 Christian M. Schürch
Christian M. Schürch- Baxter Laboratory for Stem Cell Biology, Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA, United States
Therapeutic monoclonal antibodies (mAbs) such as antibody–drug conjugates, ligand–receptor antagonists, immune checkpoint inhibitors and bispecific T cell engagers have shown impressive efficacy in the treatment of multiple human cancers. Numerous therapeutic mAbs that have been developed for myeloid neoplasms, including acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), are currently investigated in clinical trials. Because AML and MDS originate from malignantly transformed hematopoietic stem/progenitor cells—the so-called leukemic stem cells (LSCs) that are highly resistant to most standard drugs—these malignancies frequently relapse and have a high disease-specific mortality. Therefore, combining standard chemotherapy with antileukemic mAbs that specifically target malignant blasts and particularly LSCs or utilizing mAbs that reinforce antileukemic host immunity holds great promise for improving patient outcomes. This review provides an overview of therapeutic mAbs for AML and MDS. Antibody targets, the molecular mechanisms of action, the efficacy in preclinical leukemia models, and the results of clinical trials are discussed. New developments and future studies of therapeutic mAbs in myeloid neoplasms will advance our understanding of the immunobiology of these diseases and enhance current therapeutic strategies.
Introduction
Myeloid neoplasms are clonal blood cancers that arise from malignantly transformed hematopoietic stem cells (HSCs). They comprise the heterogeneous entity acute myeloid leukemia (AML), the most common acute leukemia in adults, as well as the less-frequent myeloproliferative neoplasms (MPNs), myelodysplastic syndromes (MDSs), mastocytosis, and other rare disease variants. Myeloid neoplasms are characterized by an acute or chronic accumulation of immature blasts and/or mature myeloid cells in the bone marrow (BM), blood, spleen, and other organs, which is often accompanied by a defect in blood cell maturation of one or multiple cell lineages resulting in cytopenias and BM failure. Recent advances in the molecular, cytogenetic, morphologic, and clinical understanding of myeloid neoplasms have shed light on their pathogenesis as well as improved and standardized their diagnosis and classification. These achievements are reflected in the 2016 revision of the 4th edition of the World Health Organization classification of myeloid neoplasms and acute leukemia (1, 2). However, with regards to therapy, the recent advances in the molecular understanding of myeloid neoplasms have not yet widely translated into powerful new drugs that substantially improve patient outcomes (3).
On one hand, there has been considerable progress in the treatment of some MPNs, such as chronic myeloid leukemia (CML), a previously almost uniformly fatal disease that is nowadays treated with tyrosine kinase inhibitors (TKIs). Continuous TKI treatment leads to long-term remission in the majority of CML patients, and up to 30% of patients on TKIs eventually meet the criteria to stop therapy, which is essentially synonymous to definite cure (4).
On the other hand, except for acute promyelocytic leukemia (APL), standard treatment for AML and high-risk MDS consists of highly intensive induction chemotherapy with cytarabine and an anthracycline (“3 + 7”), followed by consolidation chemotherapy and/or allogeneic hematopoietic stem cell transplantation (aHSCT). This treatment regimen has been largely unchanged for the last 30 years (5), and the 5-year overall survival (OS) rates in all age groups remains below 20%. This is mainly attributed to very poor outcomes in elderly adults >65 years of age, whereas cure rates have risen more than fivefold in younger adults during the last decades (6–9). Elderly adults in whom AML is most prevalent often have comorbidities that preclude intensive therapy (10). For (younger) patients undergoing intensive therapy, the most important reasons for treatment failure are disease relapse and primary refractoriness (11, 12).
Primary refractory AML, defined by the failure to achieve a complete remission (CR) after induction chemotherapy, is observed in 20–50% of patients and correlates with factors such as age, disease burden, secondary disease, cytogenetic risk, and molecular markers (13, 14). AML relapse, defined as recurrence after a CR, can be attributed to a rare population of therapy-resistant leukemic stem cells (LSCs) or minor blast clones with stem cell-like features that are already present in the diagnostic sample and persist during therapy as minimal residual disease (MRD) (15). As for primary refractory AML, the risk for relapse is highly correlated with age, disease burden, secondary disease, cytogenetic risk, and molecular markers. Fifty to seventy-five percent of patients who achieve a first CR will eventually relapse (16). Patients with refractory/relapsed (R/R) AML have the best chance to be cured by aHSCT after an attempt to induce a second CR. For those patients ineligible for a second round of induction chemotherapy, enrollment in a well-designed experimental clinical trial is recommended (14). In such clinical trials, novel, less-toxic treatment options, such as hypomethylating agents (HMAs), histone deacetylase inhibitors, Hedgehog signaling inhibitors, and therapeutic monoclonal antibodies (mAbs) are currently investigated (17, 18).
Therapeutic mAbs have shown impressive efficacy in the treatment of multiple human cancers, including hematological neoplasms (19). Twenty years ago, the United States Food and Drug Administration (FDA) approved the first therapeutic mAb, rituximab (anti-CD20) for the treatment of B cell malignancies [reviewed in Ref. (20)]. This milestone marked the inception of a new era of precision therapeutics and stimulated the field toward developing further mAb drugs for various human diseases. Currently, more than 60 therapeutic mAbs are approved for the treatment of cancer [e.g., lymphomas, leukemias, multiple myeloma (MM), melanoma, lung, breast, colorectal, urothelial, head and neck, and neural cancers, and sarcomas], autoimmunity and inflammation [e.g., multiple sclerosis (MS), psoriasis, inflammatory bowel disease, systemic lupus erythematosus, asthma, and rheumatoid arthritis], infections (e.g., anthrax, rabies, and respiratory syncytial virus), and other conditions (e.g., osteoporosis, hypercholesterolemia, hypercoagulability, organ transplantation, and age-related macular degeneration). In addition, more than 500 mAbs are under investigation in clinical trials or are in preclinical development (21).
Conceptually, therapeutic mAbs for myeloid neoplasms can be broadly categorized into three different classes: (1) antibodies that target leukemic blasts and LSCs directly, namely antibody–drug conjugates (ADCs) and ligand–receptor antagonists; (2) antibodies that target the interactions of blasts and LSCs with the BM microenvironment; and (3) antibodies that reinforce host immunity [immune checkpoint inhibitors and bispecific antibodies (BsAbs)]. ADCs are mAbs covalently bonded to a cytotoxic drug (e.g., calicheamicin) or a radioisotope (e.g., 90yttrium) and designed to target cell surface antigens to kill tumor cells by pinpoint delivery of a high local amount of toxin or radiation (22). Ligand–receptor antagonists are mAbs that block crucial molecules to a pathological or immunological pathway for AML blast and LSC survival, and/or exert their function via the fragment crystallizable (Fc) region by stimulating antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or complement-dependent cytotoxicity (CDC) (19). Antibodies that target the BM microenvironment interactions are designed to disrupt the molecular mechanisms that keep leukemic blasts and LSCs in their protective BM niche to render them susceptible to chemotherapy or immune attack (23–25). Furthermore, immune checkpoint inhibitors and BsAbs are used to reinforce host immunity against the malignancy.
This review addresses the use of therapeutic mAbs in the context of myeloid neoplasms, mainly AML and MDS. For each of the three conceptual classes of mAbs, examples of past and current preclinical and clinical developments and clinical trials, as well as current developments with future potential are discussed. In the coming years, therapeutic mAbs will be integrated into and will form important components of standard treatment regimens for myeloid neoplasms.
Antibodies That Target LSCs and Blasts Directly
Regarding their surface protein expression profiles, cancer cells are often significantly different from their healthy counterparts. These differences manifest either in the expression level of a certain molecule, its aberrant expression (e.g., oncofetal antigens), or the exclusive dependency of cancer cells on a certain pathway downstream of these molecules and can be exploited to directly target tumor cells using mAbs. In addition, mAbs targeting surface molecules often lead to opsonization of cancer cells, facilitating ADCC, ADCP, and CDC by the immune system. In the following section, the leukemia-associated molecules that are most promising for direct targeting, their corresponding therapeutic mAbs and ongoing clinical efforts to investigate them are described (Figure 1).
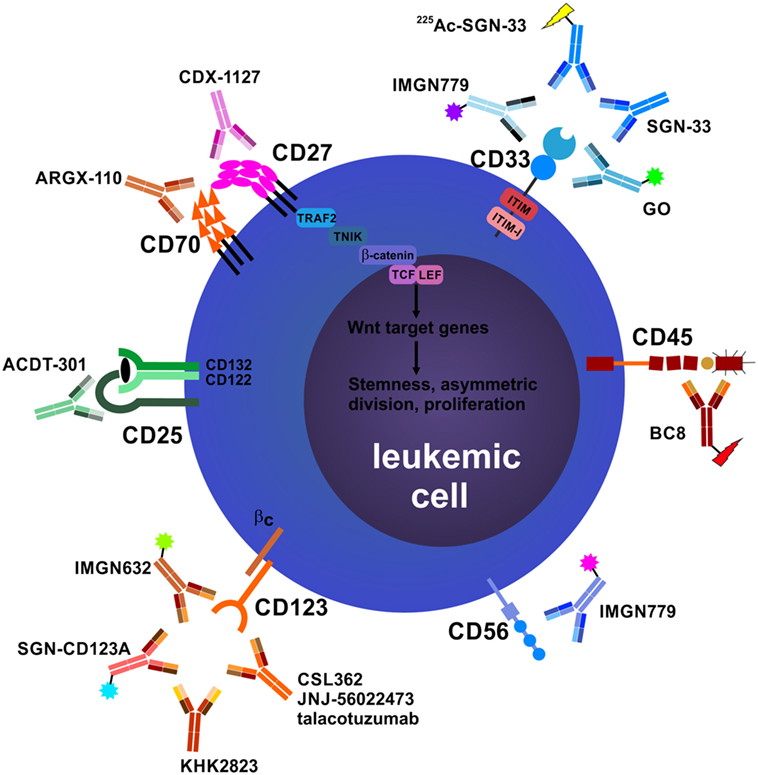
Figure 1. Antibodies that target leukemic stem cells (LSCs) and blasts directly. CD25 is exclusively expressed on LSCs in subsets of acute myeloid leukemia (AML) patients, and CD25 expression on AML blasts is an adverse prognostic marker. In addition, CD25 is highly expressed on tumor-promoting CD4+FOXP3+ regulatory T cells (Treg cells) (not depicted). Anti-CD25 monoclonal antibody (mAb) treatment may eliminate leukemic blasts, LSCs, and Treg cells, leading to enhanced host antileukemic adaptive immunity. The tumor necrosis factor superfamily members CD70 and CD27 are both expressed on AML blasts. Their interaction in an auto- and/or paracrine manner induces the Wnt pathway leading to a stem cell-like phenotype, symmetric cell division, and accumulation of blasts. Blocking the CD70/CD27 interaction induces asymmetric cell division and differentiation in AML blasts. The most well-studied antibody target in AML and myelodysplastic syndrome is CD33. Numerous unconjugated and conjugated anti-CD33 mAbs have been developed, such as gemtuzumab ozogamicin (GO). Anti-CD45 radioimmunoconjugates, such as BC8, are designed to kill CD45-expressing AML blasts and act as conditioning drugs to ablate endogenous hematopoietic and immune cells before allogeneic hematopoietic stem cell transplantation (aHSCT). They may help to reduce conditioning chemotherapy and total body irradiation doses, allowing elderly patients to undergo aHSCT. CD56 (neural cell adhesion molecule) is aberrantly expressed on AML blasts and other hematological neoplasms. High CD56 expression correlates with adverse prognosis in AML. Natural killer cells (NK cells), an important pillar in the combat against cancer, also express high levels of CD56 (not shown). IMGN779, an anti-CD56 antibody–drug conjugate (ADC), led to increased infections and infection-related deaths in a trial of small cell lung cancer and was discontinued by the manufacturer. CD123, the interleukin-3 receptor α chain, is expressed on LSCs in AML and chronic myeloid leukemia. Many anti-CD123 mAbs are currently under clinical development. Their mechanisms of action include direct toxicity (ADCs; SGN-CD123A, IMGN632) and enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) via NK cells (CSL362/JNJ-56022473/talacotuzumab, KHK2823). CD157 is another target for NK cell-mediated ADCC.
Anti-CD25
CD25, the high-affinity interleukin (IL)-2 receptor α chain (IL-2Rα), forms a trimeric receptor for IL-2 together with IL-2Rβ (CD122) and the common γ chain (γc, CD132). CD25 is upregulated on T cells upon antigen stimulation and is expressed at high baseline levels on CD4+FOXP3+ regulatory T cells (Treg cells) during steady state conditions. The cytokine IL-2 is primarily produced by activated CD4+ T cells and has a dual role in immunity: on one hand, it contributes to primary T cell responses and is essential for memory formation; on the other hand, it plays a pivotal role the development, homeostasis and function of natural and induced Treg cells, thereby preventing autoimmunity. Studies using knockout models for IL-2 or CD25 identified effects primarily linked to lymphoproliferation and increased autoimmunity; therefore, the dominant role of IL-2 signaling for Treg cell biology is now accepted (26). Initial studies on the role of CD25 in AML were conducted in the late 1980s by Davey et al. and Carron and Cawley and indicated that CD25 is expressed on blasts in a subset of AML patients and that IL-2 increases their proliferation (27, 28). Other reports demonstrated CD25 expression in lymphocytic leukemias and some lymphomas, and it was therefore reasoned that the IL-2 receptor could be a target for immunotherapy in hematological neoplasms (29). A small clinical trial with 10 patients conducted in the late 1990s (NCT00002681) addressed the effects of the anti-CD25 mAb, daclizumab, in CD25+ hematological neoplasms. Of five patients with myeloid neoplasms (three AML, two CML), one AML patient showed a biological response to daclizumab, with an impressive drop in white blood counts after the first dose, followed by relapse involving a CD25− clone and of the initial CD25+ clone after drug discontinuation. Of note, this was the patient with the highest CD25 expression per cell as indicated by flow cytometry (FACS) (30). More recently, several studies have systematically addressed CD25 expression on AML blasts in genetically defined subsets of AML patients. CD25 expression on blasts correlates with shorter OS, poorer response to induction chemotherapy, higher MRD levels after therapy, and an LSC-like gene signature (31–33). In addition, using gene expression screening followed by protein validation, Saito et al. found CD25 expression on CD34+CD38− LSCs in 25% of the AML samples analyzed (15/61 patients) (34). Importantly, in this study CD25 was exclusively expressed in LSCs but not in CD34+CD38− normal HSCs, and depletion of CD25-expressing cells before HSC xenotransplantation did not affect long-term hematopoietic engraftment—emphasizing the importance of CD25 as an exclusive LSC antibody target (35). A role for the IL-2/CD25 signaling axis was also identified for a subset of CML LSCs (36). Furthermore, anti-CD25 mAb therapy also leads to Treg cell depletion, an effect that is exploited and studied in a clinical trial of AML, using tumor vaccination against Wilms tumor protein-1 in combination with the toll-like receptor agonist polyinosinic:polycytidylic acid and the anti-CD25 mAb basiliximab (NCT01842139).
A novel anti-CD25 ADC using a DNA crosslinking agent of the pyrrolobenzodiazepine dimer (PBD) class [ADCT-301, camidanlumab tesirine (Cami-T)] showed preclinical activity in lymphoma mouse models (37), clinical activity in heavily pretreated Hodgkin lymphoma patients (38) and is currently under investigation in a clinical trial for patients with AML and acute lymphoblastic leukemia (ALL) (NCT02588092). A first interim report on 29 pretreated AML patients recruited in this study was presented at the American Society of Hematology annual meeting 2017. Single-agent Cami-T resulted in a transient CD25+ blast clearance in two patients and showed an acceptable safety profile. No dose-limiting toxicity (DLT) was reported and the maximum tolerated dose (MTD) was not reached. In the observed period, single-agent Cami-T did not lead to objective responses or remissions (39). Given that CD25 is only expressed in the hematopoietic system and absent on HSCs, targeting this receptor in the subset of CD25+ AMLs seems promising. A further advantage of targeting CD25 is its high expression on tumor-promoting Treg cells, which are depleted by anti-CD25 mAbs, resulting in a more robust host adaptive antileukemic immune response. Careful examination and follow-up of long-term effects of anti-CD25 mAb therapy, such as infections and autoimmunity will be essential.
Anti-CD27 and Anti-CD70
The costimulatory cell surface receptor–ligand pair CD27 and CD70 belongs to the tumor necrosis factor (TNF) superfamily. CD70, the unique ligand for CD27, is not present under homeostatic conditions and is only expressed on activated lymphocytes and dendritic cells (DCs) in a highly regulated manner during immune activation (40). By contrast, CD27 is constitutively expressed on lymphocytes and HSCs (40–42). Besides its immunostimulatory functions in regulating lymphocyte activation, survival, expansion, and effector function, CD27 signaling also modulates HSC self-renewal and differentiation (40, 41, 43). We have demonstrated that CD27 is expressed on LSCs from CML patients and that triggering of CD27 on LSCs in a murine CML model promotes tumor cell growth and disease progression by activating the Wnt pathway (42). Furthermore, it is inferred that CD70 expression is induced in CML LSCs by TKI treatment as a mechanism of resistance leading to compensatory Wnt pathway induction (44). Moreover, we recently found that CD70/CD27 signaling promotes a stem cell-like phenotype, proliferation in AML blasts and AML stem/progenitor cells and that mAbs blocking of the CD70/CD27 interaction induce asymmetric cell division and differentiation of AML blasts. Importantly, healthy donor (HD) hematopoietic stem/progenitor cells were negative for CD70 and CD27 and were unaffected by anti-CD70 mAb treatment (45). Based on these findings, an open-label phase 1/2 multicenter clinical trial investigating the tolerability and efficacy of the anti-CD70 mAb ARGX-110 in combination with 5-azacytidine (AZA) in untreated AML or high-risk MDS patients was recently initiated (NCT03030612). Another attractive therapeutic strategy is the stimulation of CD27 on T cells to reinforce antitumoral immune responses or directly target CD27-expressing tumors, including many lymphoma types (40). Indeed, initial results from a clinical trial (NCT01460134) investigating monotherapy with an agonistic anti-CD27 mAb (CDX-1127, varlilumab) in patients with advanced solid tumors and B cell lymphomas was recently reported (46). Several other trials with varlilumab are currently ongoing; however, this mAb has not yet been tested clinically in myeloid neoplasms, such as AML or MDS.
Anti-CD33
CD33 (siglec-3) is a member of the superfamily of sialic acid-binding I-type lectins (siglecs) that share common V-set and C2-set immunoglobulin domains. Besides the siglecs present in all mammals (siglec-1, -2, -4, and -15), humans possess a repertoire of nine CD33-related siglecs, including CD33. Siglecs are mainly expressed on myeloid cells and lymphocytes. They modulate immune cell function through recognition of microbial products and cell–cell interactions by signaling via inhibitory cytosolic motifs (47, 48). Since CD33 is frequently expressed on leukemic blasts in adult and pediatric AML and CD33 expression has not been reported outside the hematopoietic system, it is an attractive target for mAb therapy. The anti-CD33 mAb, gemtuzumab ozogamicin (GO, Mylotarg), is an antibody conjugated with a potent chemotherapeutic, the DNA intercalator calicheamicin. GO and lintuzumab, another non drug-conjugated anti-CD33 mAb, were among the first therapeutic antibodies clinically studied in AML, and GO was the first ADC to be approved by the FDA under accelerated approval regulations in 2000 (49–51). GO was intensely studied in numerous clinical trials totaling >7,800 AML patients: in elderly patients with de novo or secondary AML either as a monotherapy or combined with low-intensity chemotherapy; in patients of all ages with de novo AML in combination with standard induction therapy and as consolidation/maintenance therapy [comprehensively reviewed in Ref. (52–54)]. However, because follow-up phase 3 trials indicated no survival benefit and an increase of early deaths due to enhanced toxicity when GO was added to induction therapy, GO was voluntarily withdrawn from the market in 2010 (55). Since its withdrawal, the results of several large studies have been evaluated in a meta-analysis of 3,325 AML patients receiving GO in addition to standard induction chemotherapy. This analysis found no effect on the rate of remission, but reduced relapses at 5 years and a survival benefit particularly for low-risk, and to a lesser extent, intermediate-risk groups was reported. By contrast, no difference was observed for adverse-risk patients (56). Besides cytogenetic risk, which may impact the sensitivity of AML blasts to calicheamicin, the reasons for these differences in GO efficacy remain unclear (52). Several mechanisms have been proposed, including the expression level of CD33 on blasts and the frequency of CD33-positive blasts (57–61); CD33 expression on non-AML blood cells acting as a sink for GO (62); P-glycoprotein transporter activity in AML blasts (58); CD33 splice variants and polymorphisms (63); and other mechanisms (52, 64, 65). Therefore, CD33 remains an attractive therapeutic target for AML, which is reflected by numerous currently active clinical trials that address the effects of GO in more well-defined patient subsets (Table 1), as well as the development of novel anti-CD33 mAbs.
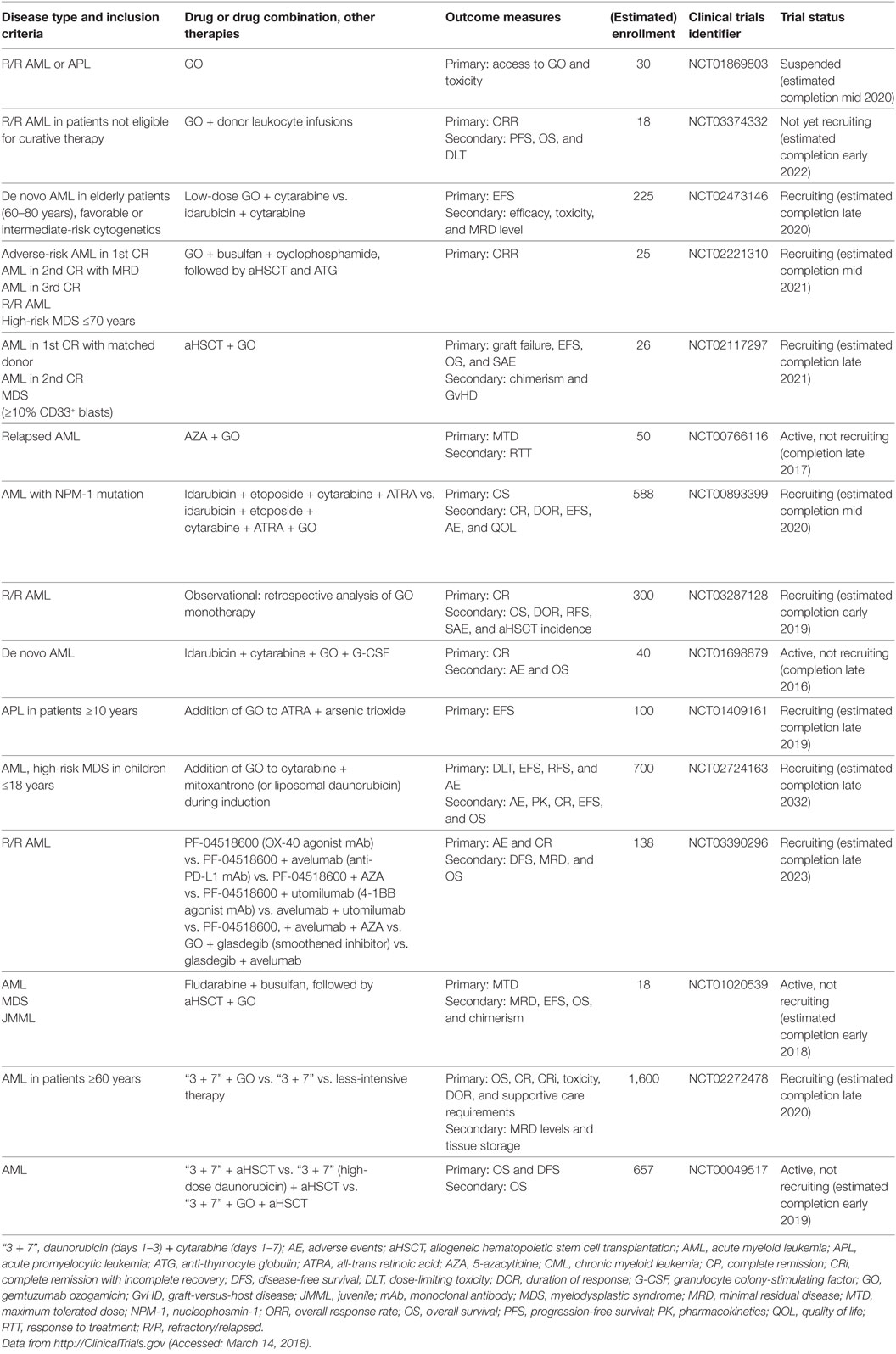
Table 1. Currently active clinical trials of GO in AML.
Lintuzumab (SGN-33, HuM195), an unconjugated anti-CD33 mAb, has been thoroughly studied as a complement to standard induction chemotherapy and as a maintenance monotherapy in R/R AML, APL, high-risk MDS, accelerated-/blast-phase CML, and chronic myelomonocytic leukemia. A total of 448 patients were enrolled in 8 clinical trials between 1994 and 2011 (NCT00002609, NCT00002800, NCT00006084, NCT00016159, NCT00283114, NCT00502112, NCT00528333, and NCT00997243). Although early studies showed promising results and low toxicity (66), subsequent larger studies were not able to demonstrate a significant survival benefit (67, 68); therefore, unconjugated lintuzumab was not further developed for these indications. Another unconjugated anti-CD33 mAb, BI 836858, has decelerated internalization kinetics after binding to CD33 on blasts and improved affinity to FcγRIIIA, with enhanced NK cell-mediated ADCC (69). BI 836858 is currently undergoing evaluation in 5 clinical trials in >800 AML and MDS patients, either as monotherapy or in combination with HMAs or other biologicals (NCT02632721, NCT03207191, NCT01690624, NCT02240706, and NCT03013998). In addition, several radioimmunoconjugates of lintuzumab were developed to overcome the limited efficacy of unconjugated lintuzumab. These mAbs include lintuzumab-90yttrium (90Y), lintuzumab-213bismut (213Bi), and lintuzumab-225actinium (225Ac) and were investigated in clinical trials (Table 2). In addition, Hagemann et al. recently reported the development of an anti-CD33 mAb linked to 227thorium (70). Moreover, anti-CD33 mAbs have been successfully conjugated to the novel cytotoxic and DNA-damaging drugs of the PBD class. Vadastuximab talirine (SGN-CD33A) is an anti-CD33 mAb coupled to a PBD that has shown superior activity compared with GO in preclinical models of drug-resistant AML (71). Early clinical studies of SGN-CD33A showed promising results (72–74); however, interim analyses of larger unblinded datasets in one study (NCT02785900) revealed a higher induction mortality rate and more infections in the SGN-CD33A verum group. Therefore, the manufacturing company, Seattle Genetics, recently halted all active clinical trials with SGN-CD33A (75). Further studies are needed to determine which AML patients optimally benefit from anti-CD33 therapy, and which mAbs ultimately prove effective, safe, and tolerable.
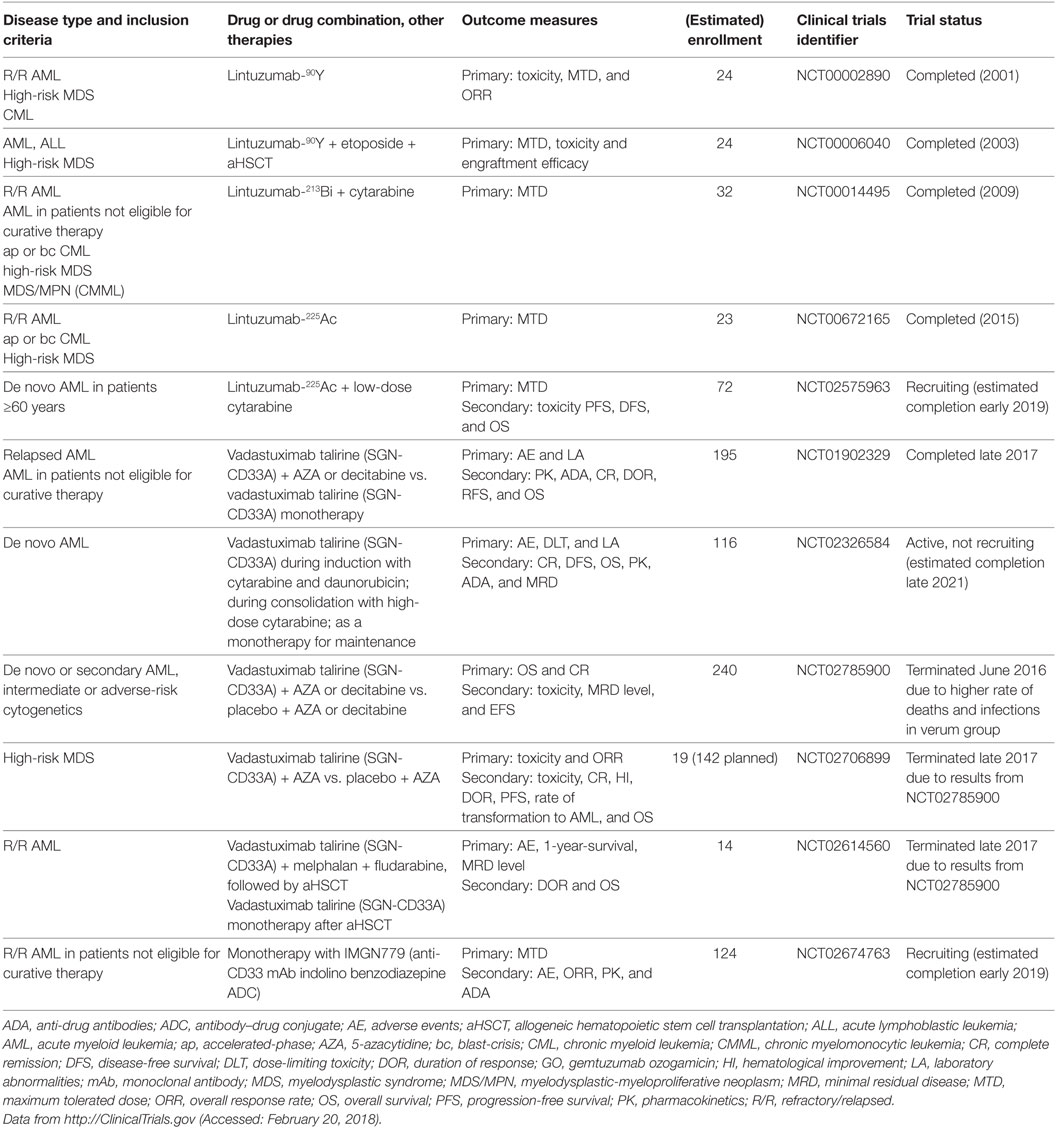
Table 2. Clinical trials of drug-conjugated anti-CD33 mAbs other than GO.
Anti-CD45
CD45 or protein tyrosine phosphatase, receptor type, C (PTPRC) is an essential regulator of signal transduction pathways in hematopoietic and immune cells. It is one of the most abundant cell surface glycoproteins and has multiple isoforms due to alternative splicing of exons 4–6 (76). Because CD45 is ubiquitously expressed in the hematopoietic and immune systems, it is not an ideal target for cancer-specific mAb treatment. Rather, anti-CD45 radioimmunoconjugates, such as BC8, are designed as adjuvants for conditioning regimens consisting of chemotherapy (fludarabine) and total body irradiation (TBI) before aHSCT. The rationale of anti-CD45 mAb in this setting is to lower the doses of or even replace conditioning chemotherapy and TBI to reduce overall toxicity and late-onset complications, such as secondary malignancies and end-organ damage. Orozco et al. showed in a mouse model of leukemia that 90yttrium-anti-CD45 mAb treatment was an effective conditioning regimen and at doses of 400 μCi led to 50% long-term OS in combination with aHSCT (77). Indeed, different radioimmunoconjugates of anti-CD45 are currently tested in clinical trials of aHSCT for AML, MDS, high-risk lymphoma and MM (Table 3) (78–80).
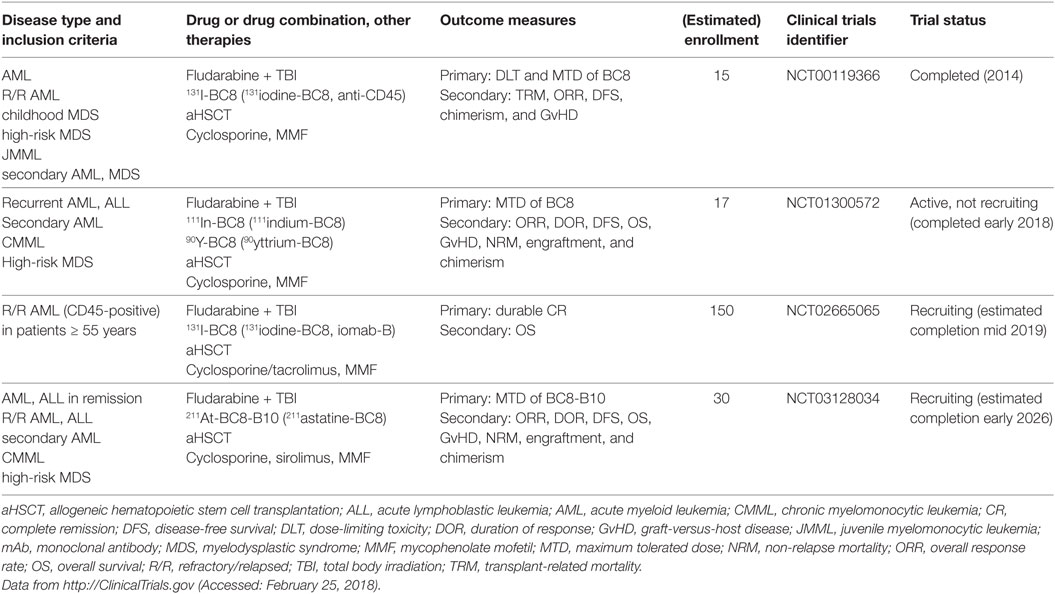
Table 3. Recent active clinical trials of anti-CD45 mAbs in myeloid neoplasms.
Anti-CD56
CD56, also known as neural cell adhesion molecule, is a member of the immunoglobulin superfamily expressed on neurons, glial cells, skeletal muscle cells, and in the hematopoietic system, mainly in natural killer (NK) cells. Aberrant expression of CD56 is seen in a wide variety of solid and hematological neoplasms, including epithelial and neural cancers, MM and leukemias (81). In AML, high CD56 expression is associated with adverse cytogenetics, extramedullary disease and poor prognosis (82–85). An anti-CD56 ADC, lorvotuzumab mertansine (IMGN901) was tested in 9 patients with hematological malignancies including AML (NCT02420873), in 2 studies in 181 patients with small cell lung cancer (NCT01237678) and in 62 patients with rare CD56-positive malignancies (NCT02452554). The lack of antitumoral efficacy in the small cell lung cancer trial and the increased incidence of infections and infection-related deaths led to discontinuation of lorvotuzumab mertansine by the company, ImmunoGen, in 2013. Given the fact that NK cells express high levels of CD56 and their important role in the control of leukemia and infections, it remains to be determined whether targeting CD56 by therapeutic mAbs is a valid treatment option for AML.
Anti-CD123
Upon biding to IL-3, the IL-3 receptor α chain (CD123) forms a heterodimer with the signal transducing beta chain (βc), which is shared among the receptors for IL-3, IL-5, and granulocyte-monocyte colony-stimulating factor. Whereas βc is widely expressed among the hematopoietic system, the expression of CD123 is more restricted and this molecule is primarily found on HSCs, monocytes, megakaryocytes, B cells, and plasmacytoid DCs (pDCs) (86). In 2000, Jordan et al. reported that CD123 is highly overexpressed on AML blasts and LSCs compared with normal hematopoietic cells and might serve as a therapeutic target for AML (87). This discovery paved the way for numerous studies dissecting the role of CD123 in leukemia biology. For example, Jin et al. reported that anti-CD123 mAb treatment of AML xenografted mice targeted LSCs, impaired their homing and engraftment in the BM microenvironment, activated innate immunity, and prolonged mouse survival (88). In addition, Nievergall et al. recently showed that CD123 is expressed on CD34+ and CD34+CD38− LSCs in chronic-phase and blast-crisis (bc) CML, and at higher levels compared with healthy HSCs. The expression of CD123 was similar on bcCML LSCs and AML LSCs. Interestingly, cpCML patients had increased serum IL-3 and TKI therapy reduced serum IL-3 to almost baseline. Administration of the anti-CD123 mAb CSL362 rendered LSCs sensitive to TKIs and promoted NK cell-induced ADCC (89). In cynomolgus monkeys, CSL362 potently reduced blood CD123+ basophils and pDCs via NK cell-mediated ADCC (90). Many anti-CD123 mAbs are currently under development and are being investigated in clinical trials (Table 4). The first anti-CD123 mAb tested in humans was CSL362. Smith et al. studied the pharmacokinetics and safety of CSL362 in a phase 1 clinical trial of 30 AML patients in first remission who were ineligible for post-induction chemotherapy or aHSCT (91). Of 11 patients with MRD-positive status, CSL362 induced MRD negativity in 4 patients at 24 weeks of follow-up, suggesting that CSL362 eradicated remaining LSCs (92). However, the surrogate endpoint MRD may not be optimal to predict long-term AML survival and antibody therapy efficacy, since different detection methods (i.e., polymerase chain reaction or FACS) and different laboratory standards complicate interpretation. The anti-CD123 mAb talacotuzumab (JNJ-56022473), which was derived from CSL362, showed potent in vitro preclinical activity against AML (93, 94). However, the two clinical trials testing JNJ-56022473 are currently suspended and further development of the drug was discontinued due to unfavorable risk/benefit profiles in vivo (95). Another anti-CD123 mAb, SGN-CD123A, is an ADC with a PBD, which induces DNA damage and apoptosis in AML cells. SGN-CD123A exhibited potent cytotoxicity in vitro against AML cell lines and primary samples and was effective in various AML murine models in vivo (96). This mAb is currently investigated in a clinical trial of R/R AML (NCT02848248). In addition, Akiyama et al. reported the generation of a non-fucosylated, fully human anti-CD123 mAb (KHK2823). This mAb was effective in suppressing the growth of the MOLM-13 AML cell line in nude rats, and depleted CD123+ cells from peripheral blood (PB) in cynomolgus monkeys and was well tolerated (97). A clinical trial testing KHK2823 in patients with R/R AML and MDS (NCT02181699) was recently completed and first results are expected soon. Furthermore, Adams et al. and Kovtun et al. generated IMGN632, a humanized anti-CD123 mAb, which is linked to the DNA-alkylating agent, DGN549. This ADC was effective against human AML cell lines and primary samples in vitro, cell line xenografts in vivo, and is currently being tested clinically (NCT03386513) (98, 99). Given the drawbacks that some of these mAbs faced in clinical trials, it remains to be determined which anti-CD123 mAbs and in which formulation will ultimately prove useful.
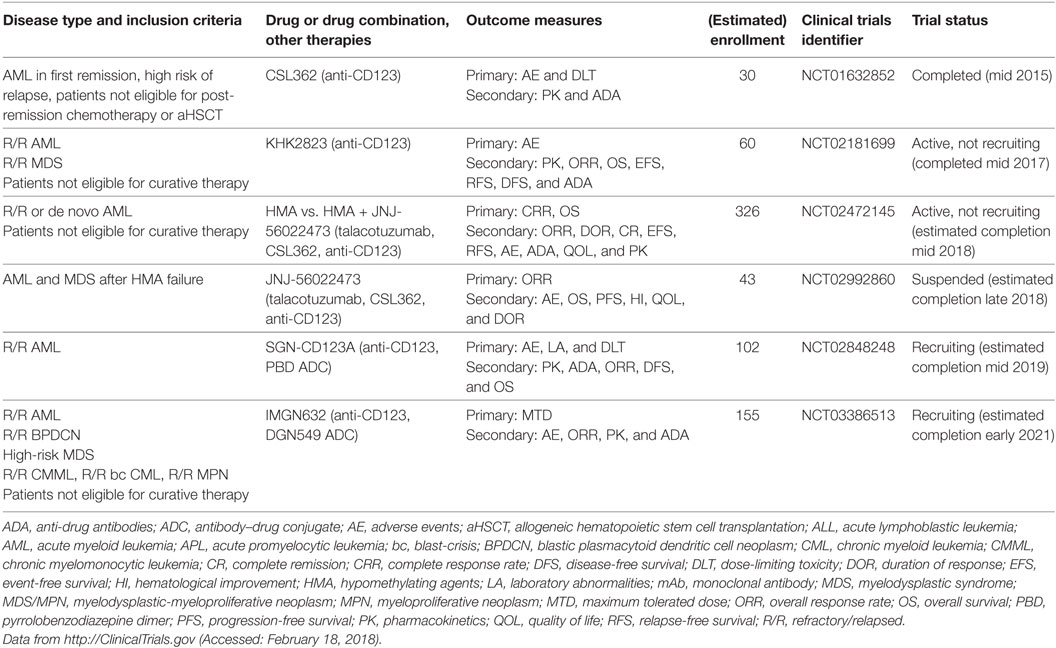
Table 4. Clinical trials of anti-CD123 mAbs in myeloid neoplasms.
Antibodies That Target the BM Microenvironment
The BM microenvironment, also known as the HSC niche, consists of both non-hematopoietic and hematopoietic cell subsets. This niche precisely regulates fundamental HSC characteristics, such as quiescence, self-renewal, asymmetric cell division, and non-exhaustive proliferative potential via direct cell–cell contacts and through soluble factors (100, 101). In recent years, the cellular elements of the HSC niche and their role in HSC function and hematopoiesis have been characterized. Non-hematopoietic cells, such as osteoblasts, adipocytes, endothelial cells (ECs), vascular pericytes, and mesenchymal stromal cells (MSCs) are derived from rare mesenchymal stem cells (102, 103). Osteoblastic niche cells (104, 105), as well as various perivascular cells, such as NG2+ pericytes ensheathing BM arterioles (106), CXCL12-abundant reticular cells (107), and leptin-receptor+ CXCL12-expressing perisinusoidal cells (108–110) regulate HSC quiescence, self-renewal, proliferation, and mobilization. In addition to the regulation of hematopoiesis during homeostasis, the BM microenvironment plays a crucial role in the process of demand-adapted hematopoiesis during stress situations, such as in systemic infections (also known as emergency hematopoiesis) (111–113), hematopoietic regeneration after injury (e.g., cytotoxic chemotherapy or irradiation) (114, 115) and in the organization of immunological memory (116). Furthermore, the BM serves as a secondary lymphoid organ that plays an important role as a priming site for T cells to blood-borne antigens (117). The BM hosts various mature immune cells such as naïve and memory T and B cells, plasma cells, DCs, and differentiated myeloid cells that contribute to the regulation of hematopoiesis by secreting various cytokines and presumably by yet undefined cell–cell interactions (118). Importantly, recent studies indicate that the BM microenvironment plays a previously underestimated role in the pathogenesis of myeloid neoplasms. In analogy to normal hematopoiesis, self-renewing LSCs are responsible for disease initiation and progression to more differentiated malignant cells that cause the clinical symptoms of leukemia. Quiescence, plasticity, the expression of drug efflux proteins, and the localization in protective niches render LSCs resistant to therapy (119, 120). Many LSC features are not only cell-intrinsic but are highly regulated by the BM microenvironment. In fact, myeloid neoplasms can create and maintain a leukemia-supporting dysfunctional niche, and vice versa, aberrant niches can contribute to leukemia development. For example, genetic ablation of the retinoic acid receptor or the retinoblastoma protein in the HSC niche is sufficient to induce an MPN (121, 122). In addition, constitutively active β-catenin in osteoblasts causes AML in mice (123) and inactivation of Dicer, a protein that processes micro-RNAs, in osteolineage cells results in MDS (124). Furthermore, BM MSC-secreted IL-33 contributes to MPN pathogenesis by inducing a cytokine- and growth factor-rich microenvironment (125). Moreover, leukemia cells disrupt the function of the sympathetic nervous system in the BM by damaging adrenergic nerve fibers and promoting apoptosis of Schwann cells and MSCs, which allows the expansion of MPN LSCs (126, 127). Interestingly, the administration of neuroprotective drugs or sympathomimietics reduced LSC accumulation and prevented MPN disease progression (126). In addition, MPN cells, via direct cell–cell contact and through soluble factors, induce the accumulation of altered osteolineage cells with an inflammatory phenotype that promote BM fibrosis and inhibit normal HSC function, while effectively supporting LSCs (128). Similar mechanisms of niche remodeling were also observed in human hematological neoplasms, such as MDS (129) and ALL (130). Importantly, these leukemic niches evolve during treatment and contribute to therapy resistance (130, 131). Therefore, targeting the interactions between LSCs, leukemic blasts and the BM microenvironment using therapeutic mAbs (Figure 2) is a rational approach to treat myeloid neoplasms and to overcome treatment resistance mechanisms (23, 25).
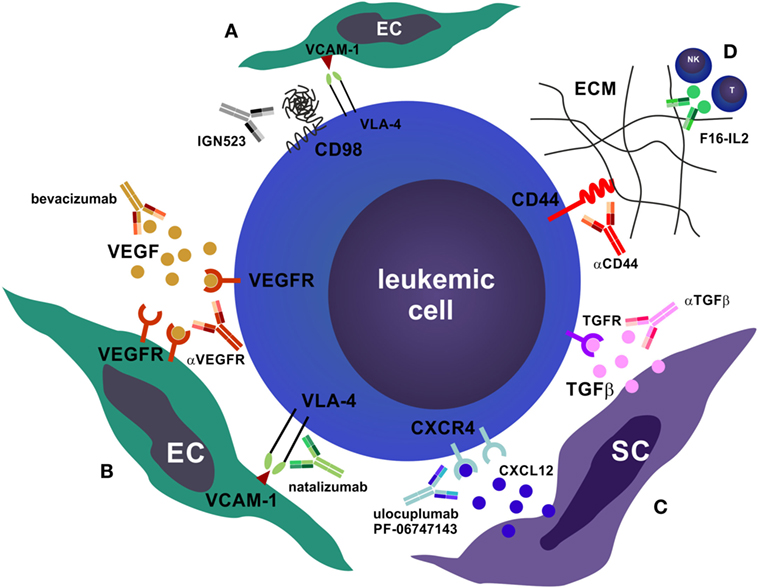
Figure 2. Antibodies that target the bone marrow (BM) microenvironment. (A) CD98 is required for the very late antigen-4 (VLA-4)/vascular cell adhesion molecule-1 (VCAM-1) mediated adhesion of leukemic cells to BM endothelial cells (ECs) (132). Blocking of CD98 by monoclonal antibodies (mAbs) disrupts this adhesion and promotes apoptosis of leukemic cells. (B) Blocking leukemic cell–EC interactions can also be realized by mAbs that target vascular endothelial growth factor (VEGF) or VEGFR (e.g., bevacizumab) as well as mAbs that target the VLA-4/VCAM-1 interaction (e.g., natalizumab). This impairs homing and niche retention of leukemic cells, leading to mobilization and chemosensitization. (C) Similarly, targeting the stromal cell (SC) niche by mAbs that block CXCR4 and prevent binding of the ligand CXCL12 (ulocuplumab, PF-06747163)—or that block transforming growth factor beta (TGFβ)—promotes quiescence exit, proliferation, mobilization, and chemosensitization. In addition, αCXCR4 mAbs induce complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and are directly cytotoxic to leukemic cells. (D) Targeting the interactions of leukemic cells with the BM extracellular matrix (ECM), e.g., with αCD44, or recruiting CTLs and natural killer cells toward the tumor site by anti-tenascin C interleukin (IL)-2 fusion proteins (F16-IL2), are further possibilities to disrupt the leukemic niche.
Anti-CXCR4
It has long been known from coculture experiments that BM-derived MSCs induce proliferation and prevent apoptosis of AML blasts (133–135). In 1994, Nagasawa and colleagues cloned a cytokine secreted by an MSC line that supported the proliferation of early B cell progenitors, which they termed pre-B cell growth-stimulatory factor (PBSF). PBSF was subsequently shown to be identical to stromal cell-derived factor 1α (SDF-1α), which is currently known as C–X–C motif chemokine ligand 12 (CXCL12) (136, 137). Its receptor, CXCR4 (CD184), is a G-protein-coupled receptor that was identified as a co-receptor for human immunodeficiency virus-1 (138, 139). CXCL12–CXCR4 signaling plays a dominant role in migration, BM retention and quiescence of HSCs and immune organ and cell development (140, 141). CXCR4 is expressed in AML (142, 143), and high expression on AML blasts is an independent adverse prognostic factor (144). Numerous studies have also indicated that inhibition of CXCR4 by small-molecule inhibitors (AMD3100/plerixafor; AMD3465) leads to peripheral mobilization of AML cells and renders them sensitive to chemotherapy (145–147). Indeed, plerixafor was safely administered to AML patients with promising results in early-phase clinical studies in a well-defined patient population. However, recent studies from more heterogeneous patient populations were mixed in terms of response rates. It is hypothesized that this effect could partly be attributed to the short half-life of plerixafor of only 4–5 h (148, 149). The recent development of humanized IgG CXCR4 blocking therapeutic mAbs (BMS-936564/MDX-1338/ulocuplumab and PF-06747143) should overcome this drawback of small-molecule inhibitors, and these mAbs have shown promising results in multiple murine models of hematologic malignancies, including AML, non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and MM. In particular, treatment with anti-CXCR4 mAbs inhibited migration, induced ADCC and CDC and led to direct cytotoxicity in malignant cells (150–156). These mAbs are currently being investigated in clinical trials, including in de novo and R/R AML, as single agents and in combination with chemotherapy (Table 5).
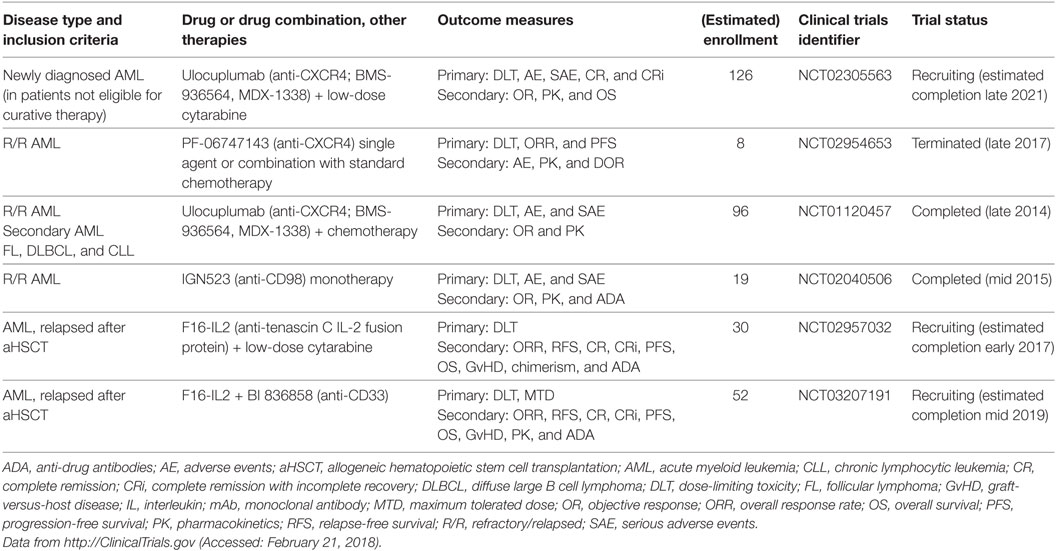
Table 5. Clinical trials of mAbs that target the microenvironment in myeloid neoplasms.
Anti-CD98
CD98, a heterodimeric transmembrane protein consisting of a heavy and a light chain, is a multifunctional molecule involved in integrin adhesion, extracellular matrix (ECM) assembly and essential amino acid transport (157–160). CD98 plays a role in various immune processes, such as lymphocyte activation and proliferation, plasma cell development and autoimmunity, and in malignant transformation of epithelial cells by promoting anchorage-independent growth and proliferation (161–165). CD98 overexpression has been demonstrated in many solid tumors and it is also implicated in the pathogenesis of hematological neoplasms (166). For example, Rosilio et al. showed that small-molecule targeting of the L-type amino acid transporter 1, one of the possible light chain subunits of CD98, induces autophagy and apoptosis in lymphoblastic leukemia cells and reduces their growth in vivo in xenografts (167). In addition, using genetic deletion of CD98 in mouse models and anti-CD98 mAb treatment of human AML xenografted NSG mice, Bajaj et al. discovered that AML LSCs and blasts depend on CD98 for their adhesion to BM ECs (132). The anti-CD98 mAb (IGN523) used by these authors was developed by Hayes et al. (168) and has already been tested in a small phase 1 clinical trial with 19 R/R AML patients, which was completed in 2015 (NCT02040506, Table 5). IGN523 showed a favorable safety profile with manageable side effects, and a modest single-agent antileukemic activity was observed in 3 of 19 patients, including improved platelet counts and reduction of BM and blood blast counts (169). The therapeutic value of adding anti-CD98 mAbs to standard chemotherapy or in combination with other drugs remains to be investigated.
Anti-VEGF
Depriving tumors of their nutrient and oxygen supply by targeting their vasculature is a reasonable strategy and has proven effective in advanced stage solid tumors. Key molecules in vascular biology and neovascularization belong to the vascular endothelial growth factor (VEGF) family of proteins, which consists of VEGF A-D and placenta growth factor. VEGFs bind to three VEGF receptor tyrosine kinases: VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4) (170). Early experimental studies of localized subcutaneous AML cell implantation and treatment with anti-VEGF and anti-VEGFR2 mAbs in mice found a dramatic effect on the tumor burden and growth of local microvasculature in mAb treated mice (171). Moreover, the addition of bevacizumab (anti-VEGFA mAb) to chemotherapy in 48 R/R AML patients resulted in a favorable CR rate and was well tolerated (172). However, single-agent bevacizumab had no antileukemic activity, despite reducing VEGF expression in the BM microenvironment (173). By combining anti-VEGF mAb therapy with another vasculature-disrupting small-molecule compound (OXi4503), Madlambayan et al. induced enhanced antileukemic activity in AML xenografts (174). Between 2001 and 2004, four clinical trials (NCT00022048, NCT00015951, NCT00096148, and NCT00023920) assessed the effects of bevacizumab in R/R AML and MDS, but currently, there are no active clinical trials on anti-VEGF mAb therapy in AML or MDS. It remains to be seen whether targeting the vasculature with therapeutic antibodies proves a clinically valid strategy for myeloid neoplasms.
Additional BM Microenvironment Targets
Tenascin C is an ECM glycoprotein that interacts with fibronectin and is expressed during development, tissue injury and disease. Tenascin C plays an important role in inflammatory and fibrotic processes, tumor angiogenesis and as an immunomodulator in cancer (175). Gutbrodt et al. demonstrated that the remodeled BM vasculature in AML can be targeted with mAbs against tenascin C. A fusion protein of anti-tenascin C and IL-2 (F16-IL2) was effective in recruiting CTLs and NK cells to the tumor site and in combination with low-dose cytarabine contributed to tumor clearance in mouse models and patients (176, 177). The efficacy of F16-IL2 is studied in two clinical trials of relapsed AML after aHSCT (Table 5). Very late antigen-4 (VLA-4) is an adhesion molecule expressed on myeloid cells and variably on AML blasts. It binds to vascular cell adhesion molecule-1 (VCAM-1), expressed on BM osteoblasts and ECs, and to fibronectin in the ECM (178). On AML blasts, VLA-4 expression itself did not have a prognostic value for OS but binding of soluble VCAM-1 to VLA-4 did (179). Therefore, Hsieh et al. treated AML xenografted mice with natalizumab, an anti-VLA-4 mAb developed for MS, which led to improved survival (180). However, due to a high rate of lethal progressive multifocal leukoencephalopathy caused by JC virus in MS patients treated with natalizumab, this mAb was not further tested in myeloid neoplasms (25). Transforming growth factor beta (TGFβ) is produced by BM MSCs and plays an important role in the quiescence of AML cells. Tabe et al. used mAb 1D11 to neutralize TGFβ, which resulted in increased AML cell proliferation, rendering the tumor cells sensitive to chemotherapy (181). Furthermore, Jin et al. showed that targeting the adhesive molecule CD44 by an activating mAb (H90) inhibits the homing and engraftment of AML LSCs in xenotransplantation assays, altering their fate and increasing their differentiation (182). Surprisingly, Ye et al. discovered that LSCs from bcCML and AML that express the fatty acid transporter CD36 can utilize a special microenvironment niche in the gonadal adipose tissue to evade chemotherapy (183). However, so far, no therapeutic anti-CD36 mAb has been generated, and neither anti-TGFβ nor anti-CD44 mAbs have been further developed for clinical use in AML.
Antibodies That Reinforce Host Immunity
The innate and adaptive immune systems are responsible for tissue homeostasis by maintaining self-tolerance and recognizing and eliminating foreign antigens. In the case of an acute immune response, costimulatory/co-inhibitory receptors and their ligands coordinate the intricate interplay of immune cells leading to activation, expansion, effector function, memory formation, and involution of T cell responses. The ultimate outcome of an adaptive immune response in terms of quality, strength and duration is directly dependent on the balance of costimulatory and co-inhibitory signals (184). If an antigenic stimulus persists over a prolonged period, such as in chronic infections or cancer, the adaptive immune system becomes non-reactive. Dysfunctional antigen-specific T cells may enter a distinct differentiation program referred to as exhaustion or anergy that is characterized by gradual loss of function and eventually culminates in physical deletion through apoptosis. Anergic antigen-specific T cells are IL-2 non-responsive and non-proliferative, do not produce pro-inflammatory cytokines and express high amounts of co-inhibitory receptors such as programmed death protein-1 (PD-1), lymphocyte activation gene-3 (LAG-3) and others (185).
Myeloid neoplasms, being cancers of the immune system, express major histocompatibility complex (MHC) class I and II molecules and costimulatory ligands and thereby possess the intrinsic capability to activate T cells (186). In addition, the majority of myeloid neoplasms have defined genetic aberrations (such as BCR-ABL1, PML-RARα, RUNX1-RUNX1T1, CBFβ-MYH11, etc.) as well as further mutations that may serve as neo-antigens and induce potent T cell responses. However, like other cancers, myeloid neoplasms have evolved numerous strategies to inhibit T cell responses and innate immunity. Examples include the expression of ligands for PD-1 (PD-L1, PD-L2) (187–190), the induction of tolerance by generating defective leukemia-derived DCs (191, 192) and the upregulation of antiphagocytic “don’t eat me” molecules, such as CD47 (193). Since myeloid neoplasms respond to immunotherapies like aHSCT [graft-versus-leukemia effect (GvL)], interferon (IFN)-α, and IL-2 (186), it seems rational to reinforce host immunity in these cancers. Therapeutic mAbs that enhance antitumoral T cell responses by blocking inhibitory signals (immune checkpoint inhibitors), BsAbs that force T cell-tumor cell interactions, and antibodies that activate antitumoral innate immunity [tumor-associated macrophages (TAMs) and NK cells] all represent potential drugs that may change current therapy standards.
Antibodies That Enhance Antitumoral T Cells: Immune Checkpoint Inhibitors
Immune checkpoint pathways are central in the maintenance of self-tolerance, regulating the duration and strength of an immune response. Immune checkpoint inhibitors that non-specifically release the brakes on anergic tumor-specific T cells are considered breakthrough therapy agents for advanced metastatic solid tumors, such as malignant melanoma. The most well-known examples of immune checkpoint inhibitors are mAbs directed against cytotoxic T lymphocyte antigen-4 (CTLA-4) and PD-1 or its ligands, mainly PD-L1. These antibodies are currently being evaluated in numerous studies for patients with AML and MDS (Table 6), as single agents and in combination with standard treatment and/or aHSCT [recently reviewed in Ref. (194, 195)].
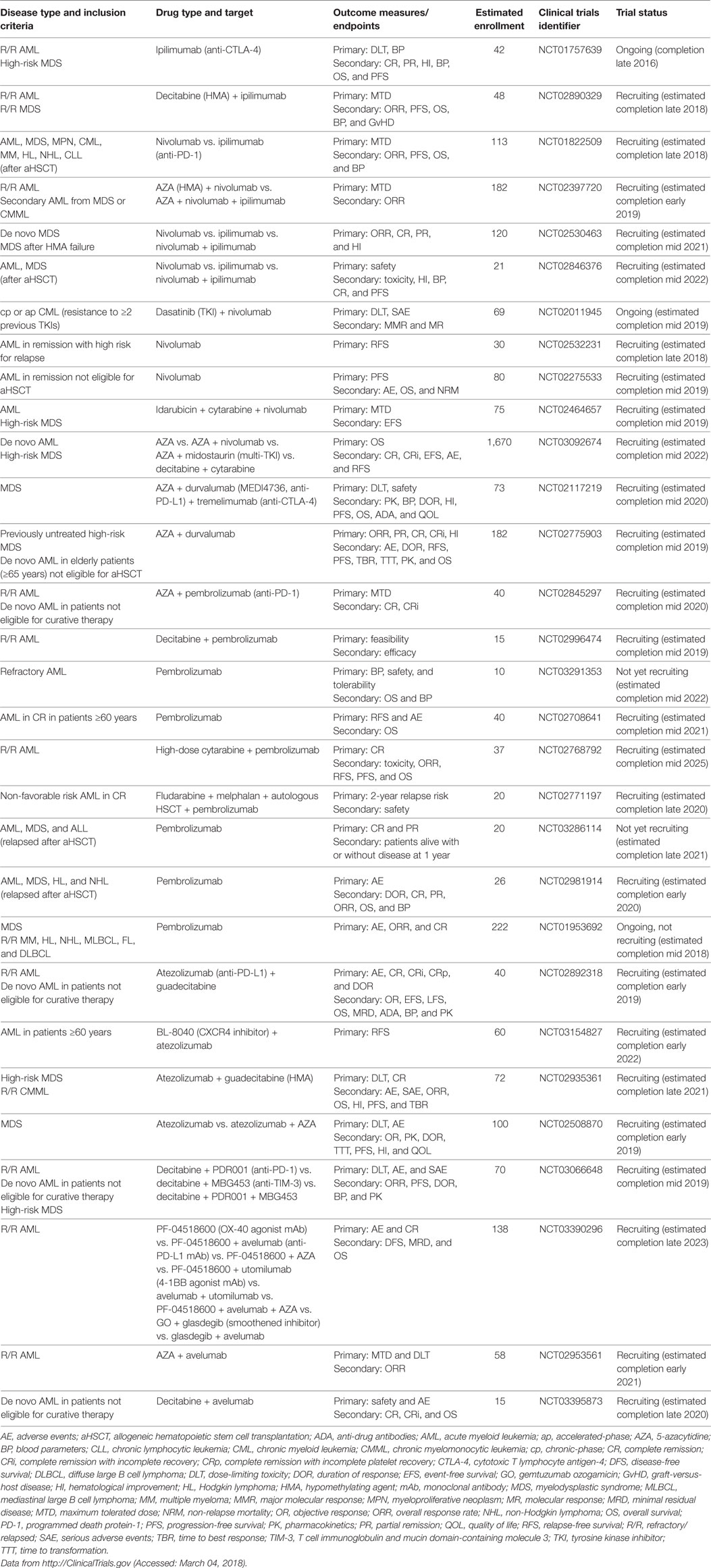
Table 6. Active clinical trials of immune checkpoint inhibitors in myeloid neoplasms.
Anti-CTLA-4
Cytotoxic T lymphocyte antigen-4 (CD152) is a co-inhibitory molecule with structural homology to the costimulator CD28. While CD28 is constitutively expressed on T cells, CTLA-4 is only upregulated 24–48 h after activation of T cells, mainly to counteract stimulatory CD28 signals in the lymph node (184). The ligands for CTLA-4 are the same as for CD28, that is, CD80 (B7-1) and CD86 (B7-2). Upon activation, both CD4+ helper T cells (TH cells) and CD8+ cytotoxic T cells (CTLs) express CTLA-4. Several mechanisms of T cell inhibition by CTLA-4 have been proposed, including the recruitment of protein phosphatases, the restriction of cellular metabolism (196), the competition for ligands with CD28, the physical disturbance of immunological synapse assembly, and the stimulation of inhibitory cytokine secretion (197). In addition, CTLA-4 is constitutively expressed on Treg cells and enhances their suppressive activity (198, 199). Ctla4-deficient mice develop a lethal lymphoproliferative disorder, underscoring the enormous importance of this molecule in restraining uncontrolled T cell activation (200, 201). The first seminal studies of CTLA-4 targeting immunotherapy were performed by Leach et al., who showed that anti-CTLA-4 mAbs resulted in tumor rejection and immunity to secondary tumor challenge in mice (202). CTLA-4 was also the first checkpoint molecule to be targeted in human cancer using mAbs, ipilimumab and tremelimumab, in advanced metastatic malignant melanoma. While both mAbs showed clinical efficacy in 10% of patients, 25–30% of patients suffered from debilitating immune-mediated toxicities, such as colitis, dermatitis, hepatitis and hypophysitis (184). The anti-CTLA-4 mAb, ipilimumab, demonstrated an OS benefit in heavily pretreated patients with advanced metastatic malignant melanoma and therefore received FDA approval in 2010 (203). In addition, anti-CTLA-4 immunotherapy promoted CTL-mediated killing of dormant AML cells in a mouse model in vivo (187). Interestingly, CTLA-4 is also constitutively expressed on blasts in 80% of AML patients and triggering via its ligands leads to apoptosis (204). Cumulatively, these results have raised interest in using anti-CTLA-4 therapy for myeloid neoplasms (Figure 3A). As in every cancer, targeting of CTLA-4 in AML or MDS requires the presence of functional T cells. In 1993, Vidriales et al. demonstrated that AML patients have a grossly normal distribution of T cell populations in the PB and BM, with an increased CD57+ CTL subset and increased NK and NKT cells in the PB (205). AML patient CD4+ and CD8+ T cells show normal immunophenotypes and can be activated to produce cytokines and proliferate under optimal anti-CD3 and anti-CD28 stimulatory conditions in vitro (206, 207). However, in vivo, these T cells demonstrate aberrant activation patterns when analyzed by gene expression profiling, indicating defective immunological synapse formation between T cells and AML blasts and ineffective T cell activation (208). A notable change in the T cell surface expression pattern was observed during AML relapse after aHSCT, where PD-1 was upregulated and T cells showed an effector memory phenotype (207). It was hypothesized that such mechanisms lead to immune escape and ineffective GvL, resulting in disease relapse after aHSCT. Therefore, Davids et al. conducted a study on ipilimumab as a single agent in 28 patients with relapsed hematological cancers after aHSCT, including 12 patients with AML, 2 with MDS, and 1 with MPN. Although the treatment was feasible and induced complete, in part durable remissions in five patients, dose-limiting toxicities and immune-related adverse events precluding further administration of ipilimumab occurred in six patients, one of whom died of colitis and pneumonitis. Increased numbers of cytotoxic CD8+ T cells (CTLs) at the site of disease were observed, as well as reduced Treg cells and increased CD4+ effector T cells in blood. Activation of the IL-8 pathway was associated with improved responses, pointing to its value as a potential predictive biomarker for anti-CTLA-4 therapy (209). These encouraging results have led to multiple follow-up clinical trials. Currently, ipilimumab is investigated in >500 patients with myeloid neoplasms in various settings, including combination therapies and after aHSCT (Table 6).
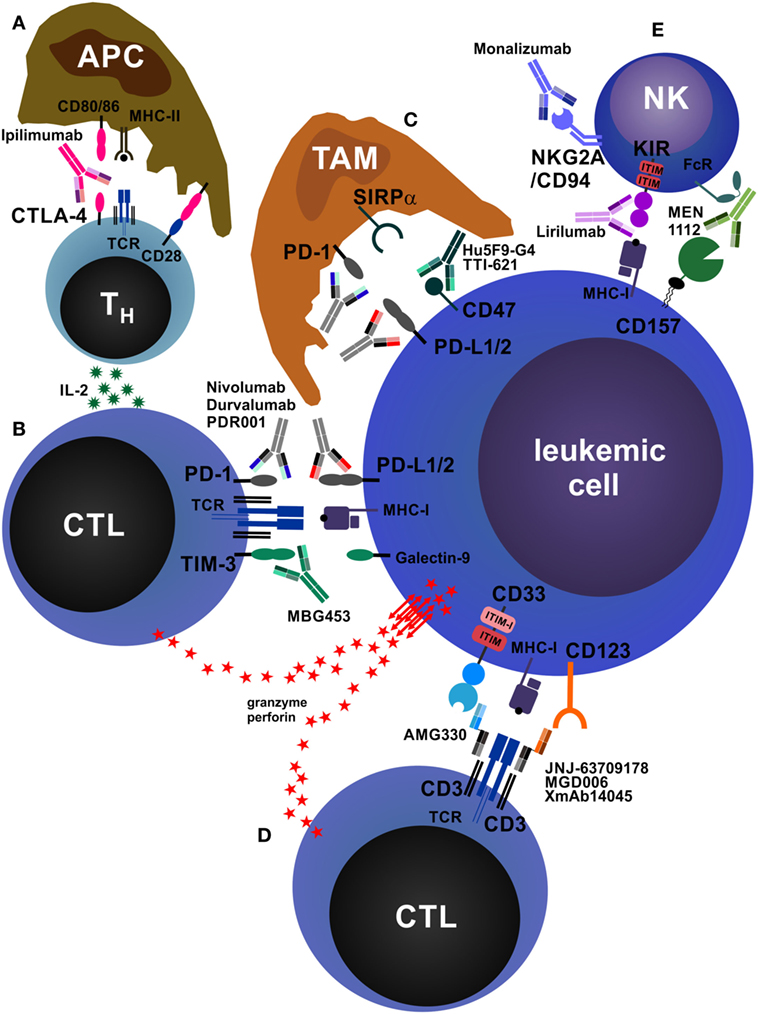
Figure 3. Antibodies that reinforce host immunity. (A) In secondary lymphoid organs, antigen-presenting cells (APCs) process and present peptides from tumor-derived (neo-)antigens to CD4+ helper T cells (TH cells) via major histocompatibility complex class II (MHC-II) molecules. The second signal required for T cell activation is CD28, which is stimulated by CD80/86 expressed on APCs. TH cells produce interleukin-2 (IL-2), increasing proliferation and survival of both TH cells and CD8+ cytotoxic T cells (CTLs). In addition, APCs may also directly activate tumor-specific CTLs via cross-presentation of tumor peptides on MHC-I (not depicted). After activation and clonal expansion, T cells upregulate the inhibitory receptor cytotoxic T lymphocyte antigen-4 (CTLA-4). The monoclonal antibodies (mAbs) ipilimumab and tremelimumab block inhibitory CTLA-4 signals on T cells and concomitantly redirect CD80/86 signals from APCs to CD28, enhancing T cell activation. (B) CTLs recognize acute myeloid leukemia (AML) cells via T cell receptor (TCR)–MHC-I interactions. AML cells express the inhibitory molecules PD-L1/2 and galectin-9, which drive programmed death protein-1 (PD-1)- and TIM-3-expressing CTLs into anergy/exhaustion. Anti-PD-1 and anti-PD-L1/2 mAbs (nivolumab, pembrolizumab, atezolizumab, durvalumab, PDR001, etc.) and anti-TIM-3 mAbs (MBG453) prevent the exhaustion of AML-specific CTLs and improve AML cell killing via perforin/granzyme. In addition, AML cells can themselves express TIM-3, which is stimulated by galectin-9 in an autocrine loop and promotes their self-renewal (not depicted) (210). In addition, PD-1 is also expressed on tumor-infiltrating CD4+FOXP3+ regulatory T cells (Treg cells), and PD-1 signaling on Treg cells enhances their proliferation (not depicted) (211). (C) Tumor-associated macrophages (TAMs) are a highly heterogeneous cell population in the tumor microenvironment that crucially influences tumor biology. TAMs can be broadly categorized into M1 (inflammatory) and M2 (pro-tumorigenic) macrophages. TAMs, especially M2 TAMs, express PD-1, which inhibits their phagocytic function. In addition, almost all tumors overexpress the “don’t eat me” molecule CD47, reinforcing the antiphagocytic state of TAMs by triggering signal regulatory protein α (SIRPα). Blocking PD-1, PD-L1/2, and CD47 by therapeutic mAbs enhances tumor cell phagocytosis. (D) Bispecific antibodies (BsAbs) are hybrid molecules with two different antigen specificities, of which one is targeted against a T cell surface molecule (most often CD3) and the other against an AML cell surface molecule, such as CD33 (AMG330), CD123 (JNJ-637079178, MGD006, XmAb14045), or C type lectin-like molecule-1/CLEC12A (MCLA-117, not depicted). BsAbs facilitate activation and killing by promoting adhesion of T cells to AML cells. (E) Natural killer (NK) cells play an important role in antitumor immunity. They can kill tumor cells directly and produce immunostimulatory cytokines. The activation of NK cells is normally tightly controlled by inhibitory receptors, such as killer cell immunoglobulin-like receptor (KIR) and NKG2A/CD94. Tumor cells lacking MHC-I and/or human leukocyte antigen-E, the ligands for NK cell inhibitory receptors, potently activate and are killed by NK cells. Blocking these inhibitory receptors by mAbs such as lirilumab (anti-KIR) or monalizumab (anti-NKG2A/CD94) mimics this effect. In addition, NK cells express Fc receptors (FcRs) and recognize antibody-opsonized tumor cells, leading to antibody-dependent cell-mediated cytotoxicity. As an example, CD157 is shown.
Anti-PD-1 and Anti-PD-L1/2
Programmed death protein-1 (CD279) is a co-inhibitory molecule and activation marker expressed on effector T cells. In contrast to CTLA-4, which primarily acts as a brake during T cell activation, PD-1 mainly acts at a late stage checkpoint to limit the function of already activated effector T cells in the periphery by inducing tolerance to prevent tissue damage and autoimmunity (212). In addition to T cells, PD-1 can also be expressed by NK cells and B cells, where it modulates lytic activity and antibody production, respectively (184). PD-1 expression is particularly high in antigen-specific, unresponsive (anergic) T cells in the presence of chronic antigen stimulation (i.e., during chronic infection and in cancer). PD-1 has two ligands, PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273), which are mainly expressed on non-hematopoietic cells, including tumor cells in numerous cancers (184). Tumor-infiltrating lymphocytes (TILs) often overexpress PD-1 and are anergic (Figure 3B). In addition, tumor-infiltrating Treg cells, which can represent a large fraction of TILs, also express high levels of PD-1. While effector T cell function is inhibited by the PD-L/PD-1 interaction, PD-1 signaling on Treg cells enhances their proliferation (211). Moreover, Gordon et al. recently reported that TAMs express high levels of PD-1, which inhibits their phagocytic capacity (Figure 3C) (213). The importance of the PD-L/PD-1 pathway in myeloid neoplasms is well established from basic research in leukemia models (188, 214, 215) and in vitro experiments with patient samples (216). High-risk MDS blasts express PD-L1 and upregulate PD-L1 in response to TNF-α and IFN-γ in an NF-κB-dependent manner (217). In addition, IFN-γ, which is produced by activated T cells, induces the expression of PD-L1 in AML blasts, PD-L1 and PD-L2 in CML LSCs, and promotes leukemia development by increasing LSC proliferation (190, 218). A combination therapy that blocked PD-1 and adoptive transfer of leukemia-specific CTLs resulted in LSC eradication and long-term survival of CML mice (219).
HMAs are the standard of care for high-risk MDS and AML patients who are not candidates for induction therapy and/or aHSCT. Importantly, HMAs were shown to increase the expression of PD-L1 and PD-L2 on CD34+ cells from 124 MDS patients (220). In addition, T cells from MDS patients express PD-1 (217), which is increased during therapy with HMAs due to PD-1 promoter DNA de-methylation (221). Furthermore, Goltz et al. recently reported that low PD-L1 promoter methylation correlates with adverse risk and poor OS in AML (222). These important findings provide a rationale for combination therapy of HMAs with PD-1 checkpoint inhibitors. Indeed, several clinical trials that address these treatment combinations are currently ongoing in >2,000 patients (Table 6).
Anti-TIM-3
T cell immunoglobulin and mucin domain-containing molecule 3 (TIM-3) is an inhibitory receptor expressed on CD4+ type 1 helper T (TH1) cells, IL-17-producing TH17 cells, Treg cells and CTLs as well as antigen-presenting cells (APCs). Its ligand, galectin-9, is widely expressed, among others, on APCs and tumor cells. TIM-3 signaling inhibits the proliferation and induces cell death of lymphocytes. Thus, blocking TIM-3 exacerbates autoimmunity in murine autoimmune disease models (223, 224). In addition, numerous studies have found that TIM-3 is co-expressed with PD-1 on exhausted CD4+ and CD8+ TILs in mice bearing solid and hematological cancers, as well as in patients with advanced malignant melanoma (215, 225, 226). TIM-3 and PD-1 co-expressing T cells were more functionally impaired and abundant than T cells only expressing PD-1, a population that also contains activated effector T cells (226). In line with this, blocking both TIM-3 and PD-1 together was more effective in controlling tumor growth in mouse models than blocking either of the two alone (227).
In addition, Kikushige et al. demonstrated that TIM-3 is selectively expressed on disease-initiating human AML LSCs, but not on normal HSCs. Only TIM-3+, but not TIM-3− AML LSCs were able to reconstitute human AML in xenotransplantation assays, and TIM-3 blocking mAb treatment inhibited AML engraftment and reduced disease burden in already established xenografts (228). Interestingly, in a subsequent study the same authors showed that soluble galectin-9 is increased in the serum of AML patients and that it binds to TIM-3 expressed on LSCs, inducing an autocrine stimulatory loop that promotes LSC self-renewal via NF-κB and β-catenin signaling (210).
Along with reinforcing host immunity by restoring exhausted T cells, these findings open up a very promising possibility to target the disease-initiating AML LSCs directly using anti-TIM-3 mAbs. A clinical trial evaluating decitabine together with either PDR001 (anti-PD-1 mAb), MBG453 (anti-TIM-3 mAb) or the combination of PDR001 and MBG453 (NCT03066648) is currently recruiting patients with AML who are R/R or not eligible for curative therapy, as well as high-risk MDS patients (Table 6).
Antibodies That Force T Cell–Tumor Cell Interactions: BsAbs
Hybrid, BsAbs are composed of two different scFv-fragments with different epitope specificities, linked by a peptide chain (229). As early as 1985, Perez et al. and Staerz et al. developed T cell-directed hybrid antibodies and demonstrated activation and directed localization of T cells to target cells (230, 231). Today, a plethora of different BsAb formats exist that are used for purposes as diverse as cytotoxic immune cell redirection, effector molecule delivery, half-life extension, and diagnostics/imaging [comprehensively reviewed in Ref. (232)]. The designated use of immunostimulatory BsAbs is to recruit and promote adhesion of T cells and NKT cells (or other effector cells) to cancer cells and to facilitate effector cell activation. Therefore, one scFv-fragment is specific for a T/NKT cell antigen, such as CD3 or T cell receptor (TCR), whereas the other scFv-fragment is specific for a cancer cell surface molecule (233). In analogy to immune checkpoint inhibitors, BsAbs are considered a seminal innovation in cancer immunotherapy and are currently investigated in myeloid neoplasms. Blinatumomab, a bispecific T cell engager (BiTE) that recognizes CD3 and CD19, gained a breakthrough therapy designation by the FDA because of significant clinical activity against therapy-resistant CD19-positive B cell ALL and was approved in late 2015 (234). Blinatumomab is currently investigated in a clinical trial of CD19+ lymphoid and myeloid neoplasms that relapsed after aHSCT. The encouraging results of blinatumomab led to the development of multiple BsAbs for myeloid neoplasms that are currently under investigation in clinical trials (Table 7; Figure 3D). The first-in-class BsAb to target AML consists of a CD3/CD33 BiTE (AMG330). In long-term in vitro cultures of peripheral blood mononuclear cells (PBMCs) from AML patients, AMG330 showed potent activation of residual endogenous T cells and subsequent lysis of AML blasts, even when effector to target (E:T) ratios were unfavorable due to low numbers of T cells (235). Interestingly, AMG330 did not lead to downregulation of CD33 on AML blasts and its binding was unaffected by CD33 polymorphisms and P-glycoprotein expression (236). AMG330 is currently being tested in a clinical phase 1 trial in patients with R/R AML (NCT02520427). A notable disadvantage of classical BiTEs is the absence of an Fc domain. This results in a short half-life due to the lack of antibody recycling by neonatal Fc receptor (FcRn) and leads to the requirement of daily intravenous infusions. Furthermore, the absence of Fc-mediated effector functions, i.e., ADCC, ADCP, and CDC, potentially hampers additional beneficial effects of the antibody (237, 238). To overcome some of the drawbacks of classical BiTEs, BsAbs were optimized to retain a modified Fc domain that allows recycling via FcRn, but lack Fc γ receptor (FcγR) activation capacity to reduce the potential for non-selective Fc-mediated effector functions and T cell activation (233). Chu et al. reported the generation of a CD3/CD123 BiTE (XmAb14045) containing a modified Fc domain for FcRn recycling. XmAb14045 is able to kill CD123-positive leukemia cell lines in vitro and has a half-life of >6 days in mice. A single injection of XmAb14045 into cynomolgus monkeys led to T cell activation and depletion of CD123-positive pDCs and basophils from blood and BM within 1 h, an effect that lasted for days to weeks (239). This Fc-containing BiTE is currently being investigated in a clinical trial in patients with CD123-positive AML, blastic plasmacytoid dendritic cell neoplasm, bc CML, and B cell ALL (NCT02730312). Another CD3/CD123 BiTE (JNJ-63709178) was developed by Gaudet et al. Similar to XmAb140445, JNJ-63709178 BiTE specifically and effectively activates T cells to kill CD123-expressing AML cell lines in vitro and in vivo in xenografts, as well as primary CD123-positive AML blasts from patient blood (240) and BM (241). JNJ-63709178 is also currently being tested in a phase 1 clinical study in patients with R/R AML (NCT02715011). C-type lectin-like molecule-1, also known as CLEC12A, was first discovered and described as a myeloid cell surface marker associated with AML in 2004 by Bakker et al. (242). Subsequently, Van Loo et al. developed a CD3/CLEC12A BiTE (MCLA-117), a full-length human bispecific IgG with a modified Fc domain defective for FcγR and complement C1q activation, but allowing for FcRn recycling (243). In ex vivo culture experiments with primary BM samples from six AML patients, MCLA-117 induced activation and up to 30-fold expansion of endogenous T cells, which resulted in potent killing of AML blasts. A clinical trial of MCLA-117 in patients with R/R AML (NCT03038230) is actively recruiting participants at present. A similar CD3/CLEC12A BiTE was developed by Leong et al.; this BiTE was well tolerated and demonstrated effective target cell depletion in cynomolgus monkeys (244). In addition to BiTEs, a plethora of BsAb classes has been developed (233), such as tandem diabody and dual affinity retargeting antibody (DART) formats. Two different DARTs (CD3/CD19 and TCR/CD19) demonstrated increased efficacy and stability in direct comparison to CD3/CD19 BiTEs (245, 246). Chichili et al. developed a CD3/CD123 DART (MGD006) that depleted CD123-positive cells from circulation and was well tolerated in monkeys (247). MGD006 is currently tested in patients with R/R AML or high-risk MDS (NCT02152956) (248).
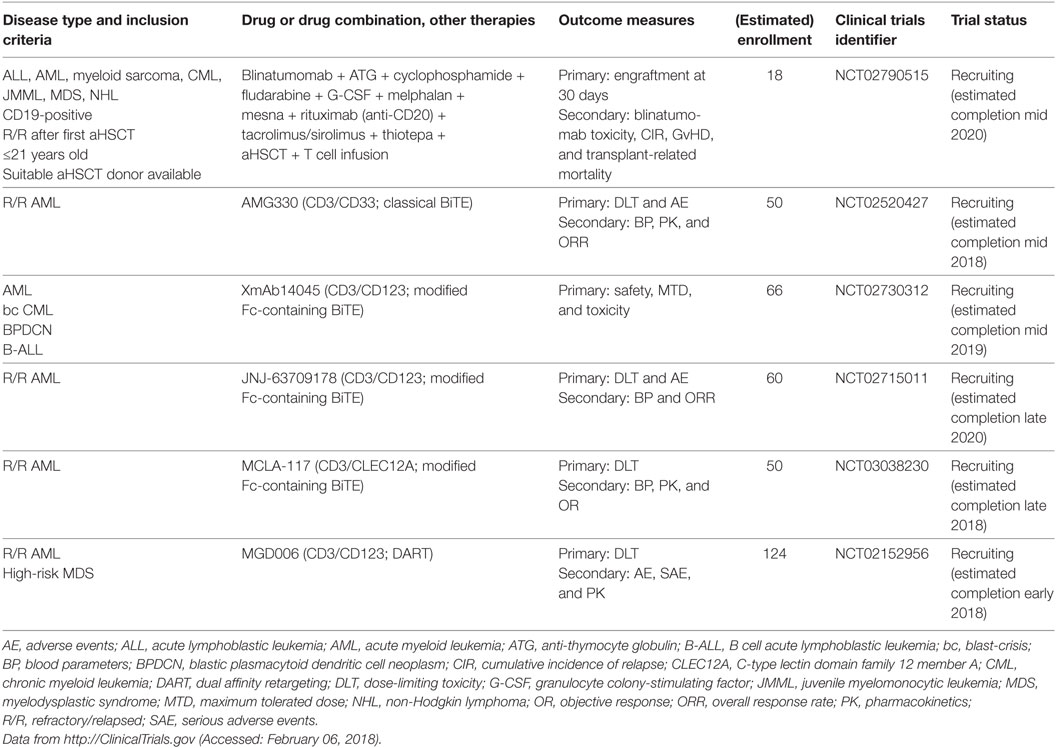
Table 7. Active clinical trials of bispecific antibodies in myeloid neoplasms.
In summary, BsAbs are powerful tools to engage endogenous antitumoral immune responses and have shown promising clinical efficacy in highly pretreated cancers. At least 23 different formats of BsAbs have been reported to date and different molecules, such as bispecific designed ankyrin repeat proteins, are also evaluated (233, 237). Moreover, patient-derived T cells are genetically engineered in vitro to express chimeric antigen receptors (CARs) that directly recognize targets on AML blasts, such as CD33, CD123, etc. Additional well-designed clinical trials are needed to determine which BsAb molecule formats and specificities will be most clinically useful to treat myeloid neoplasms and to compare their performance to CAR T cells.
Antibodies That Activate Antitumoral Innate Immunity: TAMs and NK Cells
Anti-CD47
The integrin-associated protein CD47 is a ubiquitously expressed immunoglobulin superfamily member that has various functions in cellular processes as diverse as neuronal development, cell migration and immunity (249–252). An important function of CD47 is its role as a marker of self and a “don’t eat me” signal that inhibits phagocytosis by binding to signal regulatory protein α (SIRPα) on macrophages (Figure 3C) (253–255). Overexpression of CD47 has been reported on numerous solid and hematological human cancers, and blocking CD47 promotes macrophage activation, enhances tumor cell phagocytosis and prolongs survival in murine cancer models (256–264). Importantly, CD47 is overexpressed on AML LSCs and further upregulated during their mobilization. In addition, higher CD47 mRNA expression predicted worse survival in defined subsets of AML patients (258, 265). However, CD47 protein expression on BM blasts, as analyzed by immunohistochemistry of trephine biopsies from >200 AML patients, was not significantly associated with survival or treatment response (266). At present, four clinical trials assessing the efficacy of CD47 or SIRPα blockade using mAbs are underway (Table 8). Additional anti-CD47 mAbs are currently under development, and it remains to be determined which one will be most clinically useful in the treatment of myeloid neoplasms (267).
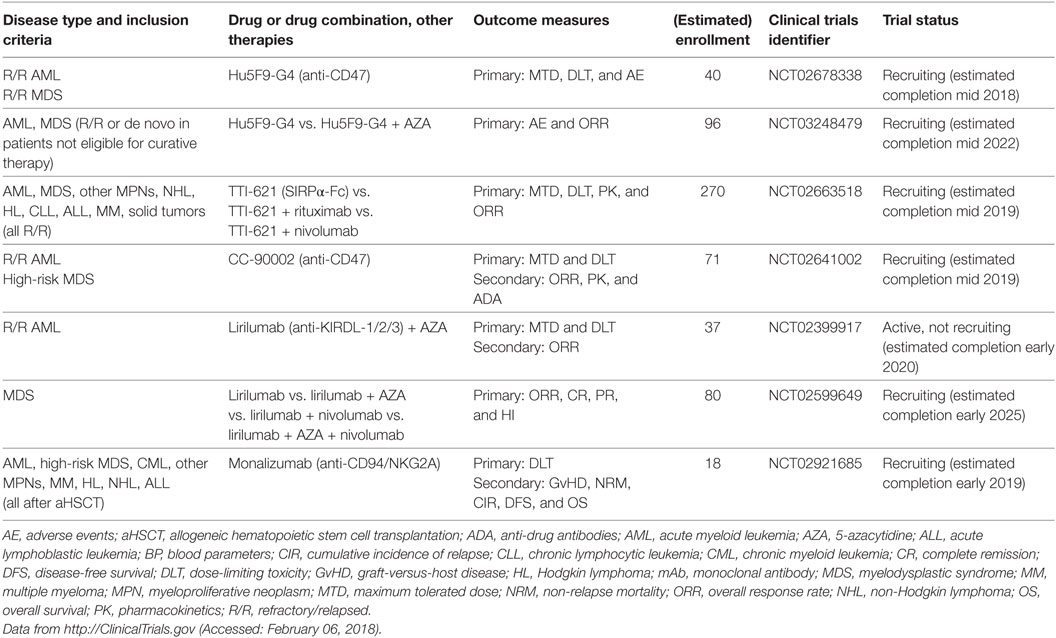
Table 8. Active clinical trials of mAbs that activate innate immunity in myeloid neoplasms.
Anti-Killer Cell Immunoglobulin-Like Receptor (KIR)
Natural killer cells are effector cells of the innate immune system that play an important role in the control of tumors and infections. Upon activation, NK cells secrete immunostimulatory cytokines, such as IFN-γ, and can directly kill target cells via perforin/granzyme B. NK cell activity is tightly regulated by the integration of stimulatory and inhibitory signals expressed on potential target cells (Figure 3E) (268). A critical negative regulator of NK cell activity is the recognition of self-MHC class I by KIRs. Tumor cells that lose the expression of MHC class I (“missing self”) are potent stimulators of NK cells. This idea has been exploited by generating anti-KIR mAbs, which show promising results in a preclinical murine leukemia model (269). In addition, clinical evidence from aHSCT for AML has shown improved outcomes in the presence of mismatches in NK receptor genes (“alloreactive” NK cells) (270–272). These findings led to the development of a therapeutic mAb that recognizes KIR2DL-1, -2, and -3 and therefore blocks the recognition of all human leukocyte antigen (HLA) class C molecules by NK cells. This antibody (1-7F9/IPH2101) was effective in a preclinical murine AML xenograft model (273). A second-generation version of this antibody, lirilumab (IPH2102/BMS-986015), was tested as maintenance therapy in a double-blind placebo-controlled phase 2 clinical trial in elderly AML patients in first CR after standard induction chemotherapy (EFFIKIR trial, NCT01687387). Therein, AML patients 60–80 years of age ineligible for alloHSCT were randomly assigned to one of three trial arms: placebo, lirilumab 0.1 mg/kg or lirilumab 1 mg/kg. Patients were treated every 4 weeks for up to 2 years (the lirilumab 0.1 mg/kg group received the verum only once every 3 months). This trial was completed in fall 2016, and first results are expected soon (274). More recently, two studies were initiated at the MD Anderson Cancer Center, Houston, TX, to further investigate the efficacy of lirilumab in myeloid neoplasms (Table 8). The first study is an open-label phase 1/2 trial that tests the safety, tolerability and efficacy of lirilumab in combination with AZA in R/R AML patients (NCT02399917). The second is a four-arm trial that investigates lirilumab alone versus lirilumab in combination with nivolumab and/or AZA in MDS patients (NCT02599649).
Anti-NKG2A/CD94
The C-type lectin receptor superfamily molecules NKG2A, B, C, E, and H form heterodimers with the invariant chain CD94 and are mainly expressed on NK cells and subsets of CTLs. NKG2A and B contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and therefore act as inhibitory receptors. By contrast, NKG2C, E and H transmit activating signals by recruiting the immunoreceptor tyrosine-based activating motif-containing adaptor molecule DAP12. NGK2/CD94 dimers recognize ubiquitously expressed non-classical HLA-E molecules. Depending on the peptide presented by HLA-E, different NKG2/CD94 heterodimer pairs are bound and activated with different affinities and dissociation kinetics (275). HLA-E molecules can be overexpressed by certain tumors and inhibit NK cell and CTL activity via NKG2A/CD94 (276). Monalizumab (IPH2201), an anti-NKG2A/CD94 blocking mAb, was developed and is currently being tested as monotherapy or in combination with an anti-epidermal growth factor receptor mAb, cetuximab, in recurrent or metastatic head and neck squamous cell carcinoma (NCT02643550) with good tolerability (277). In addition, a phase I clinical study of monalizumab in different hematologic malignancies after aHSCT is currently recruiting patients (NCT02921685).
Future Directions
Many research groups worldwide are working to further identify and characterize novel mAb targets for AML, MDS, and other myeloid neoplasms. Moreover, the functions and importance of additional immune checkpoint pathways are constantly being uncovered. In addition, sophisticated molecular engineering has allowed the design of improved mAb formulations and modifications that overcome many of the technical problems associated with drug properties such as optimal stimulation of immune effector mechanisms, stability and half-life. In the following section, an overview of emerging mAb targets, emerging mAb species and alternative approaches is provided (Figure 4).
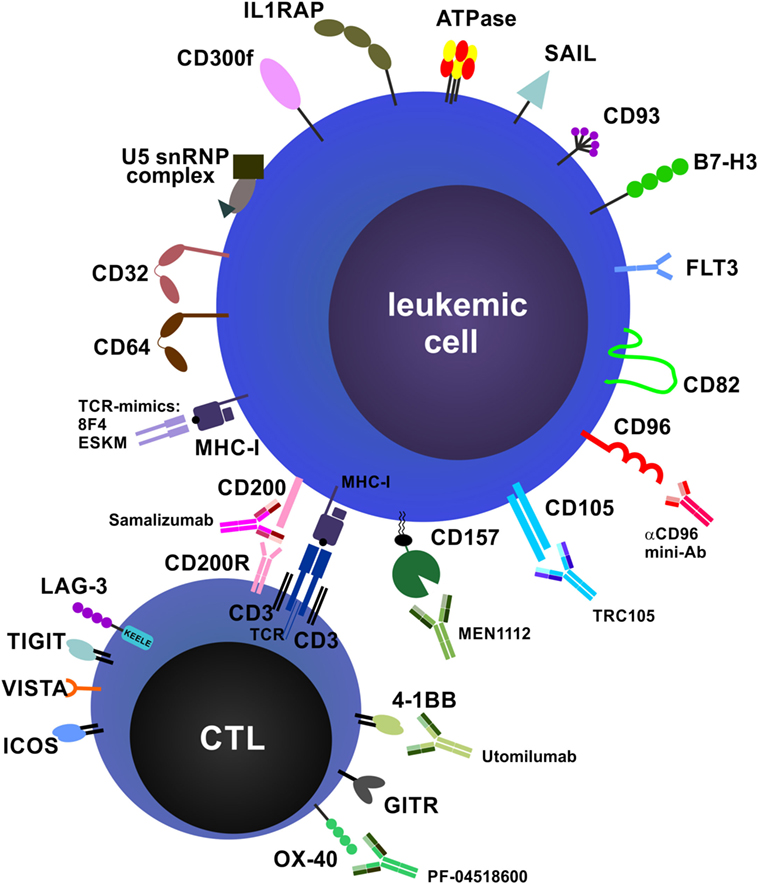
Figure 4. Future directions and potential monoclonal antibody (mAb) targets. Potential targets on leukemic cells and immune cells and examples of therapeutic mAbs that are currently in preclinical and/or clinical development are shown. Many targets, especially costimulatory and co-inhibitory molecules and their ligands, are expressed on both acute myeloid leukemia blasts and immune cells, such as T cells and natural killer cells, and can exert different functions on these cell types (not depicted).
TCR-Mimic mAbs
Many surface molecules are not exclusively expressed on AML blasts but also present at certain levels on normal HSPCs or other immune cells. Therefore, the repertoire of possible mAb targets is limited. Targeting AML-specific oncoproteins or overexpressed antigens that are present intracellularly could vastly extend the therapeutic possibilities and overcome some of the limitations of specificity. Indeed, the GvL effect in aHSCT is executed by leukemia-specific donor-derived CTLs that recognize endogenous peptides in complex with MHC class I present on AML blasts and LSCs (186). To exploit this mechanism, several groups generated TCR-mimic mAbs that recognize peptides of intracellular proteins in complex with HLA-A2. Sergeeva et al. designed a TCR-mimic mAb (8F4) that recognizes a proteinase 3- and neutrophil elastase-derived peptide (PR1) on HLA-A2. 8F4 mAb induced CDC-mediated killing of AML blasts and LSCs in vitro without affecting normal HSPCs and was effective against human AML xenografts in NSG mice in vivo (278, 279). Furthermore, Scheinberg and colleagues generated a TCR-mimic mAb (ESKM) that recognizes a peptide from Wilms tumor protein-1 (WT1) present on HLA-A2 on leukemic blasts. Therapy with ESKM mAb eradicated human ALL and AML xenografts in NSG mice, suggesting that this mAb is of potential therapeutic value (280, 281). Thus, TCR-mimics can vastly extend the repertoire of therapeutic mAbs for myeloid neoplasms.
Anti-FcγR
The IgG Fc receptor (FcγR) family consists of one high-affinity receptor (CD64/FcγRI), five low-affinity receptors (CD32A-C/FcγRIIA-C; CD16A-B/FcγRIIIA-B) and the FcRn. Fc receptors are predominantly expressed on myeloid cells including macrophages and DCs, serve to recognize monomeric or aggregated IgGs, such as immune complexes and opsonized pathogens, and play a role in antibody recycling. FcγRs constitute activating (CD64, CD32A, C, CD16) or inhibitory (CD32B) intracellular signaling motifs and therefore either activate or inhibit the expressing cell upon IgG binding. In the study by Saito et al., CD32A was also exclusively expressed in CD34+CD38− LSCs (34% of the AML samples analyzed) but not in healthy HSCs, and CD32A depletion did not affect healthy HSC engraftment (34). Similarly, Tur et al. reported the successful targeting and elimination of a CD64-expressing AML cell line (U937) in xenografts using a humanized anti-CD64 mAb conjugated to truncated Pseudomonas exotoxin A (282). However, none of these FcγR targeting approaches has made it into clinical studies yet.
Anti-IL-1 Receptor Accessory Protein (IL1RAP)
IL-1 receptor accessory protein (IL1R3) is an essential component of the IL-1 receptor complex required for signal transduction of IL-1 and IL-33 induced signaling. A soluble form of IL1RAP that is constitutively produced by the liver and neutralizes IL-1β has also been described (283, 284). IL1RAP expression is highly correlated with the BCR-ABL1 translocation in CML LSCs, can be used as a measure of LSC burden and predictor of therapy outcome, and can be targeted by therapeutic mAbs to eradicate LSCs (285–287). A current clinical trial (NCT02842320) has the purpose to prospectively assess the expression of IL1RAP on CML LSCs by FACS during TKI therapy and will be completed in 2020. Importantly, IL1RAP is also expressed on LSCs in most AML samples and at higher levels than on healthy HSCs. Therapeutic anti-IL1RAP mAbs block IL-1-mediated AML cell proliferation and induce ADCC by NK cells, leading to prolonged survival in a xenograft model (288, 289). Given the importance of IL-1 and IL-33 in the development of myeloid neoplasms (125, 290) and the expression of IL1RAP on LSCs, targeting this receptor by mAbs is a promising strategy.
Anti-CD96
CD96 (also known as TACTILE, for T cell ACTivation, Increased Late Expression) belongs to the immunoglobulin superfamily. It is expressed at low levels on T and NK cells and highly upregulated upon activation (291). The ligand for CD96 is CD155, the poliovirus receptor (292). CD96 plays a role in NK cell adhesion to target cells. It is expressed in subsets of adult and pediatric AML blasts and on the majority of AML LSCs (293–295). In a study by Jiang et al., overexpression of CD96 in AML LSCs was associated with poor chemotherapy response, increased relapse rates and worse prognosis (296). In 2012, Mohseni Nodehi et al. reported the generation of an engineered anti-CD96 single chain fragment of antibody variable region (scFv), which they genetically fused to an IgG1 Fc with enhanced FcR-binding capacity. This so-called “mini-antibody” was effective in NK cell-mediated killing of an AML cell line by ADCC in vitro (297). Given the potential for CD96-targeted mAb therapy in myeloid neoplasms, especially the capability to specifically target AML LSCs, it is surprising that no clinical trials of anti-CD96 therapy have yet been reported.
Anti-CD105
CD105 (endoglin, ENG) is a membranous glycoprotein and is part of the TGFβ receptor. CD105 is expressed on ECs and plays a role in tumor-associated angiogenesis (298). Dourado et al. identified CD105 expression on malignant blasts in most patients with AML and B cell acute lymphoblastic leukemia. They showed that CD105+ blasts have superior leukemogenic activity in xenografts and that targeting CD105 with a mAb (TRC105) abolished AML engraftment (299). CD105 may therefore emerge as a promising target for future AML mAb therapy approaches.
Anti-CD157
CD157, also known as bone marrow stromal cell antigen 1 (BST-1), is a mammalian cyclase family member with structural homology to CD38. CD157 is a glycosylphosphatidylinositol-anchored ectoenzyme with ADP ribosyl-cyclase and cyclic ADP ribose-hydrolase activities (300, 301). Besides its functions as an ectoenzyme, in antibody crosslinking experiments CD157 has also been shown to exhibit receptor and signaling capacities, modulating leukocyte adhesion, migration, and diapedesis (302–304). CD157 is expressed on a wide variety of cell types such as granulocytes, monocytes, macrophages, myeloid precursors, mast cells, ECs, as well as on BM stromal, synovial, and follicular DC lines (305). In 1985, Todd et al. first analyzed CD157 expression in AML samples from 65 patients using the mAb Mo-5. Depending on the French–American–British type, CD157 was expressed on malignant blasts in 50–74% of cases (306). More recently, Krupka et al. confirmed and expanded these results, demonstrating CD157-positivity in 7/8 AML cell lines and 98/101 patient samples, with considerable heterogeneity in expression intensity (307). These authors also demonstrated that CD157 is expressed in the LSC-containing CD34+CD38− blast compartment, albeit to a significantly lower level than in bulk AML blasts. Furthermore, CD157 was expressed on bulk CD34+ cells from HDs at comparable levels to AML blasts. In this study, the antileukemic efficacy of a novel Fc-engineered CD157 mAb (MEN1112) was tested using NK cell-mediated ADCC assays. MEN1112 was found to trigger ADCC against AML cell lines and primary AML blasts at high E:T cell ratios. ADCC was, however, also observed against HD PBMCs and CD34+ cells, a finding that may have implications on the further clinical development of MEN1112 or other mAbs targeting CD157 (307). Indeed, a phase 1 clinical trial of MEN1112 mAb in R/R AML (NCT02353143) is currently active, with an estimated study completion date in fall 2018.
Anti-CD200
CD200 (OX-2), an inhibitory ligand of the immunoglobulin superfamily, is expressed in various tissues such as brain, testis and the hematopoietic system (308). CD200 is overexpressed on AML blasts and, having no intracellular signaling domain, exerts its inhibitory function via signaling to CD200R, which is expressed on memory CD8+ T cells in AML patients (309, 310). In addition, high CD200 expression on AML blasts correlates with an increased frequency of Treg cells (311, 312). An anti-CD200 mAb, samalizumab, is currently being tested in a large biomarker-based AML treatment trial (NCT03013998).
Further promising molecules suitable for targeting by therapeutic mAbs that are in preclinical and/or clinical testing are listed in Table 9.
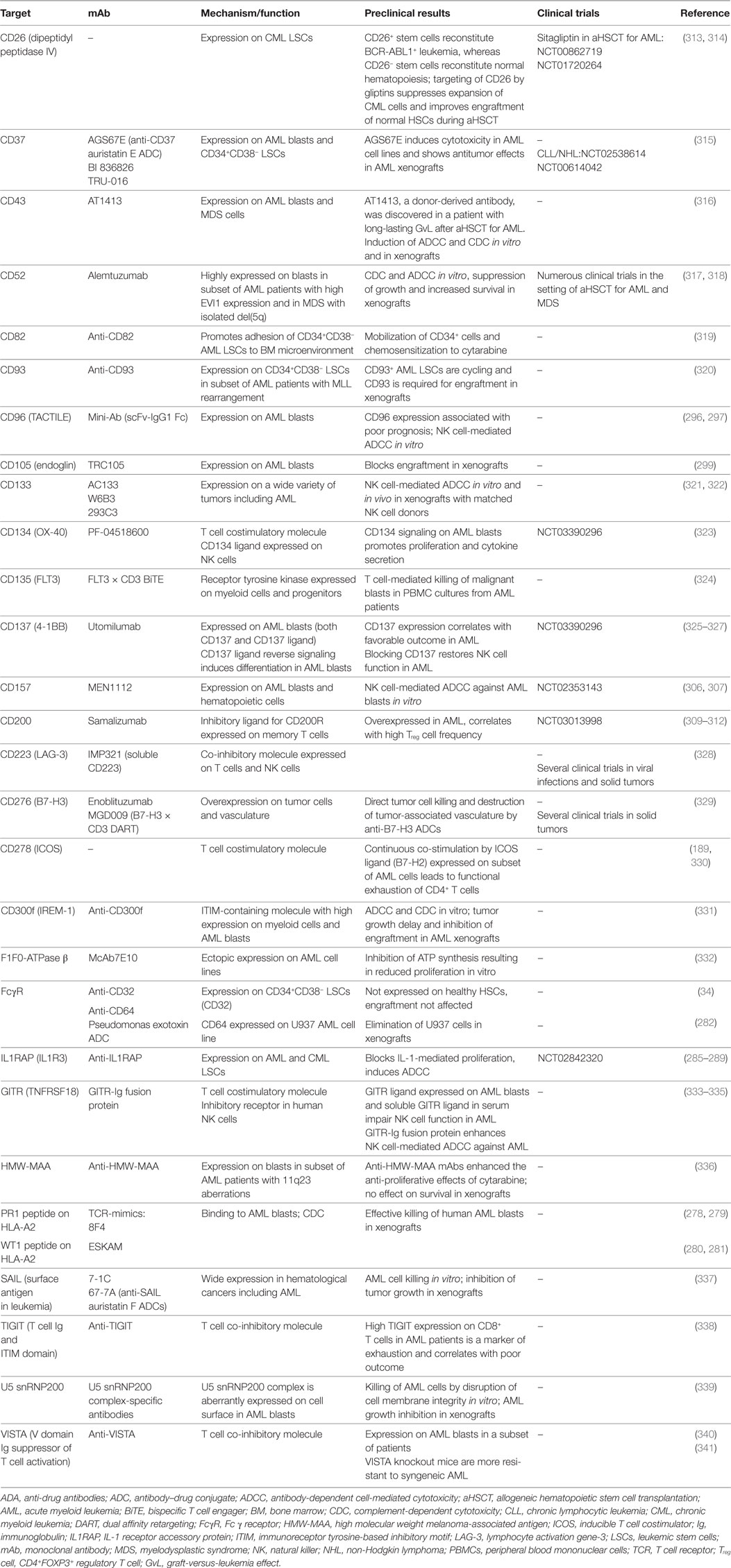
Table 9. Future mAb targets for myeloid neoplasms.
Conclusion
Gemtuzumab ozogamicin is the most well studied therapeutic mAb in AML and much can be learned from its history of accelerated approval, withdrawal, and re-approval. The heterogeneity of myeloid neoplasms requires investigators to design future studies addressing therapies in subgroups of these diseases, especially in AML. Efforts in this direction are complicated by the relative rarity of disease subgroups, so that these studies can only be conducted at large, referral centers. Long-term observation and follow-up are warranted, since an increase in early mortality might be more than compensated for years later, with reduced relapses and better long-term OS. In future trials, it is crucial to compare the standard of care or designed treatment, respectively, to the exact same treatment with the addition of a therapeutic mAb. This is because reductions in the dosage of standard chemotherapy in anticipation of suspected toxicities when adding a therapeutic mAb might mitigate the mAb effect. The combinations and timing of mAb addition, as well as its relation to aHSCT, needs to be addressed. In addition, MRD monitoring (as a surrogate marker of efficacy) must be internationally standardized. Predictive biomarkers for treatment response and prognostic biomarkers for disease-free and OS are needed. Side effects of novel therapeutic mAbs must be documented with great care and made publicly available.
We are entering an exciting era for the treatment of myeloid neoplasms. New therapeutic mAbs may finally prove a valid extension to our armamentarium and raise hope for patients and treating physicians alike.
Author Contributions
CS conceived, wrote, and revised this article and created the figures and tables.
Conflict of Interest Statement
The author is listed as a co-inventor on a U.S. patent application for combination treatment with anti-CD70 mAb and TKI, held by the University of Bern, Switzerland. No other competing interests were declared.
Acknowledgments
I thank Darci Phillips and Sizun Jiang for critically reading the manuscript.
Funding
Funded by an Advanced Postdoc Mobility Fellowship from the Swiss National Science Foundation (P300PB_171189) and an International Award for Research in Leukemia from the Lady Tata Memorial Trust, London, UK.
Abbreviations
ADC, antibody–drug conjugate; ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; aHSCT, allogeneic hematopoietic stem cell transplantation; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; APCs, antigen-presenting cells; AZA, 5-azacytidine; BiTE, bispecific T cell engager; BsAbs, bispecific antibodies; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CDC, complement-dependent cytotoxicity; CML, chronic myeloid leukemia; CTLs, CD8+ cytotoxic T cells; CTLA-4, cytotoxic T lymphocyte antigen-4; CR, complete remission; DART, dual affinity retargeting; DLT, dose-limiting toxicity; GO, gemtuzumab ozogamicin; HMAs, hypomethylating agents; HSCs, hematopoietic stem cells; IFN, interferon; IL, interleukin; LSCs, leukemic stem cells; mAb, monoclonal antibody; MDS, myelodysplastic syndrome; MHC, major histocompatibility complex; MTD, maximum tolerated dose; NK cells, natural killer cells; PBD, pyrrolobenzodiazepine dimer; PBMCs, peripheral blood mononuclear cells; PD-1, programmed death protein-1; TAMs, tumor-associated macrophages; TCR, T cell receptor; TILs, tumor-infiltrating lymphocytes; TH cells, CD4+ helper T cells; Treg cells, CD4+FOXP3+ regulatory T cells.
References
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi:10.1182/blood-2016-03-643544
2. Swerdllow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press (2017).
3. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373(12):1136–52. doi:10.1056/NEJMra1406184
4. Geelen IGP, Thielen N, Janssen J, Hoogendoorn M, Roosma TJA, Willemsen SP, et al. Treatment outcome in a population-based, ‘real-world’ cohort of patients with chronic myeloid leukemia. Haematologica (2017) 102(11):1842–9. doi:10.3324/haematol.2017.174953
5. Estey E, Döhner H. Acute myeloid leukaemia. Lancet (2006) 368(9550):1894–907. doi:10.1016/S0140-6736(06)69780-8
6. Juliusson G, Antunovic P, Derolf A, Lehmann S, Mollgard L, Stockelberg D, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood (2009) 113(18):4179–87. doi:10.1182/blood-2008-07-172007
7. Shah A, Andersson TM, Rachet B, Bjorkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol (2013) 162(4):509–16. doi:10.1111/bjh.12425
8. Schiffer CA. Prognosis of acute myeloid leukemia. In: Larson RA, Rosmarin AG, editors. UpToDate. Waltham, MA: UpToDate (2017).
9. Larson RA. Induction therapy for acute myeloid leukemia in younger adults. In: Löwenberg B, Connor RF, editors. UpToDate. Waltham, MA: UpToDate (2017).
10. Larson RA. Acute myeloid leukemia: treatment and outcomes in older adults. In: Löwenberg B, Schmader KE, Rosmarin AG, editors. UpToDate. Waltham, MA: UpToDate (2017).
11. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47. doi:10.1182/blood-2016-08-733196
12. Ferguson P, Craddock C. Allogeneic transplantation in primary refractory AML. Bone Marrow Transplant (2017) 52(7):950–1. doi:10.1038/bmt.2017.61
13. Walter RB, Othus M, Burnett AK, Lowenberg B, Kantarjian HM, Ossenkoppele GJ, et al. Resistance prediction in AML: analysis of 4601 patients from MRC/NCRI, HOVON/SAKK, SWOG and MD Anderson Cancer Center. Leukemia (2015) 29(2):312–20. doi:10.1038/leu.2014.242
14. Larson RA. Treatment of relapsed or refractory acute myeloid leukemia. In: Löwenberg B, Rosmarin AG, editors. UpToDate. Waltham, MA: UpToDate (2017).
15. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature (2017) 547(7661):104–8. doi:10.1038/nature22993
16. Sarkozy C, Gardin C, Gachard N, Merabet F, Turlure P, Malfuson JV, et al. Outcome of older patients with acute myeloid leukemia in first relapse. Am J Hematol (2013) 88(9):758–64. doi:10.1002/ajh.23498
17. Gasiorowski RE, Clark GJ, Bradstock K, Hart DN. Antibody therapy for acute myeloid leukaemia. Br J Haematol (2014) 164(4):481–95. doi:10.1111/bjh.12691
18. Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia (2014) 29(4):760–9. doi:10.1038/leu.2014.244
19. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12(4):278–87. doi:10.1038/nrc3236
20. Salles G, Barrett M, Foa R, Maurer J, O’Brien S, Valente N, et al. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther (2017) 34(10):2232–73. doi:10.1007/s12325-017-0612-x
21. Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov (2018) 17(3):197–223. doi:10.1038/nrd.2017.227
22. Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl (2014) 53(15):3796–827. doi:10.1002/anie.201307628
23. Rashidi A, Uy GL. Targeting the microenvironment in acute myeloid leukemia. Curr Hematol Malig Rep (2015) 10(2):126–31. doi:10.1007/s11899-015-0255-4
24. Cogle CR, Bosse RC, Brewer T, Migdady Y, Shirzad R, Kampen KR, et al. Acute myeloid leukemia in the vascular niche. Cancer Lett (2016) 380(2):552–60. doi:10.1016/j.canlet.2015.05.007
25. Rashidi A, DiPersio JF. Targeting the leukemia-stroma interaction in acute myeloid leukemia: rationale and latest evidence. Ther Adv Hematol (2016) 7(1):40–51. doi:10.1177/2040620715619307
26. Malek TR. The biology of interleukin-2. Annu Rev Immunol (2008) 26:453–79. doi:10.1146/annurev.immunol.26.021607.090357
27. Davey FR, Erber WN, Gatter KC, Mason DY. The use of monoclonal antibody Y1/82A in the identification of acute myeloblastic and monocytic leukemias. Am J Clin Pathol (1988) 89(1):76–80. doi:10.1093/ajcp/89.1.76
28. Carron JA, Cawley JC. IL-2 and myelopoiesis: IL-2 induces blast cell proliferation in some cases of acute myeloid leukaemia. Br J Haematol (1989) 73(2):168–72. doi:10.1111/j.1365-2141.1989.tb00248.x
29. Waldmann TA, Pastan IH, Gansow OA, Junghans RP. The multichain interleukin-2 receptor: a target for immunotherapy. Ann Intern Med (1992) 116(2):148–60. doi:10.7326/0003-4819-116-2-148
30. Koon HB, Severy P, Hagg DS, Butler K, Hill T, Jones AG, et al. Antileukemic effect of daclizumab in CD25 high-expressing leukemias and impact of tumor burden on antibody dosing. Leuk Res (2006) 30(2):190–203. doi:10.1016/j.leukres.2005.06.007
31. Terwijn M, Feller N, van Rhenen A, Kelder A, Westra G, Zweegman S, et al. Interleukin-2 receptor alpha-chain (CD25) expression on leukaemic blasts is predictive for outcome and level of residual disease in AML. Eur J Cancer (2009) 45(9):1692–9. doi:10.1016/j.ejca.2009.02.021
32. Gonen M, Sun Z, Figueroa ME, Patel JP, Abdel-Wahab O, Racevskis J, et al. CD25 expression status improves prognostic risk classification in AML independent of established biomarkers: ECOG phase 3 trial, E1900. Blood (2012) 120(11):2297–306. doi:10.1182/blood-2012-02-414425
33. Cerny J, Yu H, Ramanathan M, Raffel GD, Walsh WV, Fortier N, et al. Expression of CD25 independently predicts early treatment failure of acute myeloid leukaemia (AML). Br J Haematol (2013) 160(2):262–6. doi:10.1111/bjh.12109
34. Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med (2010) 2(17):17ra19. doi:10.1126/scitranslmed.3000349
35. Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene (2011) 30(9):1009–19. doi:10.1038/onc.2010.511
36. Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood (2014) 123(16):2540–9. doi:10.1182/blood-2013-07-517847
37. Flynn MJ, van Berkel P, Zammarchi F, Levy J-N, Tiberghien A, Masterson LA, et al. Pre-clinical activity of Adct-301, a novel pyrrolobenzodiazepine (PBD) dimer-containing antibody drug conjugate (ADC) targeting CD25-expressing hematological malignancies. Blood (2014) 124(21):4491.
38. Horwitz SM, Hamadani M, Fanale MA, Feingold J, Spira AI, Fields PA, et al. Interim results from a phase 1 study of ADCT-301 (camidanlumab tesirine) show promising activity of a novel pyrrolobenzodiazepine-based antibody drug conjugate in relapsed/refractory Hodgkin/non-Hodgkin lymphoma. Blood (2017) 130:1510.
39. Goldberg AD, Tallman MS, Solh MM, Ungar D, Rizzieri DA, Walter RB, et al. Results from an ongoing phase 1 study indicate ACDT-301 (camidanlumab tesirine) is well-tolerated in patients with relapsed or refractory CD25-positive acute leukemia. Blood (2017) 130:2662.
40. Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev (2009) 229(1):216–31. doi:10.1111/j.1600-065X.2009.00774.x
41. Wiesmann A, Phillips RL, Mojica M, Pierce LJ, Searles AE, Spangrude GJ, et al. Expression of CD27 on murine hematopoietic stem and progenitor cells. Immunity (2000) 12(2):193–9. doi:10.1016/S1074-7613(00)80172-7
42. Schürch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest (2012) 122(2):624–38. doi:10.1172/JCI45977
43. Nolte MA, Arens R, van Os R, van Oosterwijk M, Hooibrink B, van Lier RA, et al. Immune activation modulates hematopoiesis through interactions between CD27 and CD70. Nat Immunol (2005) 6(4):412–8. doi:10.1038/ni1174
44. Riether C, Schürch CM, Flury C, Hinterbrandner M, Druck L, Huguenin AL, et al. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci Transl Med (2015) 7(298):298ra119. doi:10.1126/scitranslmed.aab1740
45. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin AL, Hoepner S, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med (2017) 214(2):359–80. doi:10.1084/jem.20152008
46. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35(18):2028–36. doi:10.1200/JCO.2016.70.1508
47. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol (2007) 7(4):255–66. doi:10.1038/nri2056
48. Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annu Rev Immunol (2012) 30(30):357–92. doi:10.1146/annurev-immunol-020711-075018
49. Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res (2001) 7(6):1490–6.
50. Sievers EL, Larson RA, Stadtmauer EA, Estey E, Lowenberg B, Dombret H, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol (2001) 19(13):3244–54. doi:10.1200/JCO.2001.19.13.3244
51. Larson RA, Sievers EL, Stadtmauer EA, Lowenberg B, Estey EH, Dombret H, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer (2005) 104(7):1442–52. doi:10.1002/cncr.21326
52. Godwin CD, Gale RP, Walter RB. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia (2017) 31(9):1855–68. doi:10.1038/leu.2017.187
53. Gbolahan OB, Zeidan AM, Stahl M, Abu Zaid M, Farag S, Paczesny S, et al. Immunotherapeutic concepts to target acute myeloid leukemia: focusing on the role of monoclonal antibodies, hypomethylating agents and the leukemic microenvironment. Int J Mol Sci (2017) 18(8):E1660. doi:10.3390/ijms18081660
54. Masarova L, Kantarjian H, Garcia-Mannero G, Ravandi F, Sharma P, Daver N. Harnessing the immune system against leukemia: monoclonal antibodies and checkpoint strategies for AML. Adv Exp Med Biol (2017) 995:73–95. doi:10.1007/978-3-319-53156-4_4
55. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood (2013) 121(24):4854–60. doi:10.1182/blood-2013-01-466706
56. Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol (2014) 15(9):986–96. doi:10.1016/S1470-2045(14)70281-5
57. Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood (2005) 105(3):1295–302. doi:10.1182/blood-2004-07-2784
58. Walter RB, Gooley TA, van der Velden VH, Loken MR, van Dongen JJ, Flowers DA, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood (2007) 109(10):4168–70. doi:10.1182/blood-2006-09-047399
59. Olombel G, Guerin E, Guy J, Perrot JY, Dumezy F, de Labarthe A, et al. The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood (2016) 127(17):2157–60. doi:10.1182/blood-2016-01-689976
60. Pollard JA, Loken M, Gerbing RB, Raimondi SC, Hirsch BA, Aplenc R, et al. CD33 expression and its association with gemtuzumab ozogamicin response: results from the randomized phase III children’s oncology group trial AAML0531. J Clin Oncol (2016) 34(7):747–55. doi:10.1200/JCO.2015.62.6846
61. Khan N, Hills RK, Virgo P, Couzens S, Clark N, Gilkes A, et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia (2017) 31(5):1059–68. doi:10.1038/leu.2016.309
62. van der Velden VH, Boeckx N, Jedema I, te Marvelde JG, Hoogeveen PG, Boogaerts M, et al. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia (2004) 18(5):983–8. doi:10.1038/sj.leu.2403350
63. Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III children’s oncology group trial AAML0531. J Clin Oncol (2017) 35(23):2674–82. doi:10.1200/JCO.2016.71.2513
64. Middeldorf I, Galm O, Osieka R, Jost E, Herman JG, Wilop S. Sequence of administration and methylation of SOCS3 may govern response to gemtuzumab ozogamicin in combination with conventional chemotherapy in patients with refractory or relapsed acute myelogenous leukemia (AML). Am J Hematol (2010) 85(7):477–81. doi:10.1002/ajh.21723
65. Paubelle E, Marceau A, Zylbersztejn F, Dussiot M, Moura IC, Cornillet-Lefebvre P, et al. HFE gene mutation status predicts response to gemtuzumab ozogamicin in AML. Blood (2015) 126:1307.
66. Raza A, Jurcic JG, Roboz GJ, Maris M, Stephenson JJ, Wood BL, et al. Complete remissions observed in acute myeloid leukemia following prolonged exposure to lintuzumab: a phase 1 trial. Leuk Lymphoma (2009) 50(8):1336–44. doi:10.1080/10428190903050013
67. Feldman EJ, Brandwein J, Stone R, Kalaycio M, Moore J, O’Connor J, et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J Clin Oncol (2005) 23(18):4110–6. doi:10.1200/JCO.2005.09.133
68. Sekeres MA, Lancet JE, Wood BL, Grove LE, Sandalic L, Sievers EL, et al. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica (2013) 98(1):119–28. doi:10.3324/haematol.2012.066613
69. Vasu S, He S, Cheney C, Gopalakrishnan B, Mani R, Lozanski G, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood (2016) 127(23):2879–89. doi:10.1182/blood-2015-11-680546
70. Hagemann UB, Wickstroem K, Wang E, Shea AO, Sponheim K, Karlsson J, et al. In vitro and in vivo efficacy of a novel CD33-targeted thorium-227 conjugate for the treatment of acute myeloid leukemia. Mol Cancer Ther (2016) 15(10):2422–31. doi:10.1158/1535-7163.MCT-16-0251
71. Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood (2013) 122(8):1455–63. doi:10.1182/blood-2013-03-491506
72. Stein EM, Stein A, Walter RB, Fathi AT, Lancet JE, Kovacsovics TJ, et al. Interim analysis of a phase 1 trial of SGN-CD33A in patients with CD33-positive acute myeloid leukemia (AML). Blood (2014) 124:623.
73. Fathi AT, Erba HP, Lancet JE, Stein EM, Walter RB, DeAngelo DJ, et al. SGN-CD33A plus hypomethylating agents: a novel, well-tolerated regimen with high remission rate in frontline unfit AML. Blood (2015) 126:454.
74. Stein AS, Walter RB, Erba HP, Fathi AT, Advani AS, Lancet JE, et al. A phase 1 trial of SGN-CD33A as monotherapy in patients with CD33-positive acute myeloid leukemia (AML). Blood (2015) 126:324.
75. Robinson B. Seattle Genetics Discontinues Phase 3 CASCADE Trial of Vadastuximab Talirine (SGN-CD33A) in Frontline Acute Myeloid Leukemia. (2017). Available from: https://www.businesswire.com/news/home/20170619005466/en/Seattle-Genetics-Discontinues-Phase-3-CASCADE-Trial (Accessed: February 19, 2018).
76. Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol (2003) 21:107–37. doi:10.1146/annurev.immunol.21.120601.140946
77. Orozco JJ, Kenoyer A, Balkin ER, Gooley TA, Hamlin DK, Wilbur DS, et al. Anti-CD45 radioimmunotherapy without TBI before transplantation facilitates persistent haploidentical donor engraftment. Blood (2016) 127(3):352–9. doi:10.1182/blood-2014-12-617019
78. Mawad R, Gooley TA, Rajendran JG, Fisher DR, Gopal AK, Shields AT, et al. Radiolabeled anti-CD45 antibody with reduced-intensity conditioning and allogeneic transplantation for younger patients with advanced acute myeloid leukemia or myelodysplastic syndrome. Biol Blood Marrow Transplant (2014) 20(9):1363–8. doi:10.1016/j.bbmt.2014.05.014
79. Tuazon SA, Sandmaier BM, Orozco JJ, Gopal AK, Holmberg LA, Becker PS, et al. A phase I trial of 90Y-BC8-DOTA (anti-CD45) monoclonal antibody in combination with fludarabine and TBI as conditioning for allogeneic peripheral blood stem cell transplant to treat high risk multiple myeloma. Blood (2017) 130:910.
80. Cassaday RD, Press OW, Pagel JM, Rajendran JG, Gooley TA, Fisher DR, et al. Safety and efficacy of escalating doses of 90Y-BC8-DOTA (anti-CD45) followed by carmustine, etoposide, cytarabine, and melphalan (BEAM) chemotherapy and autologous stem cell transplantation (ASCT) for high-risk lymphoma. Biol Blood Marrow Transplant (2018) 24(3):S251–2. doi:10.1016/j.bbmt.2017.12.227
81. Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the immune system: more than a marker for cytotoxicity? Front Immunol (2017) 8:892. doi:10.3389/fimmu.2017.00892
82. Baer MR, Stewart CC, Lawrence D, Arthur DC, Byrd JC, Davey FR, et al. Expression of the neural cell adhesion molecule CD56 is associated with short remission duration and survival in acute myeloid leukemia with t(8;21)(q22;q22). Blood (1997) 90(4):1643–8.
83. Raspadori D, Damiani D, Lenoci M, Rondelli D, Testoni N, Nardi G, et al. CD56 antigenic expression in acute myeloid leukemia identifies patients with poor clinical prognosis. Leukemia (2001) 15(8):1161–4. doi:10.1038/sj.leu.2402174
84. Xu S, Li X, Zhang J, Chen J. Prognostic value of CD56 in patients with acute myeloid leukemia: a meta-analysis. J Cancer Res Clin Oncol (2015) 141(10):1859–70. doi:10.1007/s00432-015-1977-3
85. Zaidi SZ, Motabi IH, Al-Shanqeeti A. CD56 and RUNX1 isoforms in AML prognosis and their therapeutic potential. Hematol Oncol Stem Cell Ther (2016) 9(3):129–30. doi:10.1016/j.hemonc.2015.11.006
86. Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y. CD123 and its potential clinical application in leukemias. Life Sci (2015) 122:59–64. doi:10.1016/j.lfs.2014.10.013
87. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia (2000) 14(10):1777–84. doi:10.1038/sj.leu.2401903
88. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell (2009) 5(1):31–42. doi:10.1016/j.stem.2009.04.018
89. Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G, et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and stem cells. Blood (2014) 123(8):1218–28. doi:10.1182/blood-2012-12-475194
90. Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia (2014) 28(11):2213–21. doi:10.1038/leu.2014.128
91. Smith BD, Roboz GJ, Walter RB, Altman JK, Ferguson A, Curcio TJ, et al. First-in man, phase 1 study of CSL362 (anti-IL3Rα/anti-CD123 monoclonal antibody) in patients with CD123+ acute myeloid leukemia (AML) in CR at high risk for early relapse. Blood (2014) 124:120.
92. Smith BD, Roberts AW, Roboz GJ, DeWitte M, Ferguson A, Garrett L, et al. Minimal residual disease (MRD) as exploratory endpoint in a phase 1 study of the anti-CD123 mab CSL362 given as post-remission therapy in adult acute myeloid leukemia (AML). Blood (2015) 126:3819.
93. Syed K, Pietsch C, Axel A, Forslund A, Sasser K, Salvati M. Preclinical evaluation of CSL362/JNJ-56022473 single agent in in vitro assays. Blood (2015) 126:4946.
94. Syed K, Pietsch C, Axel A, Forslund A, Sasser K, Salvati M. Preclinical evaluation of CSL362/JNJ-56022473 in combination with decitabine or azacitidine in in vitro assays. Blood (2015) 126:1370.
95. Court E. Johnson & Johnson Discards Two Hoped-for Billion-Dollar Drugs. (2017). Available from: https://www.marketwatch.com/story/johnson-johnson-discards-two-hoped-for-billion-dollar-drugs-2017-10-17 (Accessed:February 18, 2018).
96. Li F, Sutherland MK, Yu C, Walter RB, Westendorf L, Valliere-Douglass J, et al. Characterization of SGN-CD123A, A potent CD123-directed antibody-drug conjugate for acute myeloid leukemia. Mol Cancer Ther (2018) 17(2):554–64. doi:10.1158/1535-7163.MCT-17-0742
97. Akiyama T, Takayanagi S-I, Maekawa Y, Miyawaki K, Jinnouchi F, Jiromaru T, et al. First preclinical report of the efficacy and PD results of KHK2823, a non-fucosylated fully human monoclonal antibody against IL-3Rα. Blood (2015) 126:1349.
98. Adams S, Wilhelm A, Harvey L, Bai C, Yoder N, Kovtun Y, et al. IMGN632: a CD123-targeting antibody-drug conjugate (ADC) with a novel DNA-alkylating payload, is highly active and prolongs survival in acute myeloid leukemia (AML) xenograft models. Blood (2016) 128:2832.
99. Kovtun Y, Jones G, Audette C, Harvey L, Gerard B, Wilhelm A, et al. A CD123-targeting antibody-drug conjugate (ADC), IMGN632, designed to eradicate acute myeloid leukemia (AML) cells while sparing normal bone marrow cells. Blood (2016) 128:768.
100. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol (2010) 10(3):201–9. doi:10.1038/nri2726
101. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505(7483):327–34. doi:10.1038/nature12984
102. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466(7308):829–34. doi:10.1038/nature09262
103. Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine. Annu Rev Immunol (2013) 31:285–316. doi:10.1146/annurev-immunol-032712-095919
104. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature (2003) 425(6960):841–6. doi:10.1038/nature02040
105. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature (2003) 425(6960):836–41. doi:10.1038/nature02041
106. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature (2013) 502(7473):637–43. doi:10.1038/nature12612
107. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25(6):977–88. doi:10.1016/j.immuni.2006.10.016
108. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature (2012) 481(7382):457–62. doi:10.1038/nature10783
109. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature (2013) 495(7440):231–5. doi:10.1038/nature11885
110. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature (2015) 526(7571):126–30. doi:10.1038/nature15250
111. Mercier FE, Ragu C, Scadden DT. The bone marrow at the crossroads of blood and immunity. Nat Rev Immunol (2012) 12(1):49–60. doi:10.1038/nri3132
112. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119(13):2991–3002. doi:10.1182/blood-2011-12-380113
113. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol (2014) 14(5):302–14. doi:10.1038/nri3660
114. Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med (2013) 19(6):695–703. doi:10.1038/nm.3155
115. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20(8):833–46. doi:10.1038/nm.3647
116. Tokoyoda K, Hauser AE, Nakayama T, Radbruch A. Organization of immunological memory by bone marrow stroma. Nat Rev Immunol (2010) 10(3):193–200. doi:10.1038/nri2727
117. Feuerer M, Beckhove P, Garbi N, Mahnke Y, Limmer A, Hommel M, et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat Med (2003) 9(9):1151–7. doi:10.1038/nm914
118. Riether C, Schürch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ (2015) 22(2):187–98. doi:10.1038/cdd.2014.89
119. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi:10.1038/35102167
120. Huntly BJP, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer (2005) 5(4):311–21. doi:10.1038/nrc1592
121. Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell (2007) 129(6):1097–110. doi:10.1016/j.cell.2007.05.014
122. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell (2007) 129(6):1081–95. doi:10.1016/j.cell.2007.03.055
123. Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature (2014) 506(7487):240–4. doi:10.1038/nature12883
124. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature (2010) 464(7290):852–7. doi:10.1038/nature08851
125. Mager LF, Riether C, Schürch CM, Banz Y, Wasmer MH, Stuber R, et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J Clin Invest (2015) 125(7):2579–91. doi:10.1172/JCI77347
126. Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature (2014) 512(7512):78–81. doi:10.1038/nature13383
127. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell (2014) 15(3):365–75. doi:10.1016/j.stem.2014.06.020
128. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell (2013) 13(3):285–99. doi:10.1016/j.stem.2013.06.009
129. Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell (2014) 14(6):824–37. doi:10.1016/j.stem.2014.02.014
130. Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell (2014) 25(6):778–93. doi:10.1016/j.ccr.2014.04.015
131. Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood (2013) 121(10):1824–38. doi:10.1182/blood-2012-02-412890
132. Bajaj J, Konuma T, Lytle NK, Kwon HY, Ablack JN, Cantor JM, et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell (2016) 30(5):792–805. doi:10.1016/j.ccell.2016.10.003
133. Scholzel C, Lowenberg B. Stimulation of proliferation and differentiation of acute myeloid leukemia cells on a bone marrow stroma in culture. Exp Hematol (1985) 13(7):664–9.
134. Bendall LJ, Daniel A, Kortlepel K, Gottlieb DJ. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia cells. Exp Hematol (1994) 22(13):1252–60.
135. Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia (2002) 16(9):1713–24. doi:10.1038/sj.leu.2402608
136. Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science (1993) 261(5121):600–3. doi:10.1126/science.8342023
137. Nagasawa T, Kikutani H, Kishimoto T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc Natl Acad Sci U S A (1994) 91(6):2305–9. doi:10.1073/pnas.91.6.2305
138. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature (1996) 381(6584):661–6. doi:10.1038/381661a0
139. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science (1996) 272(5263):872–7. doi:10.1126/science.272.5263.872
140. Karpova D, Bonig H. Concise review: CXCR4/CXCL12 signaling in immature hematopoiesis – lessons from pharmacological and genetic models. Stem Cells (2015) 33(8):2391–9. doi:10.1002/stem.2054
142. Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood (1998) 91(12):4523–30.
143. Burger JA, Spoo A, Dwenger A, Burger M, Behringer D. CXCR4 chemokine receptors (CD184) and alpha4beta1 integrins mediate spontaneous migration of human CD34+ progenitors and acute myeloid leukaemia cells beneath marrow stromal cells (pseudoemperipolesis). Br J Haematol (2003) 122(4):579–89. doi:10.1046/j.1365-2141.2003.04466.x
144. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood (2007) 109(2):786–91. doi:10.1182/blood-2006-05-024844
145. Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, Benzion K, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia (2008) 22(12):2151–5158. doi:10.1038/leu.2008.238
146. Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood (2009) 113(24):6206–14. doi:10.1182/blood-2008-06-162123
147. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood (2009) 113(24):6215–24. doi:10.1182/blood-2008-05-158311
148. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood (2012) 119(17):3917–24. doi:10.1182/blood-2011-10-383406
149. Uy GL, Rettig MP, Stone RM, Konopleva MY, Andreeff M, McFarland K, et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J (2017) 7:e542. doi:10.1038/bcj.2017.21
150. Becker PS, Foran JM, Altman JK, Yacoub A, Castro JE, Sabbatini P, et al. Targeting the CXCR4 pathway: safety, tolerability and clinical activity of ulocuplumab (BMS-936564), an anti-CXCR4 antibody, in relapsed/refractory acute myeloid leukemia. Blood (2014) 124:386.
151. Pernasetti F, Liu S-H, Hallin M, Gu Y, Ho W-H, Zhang C, et al. A novel CXCR4 antagonist IgG1 antibody (PF-06747143) for the treatment of hematological malignancies. Blood (2014) 124:2311.
152. Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai YT, Shi JT, et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep (2015) 12(4):622–35. doi:10.1016/j.celrep.2015.06.059
153. Kashyap MK, Kumar D, Jones H, Amaya-Chanaga CI, Choi MY, Melo-Cardenas J, et al. Ulocuplumab (BMS-936564/MDX1338): a fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget (2016) 7(3):2809–22. doi:10.18632/oncotarget.6465
154. Kashyap MK, Amaya-Chanaga CI, Kumar D, Simmons B, Huser N, Gu Y, et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF06747143) in chronic lymphocytic leukemia. J Hematol Oncol (2017) 10:112. doi:10.1186/s13045-017-0435-x
155. Liu SH, Gu Y, Pascual B, Yan ZM, Hallin M, Zhang C, et al. A novel CXCR4 antagonist IgG1 antibody (PF-06747143) for the treatment of hematologic malignancies. Blood Adv (2017) 1(15):1088–100. doi:10.1182/bloodadvances.2016003921
156. Zhang YY, Saavedra E, Tang RP, Gu Y, Lappin P, Trajkovic D, et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci Rep (2017) 7:7305. doi:10.1038/s41598-017-07848-8
157. Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. Complementation of dominant suppression implicates CD98 in integrin activation. Nature (1997) 390(6655):81–5. doi:10.1038/36349
158. Fenczik CA, Zent R, Dellos M, Calderwood DA, Satriano J, Kelly C, et al. Distinct domains of CD98hc regulate integrins and amino acid transport. J Biol Chem (2001) 276(12):8746–52. doi:10.1074/jbc.M011239200
159. Feral CC, Nishiya N, Fenczik CA, Stuhlmann H, Slepak M, Ginsberg MH. CD98hc (SLC3A2) mediates integrin signaling. Proc Natl Acad Sci U S A (2005) 102(2):355–60. doi:10.1073/pnas.0404852102
160. Feral CC, Zijlstra A, Tkachenko E, Prager G, Gardel ML, Slepak M, et al. CD98hc (SLC3A2) participates in fibronectin matrix assembly by mediating integrin signaling. J Cell Biol (2007) 178(4):701–11. doi:10.1083/jcb.200705090
161. Haynes BF, Hemler M, Cotner T, Mann DL, Eisenbarth GS, Strominger JL, et al. Characterization of a monoclonal antibody (5E9) that defines a human cell surface antigen of cell activation. J Immunol (1981) 127(1):347–51.
162. Hara K, Kudoh H, Enomoto T, Hashimoto Y, Masuko T. Malignant transformation of NIH3T3 cells by overexpression of early lymphocyte activation antigen CD98. Biochem Biophys Res Commun (1999) 262(3):720–5. doi:10.1006/bbrc.1999.1051
163. Henderson NC, Collis EA, Mackinnon AC, Simpson KJ, Haslett C, Zent R, et al. CD98hc (SLC3A2) interaction with beta 1 integrins is required for transformation. J Biol Chem (2004) 279(52):54731–41. doi:10.1074/jbc.M408700200
164. Cantor J, Browne CD, Ruppert R, Feral CC, Fassler R, Rickert RC, et al. CD98hc facilitates B cell proliferation and adaptive humoral immunity. Nat Immunol (2009) 10(4):412–9. doi:10.1038/ni.1712
165. Cantor J, Slepak M, Ege N, Chang JT, Ginsberg MH. Loss of T cell CD98 H chain specifically ablates T cell clonal expansion and protects from autoimmunity. J Immunol (2011) 187(2):851–60. doi:10.4049/jimmunol.1100002
166. Cantor JM, Ginsberg MH. CD98 at the crossroads of adaptive immunity and cancer. J Cell Sci (2012) 125(Pt 6):1373–82. doi:10.1242/jcs.096040
167. Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, et al. L-type amino-acid transporter 1 (LAT1): a therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia (2015) 29(6):1253–66. doi:10.1038/leu.2014.338
168. Hayes GM, Chinn L, Cantor JM, Cairns B, Levashova Z, Tran H, et al. Antitumor activity of an anti-CD98 antibody. Int J Cancer (2015) 137(3):710–20. doi:10.1002/ijc.29415
169. Bixby D, Wieduwilt MJ, Akard LP, Khoury HJ, Becker PS, Van Der Horst EH, et al. A phase I study of IGN523, a novel anti-CD98 monoclonal antibody in patients with relapsed or refractory acute myeloid leukemia (AML). Blood (2015) 126:3809.
170. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer (2008) 8(8):579–91. doi:10.1038/nrc2403
171. Reichert F, Barak V, Tarshis M, Prindull G, Tarshis E, Ben-Ishay Z. Anti-angiogenic effects and regression of localized murine AML produced by anti-VEGF and anti-Flk-1 antibodies. Eur J Haematol (2005) 75(1):41–6. doi:10.1111/j.1600-0609.2005.00436.x
172. Karp JE, Gojo I, Pili R, Gocke CD, Greer J, Guo C, et al. Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: therapy with sequential 1-beta-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res (2004) 10(11):3577–85. doi:10.1158/1078-0432.CCR-03-0627
173. Zahiragic L, Schliemann C, Bieker R, Thoennissen NH, Burow K, Kramer C, et al. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia (2007) 21(6):1310–2. doi:10.1038/sj.leu.2404632
174. Madlambayan GJ, Meacham AM, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood (2010) 116(9):1539–47. doi:10.1182/blood-2009-06-230474
175. Midwood KS, Hussenet T, Langlois B, Orend G. Advances in tenascin-C biology. Cell Mol Life Sci (2011) 68(19):3175–99. doi:10.1007/s00018-011-0783-6
176. Gutbrodt KL, Schliemann C, Giovannoni L, Frey K, Pabst T, Klapper W, et al. Antibody-based delivery of interleukin-2 to neovasculature has potent activity against acute myeloid leukemia. Sci Transl Med (2013) 5(201):201ra118. doi:10.1126/scitranslmed.3006221
177. Schliemann C, Gutbrodt KL, Kerkhoff A, Pohlen M, Wiebe S, Silling G, et al. Targeting interleukin-2 to the bone marrow stroma for therapy of acute myeloid leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Cancer Immunol Res (2015) 3(5):547–56. doi:10.1158/2326-6066.CIR-14-0179
178. Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V, et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood (2000) 95(11):3289–96.
179. Becker PS, Kopecky KJ, Wilks AN, Chien S, Harlan JM, Willman CL, et al. Very late antigen-4 function of myeloblasts correlates with improved overall survival for patients with acute myeloid leukemia. Blood (2009) 113(4):866–74. doi:10.1182/blood-2007-12-124818
180. Hsieh Y-T, Jiang E, Pham J, Kim H-N, Abdel-Azim H, Khazal S, et al. VLA4 blockade in acute myeloid leukemia. Blood (2013) 122:3944.
181. Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, et al. TGF-beta-neutralizing antibody 1D11 enhances cytarabine-induced apoptosis in AML cells in the bone marrow microenvironment. PLoS One (2013) 8(6):e62785. doi:10.1371/journal.pone.0062785
182. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med (2006) 12(10):1167–74. doi:10.1038/nm1483
183. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell (2016) 19(1):23–37. doi:10.1016/j.stem.2016.06.001
184. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
186. Schürch CM, Riether C, Ochsenbein AF. Dendritic cell-based immunotherapy for myeloid leukemias. Front Immunol (2013) 4:496. doi:10.3389/fimmu.2013.00496
187. Saudemont A, Quesnel B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood (2004) 104(7):2124–33. doi:10.1182/blood-2004-01-0064
188. Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood (2009) 114(8):1528–36. doi:10.1182/blood-2008-09-179697
189. Dolen Y, Esendagli G. Myeloid leukemia cells with a B7-2(+) subpopulation provoke Th-cell responses and become immuno-suppressive through the modulation of B7 ligands. Eur J Immunol (2013) 43(3):747–57. doi:10.1002/eji.201242814
190. Schürch C, Riether C, Amrein MA, Ochsenbein AF. Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-gamma. J Exp Med (2013) 210(3):605–21. doi:10.1084/jem.20121229
191. Mohty M, Jarrossay D, Lafage-Pochitaloff M, Zandotti C, Briere F, de Lamballeri XN, et al. Circulating blood dendritic cells from myeloid leukemia patients display quantitative and cytogenetic abnormalities as well as functional impairment. Blood (2001) 98(13):3750–6. doi:10.1182/blood.V98.13.3750
192. Mumprecht S, Claus C, Schürch C, Pavelic V, Matter MS, Ochsenbein AF. Defective homing and impaired induction of cytotoxic T cells by BCR/ABL-expressing dendritic cells. Blood (2009) 113(19):4681–9. doi:10.1182/blood-2008-05-156471
193. Chao MP, Majeti R, Weissman IL. Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer (2011) 12(1):58–67. doi:10.1038/nrc3171
194. Armand P. Immune checkpoint blockade in hematologic malignancies. Blood (2015) 125(22):3393–400. doi:10.1182/blood-2015-02-567453
195. Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma (2017) 59(4):790–802. doi:10.1080/10428194.2017.1344905
196. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
197. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol (2001) 19:565–94. doi:10.1146/annurev.immunol.19.1.565
198. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
199. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206(8):1717–25. doi:10.1084/jem.20082492
200. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
201. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
202. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
203. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
204. Laurent S, Palmisano GL, Martelli AM, Kato T, Tazzari PL, Pierri I, et al. CTLA-4 expressed by chemoresistant, as well as untreated, myeloid leukaemia cells can be targeted with ligands to induce apoptosis. Br J Haematol (2007) 136(4):597–608. doi:10.1111/j.1365-2141.2006.06472.x
205. Vidriales MB, Orfao A, Lopez-Berges MC, Gonzalez M, Hernandez JM, Ciudad J, et al. Lymphoid subsets in acute myeloid leukemias: increased number of cells with NK phenotype and normal T-cell distribution. Ann Hematol (1993) 67(5):217–22. doi:10.1007/BF01715050
206. Wendelbo O, Nesthus I, Sjo M, Paulsen K, Ernst P, Bruserud O. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother (2004) 53(8):740–7. doi:10.1007/s00262-004-0505-0
207. Schnorfeil FM, Lichtenegger FS, Emmerig K, Schlueter M, Neitz JS, Draenert R, et al. T cells are functionally not impaired in AML: increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J Hematol Oncol (2015) 8:93. doi:10.1186/s13045-015-0189-2
208. Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood (2009) 114(18):3909–16. doi:10.1182/blood-2009-02-206946
209. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med (2016) 375(2):143–53. doi:10.1056/NEJMoa1601202
210. Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell (2015) 17(3):341–52. doi:10.1016/j.stem.2015.07.011
211. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi:10.1084/jem.20090847
212. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
213. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi:10.1038/nature22396
214. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood (2009) 114(8):1545–52. doi:10.1182/blood-2009-03-206672
215. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117(17):4501–10. doi:10.1182/blood-2010-10-310425
216. Berthon C, Driss V, Liu J, Kuranda K, Leleu X, Jouy N, et al. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol Immunother (2010) 59(12):1839–49. doi:10.1007/s00262-010-0909-y
217. Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M, et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood (2010) 116(7):1124–31. doi:10.1182/blood-2009-12-255125
218. Krönig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol (2014) 92(3):195–203. doi:10.1111/ejh.12228
219. Riether C, Gschwend T, Huguenin AL, Schurch CM, Ochsenbein AF. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia (2015) 29(8):1781–5. doi:10.1038/leu.2015.26
220. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28(6):1280–8. doi:10.1038/leu.2013.355
221. Orskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD-1 and DNA methylation. Oncotarget (2015) 6(11):9612–26. doi:10.18632/oncotarget.3324
222. Goltz D, Gevensleben H, Grunen S, Dietrich J, Kristiansen G, Landsberg J, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia (2017) 31(3):738–43. doi:10.1038/leu.2016.328
223. Anderson AC, Anderson DE. TIM-3 in autoimmunity. Curr Opin Immunol (2006) 18(6):665–9. doi:10.1016/j.coi.2006.09.009
224. Su EW, Lin JY, Kane LP. TIM-1 and TIM-3 proteins in immune regulation. Cytokine (2008) 44(1):9–13. doi:10.1016/j.cyto.2008.06.013
225. Ngiow SF, Teng MW, Smyth MJ. Prospects for TIM3-targeted antitumor immunotherapy. Cancer Res (2011) 71(21):6567–71. doi:10.1158/0008-5472.CAN-11-1487
226. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol (2011) 32(8):345–9. doi:10.1016/j.it.2011.05.003
227. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi:10.1084/jem.20100643
228. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell (2010) 7(6):708–17. doi:10.1016/j.stem.2010.11.014
229. Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res (2009) 69(12):4941–4. doi:10.1158/0008-5472.CAN-09-0547
230. Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature (1985) 316(6026):354–6. doi:10.1038/316354a0
231. Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature (1985) 314(6012):628–31. doi:10.1038/314628a0
232. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs (2017) 9(2):182–212. doi:10.1080/19420862.2016.1268307
233. Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther (2017) 182:161–75. doi:10.1016/j.pharmthera.2017.08.005
234. Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol (2011) 29(18):2493–8. doi:10.1200/JCO.2010.32.7270
235. Krupka C, Kufer P, Kischel R, Zugmaier G, Bogeholz J, Kohnke T, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood (2014) 123(3):356–65. doi:10.1182/blood-2013-08-523548
236. Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood (2014) 123(4):554–61. doi:10.1182/blood-2013-09-527044
237. Sheridan C. Despite slow progress, bispecifics generate buzz. Nat Biotechnol (2016) 34(12):1215–7. doi:10.1038/nbt1216-1215
238. Chen Y, Xu Y. Pharmacokinetics of bispecific antibody. Curr Pharmacol Rep (2017) 3(3):126–37. doi:10.1007/s40495-017-0090-5
239. Chu SY, Pong E, Chen H, Phung S, Chan EW, Endo NA, et al. Immunotherapy with long-lived anti-CD123× anti-CD3 bispecific antibodies stimulates potent T cell-mediated killing of human AML cell lines and of CD123+ cells in monkeys: a potential therapy for acute myelogenous leukemia. Blood (2014) 124:2316.
240. Gaudet F, Nemeth JF, McDaid R, Li Y, Harman B, Millar H, et al. Development of a CD123xCD3 bispecific antibody (JNJ-63709178) for the treatment of acute myeloid leukemia (AML). Blood (2016) 128:2824.
241. Forslund A, Syed K, Axel A, McDaid R, Li Y, Ballesteros J, et al. Ex vivo activity profile of the CD123xCD3 Duobody® antibody JNJ-63709178 against primary acute myeloid leukemia bone marrow samples. Blood (2016) 128:2875.
242. Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res (2004) 64(22):8443–50. doi:10.1158/0008-5472.CAN-04-1659
243. Van Loo PF, Doornbos R, Dolstra H, Shamsili S, Bakker L. Preclinical evaluation of MCLA117, a CLEC12AxCD3 bispecific antibody efficiently targeting a novel leukemic stem cell associated antigen in AML. Blood (2015) 126:325.
244. Leong SR, Sukumaran S, Hristopoulos M, Totpal K, Stainton S, Lu E, et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood (2017) 129(5):609–18. doi:10.1182/blood-2016-08-735365
245. Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood (2011) 117(17):4542–51. doi:10.1182/blood-2010-09-306449
246. Rader C. DARTs take aim at BiTEs. Blood (2011) 117(17):4403–4. doi:10.1182/blood-2011-02-337691
247. Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med (2015) 7(289):289ra282. doi:10.1126/scitranslmed.aaa5693
248. Vey N, Davidson-Moncada JK, Uy GL, Rizzieri D, Khoury HJ, Foster MC, et al. A phase I, first-in-human study of MGD006/S80880 (CD123 x CD3 DART) in AML/MDS. J Clin Oncol (2017) 35(15_suppl):TS7070. doi:10.1200/JCO.2017.35.15_suppl.TPS7070
249. Lindberg FP, Bullard DC, Caver TE, Gresham HD, Beaudet AL, Brown EJ. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science (1996) 274(5288):795–8. doi:10.1126/science.274.5288.795
250. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol (2001) 11(3):130–5. doi:10.1016/S0962-8924(00)01906-1
251. Liu Y, Merlin D, Burst SL, Pochet M, Madara JL, Parkos CA. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem (2001) 276(43):40156–66. doi:10.1074/jbc.M104138200
252. Miyashita M, Ohnishi H, Okazawa H, Tomonaga H, Hayashi A, Fujimoto TT, et al. Promotion of neurite and filopodium formation by CD47: roles of integrins, Rac, and Cdc42. Mol Biol Cell (2004) 15(8):3950–63. doi:10.1091/mbc.E04-01-0019
253. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science (2000) 288(5473):2051–4. doi:10.1126/science.288.5473.2051
254. Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med (2001) 194(4):541–9. doi:10.1084/jem.194.4.541
255. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123(2):321–34. doi:10.1016/j.cell.2005.08.032
256. Rendtlew Danielsen JM, Knudsen LM, Dahl IM, Lodahl M, Rasmussen T. Dysregulation of CD47 and the ligands thrombospondin 1 and 2 in multiple myeloma. Br J Haematol (2007) 138(6):756–60. doi:10.1111/j.1365-2141.2007.06729.x
257. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138(2):271–85. doi:10.1016/j.cell.2009.05.046
258. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell (2009) 138(2):286–99. doi:10.1016/j.cell.2009.05.045
259. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142(5):699–713. doi:10.1016/j.cell.2010.07.044
260. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res (2011) 71(4):1374–84. doi:10.1158/0008-5472.CAN-10-2238
261. Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, Weissman IL. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood (2011) 118(18):4890–901. doi:10.1182/blood-2011-02-338020
262. Edris B, Weiskopf K, Volkmer AK, Volkmer JP, Willingham SB, Contreras-Trujillo H, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci U S A (2012) 109(17):6656–61. doi:10.1073/pnas.1121629109
263. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A (2012) 109(17):6662–7. doi:10.1073/pnas.1121623109
264. Schürch CM, Forster S, Brühl F, Yang SH, Felley-Bosco E, Hewer E. The “don’t eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology (2017) 7:e1373235. doi:10.1080/2162402X.2017.1373235
265. Jaiswal JK, Rivera VM, Simon SM. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell (2009) 137(7):1308–19. doi:10.1016/j.cell.2009.04.064
266. Galli S, Zlobec I, Schürch C, Perren A, Ochsenbein AF, Banz Y. CD47 protein expression in acute myeloid leukemia: a tissue microarray-based analysis. Leuk Res (2015) 39(7):749–56. doi:10.1016/j.leukres.2015.04.007
267. Pietsch EC, Dong J, Cardoso R, Zhang X, Chin D, Hawkins R, et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J (2017) 7(2):e536. doi:10.1038/bcj.2017.7
268. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
269. Koh CY, Blazar BR, George T, Welniak LA, Capitini CM, Raziuddin A, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood (2001) 97(10):3132–7. doi:10.1182/blood.V97.10.3132
270. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi:10.1126/science.1068440
271. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood (2007) 110(1):433–40. doi:10.1182/blood-2006-07-038687
272. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116(14):2411–9. doi:10.1182/blood-2010-05-283051
273. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114(13):2667–77. doi:10.1182/blood-2009-02-206532
274. Vey N, Dombret H, Ifrah N, Pigneux A, Gardin C, Buyse ME, et al. Intergroup ALFA/GOELAMS randomized phase II trial of lirilumab anti-KIR monoclonal antibody (IPH2102/BMS986015) as maintenance treatment in elderly patients with acute myeloid leukemia (EFFIKIR trial). J Clin Oncol (2013). doi:10.1200/jco.2013.31.15_suppl.tps3117
275. Borrego F, Masilamani M, Marusina AI, Tang X, Coligan JE. The CD94/NKG2 family of receptors: from molecules and cells to clinical relevance. Immunol Res (2006) 35(3):263–78. doi:10.1385/IR:35:3:263
276. Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Jimenez P, Geraghty DE, et al. Analysis of HLA-E expression in human tumors. Immunogenetics (2003) 54(11):767–75. doi:10.1007/s00251-002-0526-9
277. Cohen RB, Salas S, Even C, Kotecki N, Jimeno A, Soulié A-M, et al. Safety of the first-in-class anti-NKG2A monoclonal antibody monalizumab in combination with cetuximab: a phase Ib/II study in recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Cancer Res (2017). doi:10.1158/1538-7445.AM2017-5666
278. Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood (2011) 117(16):4262–72. doi:10.1182/blood-2010-07-299248
279. Sergeeva A, He H, Ruisaard K, St John L, Alatrash G, Clise-Dwyer K, et al. Activity of 8F4, a T-cell receptor-like anti-PR1/HLA-A2 antibody, against primary human AML in vivo. Leukemia (2016) 30(7):1475–84. doi:10.1038/leu.2016.57
280. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med (2013) 5(176):176ra133. doi:10.1126/scitranslmed.3005661
281. Dubrovsky L, Pankov D, Brea EJ, Dao T, Scott A, Yan S, et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood (2014) 123(21):3296–304. doi:10.1182/blood-2014-01-549022
282. Tur MK, Huhn M, Jost E, Thepen T, Brummendorf TH, Barth S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22(scFv)-ETA’ in a human acute myeloid leukemia xenograft tumor model. Int J Cancer (2011) 129(5):1277–82. doi:10.1002/ijc.25766
283. Subramaniam S, Stansberg C, Cunningham C. The interleukin 1 receptor family. Dev Comp Immunol (2004) 28(5):415–28. doi:10.1016/j.dci.2003.09.016
284. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi:10.1146/annurev.immunol.021908.132612
285. Jaras M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc Natl Acad Sci U S A (2010) 107(37):16280–5. doi:10.1073/pnas.1004408107
286. Zhao K, Yin LL, Zhao DM, Pan B, Chen W, Cao J, et al. IL1RAP as a surface marker for leukemia stem cells is related to clinical phase of chronic myeloid leukemia patients. Int J Clin Exp Med (2014) 7(12):4787–98.
287. Landberg N, Hansen N, Askmyr M, Agerstam H, Lassen C, Rissler M, et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia (2016) 30(1):253–7. doi:10.1038/leu.2015.135
288. Askmyr M, Agerstam H, Hansen N, Gordon S, Arvanitakis A, Rissler M, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood (2013) 121(18):3709–13. doi:10.1182/blood-2012-09-458935
289. Agerstam H, Karlsson C, Hansen N, Sanden C, Askmyr M, von Palffy S, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci U S A (2015) 112(34):10786–91. doi:10.1073/pnas.1422749112
290. Estrov Z, Kurzrock R, Estey E, Wetzler M, Ferrajoli A, Harris D, et al. Inhibition of acute myelogenous leukemia blast proliferation by interleukin-1 (IL-1) receptor antagonist and soluble IL-1 receptors. Blood (1992) 79(8):1938–45.
291. Fuchs A, Colonna M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol (2006) 16(5):359–66. doi:10.1016/j.semcancer.2006.07.002
292. Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol (2004) 172(7):3994–8. doi:10.4049/jimmunol.172.7.3994
293. Gramatzki M, Ludwig WD, Burger R, Moos P, Rohwer P, Grunert C, et al. Antibodies TC-12 (“unique”) and TH-111 (CD96) characterize T-cell acute lymphoblastic leukemia and a subgroup of acute myeloid leukemia. Exp Hematol (1998) 26(13):1209–14.
294. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A (2007) 104(26):11008–13. doi:10.1073/pnas.0704271104
295. Chavez-Gonzalez A, Dorantes-Acosta E, Moreno-Lorenzana D, Alvarado-Moreno A, Arriaga-Pizano L, Mayani H. Expression of CD90, CD96, CD117, and CD123 on different hematopoietic cell populations from pediatric patients with acute myeloid leukemia. Arch Med Res (2014) 45(4):343–50. doi:10.1016/j.arcmed.2014.04.001
296. Jiang Y, Xu P, Yao D, Chen X, Dai H. CD33, CD96 and death associated protein kinase (DAPK) expression are associated with the survival rate and/or response to the chemotherapy in the patients with acute myeloid leukemia (AML). Med Sci Monit (2017) 23:1725–32. doi:10.12659/MSM.900305
297. Mohseni Nodehi S, Repp R, Kellner C, Brautigam J, Staudinger M, Schub N, et al. Enhanced ADCC activity of affinity maturated and Fc-engineered mini-antibodies directed against the AML stem cell antigen CD96. PLoS One (2012) 7(8):e42426. doi:10.1371/journal.pone.0042426
298. Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM, et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res (2011) 31(6):2283–90.
299. Dourado KMC, Baik J, Oliveira VKP, Beltrame M, Yamamoto A, Theuer CP, et al. Endoglin: a novel target for therapeutic intervention in acute leukemias revealed in xenograft mouse models. Blood (2017) 129(18):2526–36. doi:10.1182/blood-2017-01-763581
300. Hirata Y, Kimura N, Sato K, Ohsugi Y, Takasawa S, Okamoto H, et al. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett (1994) 356(2–3):244–8. doi:10.1016/0014-5793(94)01279-2
301. Kaisho T, Ishikawa J, Oritani K, Inazawa J, Tomizawa H, Muraoka O, et al. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc Natl Acad Sci U S A (1994) 91(12):5325–9. doi:10.1073/pnas.91.12.5325
302. Funaro A, Ortolan E, Ferranti B, Gargiulo L, Notaro R, Luzzatto L, et al. CD157 is an important mediator of neutrophil adhesion and migration. Blood (2004) 104(13):4269–78. doi:10.1182/blood-2004-06-2129
303. Ortolan E, Tibaldi EV, Ferranti B, Lavagno L, Garbarino G, Notaro R, et al. CD157 plays a pivotal role in neutrophil transendothelial migration. Blood (2006) 108(13):4214–22. doi:10.1182/blood-2006-04-017160
304. Funaro A, Ortolan E, Bovino P, Lo Buono N, Nacci G, Parrotta R, et al. Ectoenzymes and innate immunity: the role of human CD157 in leukocyte trafficking. Front Biosci (Landmark Ed) (2009) 14:929–43. doi:10.2741/3287
305. Ishihara K, Hirano T. BST-1/CD157 regulates the humoral immune responses in vivo. Chem Immunol (2000) 75:235–55. doi:10.1159/000058772
306. Todd RF III, Roach JA, Arnaout MA. The modulated expression of Mo5, a human myelomonocytic plasma membrane antigen. Blood (1985) 65(4):964–73.
307. Krupka C, Lichtenegger FS, Kohnke T, Bogeholz J, Bucklein V, Roiss M, et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget (2017) 8(22):35707–17. doi:10.18632/oncotarget.16060
308. Gorczynski RM. CD200 and its receptors as targets for immunoregulation. Curr Opin Investig Drugs (2005) 6(5):483–8.
309. Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, et al. Expression of CD200 on AML blasts directly suppresses memory T-cell function. Leukemia (2012) 26(9):2148–51. doi:10.1038/leu.2012.77
310. Coles SJ, Gilmour MN, Reid R, Knapper S, Burnett AK, Man S, et al. The immunosuppressive ligands PD-L1 and CD200 are linked in AML T-cell immunosuppression: identification of a new immunotherapeutic synapse. Leukemia (2015) 29(9):1952–4. doi:10.1038/leu.2015.62
311. Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, et al. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia (2012) 26(9):2146–8. doi:10.1038/leu.2012.75
312. Memarian A, Nourizadeh M, Masoumi F, Tabrizi M, Emami AH, Alimoghaddam K, et al. Upregulation of CD200 is associated with Foxp3+ regulatory T cell expansion and disease progression in acute myeloid leukemia. Tumour Biol (2013) 34(1):531–42. doi:10.1007/s13277-012-0578-x
313. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rulicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood (2014) 123(25):3951–62. doi:10.1182/blood-2013-10-536078
314. Farag SS, Nelson R, Cairo MS, O’Leary HA, Zhang S, Huntley C, et al. High-dose sitagliptin for systemic inhibition of dipeptidylpeptidase-4 to enhance engraftment of single cord umbilical cord blood transplantation. Oncotarget (2017) 8(66):110350–7. doi:10.18632/oncotarget.22739
315. Pereira DS, Guevara CI, Jin L, Mbong N, Verlinsky A, Hsu SJ, et al. AGS67E, an anti-CD37 monomethyl auristatin E antibody-drug conjugate as a potential therapeutic for B/T-cell malignancies and AML: a new role for CD37 in AML. Mol Cancer Ther (2015) 14(7):1650–60. doi:10.1158/1535-7163.MCT-15-0067
316. Gillissen MA, de Jong G, Kedde M, Yasuda E, Levie SE, Moiset G, et al. Patient-derived antibody recognizes a unique CD43 epitope expressed on all AML and has antileukemia activity in mice. Blood Adv (2017) 1(19):1551–64. doi:10.1182/bloodadvances.2017008342
317. Saito Y, Nakahata S, Yamakawa N, Kaneda K, Ichihara E, Suekane A, et al. CD52 as a molecular target for immunotherapy to treat acute myeloid leukemia with high EVI1 expression. Leukemia (2011) 25(6):921–31. doi:10.1038/leu.2011.36
318. Blatt K, Herrmann H, Hoermann G, Willmann M, Cerny-Reiterer S, Sadovnik I, et al. Identification of campath-1 (CD52) as novel drug target in neoplastic stem cells in 5q-patients with MDS and AML. Clin Cancer Res (2014) 20(13):3589–602. doi:10.1158/1078-0432.CCR-13-2811
319. Nishioka C, Ikezoe T, Yokoyama A. Blockade of CD82 by a monoclonal antibody potentiates anti-leukemia effects of AraC in vivo. Cancer Med (2015) 4(9):1426–31. doi:10.1002/cam4.482
320. Iwasaki M, Liedtke M, Gentles AJ, Cleary ML. CD93 marks a non-quiescent human leukemia stem cell population and is required for development of MLL-rearranged acute myeloid leukemia. Cell Stem Cell (2015) 17(4):412–21. doi:10.1016/j.stem.2015.08.008
321. Koerner S, Leibold J, Grosse-Hovest L, Buehring H-J, Jung G, Kanz L, et al. Development and preclinical characterization of an Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Blood (2014) 124:2309.
322. Rothfelder K, Koerner S, Andre M, Leibold J, Kousis P, Bühring H-J, et al. Induction of NK cell reactivity against myeloid leukemia by a novel Fc-optimized CD133 antibody. Blood (2015) 126:3793.
323. Nuebling T, Schumacher CE, Hofmann M, Hagelstein I, Schmiedel BJ, Maurer S, et al. The immune checkpoint modulator OX40 and its ligand OX40L in NK-cell immunosurveillance and acute myeloid leukemia. Cancer Immunol Res (2018) 6(2):209–21. doi:10.1158/2326-6066.CIR-17-0212
324. Durben M, Schmiedel D, Hofmann M, Vogt F, Nubling T, Pyz E, et al. Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol Ther (2015) 23(4):648–55. doi:10.1038/mt.2015.2
325. Baessler T, Charton JE, Schmiedel BJ, Grunebach F, Krusch M, Wacker A, et al. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood (2010) 115(15):3058–69. doi:10.1182/blood-2009-06-227934
326. Cheng K, Wong SC, Linn YC, Ho LP, Chng WJ, Schwarz H. CD137 ligand signalling induces differentiation of primary acute myeloid leukaemia cells. Br J Haematol (2014) 165(1):134–44. doi:10.1111/bjh.12732
327. Schmohl JU, Nuebling T, Wild J, Kroell T, Kanz L, Salih HR, et al. Expression of 4-1BB and its ligand on blasts correlates with prognosis of patients with AML. J Investig Med (2016) 64(8):1252–60. doi:10.1136/jim-2016-000081
328. Teague RM, Kline J. Immune evasion in acute myeloid leukemia: current concepts and future directions. J Immunother Cancer (2013) 1:13. doi:10.1186/2051-1426-1-13
329. Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell (2017) 31(4):501–15.e508. doi:10.1016/j.ccell.2017.03.005
330. Ozkazanc D, Yoyen-Ermis D, Tavukcuoglu E, Buyukasik Y, Esendagli G. Functional exhaustion of CD4(+) T cells induced by co-stimulatory signals from myeloid leukaemia cells. Immunology (2016) 149(4):460–71. doi:10.1111/imm.12665
331. Korver W, Zhao X, Singh S, Pardoux C, Zhao J, Guzman ML, et al. Monoclonal antibodies against IREM-1: potential for targeted therapy of AML. Leukemia (2009) 23(9):1587–97. doi:10.1038/leu.2009.99
332. Wen-Li Z, Jian W, Yan-Fang T, Xing F, Yan-Hong L, Xue-Ming Z, et al. Inhibition of the ecto-beta subunit of F1F0-ATPase inhibits proliferation and induces apoptosis in acute myeloid leukemia cell lines. J Exp Clin Cancer Res (2012) 31:92. doi:10.1186/1756-9966-31-92
333. Baessler T, Krusch M, Schmiedel BJ, Kloss M, Baltz KM, Wacker A, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein ligand subverts immunosurveillance of acute myeloid leukemia in humans. Cancer Res (2009) 69(3):1037–45. doi:10.1158/0008-5472.CAN-08-2650
334. Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol (2012) 24(2):217–24. doi:10.1016/j.coi.2011.12.011
335. Schmiedel BJ, Werner A, Steinbacher J, Nuebling T, Buechele C, Grosse-Hovest L, et al. Generation and preclinical characterization of a Fc-optimized GITR-Ig fusion protein for induction of NK cell reactivity against leukemia. Mol Ther (2013) 21(4):877–86. doi:10.1038/mt.2013.11
336. Drake AS, Brady MT, Wang XH, Sait SJ, Earp JC, Ghoshal Gupta S, et al. Targeting 11q23 positive acute leukemia cells with high molecular weight-melanoma associated antigen-specific monoclonal antibodies. Cancer Immunol Immunother (2009) 58(3):415–27. doi:10.1007/s00262-008-0567-5
337. Kim SY, Theunissen JW, Balibalos J, Liao-Chan S, Babcock MC, Wong T, et al. A novel antibody-drug conjugate targeting SAIL for the treatment of hematologic malignancies. Blood Cancer J (2015) 5:e316. doi:10.1038/bcj.2015.39
338. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res (2016) 22(12):3057–66. doi:10.1158/1078-0432.CCR-15-2626
339. Gillissen MA, Kedde M, Jong G, Moiset G, Yasuda E, Levie SE, et al. AML-specific cytotoxic antibodies in patients with durable graft-versus-leukemia responses. Blood (2018) 131(1):131–43. doi:10.1182/blood-2017-02-768762
340. Lamble A, Kosaka Y, Huang F, Sasser AK, Adams H, Tognon CE, et al. Enhanced VISTA expression in a subset of patients with acute myeloid leukemia. Blood (2016) 128:4056.
Keywords: acute myeloid leukemia, myelodysplastic syndrome, gemtuzumab ozogamicin, CD33, leukemic stem cells, monoclonal antibody, targeted therapy, immune checkpoint inhibitors
Citation: Schürch CM (2018) Therapeutic Antibodies for Myeloid Neoplasms—Current Developments and Future Directions. Front. Oncol. 8:152. doi: 10.3389/fonc.2018.00152
Received: 26 March 2018; Accepted: 24 April 2018;
Published: 18 May 2018
Edited by:
Gregor Hutter, Universitätsspital Basel, SwitzerlandReviewed by:
Koichi Takahashi, University of Texas MD Anderson Cancer Center, United StatesTala Shekarian, UMR5286 Centre de Recherche en Cancerologie de Lyon (CRCL), France
Copyright: © 2018 Schürch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian M. Schürch, c2NodWVyY2hAc3RhbmZvcmQuZWR1