 Noboru Asada
Noboru Asada- Department of Hematology and Oncology, Okayama University Hospital, Okayama, Japan
Hematopoietic stem cells (HSCs) that give rise to all kinds of hematopoietic lineage cells on various demands throughout life are maintained in a specialized microenvironment called “niche” in the bone marrow (BM). Defining niche cells and unveiling its function have been the subject of intense study, and it is becoming increasingly clear how niche cells regulate HSCs in normal hematopoiesis. Leukemia stem cells (LSCs), which are able to produce leukemic cells and maintain leukemic clones, are assumed to share common features with healthy HSCs. Accumulating evidence suggests that LSCs reside in a specialized BM microenvironment; moreover, LSCs could control and rebuild the microenvironment to enhance their progression and survival. This article discusses the recent advances in our knowledge of the microenvironment supporting malignant hematopoiesis, including LSC niche.
Introduction
Hematopoiesis needs to be maintained throughout life to supply blood cells on various demands, such as infection, inflammation, blood loss, or hypoxia. Hematopoietic stem cells (HSCs) that reside at the top of hierarchy differentiate into multiple lineage hematopoietic cells through a fine-tuned differentiation process. Each step of differentiation is guided by various extrinsic factors as well as cell-autonomous intrinsic master gene regulations. In adult mammals, HSCs are known to locate in a specific microenvironment termed “niche” that orchestrates HSC function, including self-renewal and differentiation in both physiological and pathological conditions (1). Accumulating evidence reveals that various types of cells in and around the bone marrow (BM) participate in HSC function and its niche regulation (Figure 1) (2, 3).
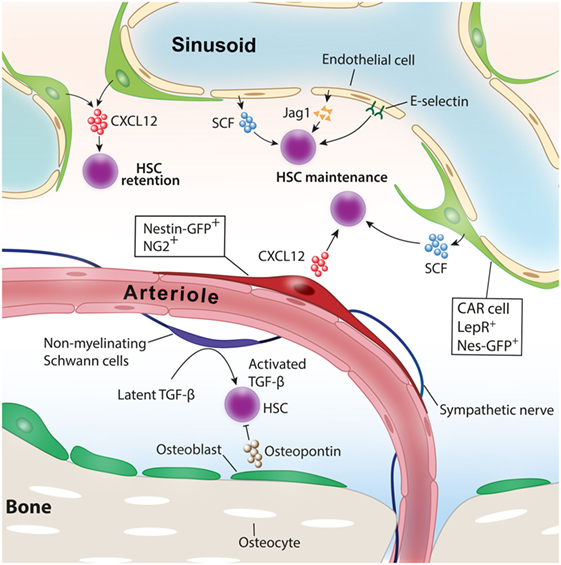
Figure 1. Niche cells for healthy hematopoietic stem cells (HSCs). Various cell types have been identified as niche cells for HSCs in steady-state bone marrow. Perivascular stromal cells such as NG2+ periarteriolar cells and LepR+ perisinusoidal stromal cells differentially regulate HSCs. Nonmyelinating Schwann cells maintain HSC quiescence by activating transforming growth factor-β (TGF-β). Adopted and modified from Ref. (4).
Cell-intrinsic genetic alterations, such as gene mutations, deletions, amplifications, or translocations and epigenetic changes have been postulated mainly as the pathogenesis of hematologic malignancies, including leukemia, myelodysplastic syndrome (MDS), and myeloproliferative neoplasms (MPNs). It is rare, however, that donor cell-derived leukemia (DCL) is a well recognized and vital entity in understanding the process of malignant transformation of hematopoietic cells (5, 6). The possible pathological mechanism of DCL is diverse, such as preleukemic changes in donor cells, oncogene transformation from residual leukemic cells, and impaired immune surveillance. Defects in the BM microenvironment (BMM) in recipient BM have also been assumed as one of the mechanisms, suggesting vital roles of cell-extrinsic factors for malignant clone emergence (1, 7, 8). Recent studies using genetically modified animals indicates that alterations in the BMM could also support the survival of malignant clones or can even be the cause of the evolution of malignant clones (9, 10). In this review, we will summarize the recent achievements uncovering the roles of the BMM for the emergence of hematological malignancies and discuss the possibility of therapeutic options targeting the BMM.
Key Players in HSC Niche in Steady-State BM
Osteolineage Cells
Since Schofield proposed the concept of the existence of a specific environment for HSCs in the BM, various cell types in the BM have been identified as niche comprising cells. Osteolineage cells, a friendly neighbor of the BM, have been assumed as niche comprising cells for healthy HSCs. Initial in vitro studies indicated that bone-forming osteoblasts have the ability to support hematopoietic stem/progenitor cell (HSPC) function (11, 12). In 2003, two reports from different groups showed that osteoblast activation in vivo increased the number of HSCs in the BM. One group pharmacologically activated osteoblasts and the other increased the number of osteoblasts by genetic manipulations, and both led to the expansion of HSCs in the BM (13, 14). Conversely, it is reported that osteopontin, a matrix glycoprotein mainly produced by osteoblasts, negatively regulates HSC number in the BM (15). Recent studies using transgenic mouse models in which the major niche factor, such as C-X-C motif chemokine ligand 12 (CXCL12) and stem cell factor (SCF), was deleted specifically in osteoblasts indicated that osteoblasts did not contribute to the maintenance of HSCs at least by producing these niche factors (16–18). The role of bone-embedded osteocytes for hematopoiesis had remained unknown for a long time. The extrinsic administration of granulocyte-colony-stimulating factor (G-CSF), a key cytokine promoting granulopoiesis, facilitates the translocation of HSPC from the BM to peripheral blood. This process is called the “mobilization” of HSPC and mobilized HSPC is collected by apheresis and used for HSC transplantation for the treatment of hematological disorders. A recent study revealed that osteocytes have critical roles in regulating HSPC mobilization by G-CSF. The depletion of osteocytes using transgenic mice in which diphtheria toxin receptor was expressed under the control of dentin matrix protein-1 (Dmp-1) promoter led to a suppression of osteoblasts, resulting in a defect of HSPC mobilization by G-CSF.
Endothelial Cells
In mammals, definitive HSCs emerge from the hemogenic endothelium within the aorta-gonado-mesonephros region during embryonic development (19, 20). Like the intimate relationship between endothelium and HSCs during development, endothelial cells lining the BM vasculature support HSC maintenance and regeneration in the BM. In vitro coculture experiments indicate that BM endothelial cells expand HSPCs by producing a variety of angiocrine factors, such as insulin growth factor binding protein 2, bone morphogenic protein (BMP) 2 and BMP4, Notch ligands, SCF, CXCL12, and wingless-type MMTV integration site (Wnt) 5a (19–22). In vivo evidence in which the functional deletion of niche factors was achieved specifically in endothelial cells revealed that SCF or CXCL12 derived from endothelial cells play an indispensable role for HSC maintenance in the BM (16, 18). Endothelial cells have also been shown to integrate HSC quiescence through surface E-selectin expression (23).
Recent studies in mice identified a distinct subset of BM endothelial cells crucial for HSC function. Endothelial cells with high expression of CD31 and endomucin, referred to as type H endothelium, which distributes in end-terminal arterioles, expressed a higher level of SCF than sinusoid endothelial cells (24). A study done by another group found that endoglin-expressing endothelial cells, referred to as human regeneration-associated endothelial cells (hRECs), are associated with BM regeneration after myelosuppression and support a subset of hematopoietic progenitors through interleukin (IL)-33. Interestingly, gene expression analysis revealed similarities between hRECs and murine type H endothelium (25). A difference of vascular permeability observed between arterioles and sinusoids provides different effects to HSC activities. Arterial vessels are less permeable and maintain HSCs in a low reactive oxygen species (ROS), keeping HSCs quiescent. On the contrary, blood plasma permeabilized from leaky sinusoids promotes a high level of ROS in HSCs, augmenting the ability of differentiation and migration (26).
Stromal Cell-Associated Vasculature
A study defining the location of HSCs in the BM by staining phenotypic endogenous HSCs revealed that HSCs are closely associated with BM vasculature (27). These findings shed light on the vasculature area as HSC niche. Stromal cells that have a potency to differentiate into trilineage mesenchymal cells have been shown to function as HSC niche and are mainly associated with sinusoids in the BM. Several studies identified different stromal cell types around sinusoids characterized by distinct surface markers or gene expression as niche comprising cells. These cells include CXCL12-abundant reticular (CAR) cells (28–30), which are cells marked by green fluorescent protein (GFP) under the elements of the nestin promoter (Nes-GFP+) (31), leptin receptor (LepR)-expressing cells (16, 17), CD144−CD146−Sca-1+ mesenchymal stromal progenitors (32), and the stromal cells targeted by Cre recombinase promoted by transcription factor osterix (Osx) (18), neural/glial antigen 2 (NG2) (33), or paired related homeobox-1 (17, 18). It has been shown that these cells expressed a high amount of niche factors supporting HSC functions, such as CXCL12, SCF, and VCAM-1, and they exhibit a significant overlap among each other (17, 18, 33, 34). Because the BM is a highly vascularized organ, as a matter of course, they have plenty of arteries and arterioles. A recent study in which the spatial distribution of endogenous HSCs in the BM was analyzed revealed that HSCs are closely and significantly associated with BM arterioles (35). The depletion of NG2-expressing pericytes in vivo led to a loss of quiescence and a reduction of HSCs and suggested the roles of periarteriolar stromal cells for HSC maintenance and quiescence. Other studies have argued that HSCs marked by α-catulin GFP and c-kit expression are randomly distributed in the BM and closely associated with sinusoids rather than arterioles (36). Another study has argued the differential contributions of sinusoids and arterioles to HSPC functions (26). Therefore, the contributions of each perivascular stromal cells to HSC niche had been controversial. To delineate the roles of perisinusoidal and periarteriolar stromal cells in HSC niche, we analyzed transgenic mice in which major niche factors, CXCL12 or SCF, were deleted specifically in either perisinusoidal or periarteriolar stromal cells. Whereas CXCL12 deletion in periarteriolar stromal cells led to a reduction of HSC number and alteration of distribution from arterioles, the deletion of CXCL12 in perisinusoidal stromal cells mobilized HSC to peripheral blood and spleen but had no impact on the HSC number or location in the BM. On the contrary, SCF deletion in perisinusoidal but not periarteriolar stromal cells impaired HSC maintenance in the BM (33). These results showed an intriguing mechanism of how different cytokines from distinct perivascular stromal cells contribute to HSC functions.
Nervous System
Bone and BM are extensively innervated by the nervous system. Catecholamine signals released from sympathetic nerve endings finely tune HSC niche functions, integrating HSC mobilization induced by cytokine G-CSF or release of HSCs under the circadian rhythm (37–39). Nonmyelinating Schwann cells wrapping the sympathetic nerves and closely associated with arterioles in the BM have been reported to maintain HSC quiescence by converting transforming growth factor-β (TGF)-β into the active form (40).
Regulatory T (Treg) Cells
It has been well known that HSCs in the BM are resistant to cytotoxic stress and recent studies revealed that Treg cells that suppress the function of effector T cells provide immunoprivileged sites to HSCs in the niche (41, 42). Intravenously transplanted HSCs in the allogeneic mouse transplantation model persisted for 1 month without immunosuppression and most of the HSCs colocalized with Treg cells in the BM. The depletion of Treg cells led to the reduction in the number of surviving donor HSCs after allogeneic transplantation, suggesting a protective function of Treg cells from immune attack to allogeneic HSCs (41). A subsequent study from the same group reported that a distinct fraction of Treg cells that highly expressed CD150 play vital roles for the maintenance of HSC quiescence and engraftment through adenosine (42).
Roles of the BMM for MPN
The clinical entity of MPNs is heterogeneous and includes four classic MPNs: polycythemia vera, essential thrombocytopenia, primary myelofibrosis, and chronic myeloid leukemia. As recent studies showed that most cases of MPNs have somatic mutations in the tyrosine kinase Janus kinase 2 (JAK2) (43–46), calreticulin gene (CALR) (47, 48), or thrombopoietin receptor (49), the pathogenesis of these neoplasms appears mostly cell intrinsic. Although the BMM originally regulates differentiation and proliferation of HSCs or immature progenitor cells without aberrant proliferation, recent evidence from mice work suggests that the defect of the BMM can be the cause of abnormal myeloproliferation. The lost of one of the major receptors for vitamin A, RARγ, in the BMM results in increased mature myeloid cells resembling MPNs, which partially depend on tumor necrosis factor-α (TNF-α) production from the BMM (50). Another report showed that the perturbation of interaction between myeloid-derived cells and the BMM by the defect of retinoblastoma protein (Rb), a vital regulator of the cell cycle, led to myeloid cell proliferation (51). The deficiency of Mindbomb-1, an essential component for Notch ligand endocytosis, in the BMM is also shown to cause enhanced myelopoiesis corresponding to MPNs through Notch signaling defects in the BMM (52). All these evidences clearly indicate that nonhematopoietic BMM cells play significant roles in promoting aberrant myelopoiesis; however, the specific cell types contributing to the enhanced myelopoiesis remain largely unknown.
Osteolineage Cells
A recent study by Fulzele et al. reported that osteocytes, which are terminally differentiated osteolineage cells embedded in the calcified bone, participate in myelopoiesis. They found that the specific deletion of Gsα in osteocytes enhanced G-CSF production, leading to the expansion of myeloid-committed cells in the BM (53). As osteocytes are also shown to regulate the BMM and control HSPC activities (54), it might be possible that osteolineage cells participate in the pathogenesis of MPNs.
Stromal Cell-Associated Vasculature and Sympathetic Nerve
A recent study done by Arranz et al. reported that, in both human MPN patients and mice expressing human JAK2 (V617F) mutation in HSCs, the number of sympathetic nerves and Schwann cells ensheathing sympathetic nerves was decreased. In the mice MPN model, the depletion of Nes-GFP+ perivascular stromal cells accelerated MPN progression. They found that abnormal HSC-derived proinflammatory cytokine IL-1β caused local neuropathy and damaged Nes-GFP+ perivascular stromal cells, leading to the progression of MPN (55). These results suggest that aberrant HSCs in the MPNs rebuild the BMM beneficial for their survival.
Cytokine Milieu
In addition to the cellular players of the BMM, non-cellular components of the BMM have significant contributions to the development or sustainment of MPNs. The increased level of various inflammatory cytokines, including IL-6, IL-8, basic fibroblast growth factor, platelet-derived growth factor, TNF-α, TGF-β, and oncostatin M, has been reported in MPNs (56–58), and these cytokines play a role in the establishment of the disease manifestations. In particular, TGF-β1 mostly produced by megakaryocytes has been implicated in the development of BM fibrosis, a major unfavorable alteration of the BMM in patients with MPNs (59, 60). JAK kinase inhibitors, including ruxolitinib, ameliorate systemic symptoms and splenomegaly in MPN patients (61–63). The reduction of proinflammatory cytokines by the inhibition of JAK-STAT signaling has been identified as one of the mechanisms of ruxolitinib (64). Moreover, a recent study identified a constitutive activation of nuclear factor-κB (NF-κB) signaling in addition to JAK-STAT pathways as a key signaling pathway leading to chronic inflammation in MPNs. Intriguingly, the combined blockade of JAK-STAT and NF-κB pathways with ruxolitinib and JQ1, the bromodomain and extra-terminal motif (BET) bromodomain inhibitor, reduced aberrant cytokine production and improved BM fibrosis in the mice MF model (65).
Roles of the BMM for the Pathogenesis of MDS
By definition, MDS are a heterogeneous group of clonal HSC diseases characterized by cytopenia, dysplasia in one or more of the major myeloid lineages, ineffective hematopoiesis, recurrent genetic abnormalities, and increased risk of developing acute myeloid leukemia (AML) (66, 67). As various types of recurrent cytogenetic abnormalities in hematopoietic aberrant clone have been identified, it is broadly accepted that the pathogenesis of MDS is mainly cell intrinsic. Some studies indicated that cultured BM stromal cells isolated from MDS patients harbor cytogenetic abnormalities distinct from hematopoietic cells (68–70). Because stromal cells analyzed in these studies were cultured in vitro and most of them were analyzed after several passages, observed abnormalities could be acquired in vitro rather than originating from primary stromal cells. Emerging evidence from sophisticated mice studies strongly suggests that defects in the BMM could promote at least a partial initiation of malignant clone or advance the disease progression.
Stromal Cell-Associated Vasculature
Genetically engineered mice in which Dicer 1, the RNase III endonuclease essential for microRNA biogenesis and RNA processing, was deleted explicitly in osteoprogenitor cells were marked by Osx-Cre-developed MDS accompanied by osteoblastic dysfunction (9). In addition to Dicer 1, the deletion of Shwachman–Diamond–Bodian syndrome (Sbds) gene in osteoprogenitor cells resulted in cytopenia and dysplastic changes in neutrophils and megakaryocytes. A subsequent study using the same mouse model done by the same group demonstrated that S100A8/9 protein, proinflammatory molecules referred to as damage-associated molecular pattern or alarmins, secreted by osteoprogenitor cells in Sbds-deficient mice induces genotoxic stress mediated by mitochondrial dysfunction, oxidative stress, and DNA damage response activation in HSPCs (71). Although Osx is one of the master regulator genes that lead mesenchymal progenitors to osteoblast lineage differentiation (72), stromal cells marked by Cre promoted by Osx showed a significant overlapping with other stromal cells that are closely associated with sinusoids, such as CAR cells (18), Nes-GFP+, or LepR-expressing stromal cells (34). Collectively, these results indicate that the dysfunction of perisinusoidal stromal cells that have osteoblastic differentiation potential might induce dysplasia in hematopoiesis through undefined mechanisms.
Cytokines and Immune Cells
It has been well recognized that both cellular and non-cellular immune systems are perturbed in MDS patients (73). The increased levels of various proinflammatory cytokines, such as IL-6, IL-8, TNF-α, TGF-β, and interferon-γ in MDS patients have been reported and implicated in the pathogenesis of MDS (74).
With regard to the roles of immune cells, immunoregulatory Treg cells might be involved in the pathogenesis of MDS. The increased number of Treg cells has been reported to correlate with unfavorable factors, such as high percentage of BM blasts, high International Prognostic Scoring System score, and disease progression (75). A recent study identified that high numbers of effector memory Treg cells that have more potent immunosuppressive function are associated with higher risk disease, increased blast percentage, and reduced overall survival (76). Although these evidences indicate crucial roles of Treg cells in the pathogenesis or the mechanism of disease progression in MDS, further studies will be necessary to determine whether Treg cells participate in the pathogenesis of MDS or a merely reactive consequence of hematological dysregulation.
Roles of the BMM for the Pathogenesis of Leukemia
Similar to the normal hematopoietic system, stem or progenitor cells reside at the top of the hierarchy and produce descendant leukemic cells and self-renew to propagate leukemia and sustain clonal tumor burden (77, 78). Although leukemia stem cell (LSC) seems to be less dependent on their niche than normal HSCs, the leukemogenic process does not completely abrogate niche dependence for LSCs. Cumulative evidence suggests that BMM influences LSC behavior in many ways similar to normal hematopoiesis (73).
Non-Cellular Component
It is shown that human AML stem cells (LSCs) expressed CXCR4, a counter-receptor for CXCL12 that is a potent chemoattractant for HSCs secreted from BM stromal cells, and the blockade of CXCR4–CXCL12 axis abrogated the homing of LSCs and propagation of leukemic cells in a xenotransplantation murine model (79). Another study reported that the level of CXCL12 in the BM with chronic myelogenous leukemia (CML) was decreased, which impaired the homing efficacy of both exogenous transplanted LSCs and healthy HSCs. Plasma isolated from the BM of CML mice BM impeded the growth of healthy HSCs but not LSCs in vitro culture, leading to a growth advantage for the leukemic clone (80). AMD3100 (plerixafor), a small-molecule inhibitor of CXCR4, have been tested in a phase 1/2 study combined with chemotherapy for relapsed/refractory AML with encouraging response rates (81). However, a subsequent trial testing the additive effect of G-CSF on AMD3100 combined with chemotherapy in AML patients failed to improve the response rate (82). More potent CXCR4 inhibitors have been developed and in vitro studies revealed that they could induce the apoptosis of AML, which is favorable to eradicate LSCs (83, 84).
In addition to CXCR4–CXCL12 interaction, the adhesion molecule CD44 on LSCs also has been documented to be involved in the crosstalk between LSCs and BMM. The ligation of CD44 by the monoclonal antibody specifically prevented LSCs to home and engraft to the BM without disturbing normal HSC function (85). The phase I study of an anti-CD44 antibody that blocks the interaction between LSCs and BMM revealed that the drug was safe and well tolerated but had limited activity to leukemia (86). These series of evidences highlighted the significant roles of the BMM for leukemia pathogenesis and LSC biology. Defining the exact cell types of LSC niche and the mechanism how niche cells regulate LSCs have been under intense study (Figure 2).
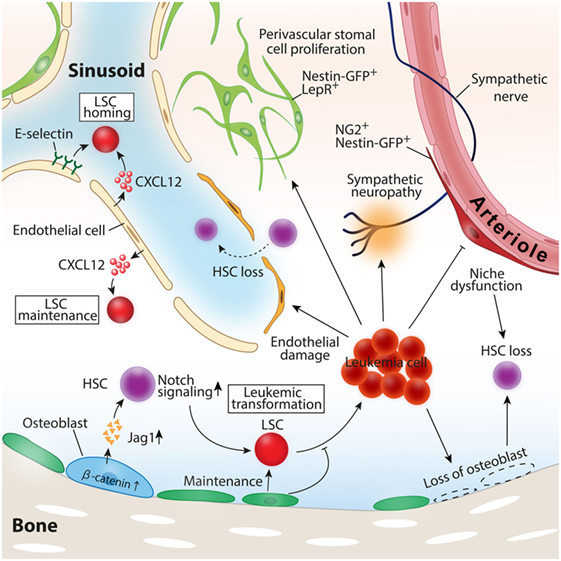
Figure 2. Roles of the BM microenvironment in leukemia pathogenesis. Constitutive activation of β-catenin in osteoblast-induced leukemia transformation in mice model. Leukemic cells induce loss of osteoblasts, vascular endothelial cells, and periarteriolar NG2+ stromal cells, leading to healthy hematopoietic stem cell (HSC) loss. CXCL12 and E-selectin expressed by vascular endothelial cells function as inducers of leukemia stem cell (LSC) homing to the bone marrow, and CXCL12 secreted by endothelial cells also contributes to LSC maintenance and the propagation of leukemia.
Endothelial Cells
Ample evidence suggests the indispensable roles for vascular endothelial cells in supporting LSCs and leukemia cell progression. Although most of the leukemia are disseminated diseases when they cause clinical symptoms, the initial clonal evolution should occur at a certain site in the BM. After the initial proliferation of aberrant clones, leukemic cells extravasate from the original BM to the bloodstream and spread to other BMs throughout the body. Similar to healthy HSCs, LSCs are required to have the ability to home and engraft to the BM for their expansion. Sipkins et al. analyzed the spatial distribution pattern of externally transplanted mice leukemic cells and revealed that leukemic cells homed and colonized around E-selectin and CXCL12 expressing BM endothelial cells, suggesting the importance of distinct vascular endothelial cells as a supporter of leukemic cell expansion (87). The deletion of CXCL12 specific from vascular endothelial cells impeded T-cell acute lymphoblastic leukemia (T-ALL) growth in both mice leukemia model and human T-ALL xenografts (88). A recent study showed that LSCs expressed a high level of CD98, an integrin binding glycoprotein, mediated adhesion of LSCs to vascular endothelial cells where LSCs were maintained. Moreover, the blockade of CD98 by monoclonal antibodies abolished leukemia engraftment and proliferation in the mice AML model, suggesting a therapeutic potential of the agents targeting CD98 (89). The antileukemic effect of anti-CD98 antibody in relapsed or refractory AML patients has also currently been under investigation.
In terms of the number of the vasculature in leukemia, the increased density of BM vasculature has been observed both in murine aggressive AML model and in leukemia patients (90, 91). However, it seems that we should take into consideration the location of vasculature rather than the magnitude of the increase of endothelial cells. Duarte et al. demonstrated that endosteal vascular endothelial cells were depleted in MLL-AF9-driven mouse AML model, which was associated with healthy HSC loss through the increase of transendothelial migration of HSCs. The prevention of endosteal endothelium impairment with a small-molecule deferoxamine or a genetic approach rescued HSC loss and prolonged the survival of the mice treated with chemotherapy (92).
Osteolineage Cells
In the human acute leukemia xenograft model, residual leukemic cells were located in the vicinity of the endosteal area after chemotherapy, implying the existence of a distinct microenvironment for chemotherapy-resistant dormant leukemic stem cells around osteolineage cells (93). Reduced numbers of mature osteoblasts and osteocalcin in the blood, one of the surrogate markers of osteoblast function, were reported in both AML patients and the MLL-AF9 mouse aggressive AML model, resulting in reduced healthy hematopoiesis (94, 95). Targeted ablation of mature osteoblasts in the mouse transgenic leukemia model representing human chronic phase CML accelerated leukemia progression possibly due to the loss of quiescence of LSCs and led to a deterioration of LSC ability to generate leukemia in the recipient mice (96). These results suggest that osteoblasts have indispensable roles to inhibit leukemia expansion and to sustain stemness of LSCs in mouse CML (96). Consistent with this idea, osteoblast activation by the treatment of parathyroid hormone decreased LSC proliferation in a transduction-transplantation model of CML (97).
A recent study by Kode et al. showed that osteoblasts are involved in not only the regulation of established leukemic cells but also the evolution of leukemia. In this study, the authors showed that the constitutive activation of β-catenin in mature osteoblasts stimulated the expression of Notch ligand jagged 1 in osteoblasts, which in turn led to the activation of Notch signaling in HSPCs, and induced malignant transformation of HSPCs to leukemic cells (10).
Perivascular Stromal Cells
As discussed in the niche cells for healthy HSCs, perivascular stromal cells in the BM have attracted much attention as a vital niche player. However, it remains elusive whether these cell types contribute to the evolution or growth of leukemia. One study showed in a transduced mouse T-ALL model that perivascular stromal cells did not contribute to leukemia propagation at least through CXCL12–CXCR4 signals between BMM and leukemia cells (88). A more recent study analyzing the dynamic interaction of T-ALL leukemic cells with the niche component across the leukemia progression demonstrated that leukemic cells had any spatial preference with any niche component including perivascular stromal cells represented by Nes-GFP+ stromal cells (98). In the BM with advanced T-ALL, the number of Nes-GFP+ cells was maintained, whereas mature osteoblasts and osteoprogenitor were completely lost (98). In contrast to the T-ALL model, the robust expansion of Nes-GFP+ cells with impaired niche factor expression for healthy HSCs has been observed in mice with transduced MLL-AF9 aggressive AML cells (91). NG2+ perivascular stromal cells closely associated with arterioles that have been shown to maintain healthy HSCs were reduced, which was consistent with the diminished number of healthy HSCs. Intriguingly, these dramatic alterations of niche components induced by AML were mediated by the disruption of sympathetic nerves in the BM induced by leukemic cells, and treatment of β2-adrenergic receptor agonist led to the reduction of LSCs in the BM and prolonged the survival of leukemic mice (91). Altogether, these evidences suggest that the roles of perivascular stromal cells in leukemia pathogenesis may vary among the subtypes of leukemia, and further studies are necessary.
In the context of leukemia evolution, although transgenic mice in which Dicer 1 or Sbds was abrogated in perivascular stromal cells presented myelodysplastic changes and subsequent evolution to leukemia (9, 71), there is, so far, no evidence clearly demonstrating that dysfunction in perivascular stromal cells causes de novo leukemia in vivo.
Immune Cells
As is the case in normal hematopoiesis, immune cells modulate BMM in leukemia. In the mice AML model, immunosuppressive Treg cells presented at the AML site and impaired the function of adoptively transferred cytotoxic T cells (CTLs). The depletion of Treg cells in turn restored CTL function and reduced leukemia progression in the mice model (99).
Concluding Remarks
Over the past decade, a significant advancement in understanding the roles of the BMM in the pathogenesis of hematologic malignancies has been achieved. Because even the mechanisms by which niche cells orchestrate healthy HSCs or hematopoiesis are not completely understood, the involvement of the BMM to malignant hematopoiesis must be diverse and complicated. For instance, the results gained thus far from murine studies indicated that a different type of leukemia interacts with a distinct BMM differently. Further studies clarifying the detailed mechanisms that underlie each type of hematopoietic malignancy will lead us to our final goal to improve therapeutic strategies and conquer hematopoietic malignancies.
Author Contributions
NA wrote the manuscript and drew the figures.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
NA is supported by a Grant-in-Aid for Young Scientists (Start-up) Grant Number JP17H06882, from the Japan Society for the Promotion of Science, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, The Kanae Foundation for the Promotion of Medical Science, and the Ryobi Teien Memory Foundation.
References
1. Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol (2016) 11:555–81. doi:10.1146/annurev-pathol-012615-044414
2. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20:833–46. doi:10.1038/nm.3647
3. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505:327–34. doi:10.1038/nature12984
4. Asada N, Takeishi S, Frenette PS. Complexity of bone marrow hematopoietic stem cell niche. Int J Hematol (2017) 106(45):45–54. doi:10.1007/s12185-017-2262-9
5. Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant (2011) 17:771–89. doi:10.1016/j.bbmt.2010.10.010
6. Shiozaki H, Yoshinaga K, Kondo T, Imai Y, Shiseki M, Mori N, et al. Donor cell-derived leukemia after cord blood transplantation and a review of the literature: differences between cord blood and BM as the transplant source. Bone Marrow Transplant (2014) 49:102–9. doi:10.1038/bmt.2013.127
7. Schepers K, Campbell TB, Passegue E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell (2015) 16:254–67. doi:10.1016/j.stem.2015.02.014
8. Korn C, Mendez-Ferrer S. Myeloid malignancies and the microenvironment. Blood (2017) 129:811–22. doi:10.1182/blood-2016-09-670224
9. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature (2010) 464:852–7. doi:10.1038/nature08851
10. Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature (2014) 506:240–4. doi:10.1038/nature12883
11. Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med (1994) 179:1677–82. doi:10.1084/jem.179.5.1677
12. Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood (1996) 87:518–24.
13. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature (2003) 425:841–6. doi:10.1038/nature02040
14. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature (2003) 425:836–41. doi:10.1038/nature02041
15. Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med (2005) 201:1781–91. doi:10.1084/jem.20041992
16. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature (2012) 481:457–62. doi:10.1038/nature10783
17. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature (2013) 495:231–5. doi:10.1038/nature11885
18. Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature (2013) 495:227–30. doi:10.1038/nature11926
19. Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell (2010) 6:251–64. doi:10.1016/j.stem.2010.02.001
20. Li W, Johnson SA, Shelley WC, Ferkowicz M, Morrison P, Li Y, et al. Primary endothelial cells isolated from the yolk sac and para-aortic splanchnopleura support the expansion of adult marrow stem cells in vitro. Blood (2003) 102:4345–53. doi:10.1182/blood-2003-03-0729
21. Rafii S, Shapiro F, Pettengell R, Ferris B, Nachman RL, Moore MA, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood (1995) 86:3353–63.
22. Kobayashi H, Butler JM, O’Donnell R, Kobayashi M, Ding BS, Bonner B, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol (2010) 12:1046–56. doi:10.1038/ncb2108
23. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med (2012) 18:1651–7. doi:10.1038/nm.2969
24. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature (2014) 507:323–8. doi:10.1038/nature13145
25. Kenswil KJG, Jaramillo AC, Ping Z, Chen S, Hoogenboezem RM, Mylona MA, et al. Characterization of endothelial cells associated with hematopoietic niche formation in humans identifies IL-33 as an anabolic factor. Cell Rep (2018) 22:666–78. doi:10.1016/j.celrep.2017.12.070
26. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) 532:323–8. doi:10.1038/nature17624
27. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell (2005) 121:1109–21. doi:10.1016/j.cell.2005.05.026
28. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25:977–88. doi:10.1016/j.immuni.2006.10.016
29. Omatsu Y, Seike M, Sugiyama T, Kume T, Nagasawa T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature (2014) 508:536–40. doi:10.1038/nature13071
30. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity (2010) 33:387–99. doi:10.1016/j.immuni.2010.08.017
31. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466:829–34. doi:10.1038/nature09262
32. Hu X, Garcia M, Weng L, Jung X, Murakami JL, Kumar B, et al. Identification of a common mesenchymal stromal progenitor for the adult haematopoietic niche. Nat Commun (2016) 7:13095. doi:10.1038/ncomms13095
33. Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol (2017) 19:214–23. doi:10.1038/ncb3475
34. Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell (2014) 29:340–9. doi:10.1016/j.devcel.2014.03.013
35. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature (2013) 502:637–43. doi:10.1038/nature12612
36. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature (2015) 526:126–30. doi:10.1038/nature15250
37. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell (2006) 124:407–21. doi:10.1016/j.cell.2005.10.041
38. Lucas D, Battista M, Shi PA, Isola L, Frenette PS. Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell (2008) 3:364–6. doi:10.1016/j.stem.2008.09.004
39. Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature (2008) 452:442–7. doi:10.1038/nature06685
40. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell (2011) 147:1146–58. doi:10.1016/j.cell.2011.09.053
41. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474:216–9. doi:10.1038/nature10160
42. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150(high) bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell (2018) 22(3):445–53.e5. doi:10.1016/j.stem.2018.01.017
43. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (2005) 365:1054–61. doi:10.1016/S0140-6736(05)74230-6
44. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434:1144–8. doi:10.1038/nature03546
45. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med (2005) 352:1779–90. doi:10.1056/NEJMoa051113
46. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell (2005) 7:387–97. doi:10.1016/j.ccr.2005.03.023
47. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369:2379–90. doi:10.1056/NEJMoa1311347
48. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369:2391–405. doi:10.1056/NEJMoa1312542
49. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood (2006) 108:3472–6. doi:10.1182/blood-2006-04-018879
50. Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell (2007) 129:1097–110. doi:10.1016/j.cell.2007.05.014
51. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell (2007) 129:1081–95. doi:10.1016/j.cell.2007.03.055
52. Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, Shin J, et al. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood (2008) 112:4628–38. doi:10.1182/blood-2008-03-148999
53. Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu X, et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood (2013) 121:930–9. doi:10.1182/blood-2012-06-437160
54. Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, Kawano H, et al. Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell (2013) 12:737–47. doi:10.1016/j.stem.2013.05.001
55. Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature (2014) 512:78–81. doi:10.1038/nature13383
56. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol (2011) 29:1356–63. doi:10.1200/JCO.2010.32.9490
57. Pourcelot E, Trocme C, Mondet J, Bailly S, Toussaint B, Mossuz P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp Hematol (2014) 42:360–8. doi:10.1016/j.exphem.2014.01.006
58. Vaidya R, Gangat N, Jimma T, Finke CM, Lasho TL, Pardanani A, et al. Plasma cytokines in polycythemia vera: phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol (2012) 87:1003–5. doi:10.1002/ajh.23295
59. Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood (2002) 100:3495–503. doi:10.1182/blood-2002-04-1133
60. Zingariello M, Martelli F, Ciaffoni F, Masiello F, Ghinassi B, D’Amore E, et al. Characterization of the TGF-beta1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood (2013) 121:3345–63. doi:10.1182/blood-2012-06-439661
61. Mesa RA, Gotlib J, Gupta V, Catalano JV, Deininger MW, Shields AL, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J Clin Oncol (2013) 31:1285–92. doi:10.1200/JCO.2012.44.4489
62. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br J Haematol (2013) 161:508–16. doi:10.1111/bjh.12274
63. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med (2010) 363:1117–27. doi:10.1056/NEJMoa1002028
64. Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov (2015) 5:316–31. doi:10.1158/2159-8290.CD-14-0736
65. Kleppe M, Koche R, Zou L, van Galen P, Hill CE, Dong L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell (2018) 33:29–43.e7. doi:10.1016/j.ccell.2017.11.009
66. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol (1982) 51:189–99. doi:10.1111/j.1365-2141.1982.tb08475.x
67. Cazzola M, Malcovati L. Myelodysplastic syndromes – coping with ineffective hematopoiesis. N Engl J Med (2005) 352:536–8. doi:10.1056/NEJMp048266
68. Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schumann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol (2007) 35:221–9. doi:10.1016/j.exphem.2006.10.012
69. Flores-Figueroa E, Arana-Trejo RM, Gutierrez-Espindola G, Perez-Cabrera A, Mayani H. Mesenchymal stem cells in myelodysplastic syndromes: phenotypic and cytogenetic characterization. Leuk Res (2005) 29:215–24. doi:10.1016/j.leukres.2004.06.011
70. Kouvidi E, Stratigi A, Batsali A, Mavroudi I, Mastrodemou S, Ximeri M, et al. Cytogenetic evaluation of mesenchymal stem/stromal cells from patients with myelodysplastic syndromes at different time-points during ex vivo expansion. Leuk Res (2016) 43:24–32. doi:10.1016/j.leukres.2016.02.007
71. Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell (2016) 19:613–27. doi:10.1016/j.stem.2016.08.021
72. Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell (2002) 108:17–29. doi:10.1016/S0092-8674(01)00622-5
73. Ghobrial IM, Detappe A, Anderson KC, Steensma DP. The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol (2018) 15(4):219–33. doi:10.1038/nrclinonc.2017.197
74. Hemmati S, Haque T, Gritsman K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front Oncol (2017) 7:265. doi:10.3389/fonc.2017.00265
75. Kordasti SY, Ingram W, Hayden J, Darling D, Barber L, Afzali B, et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood (2007) 110:847–50. doi:10.1182/blood-2007-01-067546
76. Mailloux AW, Sugimori C, Komrokji RS, Yang L, Maciejewski JP, Sekeres MA, et al. Expansion of effector memory regulatory T cells represents a novel prognostic factor in lower risk myelodysplastic syndrome. J Immunol (2012) 189:3198–208. doi:10.4049/jimmunol.1200602
77. Corces MR, Chang HY, Majeti R. Preleukemic hematopoietic stem cells in human acute myeloid leukemia. Front Oncol (2017) 7:263. doi:10.3389/fonc.2017.00263
78. Hu T, Murdaugh R, Nakada D. Transcriptional and microenvironmental regulation of lineage ambiguity in leukemia. Front Oncol (2017) 7:268. doi:10.3389/fonc.2017.00268
79. Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res (2004) 64:2817–24. doi:10.1158/0008-5472.CAN-03-3693
80. Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell (2012) 21:577–92. doi:10.1016/j.ccr.2012.02.018
81. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood (2012) 119:3917–24. doi:10.1182/blood-2011-10-383406
82. Uy GL, Rettig MP, Stone RM, Konopleva MY, Andreeff M, McFarland K, et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J (2017) 7:e542. doi:10.1038/bcj.2017.21
83. Abraham M, Klein S, Bulvik B, Wald H, Weiss ID, Olam D, et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia (2017) 31:2336–46. doi:10.1038/leu.2017.82
84. Peng SB, Zhang X, Paul D, Kays LM, Ye M, Vaillancourt P, et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS One (2016) 11:e0150585. doi:10.1371/journal.pone.0150585
85. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med (2006) 12:1167–74. doi:10.1038/nm1483
86. Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget (2016) 7:32532–42. doi:10.18632/oncotarget.8687
87. Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature (2005) 435:969–73. doi:10.1038/nature03703
88. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell (2015) 27:755–68. doi:10.1016/j.ccell.2015.05.002
89. Bajaj J, Konuma T, Lytle NK, Kwon HY, Ablack JN, Cantor JM, et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell (2016) 30:792–805. doi:10.1016/j.ccell.2016.10.003
90. Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood (2000) 95:309–13.
91. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell (2014) 15:365–75. doi:10.1016/j.stem.2014.06.020
92. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell (2018) 22:64–77.e6. doi:10.1016/j.stem.2017.11.006
93. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol (2007) 25:1315–21. doi:10.1038/nbt1350
94. Krevvata M, Silva BC, Manavalan JS, Galan-Diez M, Kode A, Matthews BG, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood (2014) 124:2834–46. doi:10.1182/blood-2013-07-517219
95. Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood (2012) 119:540–50. doi:10.1182/blood-2011-04-348151
96. Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood (2015) 125:2678–88. doi:10.1182/blood-2014-06-582924
97. Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med (2013) 19:1513–7. doi:10.1038/nm.3364
98. Hawkins ED, Duarte D, Akinduro O, Khorshed RA, Passaro D, Nowicka M, et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature (2016) 538:518–22. doi:10.1038/nature19801
Keywords: bone marrow microenvironment, niche, leukemia stem cells, hematopoietic stem cells, myelodysplastic syndrome, MPD
Citation: Asada N (2018) Regulation of Malignant Hematopoiesis by Bone Marrow Microenvironment. Front. Oncol. 8:119. doi: 10.3389/fonc.2018.00119
Received: 09 January 2018; Accepted: 03 April 2018;
Published: 23 April 2018
Edited by:
Keisuke Ito, Albert Einstein College of Medicine, United StatesReviewed by:
Federica Barbieri, Università di Genova, ItalySophie Paczesny, Indiana University Bloomington, United States
Antonio Curti, Università degli Studi di Bologna, Italy
Copyright: © 2018 Asada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noboru Asada, bmFzYWRhQG9rYXlhbWEtdS5hYy5qcA==