 Arenda D. Meedendorp
Arenda D. Meedendorp Arja ter ElstNils A. ’t Hart
Arja ter ElstNils A. ’t Hart Harry J. M. GroenEd Schuuring
Harry J. M. GroenEd Schuuring Anthonie J. van der Wekken*
Anthonie J. van der Wekken*
- University of Groningen and University Medical Center Groningen, Groningen, Netherlands
A 62-year-old man was referred to our university hospital for treatment of advanced adenocarcinoma of the lung after disease progression on two lines of EGFR TKI and one line of chemotherapy. Fluorescent in situ hybridization analysis upon progression showed an HER2 amplification. At our weekly Molecular Tumor Board (MTB), a decision was made to treat this patient with afatinib, which resulted in a partial response. However, progression was observed with a facial nerve paresis due to a metastasis in the skull. A biopsy of a location in the thorax revealed the presence of an EGFR-T790M mutation associated with acquired resistance, after which treatment with osimertinib was started. After 6 months, disease progression was observed, and a new biopsy was taken from the pelvic bone, which revealed the original amplification of HER2 together with the EGFR-L858R mutation, the EGFR-T790M mutation was not detected. The MTB decided to treat the patient with trastuzumab/paclitaxel. A partial response was observed in different bone lesions, while the skull metastasis with ingrowth in the brain remained stable for 6 months. Because of progression of the bone metastases after 6 months, a biopsy of a lesion in the thorax wall was taken. In this lesion, the EGFR-T790M mutation could be detected again. The MTB advised to start treatment with a combination of osimertinib and afatinib. This resulted in an impressive clinical improvement and a partial response of the bone metastases on the most recent 18-fluorodeoxyglucose positron emission tomography and computer tomography-scan. In conclusion, adjusting treatment to the mutational make-up of the tumor is a great challenge. For optimal treatment response multiple biopsies and re-biopsy upon progression are imperative. As more genes are investigated, treatment decision becomes increasingly difficult, therefore, expert opinions from an MTB is essential.
Introduction
Treatment of driver mutations cannot be based on large clinical trials or high levels of evidence at all times. However, a Molecular Tumor Board (MTB) can help in making treatment decisions based on databases, case reports, xenograft models, and cell lines. Here, we present such a case.
A 62-year-old man was referred to our university hospital for treatment of advanced adenocarcinoma of the lung after disease progression on two lines of EGFR TKI and one line of chemotherapy in September 2015 (Figure 1).
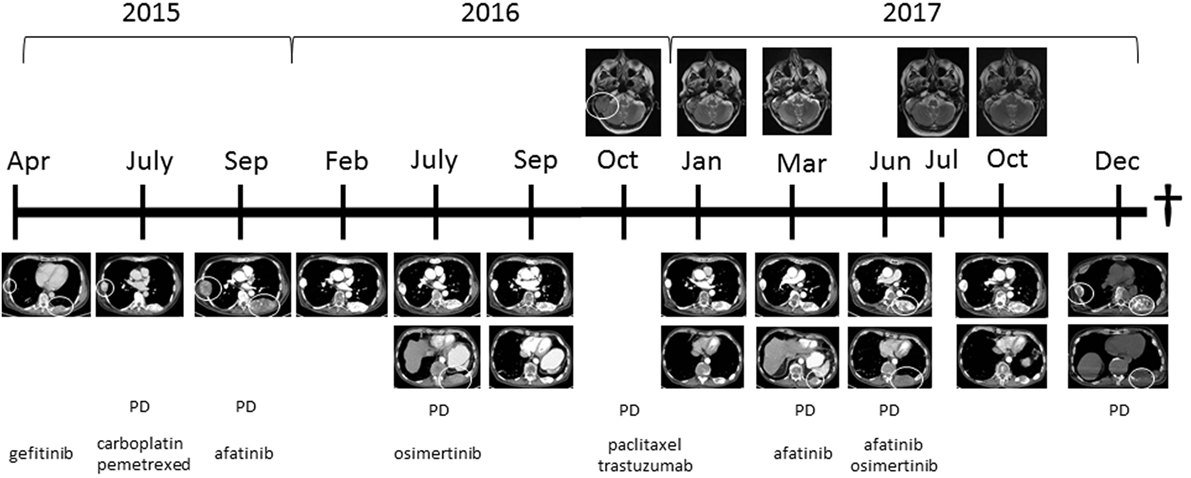
Figure 1. Computed tomography images showing the thoracic wall lesions of the adenocarcinoma of the lung and MRI images showing the metastasis in the skull with ingrowth in the brain at different time points in combination with treatment started at those time points. Abbreviation: PD, progressive disease. White circles indicate lesion, which showed progression.
Four months prior, in April 2015, he was diagnosed with an adenocarcinoma of the left lung with multiple bone metastases in sternum, ribs, and vertebrae. A biopsy from a metastasis in the left femur showed a mutation in the EGFR gene: c.2573T>G; p.(L858R). He was initially treated with gefitinib. After 2 months, the patient showed progression of bone metastases; the same EGFR mutation was found in a biopsy of a rib metastasis, without additional mutations in other mutational hotspots (e.g., BRAF, KRAS, HER2, KIT, ALK, NRAS, PDGFRA, PIK3CA, and MET). Therefore, at that time, carboplatin and pemetrexed were provided and because of pain, local irradiation of a sternal metastasis was applied. After two cycles of chemotherapy, the patient showed disease progression and was referred to our hospital. Because of lack of new treatment options, this patient was discussed in the Groningen MTB consisting of pulmonary oncologists, pathologists, clinical molecular biologists in pathology, general oncologists, and a structural biologist (www.moloncopath.nl). The MTB advised to determine the HER2-copy number status as a possible resistant mechanism for EGFR TKI. Fluorescent in situ hybridization (FISH) on a biopsy of a subcutaneous thoracic metastasis revealed HER2 amplification and treatment with afatinib (dual EGFR and HER2 inhibitor) 30 mg QD was started in October 2015. Evaluation by 18-fluorodeoxyglucose positron emission tomography and computer tomography (18-FDG-PET-CT) showed after 6 weeks a significant partial response with disappearance of the FDG activity of the bone metastases and after 4 months in the left upper lobe a single FDG-positive lesion was left. This lesion was irradiated by means of stereotactic ablative radiotherapy (1× 20 Gy), and afatinib was continued. Treatment with afatinib was well tolerated with minor skin rash; patient showed clinical improvement: he had less pain and more energy. Nine months after start of afatinib, progressive disease was again noticed. Growth of the primary tumor in the left upper lobe, a new ipsilateral pulmonary lesion and multiple new bone metastases including the skull, with ingrowth into the brain, causing paralysis of the right facial nerve (Figure 1). Sequence analysis of a new right-sided rib lesion showed the known L858R EGFR mutation and an additional T790M mutation.
Because of the novel T790M, afatinib was discontinued and replaced by osimertinib 80 mg QD (1). Eight weeks after start of osimertinib a PET-CT showed a response of most lesions except for a growing lesion in the pelvic region and the skull with ingrowth in the brain. Clinically there was, however, temporary improvement of the patient’s ability to move his right eyelid and right corner of the mouth, which had been paralyzed due to ingrowth of a skull metastasis into the brain and right facial nerve. A biopsy was performed of a growing FDG-positive lesion in the left pelvic bone that showed adenocarcinoma with the known L858R EGFR mutation, but the previously found T790M mutation was not present in this location (no biopsy of the skull metastasis available). The MTB advised to perform immunohistochemistry on Her2Neu (positive in agreement with HER2 amplification) and to determine MET amplification (negative by FISH). Based on these findings, it was decided to discontinue osimertinib because of the loss of the T790M mutation and to start a combination of paclitaxel 90 mg/m2 on days 1, 8, and 15, and trastuzumab 4 mg/kg on days 1 and 15, in cycles of 4 weeks, because trastuzumab is an HER2 antibody. Radiotherapy 1× 8 Gy was given on the pelvic lesion because of localized pain. 18-FDG-PET-scan after four cycles, paclitaxel and trastuzumab showed again a partial tumor response. No major side effects were observed although symptoms of the paralysis of the right facial nerve did not improve further; it remained stable during the course of therapy. The patient underwent plastic surgery on his right eyelid, which improved the closure of his right eye.
Two months after the fourth and last cycle, the patient presented with a subcutaneous metastasis on his forehead. Afatinib 30 mg QD was started, because this treatment worked before, pending results of a new biopsy. The biopsy, however, yielded insufficient material for mutation analysis, and re-biopsy was scheduled. In the meantime, 18-FDG-PET-scan showed multiple FDG-positive bone lesions (partly new lesions), some close to the myelum, and the patient was admitted to the hospital for radiotherapy on cervical and thoracic vertebrae. Afatinib was discontinued. Biopsy of a new lesion in the thoracic wall showed an EGFR-L858R, T790M mutation, and HER2 amplification. There were no other hotspot mutations in EGFR, BRAF, KRAS, ERBB2 (HER2), ALK, PIK3CA, or MET detected. The case was again reviewed by MTB. It was decided to treat the patient with afatinib 30 mg QD as well as osimertinib 80 mg QD at alternating days, to address the T790M mutation as well as the HER2 amplification resistance mechanism. Since the start of this latest treatment regimen, the subcutaneous skull metastasis disappeared, and the patient experienced less pain, regained his energy, and was able to walk outdoors again. The most recent 18-FDG-PET-CT-scan, 4 months after the start of this latest treatment regimen, showed again a partial response of the bone metastases again. Two months later (December 2017) progression of disease was observed, and the performance status deteriorated. Patient insisted to take a new biopsy from a new thoracic wall metastasis. Mutations analysis showed the known EGFR L858R and T790M mutations together with a new mutation in HER2: L755S. However, his condition got worse in short time, and he died in January 2018. An overview of the clinical findings, the mutational status at different time points and the given treatment regimens is provided in Table 1.
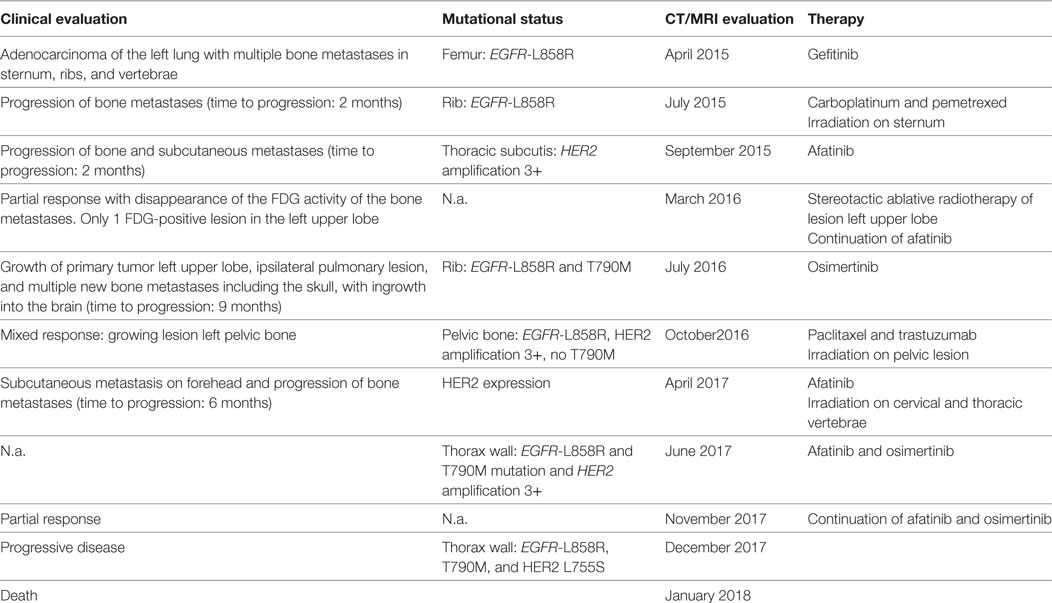
Table 1. Overview of clinical and pathological findings and subsequent therapeutic decisions.
Background
EGFR
The incidence of EGFR mutations in advanced stage adenocarcinoma of the lung in Caucasian patients is 10–15 and 40–60% in Asian patients (2). In the north of the Netherlands, the incidence is 9% (3). L858R mutation in exon 21 of the EGFR kinase domain is the main hotspot mutation in the EGFR gene and accounts for 35–45% of EGFR mutations (4, 5). L858R mutation increases the kinase activity of EGFR, leading to hyperactivation of downstream signaling pathways improving cell survival and proliferation (6). An EGFR TKI is the preferred first-line treatment in patients with activating EGFR mutation in non-small-cell lung carcinoma (NSCLC) (4). Gefitinib and erlotinib are first-generation TKI and registered as first-line treatment in patients with metastatic NSCLC with a tumor harboring an activating EGFR mutation within the European Union and according to the Dutch guideline for treatment of NSCLC (7). These small molecules bind competitively and reversibly to the adenosine triphosphate (ATP) binding site of the tyrosine kinase domain of EGFR. This prevents the autophosphorylation of the TK, blocks the activation of the EGFR signal transduction, inhibits tumor cell proliferation, and induces cell cycle arrest and apoptosis (8). The majority of patients will progress after 9–12 months of treatment due to various mechanisms of intrinsic or acquired resistance to first-generation EGFR TKIs (9).
EGFR T790M
The most common mechanism of acquired TKI resistance is the acquisition of a single recurrent missense mutation within exon 20, the T790M mutation (10). This mutation leads to the substitution of threonine by methionine at position 790, which encodes part of the kinase domain of the receptor and results in increased affinity for ATP (11). The T790M mutation can be detected in about 60% of tissue biopsy samples taken after acquired resistance (12, 13). As residue threonine at position 790 (T790) is located at the entrance in the back of the ATP binding cleft, substitution of residue threonine at position 790 with a bulky methionine (resulting in T790M) may cause steric interference with binding of TKIs (14). Irreversible inhibitors overcome this resistance simply through covalent binding (15). Osimertinib is registered for the treatment of NSCLC with an EGFR T790M mutation. It is a selective third-generation TKI which targets the ATP binding site of EGFR via irreversible covalent bond formation. In contrast to many other TKI, osimertinib penetrates the blood–brain barrier (16, 17). Osimertinib improves overall survival and progression-free survival in T790M-positive NSCLC patients with and without brain metastases (18, 19). Acquired resistance to osimertinib may be caused by primary coexistence of tumor cell populations with and without T790M mutation due to EGFR C797S mutation. Tumor progression can be explained by growth of the T790M negative population, while the tumor cells expressing T790M mutation are effectively suppressed by osimertinib (20).
HER2
Overexpression of HER2 induces cell transformation and tumorigenic growth and is clinically associated with resistance to erlotinib (21). HER2 amplification is detected in a subset of EGFR TKI resistant lung tumors. HER2 amplification and T790M mutation are thought to be mutually exclusive (22). However, in our patient HER2 amplification as well as T790M mutation appeared in the same biopsy of a new lesion in the thorax wall. Afatinib is an ATP-competitive aniline-quinazoline derivate which covalently binds to EGFR, HER2, and HER4 and irreversibly inhibits HER-family phosphorylation and signal transduction (23). As second generation TKI it is highly potent, irreversible dual EGFR/HER2 tyrosine kinase inhibitor, including the oncogenic EGFR-L858R mutation (23, 24). Afatinib is registered for advanced NSCLC with EGFR mutations. Clinical benefit of afatinib seems less in patients with de novo T790M mutations (25). Although afatinib is equally potent against wild-type EGFR and EGFR harboring the T790M mutation, in patients the dose is lower due to toxicity constraints (26).
Trastuzumab, a humanized monoclonal antibody against HER2, has been reported to be effective in HER2-positive NSCLC in vitro and in case reports (4, 27, 28).
Discussion
Here, we describe a patient with EGFR mutant advanced NSCLC with recurrent episodes of disease progression due to subsequent mutant clones. Yu et al. selected 155 patients with lung adenocarcinomas and acquired resistance to erlotinib or gefitinib. These patients underwent a re-biopsy. The most common finding was a T790M mutation. They also found transformation to small cell lung carcinoma, MET amplification and HER2 amplification (10). Sequist et al. described a wide variety of gained and lost EGFR mutations in a patient population with acquired drug resistance. They recommended reassessing cancers by taking new biopsies of growing lesions in patients with progressive disease after an initial response to TKI treatment (29). Following this strategy, we observed HER2 amplification and T790M mutation at different time points under the selective pressure of different EGFR TKI treatment. Of note, the occurrence of both aberrations at the same time has not been described earlier.
After discussion in the MTB about the most suitable therapy, as mentioned in the background, treatment was adjusted accordingly. Case evaluation by a multidisciplinary MTB is important to benefit from individualized genetic data and maximize clinical impact (30–32). MTB interprets results of routine molecular NGS-testing with those of other techniques, for example, immunohistochemistry, FISH, DNA methylation testing, and multiplex ligation-dependent probe amplification. NGS testing is not only performed on biopsies but currently also from tumor DNA in peripheral blood. The spectrum of molecular markers is constantly growing. Patients who progress after an EGFR TKI should undergo a re-biopsy to perform molecular analysis specifically looking for acquired mechanisms of resistance, such as EGFR T790M mutation. This approach can influence the next therapeutic step or reveal alternative EGFR TKI resistance mechanisms such as transformation to small cell lung cancer or bypass tracks that could potentially be addressed in clinical trials (11). Our patient responded well to subsequent treatment based on aberrations found in NGS, IHC, and FISH after discussion in the MTB. We observed that a change in treatment gave a short-lasting clinical improvement of several months and tumor response of fast growing new metastases. Osimertinib alternating with afatinib for T790M in EGFR and HER2 amplification was very effective in decreasing tumor sites. However, we expected that our patient would have immense toxicity of skin rash and diarrhea, but toxicity was not more than CTC grade 1. Long-lasting tiredness grade 1 was the most prominent side effect.
Concluding Remarks
This case report shows the importance of re-biopsy of growing lesions in lung cancer patients with metastatic progressive disease under targeted therapies. Mutation status can vary under selection pressure of these drugs, and knowledge of these changes makes it possible to adapt treatments. This patient also exemplifies the importance of having a multidisciplinary expert team (MTB) to give rational treatment advice in cancer patients with uncommon mutations or combinations of mutations causing complex resistance mechanisms.
Ethics Statement
This case report was written and offered for publication with written informed consent from the patient. The patient gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
AM: organized the relevant information about the patient; wrote all paragraphs of the article, did the literature research, and applied changes brought in by the other authors. AE, HG, and ES: supervised and corrected the manuscript. NH: supervised and gave advice. AW: main supervisor; supervised and corrected the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors AW.
Acknowledgments
Written informed consent to write and publish this case report was obtained from the patient. We thank the patient for his permission to publish this case report.
References
1. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med (2017) 376:629–40. doi:10.1056/NEJMoa1612674
2. Tan DS, Mok TS, Rebbeck TR. Cancer genomics: diversity and disparity across ethnicity and geography. J Clin Oncol (2016) 34:91–101. doi:10.1200/JCO.2015.62.0096
3. Kerner GS, Schuuring E, Sietsma J, Hiltermann TJ, Pieterman RM, de Leede GP, et al. Common and rare EGFR and KRAS mutations in a Dutch non-small-cell lung cancer population and their clinical outcome. PLoS One (2013) 8:e70346. doi:10.1371/journal.pone.0070346
4. Besse B, Adjei A, Baas P, Meldgaard P, Nicolson M, Paz-Ares L, et al. 2nd ESMO consensus conference on lung cancer: non-small-cell lung cancer first-line/second and further lines of treatment in advanced disease. Ann Oncol (2014) 25:1475–84. doi:10.1093/annonc/mdu123
5. Marchetti A, Martella C, Felicioni L, Barassi F, Salvatore S, Chella A, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol (2005) 23:857–65. doi:10.1200/JCO.2005.08.043
6. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science (2004) 305:1163–7. doi:10.1126/science.1101637
7. Felip E, Gridelli C, Baas P, Rosell R, Stahel R, Panel Members. Metastatic non-small-cell lung cancer: consensus on pathology and molecular tests, first-line, second-line, and third-line therapy: 1st ESMO Consensus Conference in Lung Cancer; Lugano 2010. Ann Oncol (2011) 22:1507–19. doi:10.1093/annonc/mdr150
8. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther (2005) 315:971–9. doi:10.1124/jpet.105.084145
9. Cortot AB, Janne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev (2014) 23:356–66. doi:10.1183/09059180.00004614
10. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res (2013) 19:2240–7. doi:10.1158/1078-0432.CCR-12-2246
11. Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG, et al. Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2016) 27:v1–27. doi:10.1093/annonc/mdw326
12. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res (2011) 17:1169–80. doi:10.1158/1078-0432.CCR-10-2277
13. Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res (2005) 65:7096–101. doi:10.1158/0008-5472.CAN-05-1346
14. Zou B, Lee VHF, Chen L, Ma L, Wang DD, Yan H. Deciphering mechanisms of acquired T790M mutation after EGFR inhibitors for NSCLC by computational simulations. Sci Rep (2017) 7:6595. doi:10.1038/s41598-017-06632-y
15. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A (2008) 105:2070–5. doi:10.1073/pnas.0709662105
16. Sullivan I, Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front Med (2017) 3:76. doi:10.3389/fmed.2016.00076
17. Ward RA, Anderton MJ, Ashton S, Bethel PA, Box M, Butterworth S, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem (2013) 56:7025–48. doi:10.1021/jm400822z
18. Santarpia M, Liguori A, Karachaliou N, Gonzalez-Cao M, Daffina MG, D’Aveni A, et al. Osimertinib in the treatment of non-small-cell lung cancer: design, development and place in therapy. Lung Cancer (2017) 8:109–25. doi:10.2147/LCTT.S119644
19. Yang JC, Ahn MJ, Kim DW, Ramalingam SS, Sequist LV, Su WC, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J Clin Oncol (2017) 35:1288–96. doi:10.1200/JCO.2016.70.3223
20. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol (2015) 26:2073–8. doi:10.1093/annonc/mdv319
21. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene (2007) 26:6469–87. doi:10.1038/sj.onc.1210477
22. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov (2012) 2:922–33. doi:10.1158/2159-8290.CD-12-0108
23. Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther (2012) 343:342–50. doi:10.1124/jpet.112.197756
24. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene (2008) 27:4702–11. doi:10.1038/onc.2008.109
25. Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol (2015) 16:830–8. doi:10.1016/S1470-2045(15)00026-1
26. van der Wekken AJ, Saber A, Hiltermann TJ, Kok K, van den Berg A, Groen HJ. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit Rev Oncol Hematol (2016) 100:107–16. doi:10.1016/j.critrevonc.2016.01.024
27. Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol (2013) 31:1997–2003. doi:10.1200/JCO.2012.45.6095
28. Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med (2006) 354:2619–21. doi:10.1056/NEJMc060020
29. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med (2011) 3:75ra26. doi:10.1126/scitranslmed.3002003
30. Tafe LJ, Gorlov IP, de Abreu FB, Lefferts JA, Liu X, Pettus JR, et al. Implementation of a molecular tumor board: the impact on treatment decisions for 35 patients evaluated at Dartmouth-Hitchcock Medical Center. Oncologist (2015) 20:1011–8. doi:10.1634/theoncologist.2015-0097
31. Petty JK, Vetto JT. Beyond doughnuts: tumor board recommendations influence patient care. J Cancer Educ (2002) 17:97–100. doi:10.1080/08858190209528807
Keywords: EGFR, HER2, non-small-cell lung carcinoma, TKI, brain metastasis, Molecular Tumor Board
Citation: Meedendorp AD, ter Elst A, ’t Hart NA, Groen HJM, Schuuring E and van der Wekken AJ (2018) Response to HER2 Inhibition in a Patient With Brain Metastasis With EGFR TKI Acquired Resistance and an HER2 Amplification. Front. Oncol. 8:176. doi: 10.3389/fonc.2018.00176
Received: 10 January 2018; Accepted: 04 May 2018;
Published: 22 May 2018
Edited by:
Lizza E. L. Hendriks, Maastricht University Medical Center, NetherlandsReviewed by:
Timothy F. Burns, University of Pittsburgh Cancer Institute, United StatesLuigi Formisano, Vanderbilt University Medical Center, United States
Copyright: © 2018 Meedendorp, ter Elst, ’t Hart, Groen, Schuuring and van der Wekken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthonie J. van der Wekken, YS5qLnZhbi5kZXIud2Vra2VuQHVtY2cubmw=