VF, Sboarina M, Falces J, Payen VL, Pellerin L, et al. Monocarboxylate transporters in the brain and in cancer. Biochim Biophys Acta (2016) 1863(10):2481–97. doi:10.1016/j.bbamcr.2016.03.013
34. Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, et al. A lactate-induced response to hypoxia. Cell (2015) 161(3):595–609. doi:10.1016/j.cell.2015.03.011
35. Kim SY. Cancer energy metabolism: shutting power off cancer factory. Biomol Ther (Seoul) (2018) 26(1):39–44. doi:10.4062/biomolther.2017.184
36. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res (2015) 34:111. doi:10.1186/s13046-015-0221-y
37. Vazquez A, Kamphorst JJ, Markert EK, Schug ZT, Tardito S, Gottlieb E. Cancer metabolism at a glance. J Cell Sci (2016) 129(18):3367–73. doi:10.1242/jcs.181016
38. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi:10.1085/jgp.8.6.519
39. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi:10.1038/nrc3038
40. Holm E, Hagmuller E, Staedt U, Schlickeiser G, Gunther HJ, Leweling H, et al. Substrate balances across colonic carcinomas in humans. Cancer Res (1995) 55(6):1373–8.
41. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest (2013) 123(9):3685–92. doi:10.1172/JCI69741
42. Fine EJ, Segal-Isaacson CJ, Feinman RD, Herszkopf S, Romano MC, Tomuta N, et al. Targeting insulin inhibition as a metabolic therapy in advanced cancer: a pilot safety and feasibility dietary trial in 10 patients. Nutrition (2012) 28(10):1028–35. doi:10.1016/j.nut.2012.05.001
43. Jadvar H, Alavi A, Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med (2009) 50(11):1820–7. doi:10.2967/jnumed.108.054098
44. Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer (2002) 2(9):683–93. doi:10.1038/nrc882
45. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol (2013) 9:712. doi:10.1038/msb.2013.65
46. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell (2016) 164(4):681–94. doi:10.1016/j.cell.2015.12.034
47. Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol (2009) 19(1):4–11. doi:10.1016/j.semcancer.2008.11.008
48. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi:10.1016/j.cmet.2006.02.002
49. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3(3):187–97. doi:10.1016/j.cmet.2006.01.012
50. Valvona CJ, Fillmore HL, Nunn PB, Pilkington GJ. The regulation and function of lactate dehydrogenase a: therapeutic potential in brain tumor. Brain Pathol (2016) 26(1):3–17. doi:10.1111/bpa.12299
51. Mazure NM, Chen EY, Yeh P, Laderoute KR, Giaccia AJ. Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res (1996) 56(15):3436–40.
52. Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res (2005) 65(3):999–1006.
53. Lee G, Won HS, Lee YM, Choi JW, Oh TI, Jang JH, et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1alpha activation. Sci Rep (2016) 6:18928. doi:10.1038/srep18928
54. Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol (2016) 7:52. doi:10.3389/fimmu.2016.00052
55. Walenta S, Salameh A, Lyng H, Evensen JF, Mitze M, Rofstad EK, et al. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am J Pathol (1997) 150(2):409–15.
56. Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol (2011) 39(2):453–63. doi:10.3892/ijo.2011.1055
57. Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol (2009) 11(4):368–80. doi:10.1215/15228517-2008-106
58. Rudrabhatla SR, Mahaffey CL, Mummert ME. Tumor microenvironment modulates hyaluronan expression: the lactate effect. J Invest Dermatol (2006) 126(6):1378–87. doi:10.1038/sj.jid.5700255
59. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi:10.1172/JCI39104
60. Micalizzi DS, Ford HL. Epithelial-mesenchymal transition in development and cancer. Future Oncol (2009) 5(8):1129–43. doi:10.2217/fon.09.94
61. Freire-de-Lima L. Sweet and sour: the impact of differential glycosylation in cancer cells undergoing epithelial-mesenchymal transition. Front Oncol (2014) 4:59. doi:10.3389/fonc.2014.00059
62. Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol (2010) 94(1):102–9. doi:10.1016/j.radonc.2009.11.007
63. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev (2011) 91(3):1071–121. doi:10.1152/physrev.00038.2010
64. Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-inducible factors and cancer. Curr Sleep Med Rep (2017) 3(1):1–10. doi:10.1007/s40675-017-0062-7
65. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi:10.1038/ni1102-991
66. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi:10.1155/2014/149185
67. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi:10.1016/j.ccr.2012.02.022
68. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72(11):2746–56. doi:10.1158/0008-5472.CAN-11-1272
69. Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res (2012) 18(5):1207–13. doi:10.1158/1078-0432.CCR-11-1591
70. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi:10.1126/science.1203486
71. Porta C, Larghi P, Rimoldi M, Totaro MG, Allavena P, Mantovani A, et al. Cellular and molecular pathways linking inflammation and cancer. Immunobiology (2009) 214(9–10):761–77. doi:10.1016/j.imbio.2009.06.014
72. Malmberg KJ, Carlsten M, Bjorklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol (2017) 31:20–9. doi:10.1016/j.smim.2017.08.002
73. Manjili MH. Revisiting cancer immunoediting by understanding cancer immune complexity. J Pathol (2011) 224(1):5–9. doi:10.1002/path.2865
74. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest (2015) 125(9):3338–46. doi:10.1172/JCI80004
75. Sistigu A, Di Modugno F, Manic G, Nistico P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev (2017) 36:67–77. doi:10.1016/j.cytogfr.2017.05.008
76. Wang M, Zhang C, Song Y, Wang Z, Wang Y, Luo F, et al. Mechanism of immune evasion in breast cancer. Onco Targets Ther (2017) 10:1561–73. doi:10.2147/OTT.S126424
77. O’Reilly E, Tirincsi A, Logue SE, Szegezdi E. The Janus face of death receptor signaling during tumor immunoediting. Front Immunol (2016) 7:446. doi:10.3389/fimmu.2016.00446
78. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21(2):137–48. doi:10.1016/j.immuni.2004.07.017
79. Schreiber RD. Cancer vaccines 2004 opening address: the molecular and cellular basis of cancer immunosurveillance and immunoediting. Cancer Immun (2005) 5(Suppl 1):1.
80. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
81. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi:10.1056/NEJMoa020177
82. van Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur J Cell Biol (2003) 82(11):539–48. doi:10.1078/0171-9335-00346
83. Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res (2012) 72(13):3125–30. doi:10.1158/0008-5472.CAN-11-4094
84. Yu P, Rowley DA, Fu YX, Schreiber H. The role of stroma in immune recognition and destruction of well-established solid tumors. Curr Opin Immunol (2006) 18(2):226–31. doi:10.1016/j.coi.2006.01.004
85. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92(11):4150–66.
86. Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett (2009) 123(2):97–102. doi:10.1016/j.imlet.2009.02.011
87. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol (2008) 18(5):349–55. doi:10.1016/j.semcancer.2008.03.004
88. Beck C, Schreiber H, Rowley D. Role of TGF-beta in immune-evasion of cancer. Microsc Res Tech (2001) 52(4):387–95. doi:10.1002/1097-0029(20010215)52:4<387:AID-JEMT1023>3.0.CO;2-W
89. He X, Stuart JM. Prostaglandin E2 selectively inhibits human CD4+ T cells secreting low amounts of both IL-2 and IL-4. J Immunol (1999) 163(11):6173–9.
90. Erdogan B, Uzaslan E, Budak F, Karadag M, Ediger D, Oral B, et al. The evaluation of soluble Fas and soluble Fas ligand levels of bronchoalveolar lavage fluid in lung cancer patients. Tuberk Toraks (2005) 53(2):127–31.
91. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICB in malignant diseases: analysis of diagnostic significance and correlation with soluble MICA. Cancer Immunol Immunother (2006) 55(12):1584–9. doi:10.1007/s00262-006-0167-1
92. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
93. Santoni M, Berardi R, Amantini C, Burattini L, Santini D, Santoni G, et al. Role of natural and adaptive immunity in renal cell carcinoma response to VEGFR-TKIs and mTOR inhibitor. Int J Cancer (2014) 134(12):2772–7. doi:10.1002/ijc.28503
94. Bernal M, Concha A, Saenz-Lopez P, Rodriguez AI, Cabrera T, Garrido F, et al. Leukocyte infiltrate in gastrointestinal adenocarcinomas is strongly associated with tumor microsatellite instability but not with tumor immunogenicity. Cancer Immunol Immunother (2011) 60(6):869–82. doi:10.1007/s00262-011-0999-1
95. Botti C, Seregni E, Ferrari L, Martinetti A, Bombardieri E. Immunosuppressive factors: role in cancer development and progression. Int J Biol Markers (1998) 13(2):51–69.
96. Gastman BR, Johnson DE, Whiteside TL, Rabinowich H. Tumor-induced apoptosis of T lymphocytes: elucidation of intracellular apoptotic events. Blood (2000) 95(6):2015–23.
97. Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother (2001) 24(5):392–407. doi:10.1097/00002371-200109000-00002
98. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
99. Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol (2006) 16(1):3–15. doi:10.1016/j.semcancer.2005.07.008
100. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5(200):200ra116. doi:10.1126/scitranslmed.3006504
101. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
102. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol (2010) 184(3):1200–9. doi:10.4049/jimmunol.0902584
103. Puig-Kroger A, Pello OM, Selgas R, Criado G, Bajo MA, Sanchez-Tomero JA, et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol (2003) 73(4):482–92. doi:10.1189/jlb.0902451
104. Girgis H, Masui O, White NM, Scorilas A, Rotondo F, Seivwright A, et al. Lactate dehydrogenase A is a potential prognostic marker in clear cell renal cell carcinoma. Mol Cancer (2014) 13:101. doi:10.1186/1476-4598-13-101
105. Sun X, Sun Z, Zhu Z, Guan H, Zhang J, Zhang Y, et al. Clinicopathological significance and prognostic value of lactate dehydrogenase A expression in gastric cancer patients. PLoS One (2014) 9(3):e91068. doi:10.1371/journal.pone.0091068
106. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol (2013) 191(3):1486–95. doi:10.4049/jimmunol.1202702
107. Dart A. Tumour metabolism: lactic acid: not just a waste product? Nat Rev Cancer (2016) 16(11):676–7. doi:10.1038/nrc.2016.109
108. Sola-Penna M. Metabolic regulation by lactate. IUBMB Life (2008) 60(9):605–8. doi:10.1002/iub.97
109. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104(49):19345–50. doi:10.1073/pnas.0709747104
110. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol (2009) 92(3):329–33. doi:10.1016/j.radonc.2009.06.025
111. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase – a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab (2014) 19(5):795–809. doi:10.1016/j.cmet.2014.03.003
112. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer (2012) 131(3):633–40. doi:10.1002/ijc.26410
113. Husain Z, Seth P, Sukhatme VP. Tumor-derived lactate and myeloid-derived suppressor cells: linking metabolism to cancer immunology. Oncoimmunology (2013) 2(11):e26383. doi:10.4161/onci.26383
114. Kato Y, Ozawa S, Tsukuda M, Kubota E, Miyazaki K, St-Pierre Y, et al. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J (2007) 274(12):3171–83. doi:10.1111/j.1742-4658.2007.05848.x
115. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11(6):393–410. doi:10.1038/nrc3064
116. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60(4):916–21.
117. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
118. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood (2006) 107(5):2013–21. doi:10.1182/blood-2005-05-1795
119. Peter K, Rehli M, Singer K, Renner-Sattler K, Kreutz M. Lactic acid delays the inflammatory response of human monocytes. Biochem Biophys Res Commun (2015) 457(3):412–8. doi:10.1016/j.bbrc.2015.01.005
120. Wei L, Zhou Y, Yao J, Qiao C, Ni T, Guo R, et al. Lactate promotes PGE2 synthesis and gluconeogenesis in monocytes to benefit the growth of inflammation-associated colorectal tumor. Oncotarget (2015) 6(18):16198–214. doi:10.18632/oncotarget.3838
121. Gottfried E, Kreutz M, Haffner S, Holler E, Iacobelli M, Andreesen R, et al. Differentiation of human tumour-associated dendritic cells into endothelial-like cells: an alternative pathway of tumour angiogenesis. Scand J Immunol (2007) 65(4):329–35. doi:10.1111/j.1365-3083.2007.01903.x
122. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology (2014) 146(7):1763–74. doi:10.1053/j.gastro.2014.03.014
123. Errea A, Cayet D, Marchetti P, Tang C, Kluza J, Offermanns S, et al. Lactate inhibits the pro-inflammatory response and metabolic reprogramming in murine macrophages in a GPR81-independent manner. PLoS One (2016) 11(11):e0163694. doi:10.1371/journal.pone.0163694
124. Selleri S, Bifsha P, Civini S, Pacelli C, Dieng MM, Lemieux W, et al. Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget (2016) 7(21):30193–210. doi:10.18632/oncotarget.8623
125. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi:10.1038/nature13490
126. Colegio OR. Lactic acid polarizes macrophages to a tumor-promoting state. Oncoimmunology (2016) 5(3):e1014774. doi:10.1080/2162402X.2015.1014774
127. Yang Z, Ming XF. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front Immunol (2014) 5:533. doi:10.3389/fimmu.2014.00533
128. Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J (2003) 17(14):2115–7. doi:10.1096/fj.03-0329fje
129. Guentsch A, Beneke A, Swain L, Farhat K, Nagarajan S, Wielockx B, et al. PHD2 is a regulator for glycolytic reprogramming in macrophages. Mol Cell Biol (2017) 37(1):e00236-16. doi:10.1128/MCB.00236-16
130. Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res (2001) 61(3):1100–6.
131. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi:10.3389/fimmu.2014.00614
132. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
133. Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol (2001) 70(4):478–90.
134. Korrer MJ, Zhang Y, Routes JM. Possible role of arginase-1 in concomitant tumor immunity. PLoS One (2014) 9(3):e91370. doi:10.1371/journal.pone.0091370
135. Dong H, Bullock TN. Metabolic influences that regulate dendritic cell function in tumors. Front Immunol (2014) 5:24. doi:10.3389/fimmu.2014.00024
136. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25(7):771–84. doi:10.1038/cr.2015.68
137. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213(1):15–23. doi:10.1084/jem.20151570
138. Campbell AR, Duggan MC, Suarez-Kelly LP, Bhave N, Opheim KS, McMichael EL, et al. MICA-expressing monocytes enhance natural killer cell Fc receptor-mediated antitumor functions. Cancer Immunol Res (2017) 5(9):778–89. doi:10.1158/2326-6066.CIR-16-0005
139. Prajapati K, Perez C, Rojas LBP, Burke B, Guevara-Patino JA. Functions of NKG2D in CD8(+) T cells: an opportunity for immunotherapy. Cell Mol Immunol (2018) 14:1–10. doi:10.1038/cmi.2017.161
140. Ding H, Yang X, Wei Y. Fusion proteins of NKG2D/NKG2DL in cancer immunotherapy. Int J Mol Sci (2018) 19(1):E177. doi:10.3390/ijms19010177
141. Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res (2012) 40(11):4794–803. doi:10.1093/nar/gks066
142. Lai CB, Mager DL. Role of runt-related transcription factor 3 (RUNX3) in transcription regulation of natural cytotoxicity receptor 1 (NCR1/NKp46), an activating natural killer (NK) cell receptor. J Biol Chem (2012) 287(10):7324–34. doi:10.1074/jbc.M111.306936
143. Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A (2014) 111(35):12823–8. doi:10.1073/pnas.1413933111
144. Kayser G, Kassem A, Sienel W, Schulte-Uentrop L, Mattern D, Aumann K, et al. Lactate-dehydrogenase 5 is overexpressed in non-small cell lung cancer and correlates with the expression of the transketolase-like protein 1. Diagn Pathol (2010) 5:22. doi:10.1186/1746-1596-5-22
145. Koukourakis MI, Giatromanolaki A, Simopoulos C, Polychronidis A, Sivridis E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin Exp Metastasis (2005) 22(1):25–30. doi:10.1007/s10585-005-2343-7
146. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol (2010) 12(1):7–13. doi:10.1093/neuonc/nop009
147. Molon B, Cali B, Viola A. T cells and cancer: how metabolism shapes immunity. Front Immunol (2016) 7:20. doi:10.3389/fimmu.2016.00020
148. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
149. Jain P, Jain C, Velcheti V. Role of immune-checkpoint inhibitors in lung cancer. Ther Adv Respir Dis (2018) 12:1753465817750075. doi:10.1177/1753465817750075
150. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
151. Alaia C, Boccellino M, Zappavigna S, Amler E, Quagliuolo L, Rossetti S, et al. Ipilimumab for the treatment of metastatic prostate cancer. Expert Opin Biol Ther (2018) 18(2):205–13. doi:10.1080/14712598.2018.1420777
152. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
153. Medina PJ, Adams VR. PD-1 pathway inhibitors: immuno-oncology agents for restoring antitumor immune responses. Pharmacotherapy (2016) 36(3):317–34. doi:10.1002/phar.1714
154. Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: emerging concepts and therapeutic opportunities. Biochim Biophys Acta (2017) 1868(1):212–20. doi:10.1016/j.bbcan.2017.04.002
155. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol (2017) 8:248. doi:10.3389/fimmu.2017.00248
156. Kareva I, Hahnfeldt P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: distinct or linked? Cancer Res (2013) 73(9):2737–42. doi:10.1158/0008-5472.CAN-12-3696
157. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
158. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27(2):173–8. doi:10.1016/j.immuni.2007.07.008
159. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33(4):168–73. doi:10.1016/j.it.2012.01.010
Keywords: cancer, metabolism, lactate, immune evasion, cytokines
Citation: Morrot A, Fonseca LM, Salustiano EJ, Gentile LB, Conde L, Filardy AA, Franklim TN, da Costa KM, Freire-de-Lima CG and Freire-de-Lima L (2018) Metabolic Symbiosis and Immunomodulation: How Tumor Cell-Derived Lactate May Disturb Innate and Adaptive Immune Responses. Front. Oncol. 8:81. doi: 10.3389/fonc.2018.00081
Received: 15 December 2017; Accepted: 09 March 2018;
Published: 23 March 2018
Edited by:
Stephen G. Maher, Trinity College, Dublin, IrelandReviewed by:
Paolo E. Porporato, Università degli Studi di Torino, ItalyPrashant Trikha, Nationwide Children’s Hospital, United States
Copyright: © 2018 Morrot, Fonseca, Salustiano, Gentile, Conde, Filardy, Franklim, da Costa, Freire-de-Lima and Freire-de-Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Morrot, YWxleGFuZHJlLm1vcnJvdEBpb2MuZmlvY3J1ei5icg==, bW9ycm90QG1pY3JvLnVmcmouYnI=;
Leonardo Freire-de-Lima, bGVvbGltYUBiaW9mLnVmcmouYnI=
†These authors have contributed equally to this work.