 Norashikin Zakaria
Norashikin Zakaria Narazah Mohd Yusoff1
Narazah Mohd Yusoff1 Darius Widera
Darius Widera Badrul Hisham Yahaya
Badrul Hisham Yahaya- 1Regenerative Medicine Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, Bertam, Penang, Malaysia
- 2Cancer Research Centre, Institute for Medical Research (IMR), Kuala Lumpur, Malaysia
- 3Stem Cell Biology and Regenerative Medicine, School of Pharmacy, University of Reading, Reading, United Kingdom
Cancer stem cells (CSCs) are a subpopulation of cancer cells that play a pivotal role in tumor development, invasion, metastasis, and recurrence. We and others have reported significant involvement of the NF-κB pathway in regulating CSCs of non-small cell lung cancer (NSCLC). In this study, we evaluated the effects of NF-κB inhibition on self-renewal, stemness, migration, and expression of genes involved in the epithelial to mesenchymal transition (EMT) and apoptosis resistance in lung CSCs. Different concentrations of the NF-κB inhibitor BMS-345541 (0.4, 4.0, and 10.0 µM), an inhibitor the NF-κB upstream kinase IKKβ, were used to treat both lung CSCs (CD166+CD44+, CD166+EpCAM+) and non-CSC NSCLC cells (CD166−CD44−, CD166−EpCAM−) in A549 and H2170 cell lines. We assessed the impact of BMS-345541 on the ability to form tumorspheres (self-renewal assay), expression of stemness genes (SOX2, OCT4, NANOG, SCA-1, and KLF4), migration, and expression of EMT and apoptosis-related genes. Inhibition of NF-κB by BMS-345541 effectively reduced the stemness, self-renewal, and migration capacity of lung CSCs. Moreover, expression of genes involved in the EMT (SNAI1 and TWIST) and apoptosis resistance (BCL-2, BAX, and BIRC5) was significantly reduced following the treatments, suggesting that NF-κB inhibition is sufficient to prevent the EMT and induce apoptosis in lung CSCs. Our findings suggest that NF-κB inhibition could reduce the capability of CSCs to maintain their population within the tumor mass, potentially decelerating cancer progression, relapse, and chemotherapy resistance.
Introduction
Emerging evidence suggests the presence of a subpopulation within tumors with the abilities to self-renew and to produce differentiated progeny. This subpopulation of cells, known as cancer-initiating cells or cancer stem cells (CSCs) (1), was first characterized in 1994 in leukemia (2). Their presence was later confirmed in many human carcinomas, including breast, brain, colon, and lung cancers (3–7). CSCs play a pivotal role in tumor development, invasion, metastasis, drug resistance, and recurrence (8). Previously, we isolated and characterized putative CSCs from non-small cell lung cancer (NSCLC) cell lines based on expression of specific cell surface markers (i.e., CD166, CD44, and EpCAM) (9). CSCs that were isolated based on the expression of combined CD166+/CD44+ and CD166+/EpCAM+ exhibited stemness in vitro and were able to form secondary tumors in vivo. The transcriptomic analysis of these populations of cells revealed a significant involvement of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in regulating the putative lung CSCs.
The NF-κB family consists of five proteins: p65 (RelA), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52) (10, 11). These NF-κB proteins share a conserved N-terminal Rel homology domain, which enables nuclear translocation, DNA binding, and formation of homo- and heterodimers (10, 11). NF-κB proteins are expressed in the cytoplasm of most cell types, including differentiated cells, stem cells, and cancer cells (12, 13). In the inactive form, NF-κB homo- and heterodimers are bound to inhibitory IκB proteins (14) that mask the nuclear localization sequences (14). Activation of canonical pathways can be induced by many stimuli, including lipopolysaccharide and pro-inflammatory growth factors and cytokines [e.g., tumor necrosis factor alpha (TNF-α) or interleukin-1] (15). Upon activation, the IkB kinase (IKK) complex (IKKα, IKKβ, and IKKγ) phosphorylates the inhibitor IκB proteins at serine residues (e.g., 32 and 36 in IκBβ) and leads to their proteasomal degradation. Subsequent nuclear translocation of NF-κB and binding to its consensus sequences activate transcription of the respective target genes that regulate cellular proliferation, migration, and preventing apoptosis process.
Although aberrant NF-κB signaling has been associated with development and progression of various cancers, there is no detailed knowledge about the effect of this pathway on CSCs in lung cancer. In this study, the impact of NF-κB inhibition on the expression of genes associated with stemness, migration, and apoptosis of putative lung CSCs was evaluated. In addition, the effects of the NF-κB inhibitor BMS-345541 on the formation of tumorspheres (self-renewal) were also assessed. Finally, the influence of the blockade of NF-κB-mediated signaling on the migratory behavior of lung CSCs was evaluated using scratch-wound healing assay.
Materials and Methods
Materials and Cell Lines
BMS-345541 purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell lines A547 (human lung carcinoma) and H2170 (human lung squamous cell carcinoma) purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA).
Cell Culture
All cells were cultured in RPMI-1640 medium (Gibco, Life Technologies, Foster City, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (Gibco) in a humidified incubator at 37°C and 5% CO2.
Isolation and Characterization of Putative Lung CSCs
The putative lung CSCs and non-CSCs used in this study were isolated based on expression of specific CSCs markers (CD166, CD44, and EpCAM) (9, 16). Cells were detached with trypsin (Life Technologies, Carlsbad, CA, USA) and washed with phosphate buffer solution (PBS) containing 2% FBS (PBS with 2% FBS). A total of 1 × 107 cells were incubated for 30 min (on ice and in the dark) with respective antibodies (Table 1). The unbound antibodies were washed away with PBS with 2% FBS through centrifugation. Prior to sorting, the labeled cell suspensions were filtered through a 40-µm cell strainer to obtain a single cell suspension. The labeled cells were sorted into CD166+/CD44+, CD166−/CD44−, CD166+/EpCAM+, and CD166−/EpCAM− subsets using a fluorescence activated cell sorter (FACSAriaII, Becton Dickinson, Franklin Lakes, NJ, USA).
Table 1. List of antibodies used in cancer stem cells isolation.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from approximately 1 × 106 cells using a Qiagen AllPrep RNA isolation kit (Qiagen, Limburg, Netherlands) according to the manufacturer’s instructions. The concentration and purity of the extracted RNA were determined using Nanodrop (Agilent Technologies, Santa Clara, CA, USA). The RNA samples were stored at −80°C for later use. Complementary DNA (cDNA) was synthesized from 3 µg of total RNA using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) using a random hexamer primer and an anchored-oligo (dT) primer. Expression of selected genes was analyzed using an ABI StepOnePlus™ Real-Time PCR machine (Applied Biosystems, Foster City, CA, USA). The qPCR reaction was prepared using a Taqman® gene expression assay (Applied Biosystems) and the primers listed in Table 2.
Table 2. List of Taqman® gene expression primers.
Sphere Assay
The effect of NF-κB inhibition on CSC self-renewal was evaluated using a sphere assay. All cells (parental, CSCs, and non-CSCs of A549 and H2170 cell lines) were treated with different concentrations (0, 0.4, 4.0, and 10.0 µM) of BMS-345541 for 24 and 48 h. After each respective time point, the cells were trypsinized and 1.0 × 103 cells/ml were cultured in 24-well ultra-low attachment plates (Corning, Corning, NY, USA) containing serum-free medium DMEM/F12 (Gibco) supplemented with 10 ng/ml fibroblast growth factor, 1% of B27 (optimized serum-free supplement), 20 ng/ml of epidermal growth factor, and 1% penicillin–streptomycin (all purchased from Invitrogen). Sphere size was assessed after 14 days. The sphere size was assessed by measuring the diameter (µM) of each formed spheres using ImageJ software.
Migration Assay
Sorted and parental NSCLC cells were seeded at a density of 3–4 × 105 cells/well in complete medium and grown overnight to a confluence of 90%. Subsequently, the cells were treated with colcemide (10 µg/ml) for 2 h. After incubation, a scratch was inflicted using a sterile 200 µl pipette tip, and gentle washing was carried out twice using PBS to remove debris. The cells were incubated with 1 ml of medium containing different concentrations of BMS-345541 for 24 and 48 h. Images of wound coverage were captured at ×10 magnification (Olympus IX 71; Olympus, Tokyo, Japan) at 0, 24, and 48 h. The coverage area was measured using ImageJ and the percentage of the migrated distance was calculated as:
Statistical Analysis
All data were expressed as the mean ± SD of three independent experiments. Comparison between treatment groups with the untreated group was performed using two-way analysis of variance (SPSS statistical package). p-Values of <0.05 were considered to be statistically significant.
Results
Inhibition of NF-κB Reduces the Expression Levels of Stemness Genes
Cancer stem cells from NSCLC lines (A549 and H2170) were isolated based on expression of surface markers CD166, CD44, and EpCAM. The isolated CSCs (A549 CD166+CD44+, A549 CD166+EpCAM+, and H2170 CD166+EpCAM+) and their non-CSC counterparts (A549 CD166−CD44−, A549 CD166−EpCAM−, and H2170 CD166−EpCAM−) were treated with 0.4, 4.0, and 10.0 µM BMS-345541. The effect of the treatment was first evaluated by measuring the expression of stem cell transcription factors SOX2, NANOG, OCT4, SCA-1, and KLF4 for both cell cancer and non-CSC populations (Figures 1A–F). Treatment with all concentrations of BMS-345541 for 48 h reduced the expression of SOX2, NANOG, OCT4, and SCA-1 in A549 CD166+CD44+ cells. However, expression levels of the genes remained unaltered in the A549 CD166−CD44− subpopulation except for SCA-1 and KLF-4. In A549 CD166+EpCAM+ cells, the expression levels of SOX2, NANOG, and OCT4 were reduced following treatment with 0.4, 4.0, and 10.0 µM BMS-345541. In the non-CSC subpopulation A549 CD166−EpCAM−, 10.0 µM BMS-345541 was needed to reduce the expression of SOX2 and SCA-1. In H2170 CD166+EpCAM+ cells and the non-CSCs subpopulation (H2170 CD166−EpCAM−), treatment with 10.0 µM reduced the expression of SOX2 and KLF4 in the cells.
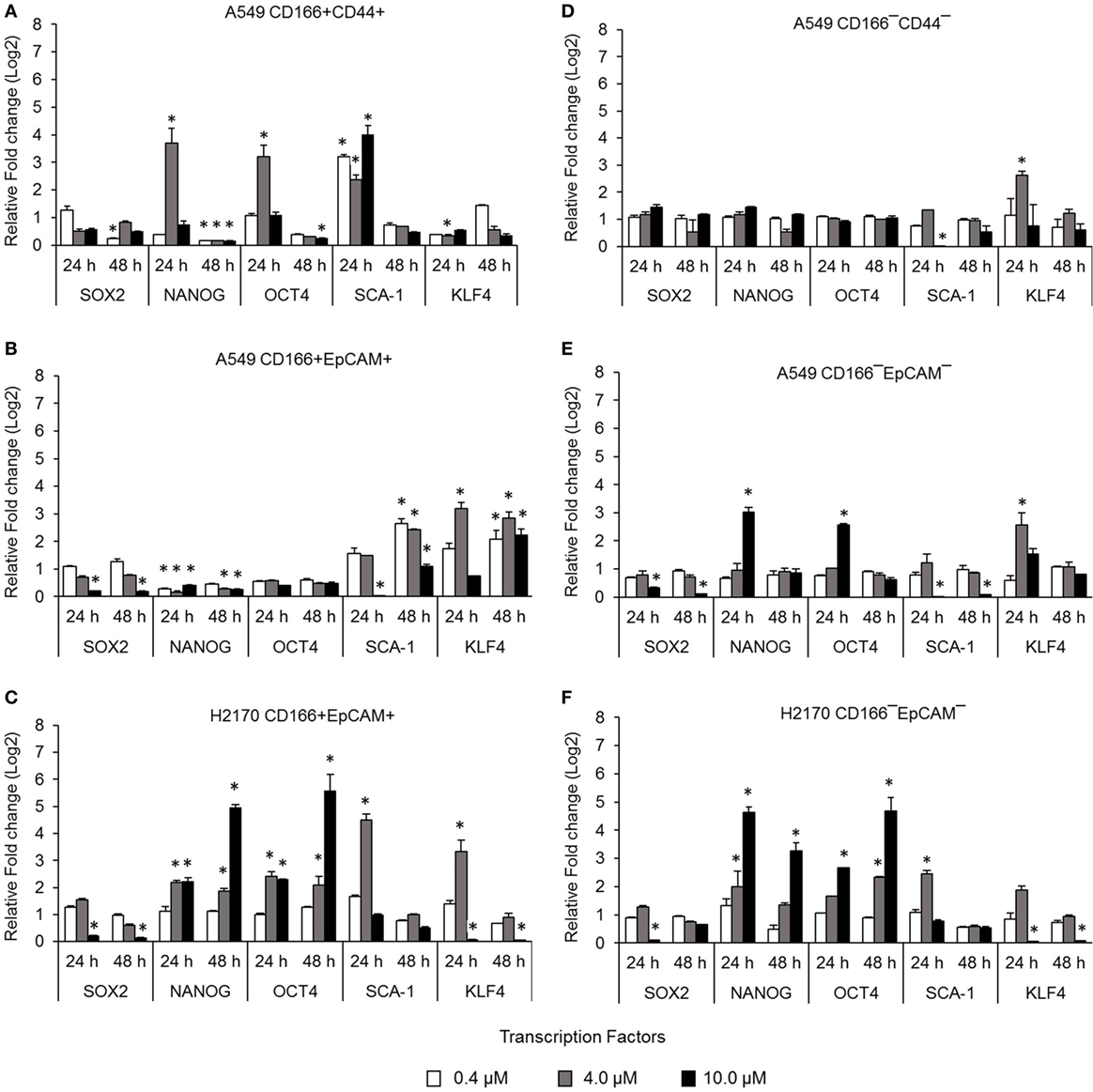
Figure 1. Effect of BMS-345541 on expression of stem cell transcription factors in lung cancer stem cells (CSCs). The graphs show the relative expression of stemness genes SOX2, NANOG, OCT4, SCA-1, and KLF4 in (A–C) lung CSC and (D–F) non-CSC populations. The fold change was calculated using the 2−ΔΔct formula, and GAPDH was used as the internal control. Graphs show fold change relative to the untreated sample. The results represent the mean ± SD of three replicates. The p-value was calculated using analysis of variance by comparing the treatment groups with the untreated group (*p < 0.05).
NF-κB Inhibits the Expression of Genes Involved in the Epithelial to Mesenchymal Transition (EMT)
Activation of NF-κB is involved in several important processes in cancer, including regulating the expression of EMT transcription factors (17), inducing apoptosis resistance (18), and regulating the angiogenesis and proliferation of cancer cells. The effectiveness of the NF-κB inhibitor BMS-345441 in targeting lung CSCs was evaluated by its ability to reduce the expression of genes involved in those processes. Treatment with 0.4, 4.0, and 10.0 µM BMS-345441 did not exert any effect on expression of SNAI1 and TWIST1 (Figure 2A), and expression levels of mesenchymal markers N-cadherin and Vimentin and the epithelial marker E-cadherin were unchanged in A549 CD166+CD44+ cells (Figure 2B). Treatment with 10.0 µM BMS-345441 downregulated the expression of TWIST1, Vimentin, and E-cadherin in A549 CD166+EpCAM+ cells when treated for 24 and 48 h (Figures 2C,D). However, treatment of the cells with the same concentration of BMS-345441 increased expression of SNAI1 when the treatment was prolonged to 48 h, but expression of N-cadherin was unchanged. In H2170 CD166+EpCAM+ cells, prolonged treatment with BMS-345541 for up to 48 h increased the expression of SNAI1 (treatment with 10.0 µM) but increased the expression of TWIST1 (treatment with 0.4, 4.0, and 10.0 µM) (Figure 2E). Expression of N-cadherin remained unchanged but expression of Vimentin increased following prolonged treatment, indicating that its expression is regulated by SNAI1 (Figure 2F).
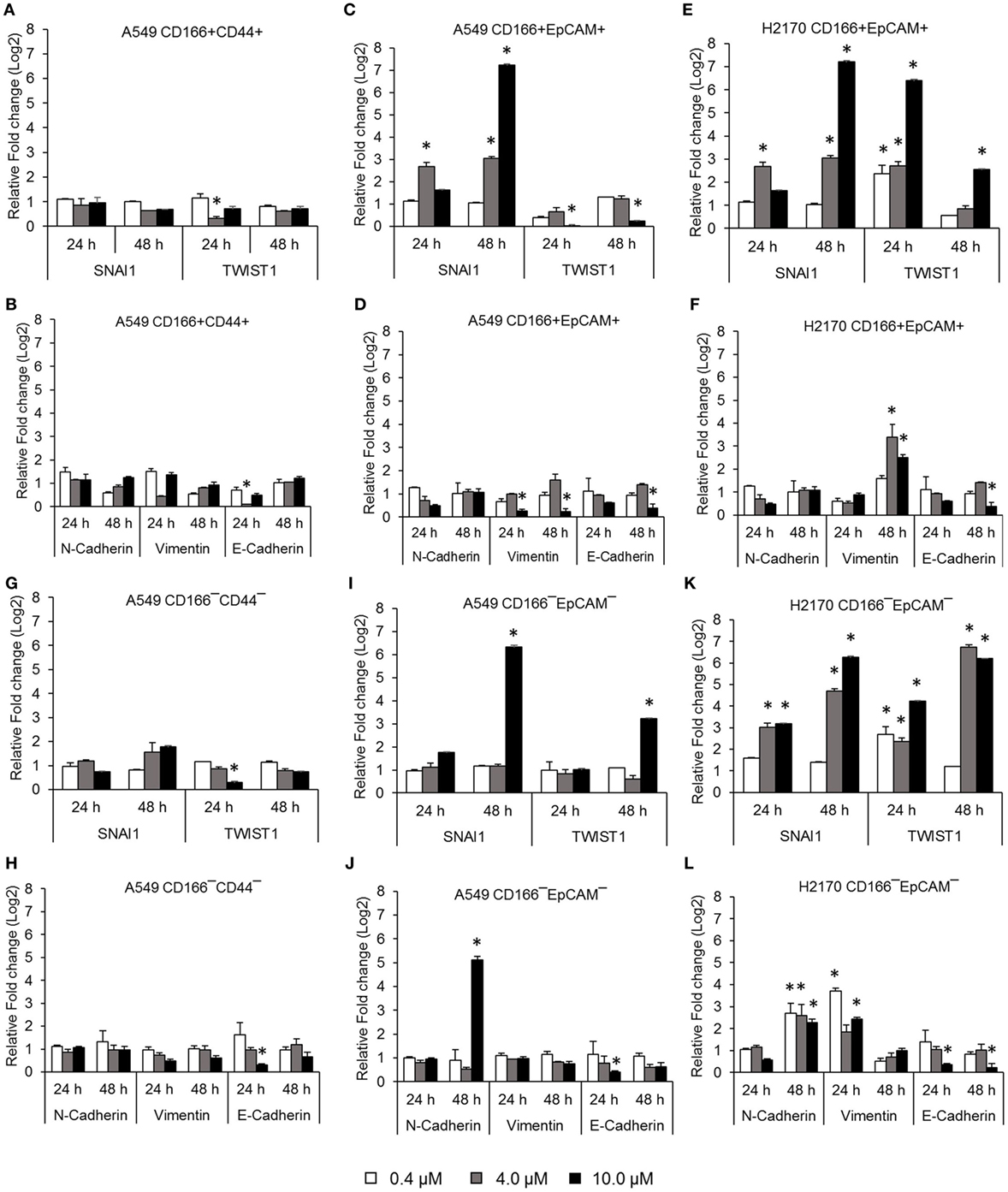
Figure 2. Quantitative real-time polymerase chain reaction results showing expression of genes involved in the epithelial to mesenchymal transition (EMT) process in lung cancer stem cells treated with different concentrations of BMS-345441. Relative expression of EMT switch genes (SNAI1 and TWIST1), mesenchymal markers (N-cadherin and Vimentin), and the epithelial marker (E-cadherin) in (A,B) A549 CD166+CD44+; (C,D) A549 CD166+EpCAM+; (E,F) H2170 CD166+EpCAM+; (G,H) A549 CD166−CD44−; (I,J) A549 CD166−EpCAM−; and (K,L) H2170 CD166−EpCAM− cells. The fold change was calculated using the 2−ΔΔct formula, and GAPDH was used as the internal control. Graphs show fold change relative to the untreated sample. The results represent the mean ± SD of three replicates. The p-value was calculated using analysis of variance by comparing the treatment groups with the untreated group (*p < 0.05).
BMS-345441 treatment also affected the expression of EMT genes in non-CSCs. In A549 CD166−CD44− cells, treatment with 4.0 and 10.0 µM BMS-345441 at 24 and 48 h slightly reduced the expression of TWIST1, but prolonged treatments increased the expression of SNAI1 (Figure 2G). However, treatment did not have a significant effect on the expression of N-cadherin, Vimentin, and E-cadherin at both time point except for the expression of E-cadherin when treated with 10 µM BMS-345441 for 24 h (Figure 2H). In A549 CD166−EpCAM− cells, treatment with 10.0 µM BMS-345441 for 48 h increased the expression of SNAI1 and TWIST1 (Figure 2I) and N-cadherin, but expression of Vimentin and E-cadherin were only slightly changed (Figure 2J). In H2170 CD166−EpCAM− cells, treatment with 4.0 and 10.0 µM increased expression of SNAI1, TWIST1, N-cadherin, and Vimentin, but expression of E-cadherin was decreased (Figures 2K,L).
NF-κB Inhibits the Expression of Genes Involved in the Apoptosis Resistance Process
The NF-κB pathway is involved in apoptosis resistance through activation of anti-apoptotic genes, including BCL-2 and BIRC5. Expression levels of BCL2, BAX, and BIRC5 in A549 CD166+CD44+ cells were downregulated when treated with 4.0 µM BMS-345541 for 24 h (Figure 3A). However, expression levels of BCL2, BAX, and BIRC5 were unchanged when treated with 0.4 and 10.0 µM BMS-345441 compared to the untreated control. Expression of BCL2 and BAX in A549 CD166+EpCAM+ cells treated with 10.0 µM BMS-345541 for 24 h was downregulated (Figure 3B). Prolonged treatment of the cells for up to 48 h led to increased expression of all three genes. Treatment with BMS-345441 reduced the expression of BCL2 and BAX in H2170 CD166+EpCAM+ cells treated with 10.0 µM for 24 and 48 h (Figure 3C). In the three non-CSC subpopulations, treatment with 10.0 µM BMS-345441 was the most effective at inducing downregulation of BCL2 and BAX (Figures 3D–F). Expression of BIRC5 in the cells remained unchanged following treatments with BMS-345441, except for A549 CD166−CD44− cells, for which treatment with 10.0 µM BMS-345441 significantly downregulated expression of the gene (p = 0.035).
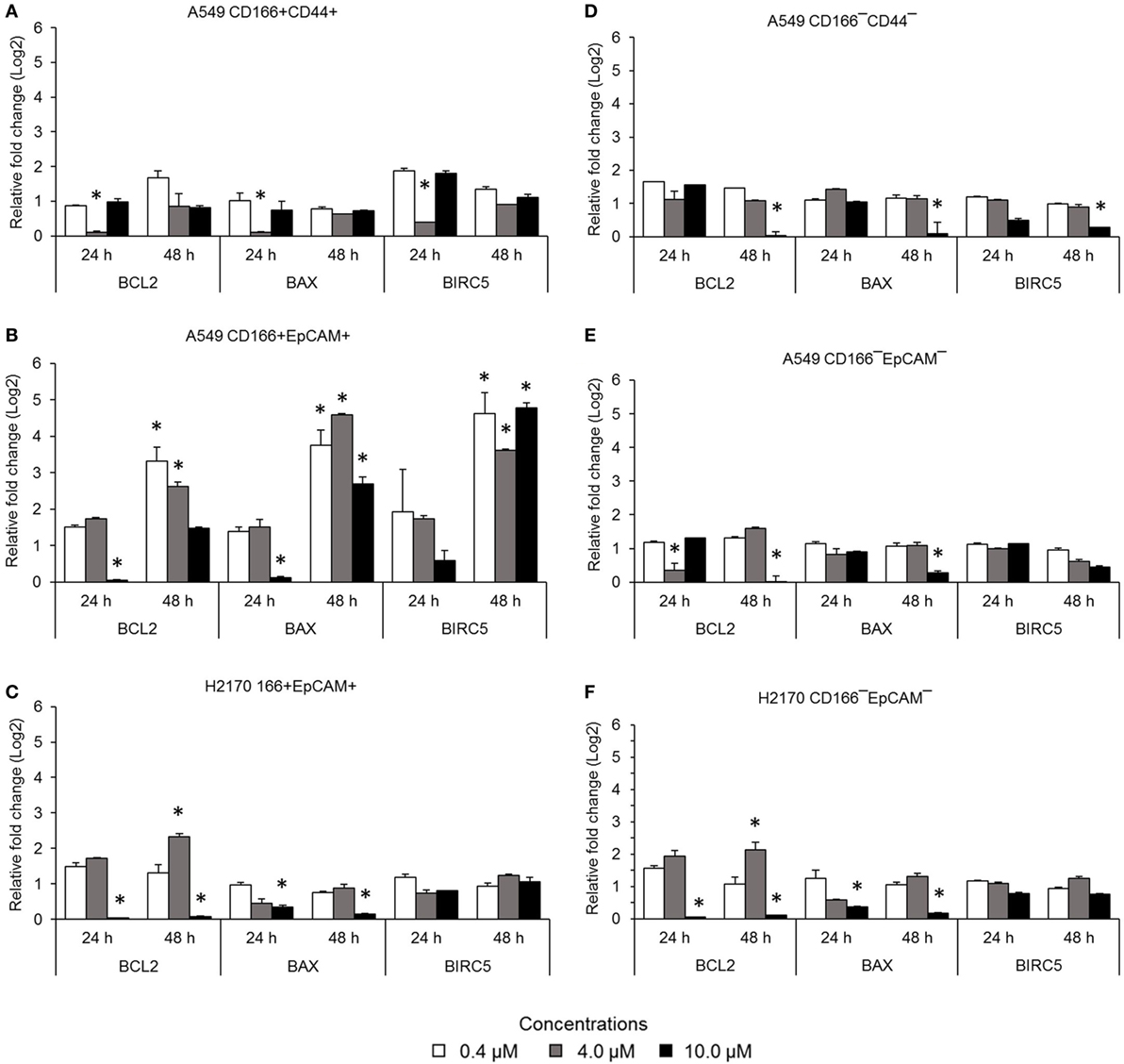
Figure 3. Expression of pro-apoptotic genes (BCL-2 and BIRC5) and the anti-apoptotic gene BAX in lung cancer stem cells following treatment with different concentrations of BMS-345541. Relative expression of BCL2, BAX, and BIRC5 in (A) A549 CD166+CD44+; (B) A549 CD166+EpCAM+; (C) H2170 CD166+EpCAM+; (D) A549 CD166−CD44−; (E) A549 CD166−EpCAM−; and (F) H2170 CD166−EpCAM− cells. The fold change was calculated using the 2−ΔΔct formula, and GAPDH was used as the internal control. The results represent the mean ± SD of three replicates. The p-value was calculated using analysis of variance by comparing the treatment groups with the untreated group (*p < 0.05).
NF-κB Inhibition Reduces Self-Renewal Capability of Parental and Lung CSCs
Self-renewal is an important characteristic of CSCs that helps maintain the CSC pool in the tumor. Therefore, the effectiveness of the NF-κB inhibitor BMS-345541 in targeting lung CSCs can be assessed by looking at its ability to reduce the sphere-forming capacity of lung CSCs. Following treatment with different concentrations of BMS-345441 for 24 or 48 h, the cells were induced to form spheres by culturing in sphere-forming medium. After 14 days, the diameter of formed spheres was measured. Regardless of the treatment status, both CSC and non-CSC populations had the ability to form spheres (Figure 4A). Treatment with BMS-345441 significantly reduced the sphere size of lung CSCs and non-CSCs in a concentration and time-dependent manner. Treatment with 0.4, 4.0, and 10.0 µM BMS-345441 for 24 h reduced the size of the spheres of A549 CD166+CD44+ cells (Figure 4B). For A549 CD166+EpCAM+ and H2170 CD166+EpCAM+ cells, the sphere size was reduced after treatment with 0.4, 4.0, and 10.0 µM BMS-345541 at 24 and 48 h (Figure 4B). The sphere size of non-CSCs was also reduced following treatment with different concentrations of BMS-345441 at different time points (Figure 4B).
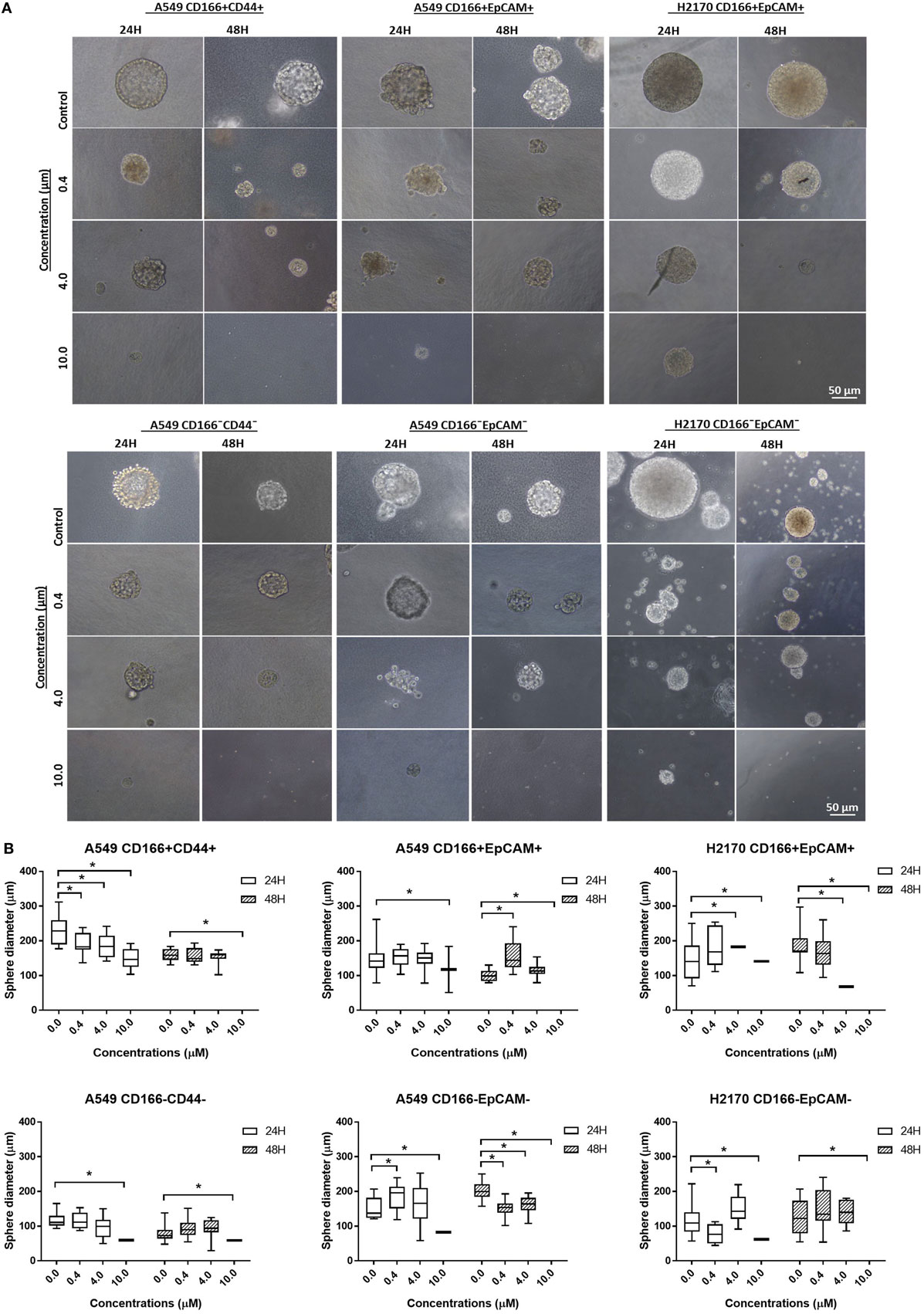
Figure 4. Effect of BMS-345541 on the sphere-forming capacity of lung cancer stem cells (CSCs). The cells were induced to form sphere following treatment with different concentrations of BMS-345541 for 24 and 48 h. (A) The images of the spheres formed following the treatments with BMS-345441 for lung CSCs and non-CSC counterparts. (B) Boxplots illustrate the sphere diameter (μm) for each cell type following the treatments. Data shown are representative of three independent experiments (n = 3). The y-axes show the sphere diameter (μm) and the x-axes show the concentration of BMS-345441 (0, 0.4, 4.0, and 10.0 µM) used in the treatment. Upper and lower boxplot margins represent the interquartile range, and the middle bar indicates the median. The whisker defines the range of values. The diameter of the spheres was measured using ImageJ analysis software. The bar above the boxes represents statistically significant differences *p < 0.05; two-way analysis of variance.
NF-κB Inhibits the Migration of Lung CSCs
The migration assay was performed to determine the effect of the NF-κB inhibitor BMS-345541 in inhibiting the migration of lung CSCs and non-CSCs. Figure 5 shows images of the scratch-wound healing assay and the graph illustrating the percentage of wound closure. Treatment with 0.4, 4.0, and 10.0 µM BMS-345441 was effective in inhibiting the migration of A549 CD166+EpCAM+ (except 4.0 µM at 48 h) and H2170 CD166+EpCAM+ cells at 24 and 48 h. However, only treatment with 10.0 µM alone reduced the migration of A549 CD166+CD44+ cells when treated for 24 and 48 h. The migration of all three non-CSCs was inhibited by treatment with 4.0 and 10.0 µM, but the effect was time dependent. Taken together, these results show that higher concentration of BMS-345441 (10.0 µM) effectively inhibited the migration of CSCs and non-CSCs.
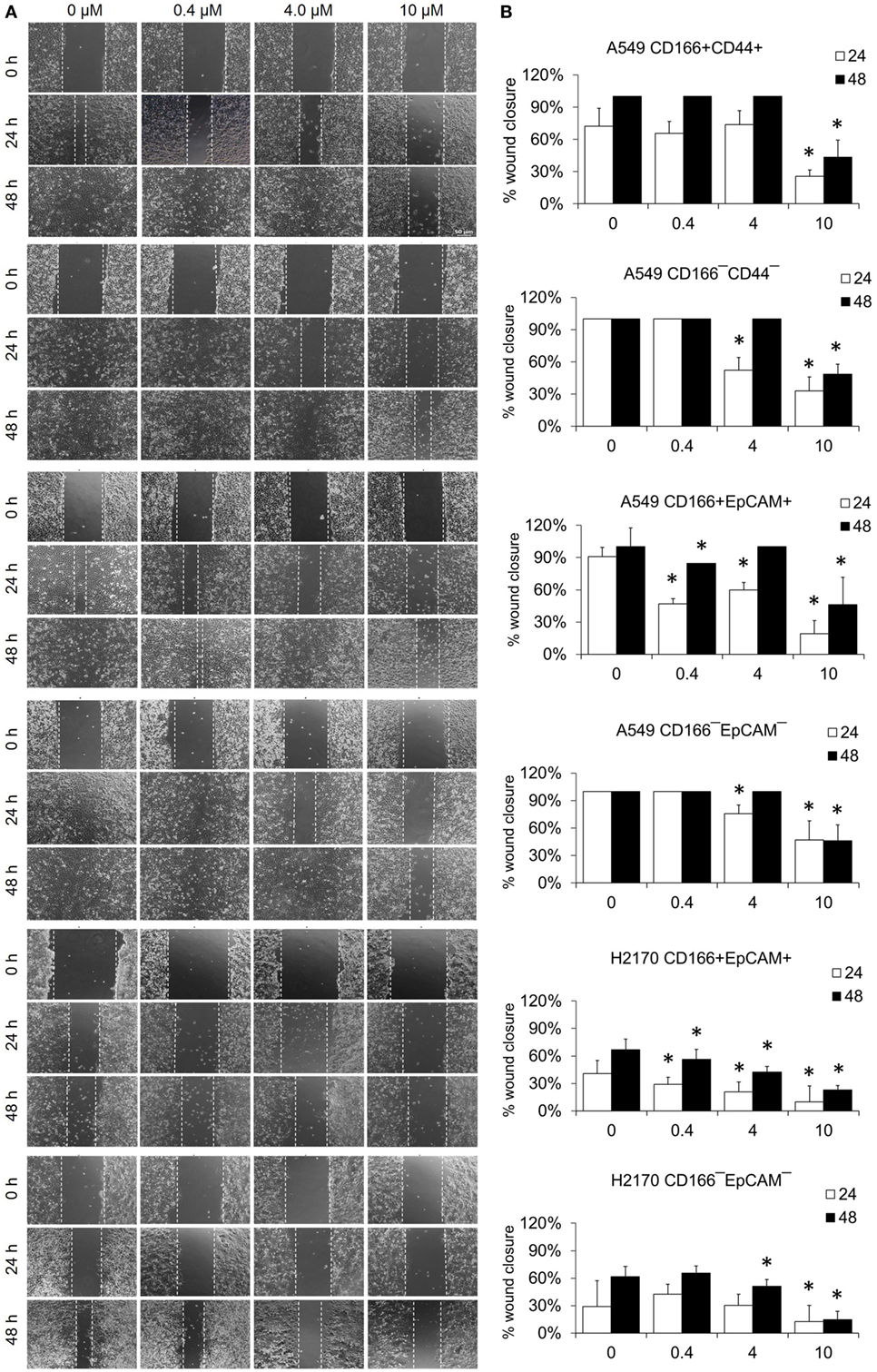
Figure 5. Effect of BMS-345541 on the migration of lung cancer stem cells of A549 and H2170 cells. Scratches were made and the cells were treated with different concentrations of BMS-345541. (A) Images of the wound area were taken at 0, 24, and 48 h after treatment. The wound areas were measured using ImageJ analysis software. (B) Percentage of migrated area [(area after 24/48 h ÷ initial area at 0 h) × 100%]. Data shown are representative of three independent experiments (n = 3). The p-value was calculated using analysis of variance by comparing the percentage of wound area for each treatment with that of the untreated group (*p < 0.05).
Discussion
The presence of CSCs population in lung tumor is the key to treatment failure (19). This cell having the ability to self-renew and differentiate, which is the key to chemotherapy resistance thus makes it very difficult to eliminate the CSCs (20). NF-κB activation has been associated with the initiation and progression of several human cancers, including breast, cervical, prostate, pancreatic, and lung cancer (21–23). In lung cancer, a high level of NF-κB activity was detected in patients with SCLC and NSCLC, and its constitutive activity was associated with advanced stage and poor prognosis of patients (22, 24). Gene expression and bioinformatics result from our previous study demonstrated that NF-κB was highly associated with the CSCs gene signature (9) and inhibition NF-κB might resulted in reduction of the lung CSCs stemness and tumorigenic properties. The NF-κB molecule is regulated by the IKK complex (23, 25, 26), hence became the most suitable candidate for a therapeutic target for cancer treatments (27). BMS-345541, an IKK complex inhibitor, has been used to deactivate of NF-κB in various types of cancer, including melanoma and leukemia (28, 29). BMS-345541 at a concentration as low as 0.4 µM and up to 10.0 µM effectively inhibited activity of NF-κB (28–31). Therefore, in our study, BMS-345441 at three different concentrations (0.4, 4.0, and 10.0 µM) was used to treat lung CSCs, and the effects were evaluated against the expression of stemness, EMT and apoptosis genes, sphere-forming capability, and migration. The results indicate that BMS-345441 shows promising results in reducing sphere size, expression of stemness genes, and migration of lung CSCs as well as parental cancer cells. Moreover, expression of genes related to EMT and apoptosis was reduced following treatment, suggesting that inhibiting the NF-κB expression may help preventing EMT activities and induce apoptosis of lung CSCs.
Activation of NF-κB is one of the mechanisms that mediates self-renewal of CSCs (32, 33). Self-renewal is one of the main features of CSCs that helps maintain CSCs population in tumor. The effectiveness of NF-κB inhibition in targeting lung CSCs is evidence by the reduction of sphere-forming capacity of lung CSCs and non-CSCs. The ability to reduce the sphere-forming capacity reflects the self-renewal capability of those cells. Our finding is in parallel with reports for other types of cancer. For instance, inhibition of NF-κB using the plant-derived agent Triptolide reduced the colony- and sphere-forming capacity of pancreatic CSCs (34). Xiang et al. reported that NF-κB induced self-renewal of CD133+ ovarian CSCs and its inhibition has reduced the self-renewal capability of the cells (35).
Expression of several stemness genes, including SOX2, OCT4, KLF4, and NANOG, is an important indicator for the multipotent characteristic of CSCs (9, 36). Our study demonstrated that inhibition of NF-κB reduced the expression of stem cell transcription factors SOX2, NANOG, and OCT4 in CSCs of A549 cells and expression of SOX2, SCA-1, and KLF4 in CSCs of H2170 cells. The roles of transcription factors in maintaining the stemness and tumourigenicity state of the CSCs have been reported previously. For example, SOX2, OCT4, and NANOG were found to be overexpressed in several cancers, including breast, prostate, and oral squamous cell carcinoma, and their expression levels were associated with tumor transformation, tumourigenicity, and tumor metastasis (37–39). Unlike in embryonic stem cells where these transcription factors mostly control differentiation of the cells, overexpression of SOX2, OCT4, and NANOG in CSCs was reported to modulate signaling pathways involved in the inhibiting apoptosis (40, 41). Therefore, reduction in the expression of these transcription factors indicates that the cells have lost their multipotent characteristics, including the self-renewal capability, and the reduction also likely induces apoptosis of the cells. The effect of NF-κB in regulating the expression of stemness genes was not consistent in every cell, which indicates differently response to the treatments. This is likely due to the heterogeneous population of CSCs as a result from asymmetric division which generates diversity within the cancer (42). NF-κB activation has been associated with invasive and metastatic capabilities of cancer and CSCs. The cancer cells invasion and metastasis process occur when the epithelial cancer cells loss its adherent properties and acquire the mesenchymal phenotype which then allow the cells to migrate (43, 44). This process is known as EMT. EMT process is activated by transcription of its genes including SNAI1 (45, 46), TWIST1 (47, 48), Slug, and ZEB1/2 (49) [reviewed in Ref. (17)] and its activation is demonstrated by the loss of epithelial marker (E-cadherin) expression and gain of mesenchymal marker (Vimentin and N-cadherin) expression (49, 50). The roles of the EMT in CSCs and cancer cell migration and metastasis suggesting that inhibiting the EMT process could result in reducing migration and metastatic potential of CSCs. Our results demonstrated that the migration capability of CSCs was reduced following the treatment with IKK kinase inhibitor, BMS-345541. This finding was in agreement with previous finding in NSCLC where inhibition of NF-κB has resulted in suppression of EMT transcription factors TWIST1, SNAI1, Slug, and ZEB2 and prevented the invasion and metastasis of the cells (51). Although our migration data show that NF-κB inhibition have significantly reduced CSCs migration, the expression of EMT genes was not in agreement with the data.
The gene expression data demonstrated that BMS-345541 treatment reduced the expression of TWIST1 in lung CSCs populations A549 CD166+CD44+, A549 CD166+EpCAM+, and H2170 CD166+EpCAM+but increased the expression of SNAI1 in A549 CD166+EpCAM+ and H2170 CD166+EpCAM+ cells. The same result was seen in the non-CSC populations A549 CD166−EpCAM− and H2170 CD166−EpCAM−. Although both SNAI1 and TWIST1 control activation of the EMT process, these genes are activated through different pathways. SNAIL family genes are activated through the NF-κB pathway, but TWIST family genes are activated through the Ras/Raf/ERK pathway (52, 53). SNAI1 upregulation via NF-κB occurs through phosphorylation of IKKα, which then activates NF-κB/p65 dimer and stimulates transcription of SNAIL (54). TheBMS-345541 molecule is specifically targets the IKKβ subunit of the IKK complex, the IKKα subunit remain free to undergo phosphorylation, activate the NF-κB/p65 dimer and increase SNAIL expression. However, the upregulation of SNAIL alone is not enough to activate the EMT because its activation is controlled by sequential activation of several activators, including TWIST, ZEB, and Slug (53).
The NF-κB activation also involved in apoptosis resistance in cancer cells (28). This event occurs through overexpression of anti-apoptotic genes (14). The anti-apoptotic proteins include BCL-2, BCL-XL, and MCL-1, and BIRC5 whereas the pro-apoptotic proteins include BAX and BAK as well as the BH3 domain molecules (e.g., Bid, Bim, Bik, Noxa, and Puma) (55). In our study, inhibition of NF-κB downregulated expression of the anti-apoptotic genes BCL-2 and BIRC5. Deactivation of NF-κB was reported to be associated with downregulation of BCL-2 via a mechanism of caspase-3 activation that mediates BCL-2 cleavage that later induces apoptosis activity (29). BMS-345541 in the low micromolar range (1–2 µM/l) has been reported to induce apoptosis in leukemia cells (56). A higher concentration (10.0 µM) of BMS-345541 was found to induce apoptosis of melanoma cells (28). A lower concentration (2–5 µM) also resulted in induction of a moderate pro-apoptotic effect (28). The ability of BMS-345541 to induce apoptosis was also seen in combination with TNF-α (28).
Conclusion
Results of this study indicate that NF-κB is a promising molecular target for lung CSCs, as the deactivation of NF-κB using the kinase inhibitor BMS-345541 effectively reduced the stemness, self-renewal, and migratory capabilities of lung CSCs. These effects limited the capability of CSCs to maintain their population, and in theory this treatment might reduce cancer resistance to therapy and limit cancer progression. This finding may be a key to solving some major challenges in cancer therapy, namely cancer relapse and chemotherapy resistance, by eliminating CSCs. Future studies are needed to further elucidate the molecular machinery responsible for the effects of the inhibitor and to determine how it effects on other pathways that regulating cancer and normal stem cells. The cytotoxicity of the kinase inhibitor also must be evaluated specifically in lung cancer patient samples because it may have an unknown adverse effect. BMS-345541 has the potential to be developed into a specific anti-CSC therapy, but its use alone did not show consistent effects in targeting all different phenotypes of lung CSCs.
Author Contributions
BHY and NZ designed, conducted the experiment and analyzed the data: NZ, BHY, DW, NMY, and ZZ contributed in writing and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by grants from the Research University Grant scheme (1001/CIPPT/811204) of Universiti Sains Malaysia and Fundamental Research Grant Scheme (FRGS), Ministry of Higher Education of Malaysia (203/CIPPT/6711509).
References
1. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi:10.1038/35102167
2. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367(6464):645–8. doi:10.1038/367645a0
3. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100(7):3983–8. doi:10.1073/pnas.0530291100
4. Singh S, Hawkins C, Clarke I, Squire J, Bayani J, Hide T, et al. Identification of human brain tumor initiating cells. Nature (2004) 19:396–401. doi:10.1038/nature03128
5. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature (2007) 445(7123):106–10. doi:10.1038/nature05372
6. Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One (2010) 5(11):e14062. doi:10.1371/journal.pone.0014062
7. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ (2008) 15(3):504–14. doi:10.1038/sj.cdd.4402283
8. Buzzeo MP, Scott EW, Cogle CR. The hunt for cancer-initiating cells: a history stemming from leukemia. Leukemia (2007) 21(8):1619–27. doi:10.1038/sj.leu.2404768
9. Zakaria N, Yusoff NM, Zakaria Z, Lim MN, Baharuddin PJN, Fakiruddin KS, et al. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer (2015) 15(1):84. doi:10.1186/s12885-015-1086-3
10. Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1(4):a000034. doi:10.1101/cshperspect.a000034
11. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys (2013) 42:443–68. doi:10.1146/annurev-biophys-083012-130338
12. Widera D, Kaus A, Kaltschmidt C, Kaltschmidt B. Neural stem cells, inflammation and NF-κB: basic principle of maintenance and repair or origin of brain tumours? J Cell Mol Med (2008) 12(2):459–70. doi:10.1111/j.1582-4934.2007.00208.x
13. Widera D, Mikenberg I, Kaltschmidt B, Kaltschmidt C. Potential role of NF-κB in adult neural stem cells: the underrated steersman? Int J Dev Neurosci (2006) 24(2):91–102. doi:10.1016/j.ijdevneu.2005.11.017
14. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12(1):1–15. doi:10.1186/1476-4598-12-86
15. Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB) – a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev (2008) 19(2):157–65. doi:10.1016/j.cytogfr.2008.01.006
16. Halim N, Zakaria N, Satar NA, Yahaya BH. Isolation and characterization of cancer stem cells of the non-small-cell lung cancer (A549) cell line. Methods Mol Biol (2016) 1516:371–88. doi:10.1007/7651_2016_326
17. Rinkenbaugh A, Baldwin A. The NF-κB pathway and cancer stem cells. Cells (2016) 5(2):16. doi:10.3390/cells5020016
18. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-[kappa]B as the matchmaker. Nat Immunol (2011) 12(8):715–23. doi:10.1038/ni.2060
19. Zakaria N, Satar NA, Abu Halim NH, Ngalim SH, Yusoff NM, Lin J, et al. Targeting lung cancer stem cells: research and clinical impacts. Front Oncol (2017) 7:80. doi:10.3389/fonc.2017.00080
20. Templeton AK, Miyamoto S, Babu A, Munshi A, Ramesh R. Cancer stem cells: progress and challenges in lung cancer. Stem Cell Investig (2014) 1(4):9. doi:10.3978/j.issn.2306-9759.2014.03.06
21. Li J, Jia H, Xie L, Wang X, Wang X, He H, et al. Association of constitutive nuclear factor-κB activation with aggressive aspects and poor prognosis in cervical cancer. Int J Gynecol Cancer (2009) 19(8):1421–6. doi:10.1111/IGC.0b013e3181b70445
22. Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol (2009) 1(5):a000141. doi:10.1101/cshperspect.a000141
23. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature (2004) 431(7007):461–6. doi:10.1038/nature02924
24. Chen W, Li Z, Bai L, Lin Y. NF-kappaB, a mediator for lung carcinogenesis and a target for lung cancer prevention and therapy. Front Biosci (2011) 16:1172–85. doi:10.2741/3782
25. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch (2005) 446(5):475–82. doi:10.1007/s00428-005-1264-9
26. Zhi Y, Duan Y, Zhou X, Yin X, Guan G, Zhang H, et al. NF-κB signaling pathway confers neuroblastoma cells migration and invasion ability via the regulation of CXCR4. Med Sci Monit (2014) 20:2746–52. doi:10.12659/MSM.892597
27. Israël A. The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb Perspect Biol (2010) 2(3):a000158. doi:10.1101/cshperspect.a000158
28. Berger A, Quast SA, Plotz M, Kammermeier A, Eberle J. Sensitization of melanoma cells for TRAIL-induced apoptosis by BMS-345541 correlates with altered phosphorylation and activation of Bax. Cell Death Dis (2013) 4:e477. doi:10.1038/cddis.2012.198
29. Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 targets inhibitor of κB kinase and induces apoptosis in melanoma: involvement of nuclear factor κB and mitochondria pathways. Clin Cancer Res (2006) 12(3):950–60. doi:10.1158/1078-0432.CCR-05-1220
30. Buontempo F, Chiarini F, Bressanin D, Tabellini G, Melchionda F, Pession A, et al. Activity of the selective IκB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia. Cell Cycle (2012) 11(13):2467–75. doi:10.4161/cc.20859
31. Agbottah E, Yeh W-I, Berro R, Klase Z, Pedati C, Kehn-Hall K, et al. Two specific drugs, BMS-345541 and purvalanol A induce apoptosis of HTLV-1 infected cells through inhibition of the NF-kappaB and cell cycle pathways. AIDS Res Ther (2008) 5(1):1. doi:10.1186/1742-6405-5-12
32. Shostak K, Chariot A. NF-κB, stem cells and breast cancer: the links get stronger. Breast Cancer Res (2011) 13(4):214–214. doi:10.1186/bcr2886
33. Armstrong L, Hughes O, Yung S, Hyslop L, Stewart R, Wappler I, et al. The role of PI3K/AKT, MAPK/ERK and NFκβ signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum Mol Genet (2006) 15(11):1894–913. doi:10.1093/hmg/ddl112
34. Liu L, Salnikov AV, Bauer N, Aleksandrowicz E, Labsch S, Nwaeburu C, et al. Triptolide reverses hypoxia-induced epithelial–mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int J Cancer (2014) 134(10):2489–503. doi:10.1002/ijc.28583
35. Xiang T, Long H, He L, Han X, Lin K, Liang Z, et al. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene (2015) 34(2):165–76. doi:10.1038/onc.2013.537
36. Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells (2015) 7(9):1150–84. doi:10.4252/wjsc.v7.i9.1150
37. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet (2008) 40(5):499–507. doi:10.1038/ng.127
38. Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res (2007) 67(10):4807–15. doi:10.1158/0008-5472.CAN-06-4608
39. Chiou S-H, Wang M-L, Chou Y-T, Chen C-J, Hong C-F, Hsieh W-J, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell–like properties and epithelial–mesenchymal transdifferentiation. Cancer Res (2010) 70(24):10433–44. doi:10.1158/0008-5472.CAN-10-2638
40. Wang P, Gao Q, Suo Z, Munthe E, Solberg S, Ma L, et al. Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS One (2013) 8(3):e57020. doi:10.1371/journal.pone.0057020
41. Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res (2008) 68(16):6533–40. doi:10.1158/0008-5472.CAN-07-6642
42. Clevers H. Stem cells, asymmetric division and cancer. Nat Genet (2005) 37(10):1027–8. doi:10.1038/ng1005-1027
43. Voulgari A, Pintzas A. Epithelial–mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta (2009) 1796(2):75–90. doi:10.1016/j.bbcan.2009.03.002
44. Thompson EW, Newgreen DF. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res (2005) 65(14):5991–5. doi:10.1158/0008-5472.CAN-05-0616
45. Barbera MJ, Puig I, Domínguez D, Julien-Grille S, Guaita-Esteruelas S, Peiro S, et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene (2004) 23(44):7345–54. doi:10.1038/sj.onc.1207990
46. Kim H-J, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-κB and Snail. Mol Cell Biol (2007) 27(8):3165–75. doi:10.1128/MCB.01315-06
47. Šošić D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-κB activity. Cell (2003) 112(2):169–80. doi:10.1016/S0092-8674(03)00002-3
48. Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C, et al. Upregulation of twist-1 by NF-κB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol (2007) 27(11):3920–35. doi:10.1128/MCB.01219-06
49. Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-[kappa]B represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene (2006) 26(5):711–24. doi:10.1038/sj.onc.1209808
50. Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E-cadherin leads to upregulation of NF[kappa]B activity in malignant melanoma. Oncogene (2004) 23(52):8509–19. doi:10.1038/sj.onc.1207831
51. Kumar M, Allison DF, Baranova NN, Wamsley JJ, Katz AJ, Bekiranov S, et al. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS One (2013) 8(7):e68597. doi:10.1371/journal.pone.0068597
52. Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, Van Roy F, et al. Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene (2007) 26(53):7445–56. doi:10.1038/sj.onc.1210546
53. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (2014) 5:10. doi:10.3389/fendo.2014.00010
54. Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets (2013) 13(9):963–72. doi:10.2174/15680096113136660102
55. Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res (2009) 15(4):1126–32. doi:10.1158/1078-0432.CCR-08-0144
56. López-Guerra M, Roué G, Pérez-Galán P, Alonso R, Villamor N, Montserrat E, et al. p65 Activity and ZAP-70 status predict the sensitivity of chronic lymphocytic leukemia cells to the selective IκB kinase inhibitor BMS-345541. Clin Cancer Res (2009) 15(8):2767–76. doi:10.1158/1078-0432.CCR-08-2382
Keywords: NF-κB signaling, lung cancer, cancer stem cells, cancer migration, self-renewal
Citation: Zakaria N, Mohd Yusoff N, Zakaria Z, Widera D and Yahaya BH (2018) Inhibition of NF-κB Signaling Reduces the Stemness Characteristics of Lung Cancer Stem Cells. Front. Oncol. 8:166. doi: 10.3389/fonc.2018.00166
Received: 22 February 2018; Accepted: 30 April 2018;
Published: 17 May 2018
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Dhiraj Kumar, University of Texas MD Anderson Cancer Center, United StatesFrank Arfuso, Curtin University, Australia
Copyright: © 2018 Zakaria, Mohd Yusoff, Zakaria, Widera and Yahaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Badrul Hisham Yahaya, YmFkcnVsQHVzbS5teQ==