 Tatjana Abaffy1*
Tatjana Abaffy1* James R. Bain2Michael J. Muehlbauer2Ivan Spasojevic3Shweta Lodha4Elisa Bruguera4Sara K. O’Neal2
James R. Bain2Michael J. Muehlbauer2Ivan Spasojevic3Shweta Lodha4Elisa Bruguera4Sara K. O’Neal2 So Young Kim5
So Young Kim5 Hiroaki Matsunami6
Hiroaki Matsunami6
- 1Department of Molecular Genetics and Microbiology, Duke Cancer Institute, Duke University School of Medicine, Durham, NC, United States
- 2Duke Molecular Physiology Institute, Duke University School of Medicine, Durham, NC, United States
- 3Department of Medicine, Duke University School of Medicine, Durham, NC, United States
- 4Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC, United States
- 5Department of Molecular Genetics and Microbiology, Functional Genomics Shared Resource, Duke University School of Medicine, Durham, NC, United States
- 6Department of Molecular Genetics and Microbiology, Department of Neurobiology, Duke Institute for Brain Sciences, Duke Cancer Institute, Duke University School of Medicine, Durham, NC, United States
Olfactory receptor OR51E2, also known as a Prostate Specific G-Protein Receptor, is highly expressed in prostate cancer but its function is not well understood. Through in silico and in vitro analyses, we identified 24 agonists and 1 antagonist for this receptor. We detected that agonist 19-hydroxyandrostenedione, a product of the aromatase reaction, is endogenously produced upon receptor activation. We characterized the effects of receptor activation on metabolism using a prostate cancer cell line and demonstrated decreased intracellular anabolic signals and cell viability, induction of cell cycle arrest, and increased expression of neuronal markers. Furthermore, upregulation of neuron-specific enolase by agonist treatment was abolished in OR51E2-KO cells. The results of our study suggest that OR51E2 activation results in neuroendocrine trans-differentiation. These findings reveal a new role for OR51E2 and establish this G-protein coupled receptor as a novel therapeutic target in the treatment of prostate cancer.
Introduction
G-protein coupled receptors (GPCRs) have emerged as important factors in tumor growth and metastasis (1). Several GPCRs, such as the 5HT1c serotonin receptor (2), the M1, M3, and M5 muscarinic receptors (3), and the α1B-ADR adrenergic receptor (4), can function as oncogenes when persistently activated. These GPCRs, which are normally expressed in fully differentiated, post-mitotic neuronal cells, are able to induce cellular oncogenic transformation when introduced to an ectopic environment of proliferating cells and activated by agonist (5). In addition to oncogenes and tumor-suppressor genes essential for cancer initiation and progression, autocrine and/or paracrine secretion of GPCR-activating molecules and their downstream signaling events affect tumor growth, survival, and metastasis (6).
Olfactory receptors (ORs) are the largest family of GPCRs present in the olfactory epithelium but are also found in various ectopic or non-olfactory locations such as prostate, heart, placenta, embryo, erythroid cells, spleen, kidney, gut, tongue, and carotid body (7). Some ectopic ORs also play roles in chemotaxis (8), muscle regeneration (9), blood pressure regulation (10), and hypoxia response (11).
OR51E2, or prostate-specific G-protein receptor (PSGR), is one of the most highly conserved and broadly expressed ectopic ORs (12–14). OR51E2 is present in healthy prostate tissue and shows significantly increased expression in prostate intraepithelial neoplasia (PIN), prostate adenocarcinoma (PC), and castration-resistant prostate cancer (CRPC) (15–22).
The majority of prostate tumors start as androgen-dependent adenocarcinomas. As localized cancer progresses to a metastatic state, the number of neuroendocrine (NE)-like cells increases, contributing to the development of a highly aggressive form of castrate-resistant prostate cancer (CRPC) known as neuroendocrine prostate cancer (NEPC) (23). Many clinical studies have demonstrated a correlation between neuroendocrine trans-differentiation (NEtD) and PC progression with poor prognosis (24). Tumor-derived NE-like cells are localized in tumor foci and are non-proliferating, terminally differentiated cells rich in serotonin and positive for NE markers, including neuron-specific enolase (NSE) and chromogranin A (CGA) (24).
The molecular mechanism underlying development of a neuroendocrine phenotype in PC is not fully understood. In the androgen-dependent LNCaP cell line, serum deprivation and agents that increase cAMP also increase expression of NEtD markers and genes indicative of neuronal phenotype (25).
In the olfactory system, ORs signal via the canonical cAMP pathway (26), and several reports have indicated cAMP-mediated signaling for ectopic ORs (27–29). Furthermore, high expression of the OR downstream targets adenylate cyclase 3 and Gαolf was recently identified in prostate tissue, supporting the role of cAMP-mediated pathway in ectopic OR activation (12). We hypothesized that agonist-mediated activation of OR51E2 increases cAMP and facilitates cellular transformation, resulting in NEtD. Thus, ectopic expression of this GPCR in proliferating cells and ligand-dependent activation could enable this receptor to function as an oncogene.
Recently, it has been demonstrated that overexpression of OR51E2/PSGR in a PSGR-Pten (Δ/Δ) mouse model accelerates PC development and progression (30). Furthermore, β-ionone, an agonist for OR51E2, decreased proliferation and increased invasiveness of human PC cells (31–33).
In this paper, we aimed to identify biologically relevant OR51E2 ligands using a combination of in silico investigations and experimental validation, and we also set out to study the effects of these ligands on androgen-dependent LNCaP cells. Currently identified OR51E2 agonists include the short-chain fatty acids acetate and propionate (10, 34), steroid derivatives, β-ionone (31), and lactate (11). However, it is not known whether activation of OE51E2 by endogenous ligands is involved in PC pathogenesis.
Here, we virtually screened >2,500 metabolites, experimentally validated 55 of these candidates in vitro, and ultimately identified 24 new agonists and 1 antagonist for the human OR51E2 receptor. Among the agonists, we identified 19-hydroxyandrostenedione (19-OH AD)—which is synthesized by aromatase, an enzyme highly expressed in NEPC and CRPC—and N-acetyl-N-formyl-5-methoxykynurenamine (AFMK), a tryptophan and melatonin metabolite. We detected endogenous production of 19-OH AD in the LNCaP cells stimulated with AFMK agonist. The identity of the newly discovered agonists, as well as significant differences in metabolomics signatures in agonist-stimulated PC cells indicative of non-proliferation, prompted us to further investigate their effects on cell viability, cell cycle status, and several NE markers to examine if OR51E2 receptor activation drives NEtD.
Materials and Methods
Homology Modeling
Amino acid sequences of bovine rhodopsin, the human adrenergic beta-2-receptor β 2AR, the mouse olfactory receptor MOR42-3, and the human olfactory receptor OR51E2, were initially aligned using an MAFFT program. Our model was built based on the crystal structure of β2AR (4LDO). We used BioEdit Sequence Alignment Editor1 to manually remove gaps in the β2AR and OR51E2 sequences and re-align Cys178 in OR51E2 (EC2) with Cys191 from the β2AR sequence (Figure S1 in Supplementary Material). The β2AR has a disulfide bond between the conserved Cys106 residue at the N-terminal end of TM3 and Cys191 from EC2. Thus, to introduce a disulfide bond, we re-aligned equivalent cysteines in OR51E2 (Cys96-Cys178). In addition, the most conserved residues were also aligned (Asp41, Leu54, Cys96, Leu114, Asp120, Arg121, Tyr122, Pro128, Pro159, Tyr217, Asp287, Pro288, Ile290, and Tyr290). We used Modeller v.9.14 to create homology models (35)2, and 20 homology models were made using an automodel script with the default optimization and refinement. Each model was assessed using a DOPE score (36). The best model, which had a DOPE score of −37275, was chosen for further analysis. The .pdb file was imported into ICM Software (MolSoft v.3.8, LLC, La Jolla, CA, USA) (37), and the protein structure was analyzed using Ramchandran Plot Analysis. Residues that were out of place (Glu90, Arg222, His180, Leu81, and Lys294) were optimized. The model was transformed into an ICM object and subjected to regularization and minimization using MMFF Cartesian minimization (300 steps). The minimized model was imported into Modeller and again evaluated to calculate DOPE score (new DOPE = −38897). This final, minimized model was used for docking and Virtual Library Screening (VLS) in the ICM program.
Virtual Library Screening
The library used for VLS was selected from the HMDB3 and consisted of metabolites detected in human blood, tissue, urine, and saliva. VLS was performed with the ICM software as described previously (38). Briefly, the potential energy maps of the receptor were made in a box with a 0.5-Å grid and a size of 53 Å × 50 Å × 45 Å. The initial position of the ligand was set within the center of this box, which extends to the receptor interior [above the center positions of Ser107 and Ala108, which are equivalent positions to Ile112 and Val113 in MOR42-3 that are known to be part of the ligand binding pocket (39)]. The docking stimulation was set to 1 and the maximal number of conformations to 10. The remainder of the docking parameters was set at the default values. The VLS result lists ligand–metabolite pairs according to their scores, and lower scores indicate ligands that are more likely to bind to the receptor. Binding of the ligand to the receptor is described using Score and mfScore functions. The Score function is based on a theoretical calculation of receptor–ligand binding energy.
The mfScore function is a knowledge-based potential derived from the frequencies of occurrence of various atom pairs within the experimental ligand/receptor complex structures deposited in the Protein Data Bank (PDB) (40). It represents a measure of statistical probability of interaction between the ligand and receptor. Our previous results indicate that both scoring functions were equally successful in predicting ligands (38). The top 50 hits from each scoring list are presented in Tables S1 and S2 in Supplementary Material.
Cell Culture
Prostate cancer LNCaP-FGC cells derived from lymph node metastatic site (passage 30–32) were obtained from the American Type Culture Collection (ATCC). Cells were maintained at 37°C in RPMI-1640 medium (Sigma R8758) enriched with 0.5% glucose (45%, Sigma G8769), 1% 1 M HEPES (Gibco-Thermo 15630), 1% 100 mM Na-pyruvate (Gibco-Thermo 11360), 10% FBS (HyClone, SH30071.03), 100 U/mL penicillin, and 0.1 mg/mL streptomycin. At 4 to 5 days after seeding, cells were 70% to 80% confluent and the stimulus was applied at the indicated concentration for the indicated time. The medium was replaced every fourth day.
Luciferase Assay
Briefly, the OR51E2 plasmid was transfected into HANA3A cells along with a CREB-dependent luciferase (firefly) and a constitutively active luciferase (Renilla) (34). Upon ligand binding, an increase in cAMP drives the expression of firefly luciferase and increases the signal. To control for variation in cell number and transfection efficiency, the luciferase signal was normalized to the activity of the Renilla luciferase in the same cells. All stock solutions of chemicals (Table S11 in Supplementary Material) were prepared either in dimethylsulfoxide (DMSO) or ethanol. Final dilutions were made in M10d medium. M10d is MEM medium enriched with 5% dialyzed FBS serum, which is devoid of small molecular weight compounds (<10,000 Da), since OR51E2-transfected cells gave a high luciferase signal in the CD293 medium (Gibco 11913-019, supplemented with 30 µM CuCl2 and 2 μM l-glutamine) even in the absence of chemical stimulation and when compared to the basal activity of the control OR2W1 receptor-expressing cells (data not shown). All compounds that did not show agonist activity in M10d were later diluted in CD293 medium and tested for antagonist activity. The rest of the protocol was performed as previously published (34). Cells were exposed to candidate ligands for 3.5 h at various concentrations. For each compound that showed a response >2 SD of the baseline response (no chemical applied), the EC50 or IC50 was determined from a sigmoid dose–response curve using a Graph-Pad Prism (Graph-Pad Prism Software, San Diego, CA, USA). Data were fitted to the equation: Y = Bottom + (Top−Bottom)/(1 + 10^((LogIC50−X) * HillSlope)).
19-OH AD Measurements Using LC/MS
LNCaP cells were first grown in T-75 flasks until just fully confluent. Cells were then split into six T-25 flasks. Cells were exposed to 250 µM AFMK, or to the medium only, for 3 days. We used phenol-red free RPMI 1640 with 10% CD-serum (Hyclone, SH30068) or RPMI 1640 with 10% FBS, as described in cell culture protocol. After 3 days, medium was collected and frozen at −80°C until LC/MS measurements.
In 2 mL polypropylene vial, 800 µL of cell media sample was vigorously mixed with 1 mL of ethyl acetate, centrifuged, and 900 µL of the organic (top) layer evaporated under a stream of nitrogen at room temperature. To the dried residue, 20 µL of the following reagents was added for picolinoyl ester derivatization: 40 mg of picolinic acid, 20 mg of 2-methyl-6-nitrobenzoic anhydride (MNBA), 10 mg of 4-dimethyl-laminopyridine, 1 mL acetonitrile, and 20 µL trimethylamine. After 20 min of incubation at 50°C, 30 µL of 0.1% formic acid in water was added and 25 µL of the sample was injected into the LC-MS/MS system [slightly modified from Ref. (41)]. LC conditions (Shimadzu 20 A series HPLC): Agilent Eclipse PLUS, C18, 50 × 4.6 mm, 1.8 µm particle size column; mobile phases A/B: 0.1% formic acid in water/acetonitrile; elution gradient: 0–1 min 20–90%B, 1–1.5 min 90%B, 1.5–1.7 min 90–20%B; run time: 5 min. The following mass spectrometer conditions were used for quantification (AB/Sciex API5500 QTrap): after optimization of the electrospray and quadrupoles parameters by infusion of 100 ng/mL 19-OHAD at 10 µL/min rate, 408.2/267.2 MS/MS transition.
Metabolomics GC-MS Analysis
LNCaP cells were first grown in T-75 flasks until just fully confluent. Cells were then split into six T-25 flasks and exposed to agonists the following day: 250 µM AFMK, 100 nM 19-OH AD, or 1 mM propionic acid (PA), with six biological replicates in each group. After agonist treatment for 3 days, CM was removed and banked at −80°C, and the cells were rinsed with 10 mL ice-cold PBS. Next, 3 mL of ice-cold 0.9% NaCl was added, and the cells were scraped off the plates and transferred to 5-mL tubes that were previously cleaned with acetonitrile. Each flask was rinsed with an additional 2 mL of ice-cold 0.9% NaCl, and the cell suspensions were transferred to the 5-mL tubes and centrifuged at 1000 rpm for 5 min at 4°C. Supernatant was removed, and 200 µL of 0.6% formic acid in ice-cold dH2O was added to the cell pellets. Then, 20 µL of resuspended cell pellets were separated for protein measurements (Qubit Protein Assay kit, Cat. # Q33211, Thermo Fisher Scientific), and 180 µL of acetonitrile was added to the remaining pellet. Prepared cell lysates were maintained at −80°C until metabolomic analysis.
For exploratory, non-targeted metabolomics via gas chromatography/mass spectrometry (GC/MS), metabolites were extracted from cell lysates with methanol, methoximated in dry pyridine, and then silylated with N-methyl-N-(trimethylsilyl) trifluoroacetamide. Samples were analyzed on a 6890 N GC connected to a 5975 MS (Agilent Technologies, Santa Clara, CA, USA) equipped with two wall-coated open-tubular (WCOT) GC columns connected in series (Agilent part 122-5512, DB5MS, 15 m in length, 0.25 mm in diameter, with an 0.25-µm luminal film) separated by a microfluidic flow splitter to enable hot back-flushing at the end of each run. Data were acquired by scanning from m/z 600 to 50 as the oven ramped from 70 to 325°C. Data were deconvoluted using AMDIS software (42). Metabolites were identified using our retention time-referenced spectral library, which is based in part on that of Kind et al. (43). Reported data are log2 transforms of the areas of deconvoluted peaks.
Data were normalized to the protein content in each sample. MetaboAnalyst 3.0 was used for statistical analysis (44). Briefly, peak intensity data were presented in columns and log2-normalized. We used unpaired analysis, and data were auto-scaled (mean-centered and divided by the standard deviation of each variable). For pathway analysis, we used a Globaltest pathway-enrichment analysis algorithm in MetaboAnalyst 3.0.
RT-PCR Analysis
LNCaP cells were grown in T-75 flasks until fully confluent. A split ratio of 1:6 was used to subculture the cells in T-25 flasks for 24 to 48 h before stimulation. Stimulation with agonists lasted 3 or 12 days. Medium was changed every 4 days in the 12-day experiment. Total RNA was extracted using Trizol reagent (Invitrogen) and cleaned using the RNeasy kit (Qiagen) following the manufacturer’s protocol. Integrity of total RNA was confirmed by agarose gel electrophoresis. cDNA was generated by reverse transcription using SuperScript II Reverse Transcriptase (Invitrogen). Primers for the following genes were designed with the Primer 3.0 program: OR51E2, NSE, α-methylacyl-CoA racemase (AMACR), α1H T-type calcium channel (Cav3.2), androgen receptor (AR), GAPDH, keratin 5, keratin 8, and keratin 18 (Table S3 in Supplementary Material). PCR amplification was performed with HotStart Taq Polymerase (Qiagen) using the following protocol: 95°C for 15 min followed by 30 cycles at the following times and temperatures: 95°C for 15 s, 55°C for 15 s, and 72°C for 30 s. Final annealing was done at 72°C for 5 min. Expression levels of the test genes were normalized to GAPDH.
Cell Proliferation Assay
Cells were plated in five 96-well plates (100 µL cell suspension per well) and after overnight attachment when cells were 15% to 20% confluent, cells were stimulated with increasing concentrations of selected agonists. Cell growth and viability were estimated each day for the next 4 days by using Cell Titer-Glo Luminescent Cell Viability Assay (Promega, Cat. No. G7570) according to the manufacturer’s instructions.
Cell Cycle Analysis
LNCaP cells were plated in 100-mm Petri dishes. After overnight attachment, when cells were 40% to 50% confluent, cells were stimulated once with 100 nM 19-OH AD and 250 µM AFMK for the next 4 days. Cells incubated in the regular RPMI-1640 medium served as controls. After treatment, cells were trypsinized with 0.05% trypsin-EDTA (Gibco) and fixed with 70% ethanol in PBS for 15 min at 4°C. Fixed cells were washed and incubated with 50 µg/µL RNase A and 20 µg/µL propidium iodide and subjected to cell cycle analysis using a Becton Dickinson FACSCalibur cytometer. Data were analyzed with BD CellQuest software. Experiments were performed three times in triplicate (three biological and three technical replicates).
LNCaP OR51E2 Knockout Cell Lines
To generate CRISPR/Cas9-mediated knockout LNCaP cells, we cloned sgRNAs targeting exon 2 of OR51E2 into LentiCRISPR.v2 (Addgene #52961) for coexpression of sgRNA with Streptococcus pyogenes Cas9. We prepared lentiviral particles for each sgRNA vector by cotransfecting HEK293T cells with the LentiCRISPR, vectors psPAX2 and pMD2.g using TransIT-LT1 (Mirus). LNCaP cells were transduced with this virus at a multiplicity of infection (MOI) of <1 in the presence of 8 µg/ml polybrene. At 24 h post-infection, cells were selected with 2 µg/mL puromycin for 72 h and then expanded for verification of gene editing and experimental analysis. Cells were harvested for genomic DNA and PCR amplification of the OR51E2 locus for the Surveyor Assay (Integrated DNA Technologies) 1 week after infection. Additionally, for stable cells harboring OR51E2 sgRNA #1, PCR products were cloned into a TOPO-TA cloning vector (ThermoFisher Scientific) and Sanger sequenced to assess the rate of mutation.
Sequences:
sgRNA #1: CGTGGTCTTCATCGTAAGGA
sgRNA #2: AGGCCTCAAAGCTAATCTCT
sgRNA #3: CATTGAATCCACCATCCTGC
OR51E2 forward: ACGAAGGTATGGACCAGTAGGA
OR51E2 reverse: AAGACCATATACCACATTGGGC
Quantification and Statistical Analysis
All results are expressed as mean ± SEM. Computations assumed that all groups were samples from populations with the same scatter. The investigators involved in this study were not completely blinded during sample collection or data analysis. Significance was determined by (multiple) two-tailed unpaired t-test using Prism 7 software. A P value of 0.05 was considered significant.
Results
In Vitro Validation of Ligands Predicted In Silico
A structural model of OR51E2 was made in silico using Modeller (see Materials and Methods for details). To experimentally validate our VLS predictions, we used an in vitro heterologous expression system (45) in which Hana3A cells transfected with the OR51E2 receptor were stimulated with the top candidate ligands. Responses were subsequently measured with a cAMP-mediated luciferase reporter gene assay. First, a small library of 33 compounds identified as PC-associated metabolites was selected from the Human Metabolome Database HMDB (46). These compounds were identified from previous reports (47–49). Additional compounds previously identified as OR51E2 agonists were also included: 1,4,6-androstatriene-17-β-ol-3-one; 1,4,6-androstatriene-3,17-dione; 6-dehydrotestosterone; and β-ionone (31). Thus, a total of 37 compounds were used for the initial, small-scale VLS (Table S4 in Supplementary Material). Two scoring functions, Score and mfScore, were used to predict the best binders. The top nine compounds from each score list (italicized in Table S4 in Supplementary Material) were then tested in vitro, and the following metabolites were identified as novel agonists for OR51E2 (bold and italics): bradykinin, kojibiose, glycylglycine, l-histidinol N-acetylglutamic acid, and d-alanyl-d-alanine. We also confirmed the previously reported agonist 1,4,6-androstatriene-3,17-dione (Figure S2 in Supplementary Material) (31).
Next, a larger library of 2,511 human metabolites from the HMDB was selected for virtual screening and docking into the receptor pocket (Figures 1A,B). Here, the top potential ligands (in italics, Tables S1 and S2 in Supplementary Material) were tested in vitro using a biologically relevant concentration range reported in the HMDB, and concentration–response curves were subsequently produced. In total, 55 compounds were tested (9 and 46, from the smaller and larger screens, respectively), and 24 agonists (Figure 1C; Figures S2 and S3 in Supplementary Material) and 1 antagonist for OR51E2 (Figure 1D) were identified. In each experiment, OR51E2-expressing Hana3A cells were also stimulated with a known agonist, 1 mM PA, so we were able to compare the efficacy of each metabolite relative to PA. Furthermore, potency values (EC50) were determined (Table 1). Glycylglycine was the most efficient agonist, while l-histidinol was the most potent. Diverse metabolites were discovered as novel agonists for OR51E2. Concentration–response curves for metabolites from the large VLS screen that did not activate the receptor are presented in Figure S4 in Supplementary Material.
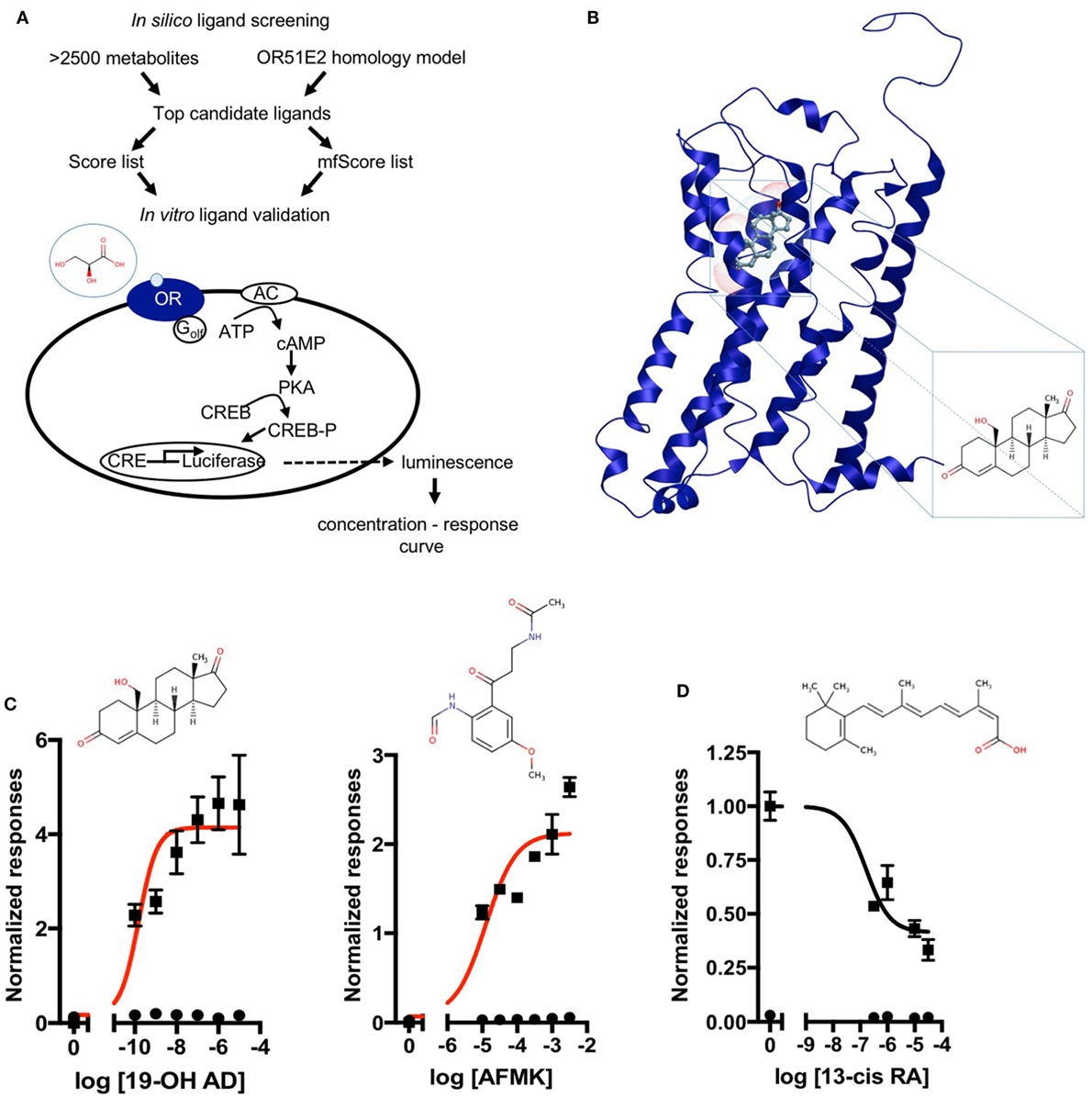
Figure 1. Discovery of novel endogenous metabolite-ligands for OR51E2. (A) Study design. (B) Homology model of OR51E2 with 19-hydroxyandrost-4-ene-3,17-dione (19-OH AD) docked into the receptor pocket. (C) Concentration–response curves for 19-OH AD and acetyl-N-formyl-5-methoxykynurenamine (AFMK) (red) and their structures. Responses are obtained in the M10d medium and normalized to a no-metabolite control (0 = no metabolite stimulation). (D) Concentration–response curve for antagonist 13-cis retinoic acid (13-cis RA) obtained in the CD293 medium. Response is normalized to a no-metabolite control (1 = no metabolite stimulation) (N = 3, mean ± SEM).
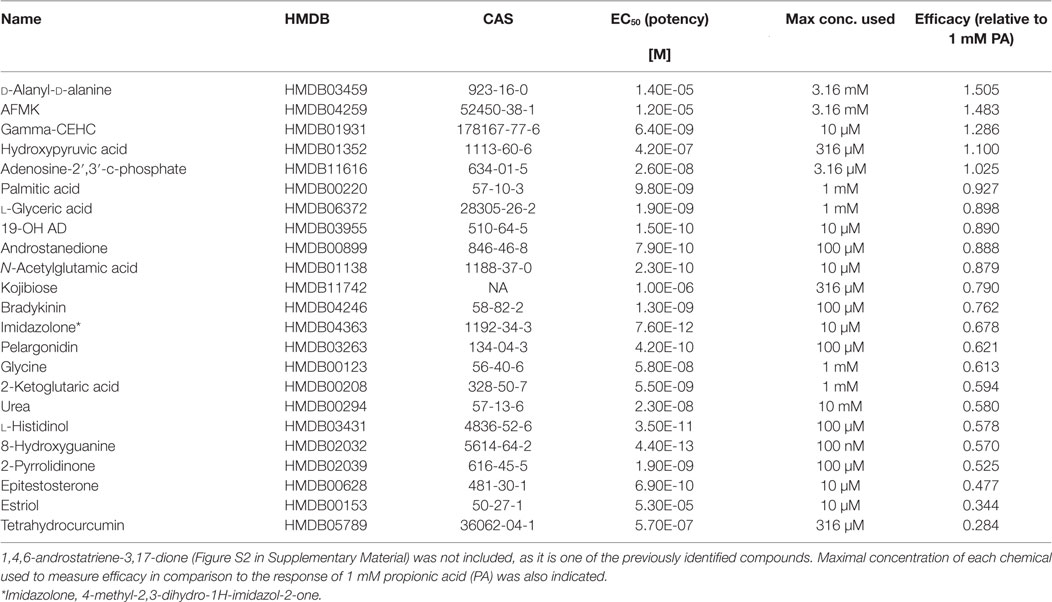
Table 1. Potency and efficacy of 24 newly discovered OR51E2 agonists with their associated HMDB and CAS identifiers.
Some of the newly discovered agonists are: 19-hydroxyandrostenedione (19-OH AD), a hypertensive steroid (vasopressor) secreted by the adrenal gland (50–52), an intermediate in estrogen synthesis from testosterone (53) also found in porcine testes (54), and rat ovarian granulosa cells (55).
Acetyl-N-formyl-5-methoxykynurenamine, a melatonin and kynurenamine metabolite, was previously reported to be abundant in aggressive PC (56); estradiol (57); adenosine-2,3-cyclic phosphate; 8-hydroxyguanine; α-ketoglutaric acid; urea; glycine; and palmitic acid.
Because in situ estrogen production is an important factor in prostate carcinogenesis, and since the expression of aromatase, the enzyme that synthetizes estrogens from androgens, is increased 30-fold in PC, we decided to further examine the presence and production of 19-OH AD in LNCaP cells (58, 59).
Endogenous 19-OH AD Production Upon OR51E2 Activation With AFMK
We have developed a liquid chromatography/mass spectrometry (LC-MS/MS) assay for the measurements of 19-OH AD in the cell media. LNCaP cells were stimulated with newly discovered agonist, 250 µM AFMK, for 3 days, and the 19-OH AD was measured in the cell medium (Figure 2). We used CD-phenol free RPMI1640 medium (Figure 2A) and regular RPMI1640 medium (Figure 2B) to estimate the production of 19-OH AD. Three times more 19-OH AD was detected when cells were stimulated in CD-phenol free medium (0.83 vs. 0.27 ng/mL). No 19-OH AD was detected in unstimulated cells (dotted lines in Figures 2A,B).
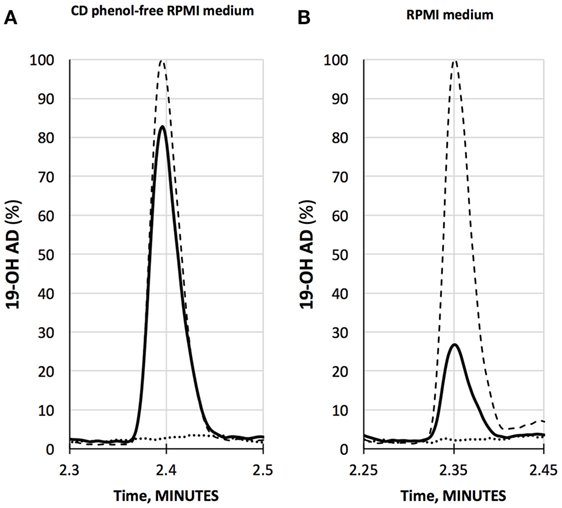
Figure 2. Quantification of 19-OH AD by LC-MS/MS in the medium from AFMK-stimulated LNCaP cells. (A) CD phenol-free RPMI medium with 250 μM acetyl-N-formyl-5-methoxykynurenamine (AFMK) (solid line), 1 ng/mL of 19-OH AD- standard spike (dashed line); estimated concentration of 19-OH AD is 0.83 ng/mL medium. (B) RPMI medium + 250 μM AFMK (solid line), 1 ng/mL 19-OH AD of standard spike (dashed line); estimated concentration of 19-OH AD is 0.27 ng/mL. Dotted lines at the bottom in A and B are media only, with no AFMK. Curves are normalized to 1 ng/mL 19-OH AD.
Metabolomic Signatures of LNCaP Cells Treated With Selected OR51E2 Agonists
In addition to the newly discovered agonists 19-OH AD and AFMK, we also selected the previously identified OR51E2 agonist PA for metabolomic analysis (34). The cells were incubated with 100 nM 19-OH AD, 250 µM AFMK, and 1 mM PA for 72 h, and non-targeted metabolomics analysis was performed to identify metabolites that differed significantly in the cell lysate and conditioned medium (CM). Agonist treatment resulted in pronounced intra- and extra-cellular changes in metabolomic signatures, as seen in heat maps (Figures S5 and S6 in Supplementary Material, respectively). Differentially expressed features/metabolites identified in each group using the t-test (P < 0.05) and fold change (2-fold or greater) are presented in Figures 4A–E; Figures S5–S10 and Tables S5–S10 in Supplementary Material.
The top 15 differentially expressed extracellular metabolites are presented in Figures 3A–C. All three agonists also produced robust intracellular decreases in amino acids, especially serine and threonine, and also two glycolytic intermediates: glucose-6-P and fructose-6-P (Figures 3D–F). Furthermore, agonist treatment resulted in decreases in both methionine and glycine levels. In addition to decreased lactic acid, significant decreases were noted in fumaric, malic, and succinic acids, all intermediates of the TCA. Interestingly, we detected an increased level of phosphoenolpyruvate (PEP) in all agonist-treated samples (fold change analysis, Figures S7B, S8B and S9B in Supplementary Material). We also observed decreased levels of myoinositol, inosine, adenosine, asparagine, aspartate, and guanosine, which have been previously reported as being depleted in metastatic PC tissue (47), indicating that activation of OR51E2 by agonists in LNCaP cells produces metabolic signatures similar to those observed in human metastatic tissues. Agonist treatment also reduced levels of intracellular urea, spermine/spermidine, and ornithine.
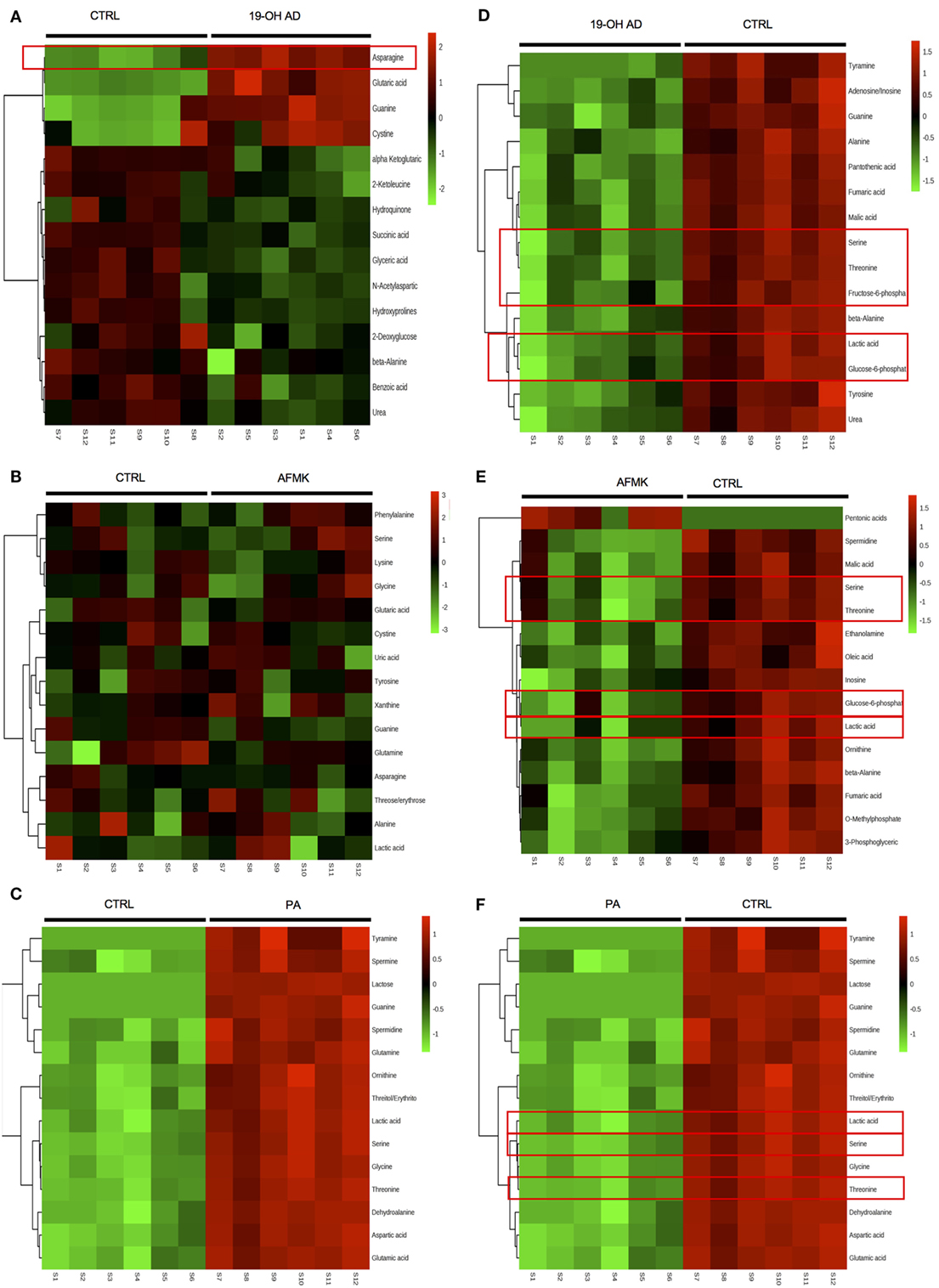
Figure 3. Agonist treatment of LNCaP cells results in robust metabolomic signatures. The top 15 extracellular metabolites were identified after stimulation with (A) 19-OH AD, (B) acetyl-N-formyl-5-methoxykynurenamine (AFMK), and (C) propionic acid (PA). The top 15 intracellular metabolites were identified after stimulation with (D) 19-OH AD, (E) AFMK, and (F) PA. Heatmaps are based on the Pearson correlation analysis (Ward) and indicate annotated metabolites identified by t-test (P < 0.05, FDR < 0.1, n = 6). Columns correspond to the samples treated with agonists (S1-6) and control (S7-12), and rows correspond to annotated metabolites.
Fold change analysis of the CM revealed significantly increased glutamine (25-, 11-, and 51-fold, for 19-OH AD, AFMK, and PA, respectively), indicating that agonist-treated cells do not have an increased demand for glutamine as highly proliferating cancer cells usually do (Figures S10B, S11B, and S12B in Supplementary Material). Thus, these results argue for a non- or a low-proliferative phenotype. Decreased levels of docosanoic and decanoic acid and increased levels of asparagine were also prominent in the CM in all three treatments (Figure 4G). The pathways most affected, as identified by MetaboAnalyst, were the serine, threonine, and glycine; alanine, aspartate, and glutamine; ketogenesis; arginine and proline; and beta-alanine metabolic pathways (Figure 4F).
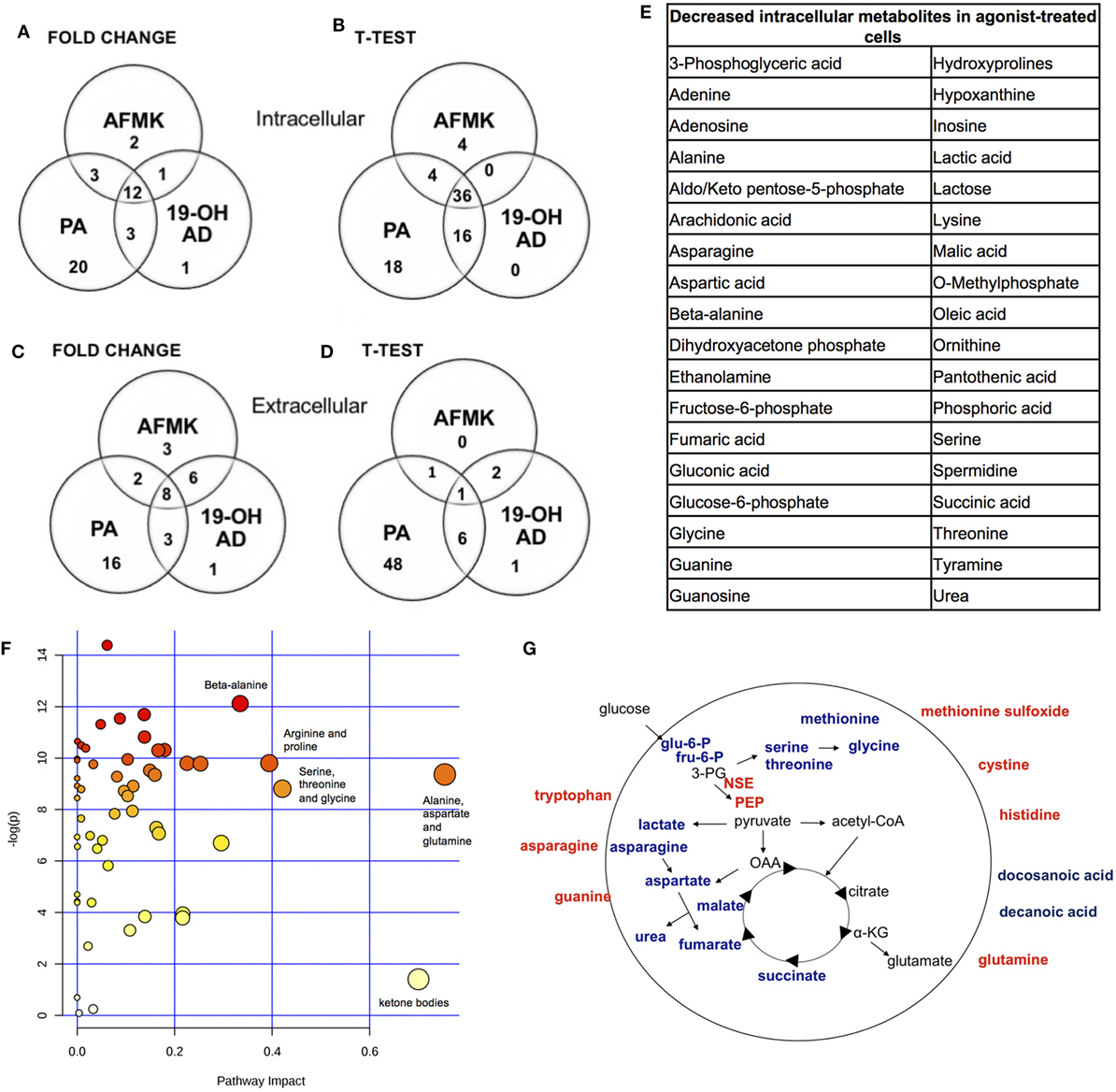
Figure 4. Most pronounced metabolic changes induced by activation of OR51E2 with selected agonists. Venn diagrams show the set of annotated intracellular metabolites differentially expressed by (A) fold change and (B) t-test analysis, and the set of extracellular metabolites identified by (C) fold change and (D) t-test. (E) In total, 36 annotated intracellular metabolites significantly decreased in all three treatments (see Figure 3B). (F) Pathway analysis. The most pronounced pathways in 19-OH AD-treated cells. Pathways are displayed as circles, and the color and size of each circle are based on its P value and pathway impact value, respectively. The top-right area indicates the most significant changes in metabolites. (G) Schematic representation of cellular metabolites. Significantly increased metabolites are in bold red, and significantly decreased are in bold blue (PEP, phosphoenol pyruvate; OAA, oxalacetate; 3PG, 3-phosphoglycerate; α-KG, α-ketoglutarate). Increased neuron-specific enolase (NSE) catalyzes the formation of PEP and is also indicated in red.
Time-Dependent Modulation of Cellular Proliferation
Our metabolomics results indicated reduced capacity for anabolic reactions in LNCaP cells following receptor activation, which prompted us to further analyze the effects of OR51E2 agonists on cellular proliferation. Moreover, because melatonin reduces cell proliferation, we examined whether AFMK, a melatonin metabolite, also reduces or inhibits cell proliferation (60). LNCaP cells were stimulated with various concentrations of 19-OH AD and AFMK for 4 days and analyzed every 24 h using an ATP viability assay. At day 4, both agonists significantly decreased the number of viable cells when compared to the control, non-stimulated cells (Figure 5A).
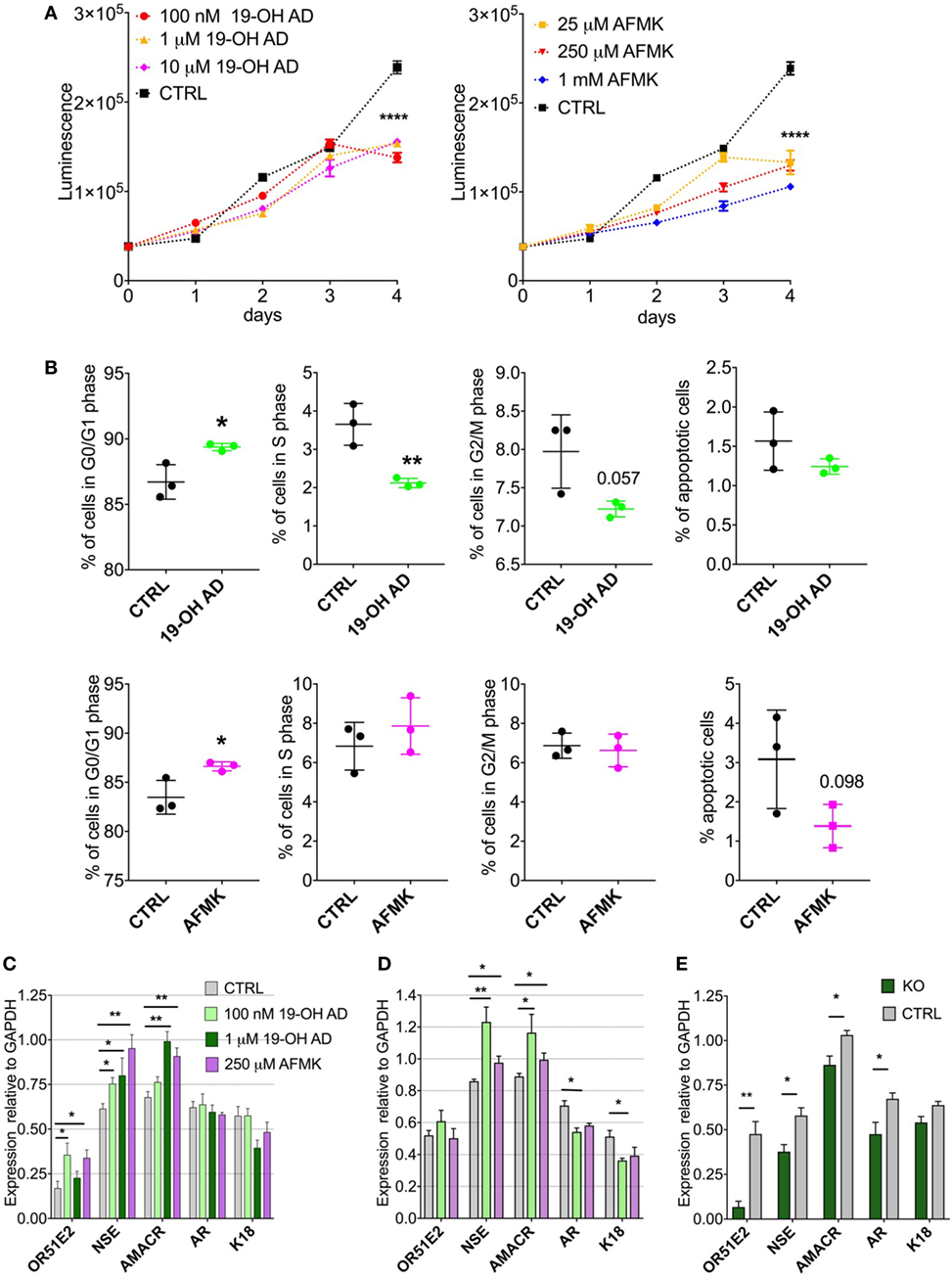
Figure 5. Activation of OR51E2 receptor by selected agonists induces a neuroendocrine phenotype. (A) Cell viability assay at various indicated concentrations of 19-OH AD and acetyl-N-formyl-5-methoxykynurenamine (AFMK). Cell viability correlates with luminescence signal. Statistical significance at day 4 with 100 nM 19-OH AD and 250 µM AFMK is ****P < 0.0001. (B) Cell cycle analysis after incubation with 100 nM 19-OH AD and 250 µM AFMK for 7 and 3 days, respectively. (C) Transcript levels of markers after agonist stimulation for 3 days; N = 3–6, unpaired t-test, **P < 0.01. *P < 0.05. (D) Transcript levels of makers after stimulation with agonists for 12 days, N = 3–6, unpaired t-test, **P < 0.01. *P < 0.05. (E) Transcript levels of markers relative to GAPDH after 3 days of stimulation with 1 µM 19-OH AD in OR51E2-KO LNCaP cells. N = 4, mean ± SEM. **P < 0.01. *P < 0.05.
Cell Cycle Arrest
Next, we determined if the decrease in cell viability during agonist stimulation is attributable to increased apoptosis or cell-cycle arrest. Cells were treated with various concentrations of agonists for 3 and 7 days. Both agonists increased the fraction of cells in the G0/G1 phase and decreased the number of apoptotic cells (Figure 5B). Our results are in agreement with previous reports that the majority of cells with a neuroendocrine-like phenotype show signs of resistance to apoptosis (61).
Neuroendocrine Markers
To assess the effect of selected OR51E2 agonists on NEtD, we analyzed transcript levels after 3 and 12 days of stimulation of the following neuroendocrine, epithelial, and receptor genes: NSE; AMACR; keratins 5, 8, and 18; voltage-gated Ca channel α1 H (Cav3.2); AR; and OR51E2 receptor. NSE was used to specifically identify NEtD status, and although LNCaP (being a cell line established from lymph-node metastatasis) cells already express low levels of NSE, treatment with OR51E2 agonists significantly increase levels of the NSE transcript (Figures 5C,D; Figure S13A in Supplementary Material). These results correlate well with the increased intracellular level of PEP detected in the metabolomic analysis (Figures S7B, S8B and S9B in Supplementary Material), since the glycolytic enzyme enolase catalyzes PEP synthesis. AMACR is an enzyme essential for isomerization of branched-chained fatty acids and is present at very low levels in healthy prostate and increased in PC and NE-like cells (62). Since NE-like tumor cells express AMACR, we investigated whether activation of OR51E2 also increased AMACR levels. Indeed, AMACR levels were significantly increased after 3 and 12 days of agonist stimulation (Figures 5C,D, and S13A).
Furthermore, 19-OH AD decreased the AR transcript after 12 days, and although AFMK also showed a similar trend, it did not reach statistical significance (Figure 5D). Normal basal prostate epithelial cells are positive for K5, and expression of K5 is also associated with the epithelial-to-mesenchymal transition during tumor progression and metastasis (63). K5 and K8 were not detected in the agonist-stimulated cells (data not shown). A significant decrease in K18, a luminal secretory marker, was found after 12 days of stimulation with 100 nM 19-OH AD (Figure 5D). Although Ca+2 entry through the voltage-gated calcium channel α1 H (Cav3.2) was previously reported to be involved in NEtD of LNCaP cells when cultured in steroid-free conditions, we did not detect changes in its transcript levels (data not shown) (64).
OR51E2 Knock-Out Confirms NE-Like Phenotype Upon Receptor Activation
To confirm receptor involvement in the agonist-mediated increase of NSE, OR51E2 was deleted using a CRISPR-Cas9 method (Figure 5E). We designed three gRNAs to target Cas9 to the OR51E2-gene and generated a lentiviral sgRNA-Cas9 vector to deliver gRNA into the cells. The efficiency of each gRNA was measured, and we observed that by using sgRNA #1, OR51E2 was abrogated in 80% of cells. These OR51E2-knockout cells were exposed to 1 µM 19-OH AD for 3 days and analyzed for the presence of specific markers (Figure 5E; Figure S13C in Supplementary Material). The summary of four independent experiments is presented in Figure 5E. Expression of markers is normalized to GAPDH levels. Representative gel is presented in S13B. A statistically significant decrease of NSE in OR51E2-knockout cells in comparison with control cells was observed (from 0.579 ± 0.043 in control to 0.377 ± 0.04, P < 0.05, n = 4 biological replicates, Figure 5E), confirming that increased NSE during stimulation with agonists is at least partially a receptor-mediated phenomenon (Figure 5E; Figure S13C in Supplementary Material).
Discussion
As the number of ectopic olfactory receptors associated with diverse pathological states continues to increase, the implications and significance of these receptors will be greatly enhanced by receptor “deorphanization” (i.e., defining the ligands). Previously, we successfully identified novel ligands for mouse OR using a similar in silico approach with VLS (38). Here, we present a highly successful approach of combining in silico and in vitro analyses to identify novel biologically relevant ligands for the human ectopic OR, OR51E2. This method can be used to elucidate ligand specificities of other ectopic ORs. Once identified, these new ligands can help define the role and function of ORs in cancer and other diseases.
Among the newly discovered metabolites identified as OR51E2 agonists, several were previously reported to be associated with PC, including bradykinin, kojibiose, glycylglycine, N-acetylglutamic acid, and d-alanyl-d-alanine (47). Thus, our results indicate that these metabolites are likely endogenous agonists. New agonists with known biological roles were also discovered: epitestosterone, known to be a major metabolite of androstenedione and testosterone (65) and androstanedione (also known as 5α-androstane-3,17-dione), an intermediate in steroid synthesis (66).
In addition to these agonists, we also identified a previously under-reported metabolite of the complex steroid biosynthetic network, 19-hydroxyandrost-4-ene-3,17-dione (19-OH AD) (53, 67, 68). It is produced by aromatase P450 (CYP19A1), which catalyzes the irreversible aromatization of the androgens androst-4-ene-3,17-dione and testosterone and their consequent conversion to estrogens.4 We detected this testosterone metabolite in agonist-stimulated prostate cancer cells. These results demonstrate that 19-OH AD is actively produced by cancer cells when the OR51E2 receptor is activated. Thus, we demonstrate that 19-OH AD is an endogenous agonist produced by activation of OR51E2 in prostate cancer cells.
Aromatase is increased 30-fold in metastatic PC (59), and aromatase-knockout mice have a reduced incidence of PC following exposure to testosterone and estrogen, indicating that aromatase metabolites, mainly 19-OH AD and estradiol, are likely involved in prostate carcinogenesis. Results from our study demonstrate that 19-OH AD is a potent OR51E2 agonist (EC50 = 1.5−10 M) and support the notion that increased in situ estrogen production via 19-OH AD is an important factor in PC (58).
Acetyl-N-formyl-5-methoxykynurenamine is a metabolite of melatonin (69). Previous studies demonstrated that melatonin reduces proliferation of LNCaP cells, leading to NEtD, and the phenotype was not reversed by melatonin receptor antagonists, suggesting that additional receptors may be mediating this process (60). An additional source of AFMK might be a tryptophan metabolic pathway (70). Tumors produce high levels of tryptophan and kynurenic acid metabolites (71). Significant amplification of tryptophan-2,3-dioxygenase (EC 1.13.11.11) TDO2, which catalyzes the oxidation of l-tryptophan to N-formyl-l-kynurenamine, was observed in NEPC and PC (72, 73). Thus, in more advanced stages of PC, AFMK production may be increased via this tryptophan metabolic pathway.
We also identified bradykinin as an agonist for OR51E2. Prostatic secretions of PC patients have elevated levels of human kallikrein 2 (74), which produces bradykinin and thus stimulates proliferation of androgen-independent PC cells in later stages of PC (75).
The OR51E2 antagonist 13-cis RA is an endogenous component of human serum, and many of its actions can be explained by isomerization to all-trans RA and 9-cis RA, which both act via retinoid receptors. However, since 13-cis RA does not have potent gene regulatory activities, additional pathways via membrane receptors have been proposed to explain its pharmacological and anti-inflammatory actions (76). Our results demonstrate that 13-cis RA acts via the OR51E2 receptor when expressed heterologously.
OR51E2 receptor activation by 19-OH AD, AFMK, and PA induced pronounced metabolic reprograming of LNCaP cells, with the most significant changes being decreased intracellular serine and threonine levels. Because metabolism of these amino acids includes one-carbon metabolism, which provides cofactors for biosynthetic reactions in highly proliferating cells, intracellular depletion may indicate a general decrease in anabolic reactions (77). Furthermore, an intracellular decrease in aspartate, which is normally required for protein, purine, and pyrimidine synthesis, and an increase in the CM indicate that agonist-activated LNCaP cells are not preparing for proliferation. We also detected decreased intermediates of glycolysis (glucose-6-phosphate and fructose-6-phosphate) in activated cells. Agonist treatment decreased intracellular lactate, suggesting a slower rate of glycolysis. An intriguing result was the increased intracellular level of PEP. We also found increased NSE transcription for the glycolytic enzyme enolase, which catalyzes the formation of PEP, indicating predominance of the PEP-forming reaction.
Neuron-specific enolase is not only a marker of neuronal differentiation and maturation characteristic of neurons and neuroendocrine cells (78), but it also has an important role in synaptogenesis (79) and is reported to be stable in the high-chloride environment characteristic of neurons, in which it reaches a concentration of 2% to 3% of the soluble protein (80). Taken together, these results indicate that receptor activation results in a neuronal-like phenotype of LNCaP cells.
Cystine was increased in the medium after 19-OH AD and AFMK treatments (Tables S12 and S13 in Supplementary Material). In healthy cells, cystine is transported into cells and reduced to cysteine, which can then be utilized for synthesis of gluthatione, a protective antioxidant. As a consequence of rapid cell growth during tumorigenesis, the production of reactive oxygen species increases, providing a proliferative signal for glutamine to enter the cell and, after deamidation, condense with cysteine to form a precursor of glutathione. However, in our experiments, the medium, but not the cells, showed increased levels of glutamine and cysteine, indicating a reduction in protective oxidative and proliferative signals in agonist-stimulated cells. The alanine/aspartate/glutamine pathway is the most affected biochemical pathway during NEtD of LNCaP cells induced by steroid-reduced medium, which corroborates our results (Figure 4F) (81). In cancer-associated fibroblasts, asparagine and aspartate are involved in glutamine synthesis (82), and our experiments showed decreased intracellular levels of these amino acids, suggesting increased use for intracellular synthesis of glutamine. These results might also indicate a decreased cellular influx of asparagine, since it is abundant in CM. Flux studies will be necessary to determine the exact relationship between glutamine synthesis and transport in PC cells upon receptor activation.
To explain the role of OR51E2 in PC, we propose the following model: agonist stimulation generates new cells through asymmetric division and gradually increases the subpopulation of terminally differentiated cells expressing neuroendocrine markers.
NE-like cells from PC are characterized by increased expression of NSE and AMACR and decreased expression of K18 and AR (83). Increased expression of NSE and AMARC and decreased expression of AR and K18 following 19-OH AD and AFMK treatment demonstrate that these OR51E2 agonists induce a neuroendocrine phenotype. We confirmed that this effect is indeed receptor-mediated, as treatment of OR51E2-knockout LNCaP cells significantly reduced the NSE and AMACR transcript levels (Figure 5; Figure S13 in Supplementary Material).
Cell proliferation and differentiation have an inverse relationship, and terminal differentiation coincides with proliferation arrest and exit from the division cycle. Our results demonstrate that agonist treatment during the first 3 days induces cell proliferation at a rate similar to control cells, but after 4 days the viability of these cells, as measured by ATP content, was significantly reduced. Our results also demonstrate that receptor activation results in a new subpopulation of cells that undergoes G0/G1 cell cycle arrest and has decreased DNA synthesis, which is concordant with the results from our metabolomics analysis. Cellular senescence is an irreversible growth arrest, and senescent cells actively suppress apoptosis. We found that agonist treatment decreases the fraction of apoptotic cells, indicating that growth arrest likely induces cellular senescence. Future studies are needed to confirm the irreversibility of this process.
Furthermore, recent whole-exome sequencing of NEPC and CRPC showed an overlap in genomic alterations, and in both demonstrated increased amplification of the OR51E2 gene, supporting our hypothesis that this receptor contributes to the NE-phenotype of PC (72, 73).
Prostatic adenocarcinomas typically contain foci of non-proliferating NE-like cells that increase in number as cancer progresses (84). Although these cells are non-mitotic, proliferating carcinoma cells have been found in their proximity, suggesting that the non-proliferating NE-like cells likely provide paracrine stimuli for growth of the surrounding carcinoma cells. Our results demonstrate that activation of OR51E2 by newly discovered PC-relevant agonists induces and/or facilitates cellular transformation, resulting in NEtD, a characteristic phenotype of CRCP. This indicates that activation of OR51E2 in PC might contribute to development of non-proliferating foci. Our data demonstrate that activation of OR51E2 results in NEtD and establish this GPCR as a novel and therapeutic target for NEPC and CRPC.
Author Contributions
Conceptualization: TA; methodology: TA, JB, MM, IS, EB, and SK; investigation: TA, JB, MM, SK, IS, and SL; writing: TA; writing, review, and editing: TA, JB, MM, SL, and HM; resources: HM; supervision: TA and HM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would also like to thank Professors J. Heitman, J. Huang, M. P. Dewhirst and J. A. Chi, and Drs. C. de March and T. Zhou for their critical review on the manuscript. We thank Mengjue Ni for expert technical assistance. This study was supported by grants from NIH (DC14423 and DC016224). The authors declare no conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fonc.2018.00162/full#supplementary-material.
Footnotes
- ^http://www.mbio.ncsu.edu/BioEdit/bioedit.html (Accessed: May 15, 2018).
- ^https://salilab.org/modeller/ (Accessed: May 15, 2018).
- ^www.HMDB.ca (Accessed: May 15, 2018).
- ^http://www.brenda-enzymes.org/enzyme.php?ecno=1.14.14.14 (Accessed: May 15, 2018).
References
1. Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer (2007) 7(2):79–94. doi:10.1038/nrc2069
2. Julius D, Livelli TJ, Jessell TM, Axel R. Ectopic expression of the serotonin 1c receptor and the triggering of malignant transformation. Science (1989) 244(4908):1057–62. doi:10.1126/science.2727693
3. Gutkind JS, Novotny EA, Brann MR, Robbins KC. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc Natl Acad Sci U S A (1991) 88(11):4703–7. doi:10.1073/pnas.88.11.4703
4. Allen LF, Lefkowitz RJ, Caron MG, Cotecchia S. G-protein-coupled receptor genes as protooncogenes: constitutively activating mutation of the alpha 1B-adrenergic receptor enhances mitogenesis and tumorigenicity. Proc Natl Acad Sci U S A (1991) 88(24):11354–8. doi:10.1073/pnas.88.24.11354
5. Gutkind JS. Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene (1998) 17(11 Reviews):1331–42. doi:10.1038/sj.onc.1202186
6. O’Hayre M, Degese MS, Gutkind JS. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol (2014) 27:126–35. doi:10.1016/j.ceb.2014.01.005
7. Abaffy T. Human olfactory receptors expression and their role in non-olfactory tissues – a mini review. J Pharmacogenomics Pharmacoproteomics (2015) 6(4):152. doi:10.4172/2153-0645.1000152
8. Spehr M, Gisselmann G, Poplawski A, Riffell JA, Wetzel CH, Zimmer RK, et al. Identification of a testicular odorant receptor mediating human sperm chemotaxis. Science (2003) 299(5615):2054–8. doi:10.1126/science.1080376
9. Pavlath GK. A new function for odorant receptors: MOR23 is necessary for normal tissue repair in skeletal muscle. Cell Adh Migr (2010) 4(4):502–6. doi:10.4161/cam.4.4.12291
10. Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A (2013) 110(11):4410–5. doi:10.1073/pnas.1215927110
11. Chang AJ, Ortega FE, Riegler J, Madison DV, Krasnow MA. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature (2015) 527(7577):240–4. doi:10.1038/nature15721
12. Flegel C, Manteniotis S, Osthold S, Hatt H, Gisselmann G. Expression profile of ectopic olfactory receptors determined by deep sequencing. PLoS One (2013) 8(2):e55368. doi:10.1371/journal.pone.0055368
13. Niimura Y, Matsui A, Touhara K. Extreme expansion of the olfactory receptor gene repertoire in African elephants and evolutionary dynamics of orthologous gene groups in 13 placental mammals. Genome Res (2014) 24(9):1485–96. doi:10.1101/gr.169532.113
14. Xu LL, Stackhouse BG, Florence K, Zhang W, Shanmugam N, Sesterhenn IA, et al. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res (2000) 60(23):6568–72.
15. Xu LL, Sun C, Petrovics G, Makarem M, Furusato B, Zhang W, et al. Quantitative expression profile of PSGR in prostate cancer. Prostate Cancer Prostatic Dis (2006) 9(1):56–61. doi:10.1038/sj.pcan.4500836
16. Weng J, Wang J, Cai Y, Stafford LJ, Mitchell D, Ittmann M, et al. Increased expression of prostate-specific G-protein-coupled receptor in human prostate intraepithelial neoplasia and prostate cancers. Int J Cancer (2005) 113(5):811–8. doi:10.1002/ijc.20635
17. Xia C, Ma W, Wang F, Hua SB, Liu M. Identification of a prostate-specific G-protein coupled receptor in prostate cancer. Oncogene (2001) 20(41):5903–7. doi:10.1038/sj.onc.1204803
18. Vaarala MH, Hirvikoski P, Kauppila S, Paavonen TK. Identification of androgen-regulated genes in human prostate. Mol Med Rep (2012) 6(3):466–72. doi:10.3892/mmr.2012.956
19. Ashida S, Nakagawa H, Katagiri T, Furihata M, Iiizumi M, Anazawa Y, et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res (2004) 64(17):5963–72. doi:10.1158/0008-5472.CAN-04-0020
20. Cunha AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens: comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett (2006) 236(2):229–38. doi:10.1016/j.canlet.2005.05.021
21. Weigle B, Fuessel S, Ebner R, Temme A, Schmitz M, Schwind S, et al. D-GPCR: a novel putative G protein-coupled receptor overexpressed in prostate cancer and prostate. Biochem Biophys Res Commun (2004) 322(1):239–49. doi:10.1016/j.bbrc.2004.07.106
22. Rodriguez M, Siwko S, Liu M. Prostate-specific G-protein coupled receptor, an emerging biomarker regulating inflammation and prostate cancer invasion. Curr Mol Med (2016) 16(6):526–32. doi:10.2174/1566524016666160607091333
23. Hu CD, Choo R, Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Front Oncol (2015) 5:90. doi:10.3389/fonc.2015.00090
24. Komiya A, Suzuki H, Imamoto T, Kamiya N, Nihei N, Naya Y, et al. Neuroendocrine differentiation in the progression of prostate cancer. Int J Urol (2009) 16(1):37–44. doi:10.1111/j.1442-2042.2008.02175.x
25. Farach A, Ding Y, Lee M, Creighton C, Delk NA, Ittmann M, et al. Neuronal trans-differentiation in prostate cancer cells. Prostate (2016) 76(14):1312–25. doi:10.1002/pros.23221
26. Firestein S. How the olfactory system makes sense of scents. Nature (2001) 413(6852):211–8. doi:10.1038/35093026
27. Massberg D, Simon A, Haussinger D, Keitel V, Gisselmann G, Conrad H, et al. Monoterpene (-)-citronellal affects hepatocarcinoma cell signaling via an olfactory receptor. Arch Biochem Biophys (2015) 566:100–9. doi:10.1016/j.abb.2014.12.004
28. Manteniotis S, Wojcik S, Brauhoff P, Mollmann M, Petersen L, Gothert JR, et al. Functional characterization of the ectopically expressed olfactory receptor 2AT4 in human myelogenous leukemia. Cell Death Discov (2016) 2:15070. doi:10.1038/cddiscovery.2015.70
29. Gelis L, Jovancevic N, Veitinger S, Mandal B, Arndt HD, Neuhaus EM, et al. Functional characterization of the odorant receptor 51E2 in human melanocytes. J Biol Chem (2016) 291(34):17772–86. doi:10.1074/jbc.M116.734517
30. Rodriguez M, Siwko S, Zeng L, Li J, Yi Z, Liu M. Prostate-specific G-protein-coupled receptor collaborates with loss of PTEN to promote prostate cancer progression. Oncogene (2016) 35(9):1153–62. doi:10.1038/onc.2015.170
31. Neuhaus EM, Zhang W, Gelis L, Deng Y, Noldus J, Hatt H. Activation of an olfactory receptor inhibits proliferation of prostate cancer cells. J Biol Chem (2009) 284(24):16218–25. doi:10.1074/jbc.M109.012096
32. Sanz G, Leray I, Dewaele A, Sobilo J, Lerondel S, Bouet S, et al. Promotion of cancer cell invasiveness and metastasis emergence caused by olfactory receptor stimulation. PLoS One (2014) 9(1):e85110. doi:10.1371/journal.pone.0085110
33. Cao W, Li F, Yao J, Yu J. Prostate specific G protein coupled receptor is associated with prostate cancer prognosis and affects cancer cell proliferation and invasion. BMC Cancer (2015) 15:915. doi:10.1186/s12885-015-1921-6
34. Saito H, Chi Q, Zhuang H, Matsunami H, Mainland JD. Odor coding by a mammalian receptor repertoire. Sci Signal (2009) 2(60):ra9. doi:10.1126/scisignal.2000016
35. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol (1993) 234(3):779–815. doi:10.1006/jmbi.1993.1626
36. Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci (2006) 15(11):2507–24. doi:10.1110/ps.062416606
37. Abagyan R, Totrov M, Kuznetsov D. ICM-A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem (1994) 15(5):488–506. doi:10.1002/jcc.540150503
38. Bavan S, Sherman B, Luetje CW, Abaffy T. Discovery of novel ligands for mouse olfactory receptor MOR42-3 using an in silico screening approach and in vitro validation. PLoS One (2014) 9(3):e92064. doi:10.1371/journal.pone.0092064
39. Abaffy T, Malhotra A, Luetje CW. The molecular basis for ligand specificity in a mouse olfactory receptor – a network of functionally important residues. J Biol Chem (2007) 282(2):1216–24. doi:10.1074/jbc.M609355200
40. Neves MAC, Totrov M, Abagyan R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des (2012) 26(6):675–86. doi:10.1007/s10822-012-9547-0
41. Rege J, Nakamura Y, Satoh F, Morimoto R, Kennedy MR, Layman LC, et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. J Clin Endocrinol Metab (2013) 98(3):1182–8. doi:10.1210/jc.2012-2912
42. Halket JM, Przyborowska A, Stein SE, Mallard WG, Down S, Chalmers RA. Deconvolution gas chromatography/mass spectrometry of urinary organic acids – potential for pattern recognition and automated identification of metabolic disorders. Rapid Commun Mass Spectrom (1999) 13(4):279–84. doi:10.1002/(SICI)1097-0231(19990228)13:4<279::AID-RCM478>3.0.CO;2-I
43. Kind T, Wohlgemuth G, Lee DY, Lu Y, Palazoglu M, Shahbaz S, et al. FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal Chem (2009) 81(24):10038–48. doi:10.1021/ac9019522
44. Xia J, Wishart DS. Using metaboanalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformatics (2016) 55:14.10.1–14.10.91. doi:10.1002/cpbi.11
45. Zhuang H, Matsunami H. Evaluating cell-surface expression and measuring activation of mammalian odorant receptors in heterologous cells. Nat Protoc (2008) 3(9):1402–13. doi:10.1038/nprot.2008.120
46. Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, et al. HMDB: the human metabolome database. Nucleic Acids Res (2007) 35(Database issue):D521–6. doi:10.1093/nar/gkl923
47. Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature (2009) 457(7231):910–4. doi:10.1038/nature07762
48. Key TJ, Appleby PN, Allen NE, Travis RC, Roddam AW, Jenab M, et al. Plasma carotenoids, retinol, and tocopherols and the risk of prostate cancer in the European prospective investigation into cancer and nutrition study. Am J Clin Nutr (2007) 86(3):672–81. doi:10.1093/ajcn/86.3.672
49. Nyman DW, Suzanne Stratton M, Kopplin MJ, Dalkin BL, Nagle RB, Jay Gandolfi A. Selenium and selenomethionine levels in prostate cancer patients. Cancer Detect Prev (2004) 28(1):8–16. doi:10.1016/j.cdp.2003.11.002
50. Sekihara H. 19-Hydroxyandrostenedione: evidence for a new class of sodium-retaining and hypertensinogenic steroids. Endocrinology (1983) 113(3):1141–8. doi:10.1210/endo-113-3-1141
51. Sekihara H. 19-Hydroxyandrostenedione: a potent hypertensinogenic steroid in man. J Steroid Biochem (1983) 19(1A):353–8. doi:10.1016/S0022-4731(83)80047-8
52. Sekihara H. Evidence that 19-hydroxyandrostenedione is secreted by the adrenal cortex and is under the control of ACTH and the renin-angiotensin system in man. Biochem Biophys Res Commun (1982) 105(2):610–4. doi:10.1016/0006-291X(82)91478-4
53. Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman MR, Kitagawa T. Raman evidence for specific substrate-induced structural changes in the heme pocket of human cytochrome P450 aromatase during the three consecutive oxygen activation steps. Biochemistry (2006) 45(17):5631–40. doi:10.1021/bi060094a
54. Raeside JI, Renaud RL, Friendship RM, Khalil MW. Secretion of 19-hydroxyandrostenedione and 19-hydroxytestosterone by porcine Leydig cells in vitro and in vivo. J Endocrinol (1993) 137(2):281–9. doi:10.1677/joe.0.1370281
55. Norton BI, Miyairi S, Fishman J. 19-Hydroxylation of androgens by rat granulosa cells. Endocrinology (1988) 122(3):1047–52. doi:10.1210/endo-122-3-1047
56. McDunn JE, Li Z, Adam KP, Neri BP, Wolfert RL, Milburn MV, et al. Metabolomic signatures of aggressive prostate cancer. Prostate (2013) 73(14):1547–60. doi:10.1002/pros.22704
57. Carruba G. Estrogen and prostate cancer: an eclipsed truth in an androgen-dominated scenario. J Cell Biochem (2007) 102(4):899–911. doi:10.1002/jcb.21529
58. Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, Risbridger GP. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J (2008) 22(5):1512–20. doi:10.1096/fj.07-9526com
59. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res (2008) 68(11):4447–54. doi:10.1158/0008-5472.CAN-08-0249
60. Sainz RM, Mayo JC, Tan DX, Leon J, Manchester L, Reiter RJ. Melatonin reduces prostate cancer cell growth leading to neuroendocrine differentiation via a receptor and PKA independent mechanism. Prostate (2005) 63(1):29–43. doi:10.1002/pros.20155
61. Fixemer T, Remberger K, Bonkhoff H. Apoptosis resistance of neuroendocrine phenotypes in prostatic adenocarcinoma. Prostate (2002) 53(2):118–23. doi:10.1002/pros.10133
62. Huang J, Yao JL, di Sant’Agnese PA, Yang Q, Bourne PA, Na Y. Immunohistochemical characterization of neuroendocrine cells in prostate cancer. Prostate (2006) 66(13):1399–406. doi:10.1002/pros.20434
63. Verhagen AP, Aalders TW, Ramaekers FC, Debruyne FM, Schalken JA. Differential expression of keratins in the basal and luminal compartments of rat prostatic epithelium during degeneration and regeneration. Prostate (1988) 13(1):25–38. doi:10.1002/pros.2990130104
64. Mariot P, Vanoverberghe K, Lalevee N, Rossier MF, Prevarskaya N. Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J Biol Chem (2002) 277(13):10824–33. doi:10.1074/jbc.M108754200
65. Starka L. Epitestosterone. J Steroid Biochem Mol Biol (2003) 87(1):27–34. doi:10.1016/S0960-0760(03)00383-2
66. Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. Br J Cancer (2014) 111(7):1249–54. doi:10.1038/bjc.2014.268
67. Harkonen P, Torn S, Kurkela R, Porvari K, Pulkka A, Lindfors A, et al. Sex hormone metabolism in prostate cancer cells during transition to an androgen-independent state. J Clin Endocrinol Metab (2003) 88(2):705–12. doi:10.1210/jc.2002-020236
68. Fishman J, Goto J. Mechanism of estrogen biosynthesis. Participation of multiple enzyme sites in placental aromatase hydroxylations. J Biol Chem (1981) 256(9):4466–71.
69. Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res (2007) 42(1):28–42. doi:10.1111/j.1600-079X.2006.00407.x
70. Ximenes VF, Catalani LH, Campa A. Oxidation of melatonin and tryptophan by an HRP cycle involving compound III. Biochem Biophys Res Commun (2001) 287(1):130–4. doi:10.1006/bbrc.2001.5557
71. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478(7368):197–203. doi:10.1038/nature10491
72. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22(3):298–305. doi:10.1038/nm.4045
73. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi:10.1158/2159-8290.CD-12-0095
74. Charlesworth MC, Young CY, Miller VM, Tindall DJ. Kininogenase activity of prostate-derived human glandular kallikrein (hK2) purified from seminal fluid. J Androl (1999) 20(2):220–9.
75. Raj GV, Barki-Harrington L, Kue PF, Daaka Y. Guanosine phosphate binding protein coupled receptors in prostate cancer: a review. J Urol (2002) 167(3):1458–63. doi:10.1016/S0022-5347(05)65345-1
76. Blaner WS. Cellular metabolism and actions of 13-cis-retinoic acid. J Am Acad Dermatol (2001) 45(5):S129–35. doi:10.1067/mjd.2001.113714
77. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino Acid. J Biol Chem (2012) 287(24):19786–91. doi:10.1074/jbc.R112.357194
78. Isgro MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol (2015) 867:125–43. doi:10.1007/978-94-017-7215-0_9
79. Whitehead MC, Marangos PJ, Connolly SM, Morest DK. Synapse formation is related to the onset of neuron-specific enolase immunoreactivity in the avian auditory and vestibular systems. Dev Neurosci (1982) 5(4):298–307. doi:10.1159/000112689
80. Marangos PJ, Schmechel DE. Neuron specific enolase, a clinically useful marker for neurons and neuroendocrine cells. Annu Rev Neurosci (1987) 10:269–95. doi:10.1146/annurev.ne.10.030187.001413
81. Cerasuolo M, Paris D, Iannotti FA, Melck D, Verde R, Mazzarella E, et al. Neuroendocrine transdifferentiation in human prostate cancer cells: an integrated approach. Cancer Res (2015) 75(15):2975–86. doi:10.1158/0008-5472.CAN-14-3830
82. Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, et al. Targeting Stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab (2016) 24(5):685–700. doi:10.1016/j.cmet.2016.10.011
83. Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer (2007) 14(3):531–47. doi:10.1677/ERC-07-0061
Keywords: olfactory receptor, OR51E2, PSGR, prostate cancer, neuroendocrine trans-differentiation, neuron-specific enolase, agonists, 19-hydroxyandrostenedione
Citation: Abaffy T, Bain JR, Muehlbauer MJ, Spasojevic I, Lodha S, Bruguera E, O’Neal SK, Kim SY and Matsunami H (2018) A Testosterone Metabolite 19-Hydroxyandrostenedione Induces Neuroendocrine Trans-Differentiation of Prostate Cancer Cells via an Ectopic Olfactory Receptor. Front. Oncol. 8:162. doi: 10.3389/fonc.2018.00162
Received: 16 January 2018; Accepted: 30 April 2018;
Published: 28 May 2018
Edited by:
Masaki Shiota, Kyushu University, JapanReviewed by:
Elena Monica Borroni, Humanitas Research Hospital, ItalyMomoe Itsumi, Tokyo Medical and Dental University, Japan
Copyright: © 2018 Abaffy, Bain, Muehlbauer, Spasojevic, Lodha, Bruguera, O’Neal, Kim and Matsunami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatjana Abaffy, dGF0amFuYS5hYmFmZnlAZHVrZS5lZHU=