 Mihnea Dragomir1,2,3
Mihnea Dragomir1,2,3 George A. Calin1,4*
George A. Calin1,4*
- 1Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Research Center for Functional Genomics, Biomedicine and Translational Medicine, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 3Department of Surgery, Fundeni Clinical Hospital, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania
- 4Center for RNA Interference and Non-Coding RNAs, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Circular RNAs (circRNA) are RNA molecules built from fragments of linear pre-messenger RNAs and other linear RNA species through a process termed “back-splicing” in which the 3′ and 5′ ends are joined together giving rise to a covalently uninterrupted loop. circRNAs are not new members of the RNA world; they were first discovered in the early 1990s. The novelty is their abundance in the mammalian cells, as recently thousands of circRNAs were discovered and annotated. The biogenesis of circRNAs is a partially characterized process, regulated by three different mechanisms: exon skipping, intron pairing, and RNA-binding proteins. On the other hand, the function of circRNAs remains largely unknown and only a handful of singular reports describe in detail the biological roles of some circular transcripts. In a very short period of time, numerous circRNAs were associated with various cancer types and were also identified in bodily fluids with the potential of being disease-specific biomarkers. In this review, we briefly describe the biogenesis and function of circRNAs and present the circular transcripts that were more than once reported in literature to be associated with cancer. Finally, we point out some of the difficulties encountered in the study of circRNAs in cancer, as we consider that taking these into account could accelerate and improve our understanding of the biologic and translational use of circRNAs in human diseases.
Generalities about Circular RNA
Circular RNAs (circRNAs) are among the last addition to the ever-growing world of non-coding RNA (ncRNA) molecules. CircRNAs are built from the exons of linear pre-messenger RNAs (mRNAs) through a process termed “back-splicing” in which the 3′ and 5′ ends of the exon are joined together giving rise to a covalently uninterrupted loop. This classical definition of circRNA is challenged by numerous exceptions making this transcript so hard to characterize.
First, circRNAs are not new members of the ncRNA world. CircRNAs were first discovered in the 1990s when Nigro et al. observed that the exons of the tumor suppressor DCC after being spliced are joined in a different order than their genomic sequence. The upstream 5′ exons were moving downstream of 3′ exons where they were binding in a non-sequential manner, resulting in circular isoforms of DCC. They termed this novel RNA transcripts as scrambled exons (1). Two years later, it was discovered that in adult mice, the Sry gene is expressed as a circular transcript and not as a linear mRNA, which plays a crucial role in the determination of the sex during embryogenesis. The circular Sry does not bind polysomes and most probably is not translated (2). In the next decades, some anecdotal accounts describing the circularization of endogenous RNA followed (3–6), but none were convincing enough to change the perception of the scientific world regarding this topic. The breakthrough came in 2012, when Salzman et al. underlined the abundance of circRNA species in the mammalian cells, discovering and annotating thousands of circular transcripts (7).
Second, circRNAs can originate from several types of RNA molecules. Not only exons are susceptible to circularization, but also introns, long non-coding RNAs (lncRNAs), antisense transcripts, or intergenic regions. Additionally, circRNAs originating from coding regions can be assembled from multiple exons and sometimes they also include intronic regions (8) (Figure 1). A possible generalization regarding the source of circRNA is that most of them originate from two or three pre-mRNA exons that exceed the average length and that the flanking introns are likewise unusually long (9).
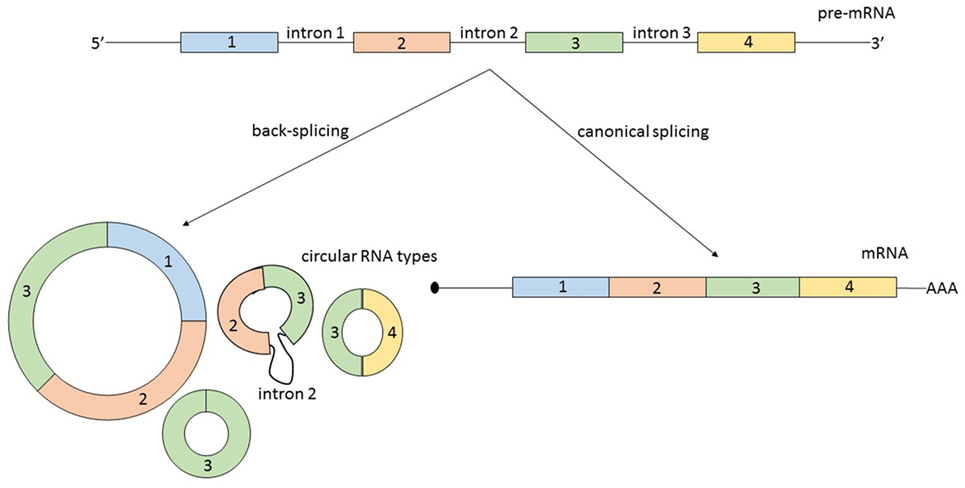
Figure 1. From the same linear pre-mRNA, multiple types of circular RNA (circRNAs) can be generated via back-splicing, a form of alternative splicing. CircRNAs can be composed by one or more exons and some circular transcripts are containing also intronic segments. As a generalization: circRNAs contain two or three exons, exceeding the average length, and the flanking introns are likewise longer. The relationship between the expression level of a mature messenger RNA (mRNA) and circRNA originating from the same pre-mRNA is not always correlated and is not predictable.
Third, some circRNAs are translated into proteins (10, 11). The first circRNA which was described to associate with polysomes and to translate is circ-ZNF609 (10). Therefore, one can ask if all circRNAs are in fact ncRNA transcripts.
In conclusion, this new class of transcripts cannot be yet very precisely defined. CircRNAs can be perceived as transcripts that mainly originate from the coding region of the DNA, which occasionally prove to have a translational potential, but usually are non-coding.
Biogenesis of circRNA
The unanswered questions regarding circRNA biogenesis does not concern the mechanism which is mostly explained, but the factors that regulate the formation of the circRNA loop. Very intriguing, circRNAs are expressed differently in various cell types of an organism and during the stages of ontogenesis, but from a phylogenetic point of view, a similar expression level between the same cell types is conserved (12). Hence, the synthesis of circRNAs seems to be a regulated process. Additionally, the relationship between the expression of an mRNA and circRNA originating from the same pre-mRNA is not linear and is not predictable. There are several publications revealing that some circRNAs are more abundantly expressed than their associated linear mRNA isoform (8, 13). Therefore, circRNAs are most probably not transcriptional noise and most likely have a biological function.
The term that summarizes the circRNA biogenesis is back-splicing, in which the head-to-tail splice junctions are joined together and form a circular transcript. But what makes this non-canonical splicing possible? There are three known processes that can lead to back-splicing (Figure 2).
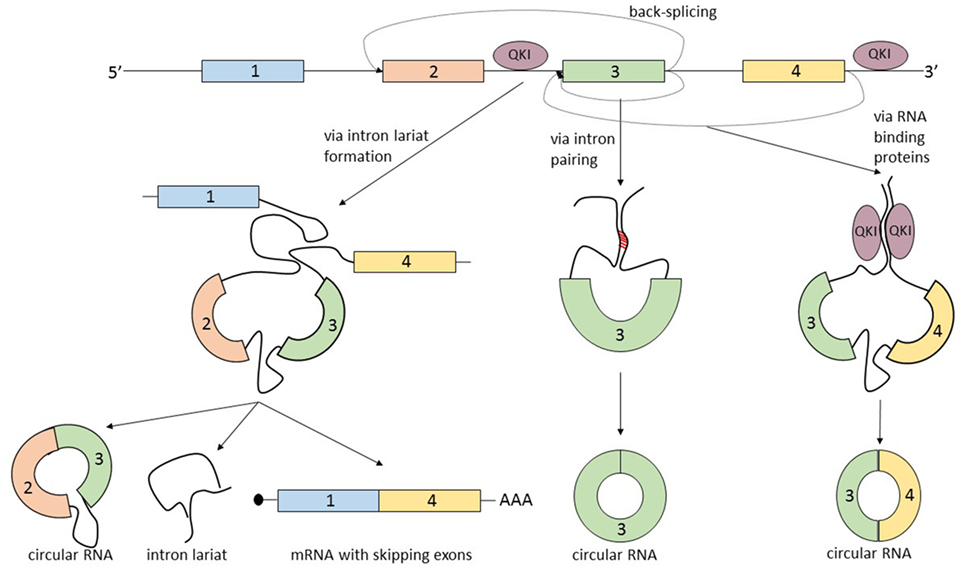
Figure 2. The biogenesis of circular RNAs (circRNAs) is summarized by the term “back-splicing.” Until now, three different mechanisms that promote this process were described. The first mechanism is characterized by exon skipping and intron lariat formation. In order to bring distant exons together (jumping exons), the introns build a circular lariat. The lariat formation makes the circularization of the “skipped” exons possible. This process leads to the formation of three different types of RNA molecules: circRNA, intron lariat, and mRNA with skipping exons. In the second mechanism, the circularization of the exon/exons is promoted by the complementary pairing of the flanking introns. A common hallmark of the introns that are prone to pair is the inverted ALU repeats. The third mechanism is controlled by RNA-binding proteins, which bind the neighboring introns of the future circular exon and dimerize, creating an RNA loop. The most studied proteins able to induce circularization are Quaking (QKI) and Muscleblind (MBL).
The first mechanism that promotes back-splicing is exon skipping. A pre-mRNA is spliced and two transcripts are generated: a mRNA from which one or more exons are missing, and a lariat containing the skipped exons which makes the circularization possible. The exon lariat is one more time spliced (process termed intralariat splicing) and two other molecules are produced: a circRNA and an intron lariat (4). In order to confirm this mechanism, Jeck et al. researched the association of circRNAs and exon skipping transcripts. For 45% of tested circRNAs, they identified a corresponding exon skipping mRNA (9). Furthermore, these observations are supported by another group which found a linear correlation between circularization and the exon skipping phenomenon (14).
A second mechanism that leads to the circularization of exons is intron pairing: the two introns that flank the exon/exons of the nascent circRNA have a complementary structure able to bind to each other. The pairing of the flanking introns brings the splice sites close to each other creating a secondary structure that makes back-splicing possible (15–18). Computational analysis revealed that a common feature of this flanking introns are the inverted ALU repeats (9). This feature of circRNA makes it possible to predict the sites of circularization by using bioinformatic methods.
The third mechanism of circRNA formation is directed by RNA-binding proteins (RBP). The simplest example of this model of biogenesis is the interaction between the RBP Quaking (QKI) and the flanking introns of the forming circRNA. QKI proteins bind to each of the flanking introns and dimerize creating a bridge between them. This process leads to the formation of a closed RNA loop allowing back-splicing. This mechanism was demonstrated in the case of the SMARCA5 gene, where only in the presence of QKI, a circular transcript can be generated (19). A similar mechanism was described by Ashwal-Fluss et al., which showed that the biosynthesis of the circRNA, circMbl, is controlled by the Muscleblind protein (MBL). High levels of MBL bind to its own pre-mRNA and determine the back-splicing of it, leading to the inhibition of the canonical splicing and decreasing the levels of MBL, and increasing the levels of circMBL. The authors describe the circRNA biogenesis as a process that competes with canonical splicing, and as a post-transcriptional regulator of protein synthesis (20). It is not yet clear how broad the RBP mechanism of generating circRNA is and in which way it can be used to create computational predictions regarding the exons prone to circularization. There is an additional mechanism that describes the biogenesis of circRNA which actually combines two of the previous described mechanisms: intron pairing and RBP. The RBP ADAR is responsible for A-to-I RNA editing and was recently linked to the circRNA biogenesis. Because it was well known that ADAR interacts with double strand ALU repeats (21, 22), Ivanov et al. hypothesized that the ADAR enzyme by decreasing the complementary of the inverted ALU repeats of introns can lead to a downregulation of the synthesis of circRNA. These observations were confirmed by knockingdown ADAR in vitro. The authors detected a two-fold upregulation of 84 circRNA and the downregulation of the corresponding linear transcripts (23).
Most probably the biogenesis of circRNA is not yet fully deciphered and the above described mechanisms represent only a limited view. Ebbesen et al. consider that the three mechanisms of circRNA biogenesis can be overlapping processes and what we currently know are three perspectives on the same phenomenon (24). Future research is needed, because our understanding of the biogenesis of circRNA will enable the development of computational tools that can predict the genomic sights susceptible to circularization.
Function of circRNA
Research regarding the function of circRNAs is still limited and challenging. Conservation of a transcript between species is always a powerful argument that supports the functionality of a molecule. Even the conservation of circRNA is disputed. The first study that affirms the conservation of circRNA mentions that there are hundreds of analogous transcripts in the brain of mouse and humans but no details exist about the similarity of the primary structure of the molecules (7). Another study that compares the neuronal circRNAs in the brain of mice and humans (separated by about 80 million years of evolution) reports that 4,522 circRNAs out of 15,849 are highly conserved between the two species (12). By comparing the number of human genes that produce circRNAs to murine genes that code circular loops, it was detected an overlap of 22%, but only 69 circular molecules share the same start and stop points (9). Guo et al. compared the same species and observed that if a mouse gene can code for a circular transcript, then in 66% of the cases the ortholog human gene can also code circRNAs. Of these genes, only one-third shared the same splicing sites for circRNA in humans and mice. This discovery led to the conclusion that the pre-mRNA fragments that give rise to circRNAs do not have a higher degree of conservation than their neighboring exons (25). Additionally, another research group studied the conservation of circRNA from the brain of pig and mice and discovered that the splice site of circularized exon loops matches between the two species at a proportion of 20.4% (26). The results between studies are divergent and the cause is easily identifiable: the conservation is higher if comparing the expression of circRNA between the same organs or tissues (i.e., brain), and if comparing the conservation of highly expressed circRNAs.
A second argument for the functionality of circRNAs is that these transcripts are expressed in a cell-specific manner and have a determined subcellular localization. Regarding their abundance, circRNAs have the highest level in brain and this feature is highly conserved (12, 26–28). In regard to their diversity, Maass et al. analyzed the expression of circRNAs in 20 different human tissues and discovered 5,225 transcripts, with the highest expression of circRNAs in platelets (3,324 circRNAs, out of which 2,339 unique to this cell type) (8). Recently, circRNAs were discovered also in bodily fluids with the potential of being disease-related biomarkers. The first account of circRNAs in the extracellular environment described over 400 different circRNAs in the saliva of healthy donors (29). Subsequently, circRNAs were detected in whole blood; over 2,400 circRNAs were discovered, which had expression levels similar to neuronal tissues (30). The world of circulating circRNAs was further characterized: circRNAs are abundant in exosomes and can be used to diagnose colorectal cancer (CRC). Compared to healthy controls, exosomes from CRC patients contain 257 new species of circRNAs, while other 67 circRNAs are missing (31). It appears that a precise mechanism controls the sorting and exporting of circRNAs into circulation via exosomes exists and needs to be revealed.
A third indirect argument of the functionality of circRNAs is their high stability compared to other RNA species. It was reported that a circRNA has a half-life of approximately 48 h (9), probably due to the resistance to RNA exonuclease. The half-life of a circRNA is two to four times longer than a mRNA. This difference in kinetics makes it difficult to interpret the relationship between the two transcripts (circRNA and corresponding mRNA) that originate from the same pre-mRNA.
Concrete information of the function of circRNA is scarce. Data about the mechanism of some individual circRNAs are available. But in all cases, the properties that confer functional potential cannot be extrapolated to other circRNAs. Simultaneously, two articles reported that the circRNA, antisense to cerebellar degeneration-related protein 1 transcript (CDR1as) has more than 70 binding sites for miR-7. MiR-7, associated with Argonaute proteins, binds to CDR1as, but the RISC complex does not degrade the circRNA (32, 33). CDR1as, a circRNA abundant in the brain of mammals (34), seems to act as a sponge for miR-7, which in the presence of CDR1as is strongly suppressed (Figure 3A). In vivo, overexpression of CDR1as leads to a phenotype characterized by impaired midbrain development, comparable to miR-7 knockdown models (32). Similarly, SRY, one of the first described circRNAs (2), has 16 binding sites for miR-38, also appearing to be an endogenous sponge (33). In order to check if other circRNAs also have sponging potential, Guo et al analyzed the interaction of 7,112 circRNAs with 87 microRNA (miRNA) families. The authors concluded that CDR1as which contains 71 binding sites for miR-7 is an exception and the next best miRNA sponge is circRNA ZNF91 which has 24 binding sites for miR-23 (25).
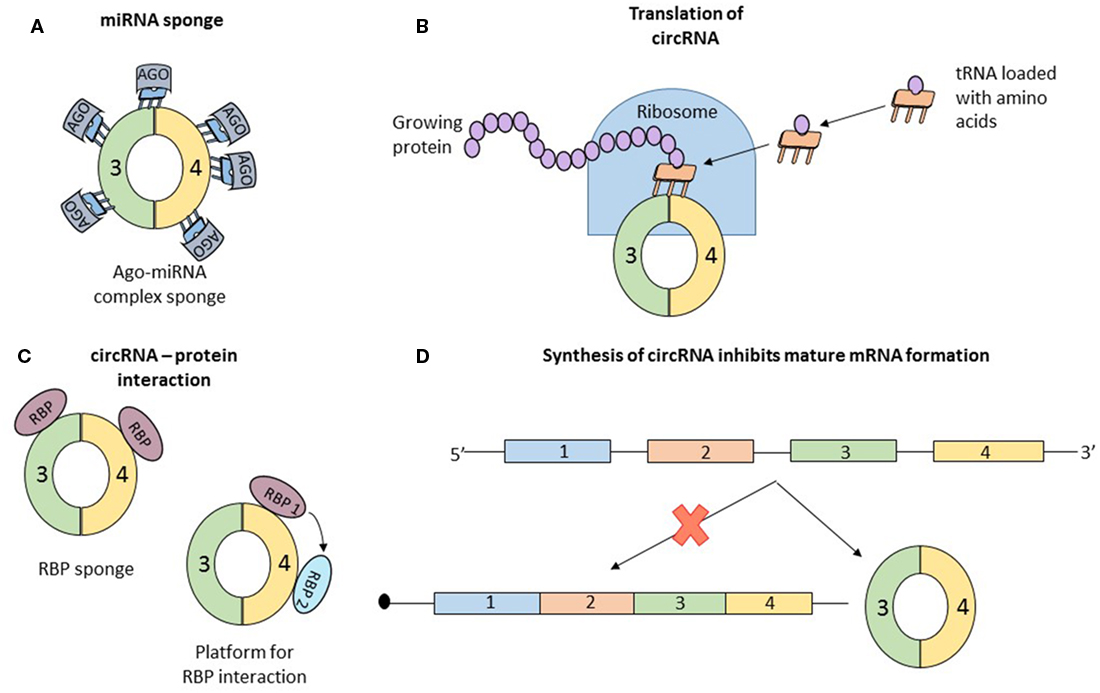
Figure 3. Data about the function of circular RNAs (circRNAs) is limited; until now, only four roles of circRNAs were studied in detail, and the observations regarding the function cannot be generalized to all circular transcripts. (A) The most studied function of circRNAs is microRNA (miRNA) sponging. By sequestering miRNA molecules that downregulate mRNAs, circRNAs indirectly increase the expression of mRNAs. Initially proposed as potent miRNA sponges which are able to bind to dozens of short RNAs, circRNAs are considered now to have the same number of interaction sites with miRNAs as other molecules. (B) Recently, two papers reported that circRNAs can associate with ribosomes and be translated into short proteins, long enough to contain a functional domain. (C) Similar to miRNA sponging, circRNAs also bind to proteins. The exact function of this interaction is not clear and numerous speculations exist: circRNA are a transport vehicle for proteins; circRNAs are sequestering proteins; circRNAs are a platform for protein interactions; or circRNAs bind to proteins and induce allosteric changes, regulating their function. (D) Finally, simply the formation of circRNAs can be perceived as a function of circular transcripts. It was reported that synthesis of a circRNA from a pre-mRNA competes with the formation of a linear, functional mRNA. Therefore, circRNA formation is another form of post-translational regulation.
Recently, two papers proved that circRNAs can be translated into proteins (Figure 3B). Circ-ZNF609 has the ability to associate with polysomes and because of its primary structure containing “start” and “stop” codons, this transcript is translated in a cap-independent manner (10). One of the papers also confirmed that several circRNAs, in vitro and in vivo, associate with translating ribosomes and produce small proteins in a cap-independent way. Furthermore, the authors present the necessary characteristics of a circRNA to be translated: frequently contain the start codon of the host gene, are longer than other circRNAs, and contain a stop codon highly conserved and located near the splice site. The translated proteins originating from circular transcripts are long enough and contain a full protein domain, suggesting functional potential (11). The activity of the proteins encoded by these circRNAs is unknown and remains to be elucidated.
Several research papers report that circRNAs associate with RBP (Figure 3C). Maybe the best known example is that of circMBL. The mature MBL protein not only binds to the introns flanking the circularized exon but also interacts directly with the exons. The interaction with the exon does not affect the biogenesis of circRNA and the authors assume that circMBL can sponge excessive MBL and play a role in subcellular delivery of the mature protein (20). In another paper, it was proven that circRNAs form cytoplasmic complexes with proteins. 12 circRNAs were tested, and all of them can build complexes with proteins. A more detailed analysis using RNA-seq and immunoprecipitation showed that the RBP, insulin-like growth factor 2-binding protein 3 (IGF2BP3) can interact with three distinct circRNAs (CDYL, NFATC3, and ANKRD17) (35). These functional details open more questions than provide answers: (1) Why do circRNAs bind to proteins? (2) Are all circRNAs able to interact with RBP? and (3) What characteristics should a circRNA have to bind to a specific type of protein? Hentze and Preiss provide some hypothetical answers for these questions but experimental confirmation is still missing. The two authors propose multiple scenarios: (a) circRNAs are vehicles for RNP, delivering these molecules in a specific subcellular localization; (b) similar to the circRNA–miRNA interaction, circRNAs can be sponges for RNP, sequestering the proteins which will be unable to perform their functions; (c) circRNAs can be a platform for multiple RBPs, which can interact with each other by binding to a circularized transcript; and (d) circRNAs can bind RBPs and induce allosteric changes, regulating the function of the proteins (36). Like in the case of circRNAs acting as miRNA sponge also the hypothesis that circRNAs can interact with RBPs is disputed. You et al. predicted the capacity of circRNA to bind RBP and concluded that circular transcripts possess a lower affinity to bind RBPs than coding regions and 3′UTR of linear transcripts (28). Future research is necessary to clarify the role of the interaction of circRNAs with RBPs.
Finally, the circRNAs may not have a function, but simply the assembly of the circular transcripts can be perceived as a translational brake, a different type of post-transcriptional regulation (Figure 3D). Every time a circRNA is synthesized, the formation of the mature linear mRNA is impeded, the two transcripts (mRNA and circRNA) compete for their production. The already well-known example of muscleblind gene is a model of this phenomenon. Conditioned by the presence of the mature MBL protein, the pre-mRNA of MBL can be spliced canonical or back-spliced. If the MBL protein is abundant, it binds to the flanking introns of exon 2 of the pre-mRNA and induces its circularization generating a circRNA; if the expression of MBL is low, canonical splicing takes place and a mature mRNA is synthesized, which will be transcribed into MBL proteins. This process is highly conserved, from flies to humans, and can be perceived like an auto-feedback mechanism that controls the expression level of MBL. Exogenous MBL overexpression leads to an increased circularization of the second exon, whereas a low level of MBL induces canonical splicing (20). But once again, this process remains singular and may be an exception.
Cancer and circRNA
The same enthusiasm observed after the discovery of the association of miRNA with cancer (37) is happening now with circRNA. In a very short period of time, numerous circRNAs were associated with various solid and hematologic malignancies. Complete reviews of the circRNAs down-/upregulated in cancers can be found elsewhere (38–40). We included in this review only circRNAs that were associated with the same type of cancer/different types of cancer in at least two publications (Table 1). Only a negligible number of circRNAs fulfill these conditions. By including circRNAs that were linked with multiple types of cancer, we observe a lack of specificity of a circRNA for a given type of malignancy.
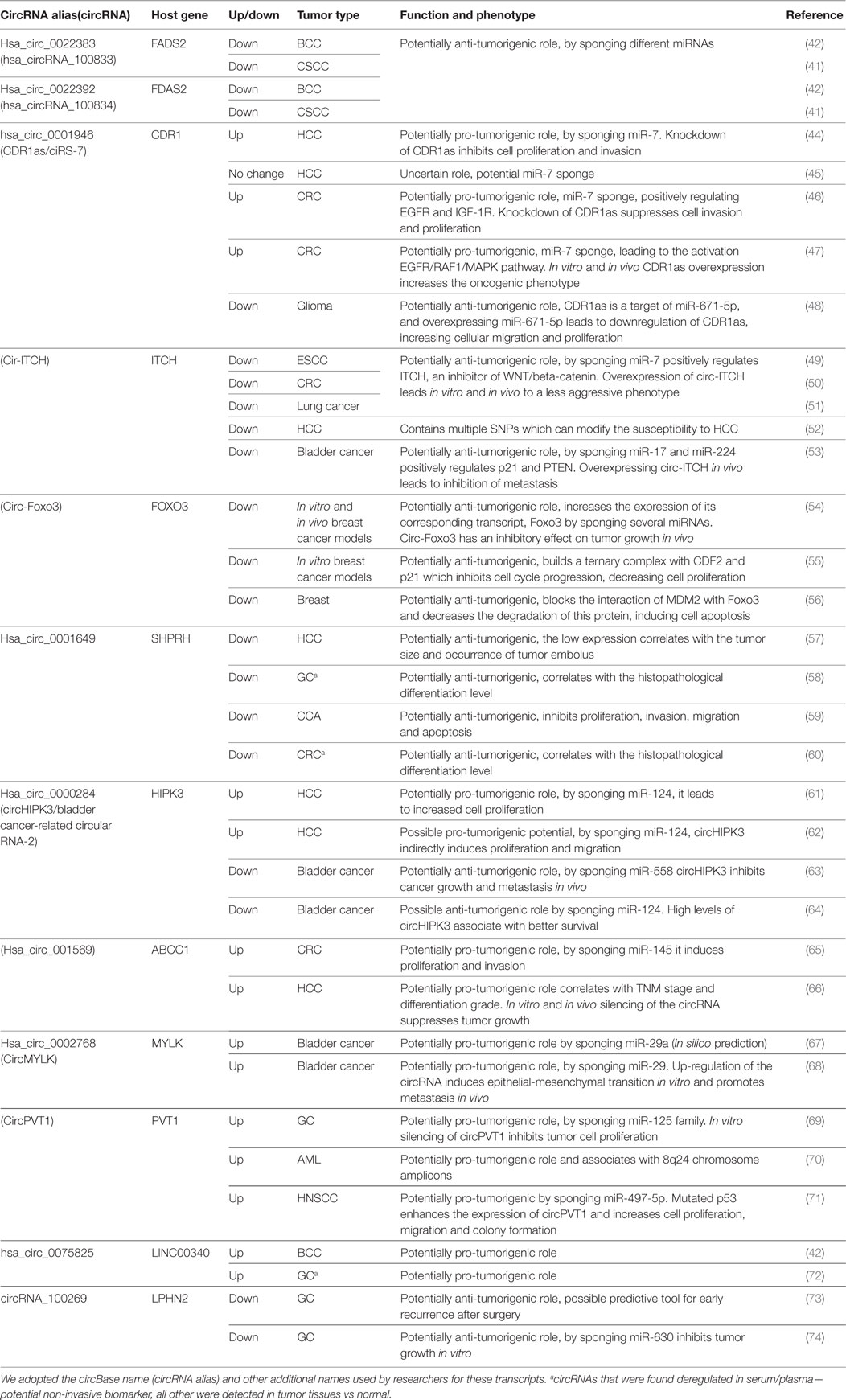
Table 1. CircRNAs that were reported by two or more studies to be associated with the same/different types of cancers.
Circular RNAs were linked to dermatologic malignancies. By performing microarray and confirming the data by qRT-PCR, Sand et al. discovered 143 up- and 179 downregulated circular transcripts in tissue samples of cutaneous squamous cell carcinoma (CSCC) compared to controls. The top 2 up- and downregulated circRNAs were hsa_circ_0070933, hsa_circ_0070934 and hsa_circ_0022392, hsa_circ_0022383, respectively (41). By performing the same analysis, the same research group reported that the top two downregulated circRNAs in CSCC have also a lower expression level in basal cell carcinoma (BCC) compared to normal tissue (42). To our knowledge, only one circRNA was linked to melanoma, the circular isoform of the lncRNA ANRIL, circANRIL. It was observed that numerous isoforms of circANRIL are present in the cytoplasm of melanoma cell lines, while the linear lncANRIL is abundant in the nucleus (43).
Most of the cancer related circRNAs reported by now are associated with malignancies of the gastrointestinal tract. Yu et al. detected CDR1as to be upregulated in hepatocellular carcinoma (HCC) compared to normal adjacent tissue and that the expression of miR-7 anticorrelates with that of CDR1as. In vitro experiments showed that CDR1as is an oncogene by sponging miR-7 and increasing cancer cell proliferation and invasion potential (44). More recently, another group analyzed the level of CDR1as in HCC tissue versus paired neighboring normal tissue and observed that the circular transcript has the tendency for a lower expression in malignant tissue. Curiously, a high expression of CDR1as was associated with microvascular invasion and acted as a miR-7 sponge (45). The two publications contradict to some extent one another; thus, supplementary research is needed to elucidate the real function of CDR1as in HCC, if any function exists. Furthermore, other two publications reported that CDR1as is upregulated in CRC tissue compared to paired healthy mucosa, and also both papers show independently that a high level of this circular transcript is associated with poor overall survival. Mechanistically, the overexpressed CDRas1 sponges the tumor suppressor miR-7 and the downstream target of miR-7 – EGFR – is upregulated (46, 47). CDR1as was also associated with glioblastoma, since tumor biopsies had a lower level of CDR1as compared to biopsies from normal controls. The expression of CDR1as negatively correlates with that of miR-671-5p in glioblastoma. Because this miRNA can degrade CDR1as, it can be hypothesized that miR-671-5p directly controls the expression of a circular transcript in cancer (48).
Cir-ITCH is another well-known circRNA already linked with five types of cancer. Three papers reported that cir-ITCH is downregulated in esophagus squamous cell carcinoma (ESCC), CRC, and lung cancer. The proposed mechanism of action was similar in all three articles: low levels of cir-ITCH are not able to sponge miR-7 and miR-214 which will target ITCH, an inhibitor of WNT/beta-catenin pathway. Cir-ITCH positively regulates the level of its host gene (49–51). In HCC, cir-ITCH seems similarly to be downregulated and low cir-ITCH is an independent prognostic factor. Moreover, cir-ITCH displays six different single-nucleotide polymorphism (SNPs) regions which can change the susceptibility to HCC (52). Recently, low cir-ITCH was associated with bladder cancer, and cir-ITCH plays a tumor suppressing role by directly binding miR-17 and miR-224, which if sponged lead to the inhibition of PTEN and p21 (53).
By using in vitro and in vivo models of breast cancer it has been shown that the circ-Foxo3 has a tumor suppressor potential. The authors demonstrated that high levels of circ-Foxo3 and Foxo3 pseudogene can sponge multiple miRNAs that target the mRNA Foxo3, which is transcribed in a protein with anti-neoplastic effect (54). The same study group additionally showed that circ-Foxo3 has a tumor suppressor function, independently of controlling the expression level of its host gene. Circ-Foxo3 can build together with p21 and CDK2, a complex that blocks the cell cycle progression by inhibiting the interaction of CDK2 with cyclin A and cyclin E (55). In a third paper, the authors confirmed that circ-Foxo3 is downregulated in breast tumor samples compared to benign mammary lesions. Circ-Foxo3 is additionally able to block the interaction of MDM2 with Foxo3 and rise the expression level of Foxo3, increasing the apoptosis of tumor cells (56).
Hsa_circ_0001649 is a circular transcript linked to multiple types of gastrointestinal tract cancers. In HCC, it was shown that the expression of circ_0001649 is downregulated in tumor tissue compared to paired healthy tissue (57). In gastric cancer (GC), hsa_circ_0001649 is downregulated in tumor tissue, consequently leading to expression drops in serum of GC patients after surgery, being a potential recurrence biomarker (58). The same circRNA is downregulated in cholangiocarcinoma (CCA) tissue and regulates cell proliferation, apoptosis, migration, and invasion (59). Ultimately, this circRNA is downregulated in CRC tumor tissue and in serum of CRC patients (60). Very curiously, no in vitro or in silico data exists regarding the molecular mechanism of hsa_circ_0001649, a transcript that seems to be implicated in multiple cancer types.
CircHIPK3 is the circular transcript formed from the second exon of HIPK3, a tumor suppressor gene with protein kinase activity. In HCC, circHIPK3 is overexpressed and sponges 9 different miRNAs displaying 18 miRNA interacting binding sites. Knockdown of circHIPK3, but not of HIPK3, leads to an inhibition of cellular proliferation, proving that the function of the circRNA is independent from that of the linear transcript (61). In a more recent study, it was confirmed that circHIPK3 is upregulated in HCC tissue compared to normal tissue and the level of the circular transcript anticorrelates with miR-124. The sponging of miR-124 by circHIPK3 leads to the overexpression of aquaporin 3, a transmembrane channel with tumorigenic function (62). On the contrary, circHIPK3 was reported to be downregulated in bladder cancer tissue compared to normal adjacent epithelium. Mechanistically, a low expression level of circHIPK3 cannot efficiently sponge miR-558, a miRNA which promotes angiogenesis by targeting heparanase. In vivo overexpression of the circRNA inhibits cell growth and lung metastasis (63). Furthermore, in a fourth study, it has been demonstrated that low levels of circHIPK3 are associated with low rate of progression free survival in bladder cancer, confirming the results of a previous research group (64). Hence, it is possible to hypothesize that circHIPK3 plays opposite roles in HCC and bladder cancer, being an oncogene and a tumor suppressor gene, respectively.
CircRNA_001569 was identified as an oncogene in CRC and HCC. In CRC, Xie et al. found that circ_001569 is upregulated and directly correlates to the T stage, N stage, M stage, and histopathological differentiation grade. The authors demonstrated that the circRNA regulates the proliferation and invasion of neoplastic cells by sponging miR-145 – a miRNA that downregulates multiple oncogenes: E2F5, BAG4, and FMNL2 (65). In HCC, the same circ_001569 is overexpressed and also correlates with the TNM stage and tumor differentiation grade, but data regarding the function of the circRNA were not provided (66).
CircMYLK is an additional circular transcript linked to bladder cancer. Huang et al. discovered circMYLK overexpressed in bladder cancer, and by using in silico methods, predicted that this circRNA with the lncRNA H19 could sponge miR-29a-3p leading to the overexpression of DNMT3B, VEGFA, and ITGB1 (67). In a subsequent study, the same research group confirmed their in silico data: circMYLK is overexpressed in bladder cancer and the expression level correlates with progression level. Additionally, circMYLK can directly bind to miR-29a-3p and control the expression level of VEGFA (68).
CircPVT1, due to the amplification of its DNA locus, is overexpressed in GC. The expression of circPVT1 and its equivalent not circularized RNA, PVT1, are inversely correlated. The association of high circular transcript and low lncRNA is an accurate overall survival prognosis marker in GC. The function of circPVT1 in GC is described only partially: this circRNA sponges the members of the tumor suppressor miR-125 family (69). Afterward, it was discovered that the same amplification of the 8q24 region containing the PVT1 gene leads to the overexpression of circPVT1 and in a lesser amount of the lncRNA PVT1 in acute myeloid leukemia (AML) patients with cytogenetic abnormalities. The phenotype of 8q24 amplification in AML is known, but remains unclear how circPVT1 contributes to this genomic alteration (70). A third article confirms the oncogenic potential of circPVT1: the circularized transcript is high in head and neck squamous cell carcinoma (HNSCC), especially in patients with TP53 mutations, and has the ability to sponge miR-497-5p (71).
One of the top overexpressed circRNAs in BCC (42), has_circ_0075825, was also found by RT-droplet digital PCR to have a high expression level in the plasma of GC patients (72).
In stage III GC, it was observed that 46 different circRNAs are deregulated, the top downregulated circular transcript being circRNA_100269 (73). In a subsequent paper from the same research group, it was confirmed that circRNA_100269 and its corresponding mRNA, LPHN2 are downregulated in GC. CircRNA_100269 has an anti-neoplastic activity by directly targeting miR-630, but further details are still lacking regarding the downstream targets of miR-630 (74).
Understanding circRNAs Lessons Learned from the Study of microRNAs
Shortly after the discovery of miRNAs implication in cancer, these transcripts were divided in oncomiRs and tumor suppressor miRNAs. Later, a more in depth research made it clear that a miRNA, which has the ability to target hundreds of different mRNAs, is too versatile to be classified as “good” or “bad.” The role of a miRNA in tumorigenesis is largely context dependent (i.e., depending on genomic features of the tumor). The same miRNA can be an oncogene in some tumor types and a tumor suppressor gene in other cancers. A similar tendency to divide circRNAs in oncogenes and tumor suppressor genes can be now remarked. We presented in this review only circRNAs which are reported by more than one paper. Already two circRNAs, CDRas1 and circHIPK3, were defined to have tumor suppressor potential by some researchers and oncogenic potential by others (44–48, 61–64).
There are hundreds of papers defining circRNAs as cancer biomarkers or tools that can aid the stratification and predict the outcomes of cancer. Already in some cases, the same circRNA was proposed by different groups as a diagnostic tool for multiple types of cancers. We selected for this review only the circRNAs which were linked more than once with neoplastic disease. Twelve different circRNAs fulfill this condition and nine of these are linked to more than one cancer type. Only three circRNAs are, until now, cancer type specific (Table 1). Each of these three specific circRNAs is supported by publications that originate from the same research group and have common authors. The example that best depicts the lack of specificity from our list is cir-ITCH, a circRNA associated with five different types of cancers: ESCC, CRC, lung cancer, HCC, and bladder cancer. This low level of specificity reminds us of the miRNA world, where the same miRNA is “specific” for multiple non-neoplastic and neoplastic diseases. This raises the question of how could these non-specific molecules find their way to the clinic. Two possible interpretations can be given to these observations: (a) circRNAs are a common driving mechanism of oncogenesis or (b) circRNAs are a common byproduct/end-product of oncogenesis. A very important aspect of future research is to determine where circRNAs are localized in the molecular pathogenic pathway.
The discovery of circRNAs in serum/plasma and their abundant presence in blood cells should be treated with caution. CircRNAs could be potential non-invasive biomarkers, and same pitfalls already proven for circulating miRNAs could be also true for circRNAs. Relatively late it has been proven that 58% of serum/plasma circulating miRNAs, which were proposed as cancer-specific biomarkers, are highly expressed in sub-populations of blood cells and only reflect the level of the circulating cells (75). The same can be true with circRNAs, since it was described that circRNAs are abundant in blood cells, predominantly in platelets and erythrocytes (8); hence, the high level of circRNAs from serum/plasma echoing the number of cells. Therefore, a precise characterization of the abundance of circRNAs in different blood cells, a good description of the in serum/plasma transportation mechanism, and the employment of methods that can predict the origin of the circulating circRNAs should be considered as preliminary steps before proposing that circRNA can solve the problem of “liquid biopsy.” Additionally, important steps like the pre-analytical and analytical processes, which can be the cause of supplementary errors and data misinterpretation, should be considered and analyzed before claiming that a circRNA is a circulating cancer specific biomarker.
The majority of the papers providing evidence that circRNAs play a role in cancer offer only limited information about their function. Most of the studies suggest that circRNAs fulfill their function by miRNA sponging. The concept of miRNA sponging became attractive after the competitive endogenous RNA (ceRNA) hypothesis was proposed: ncRNAs interfere with mRNA translation by binding (sponging) miRNAs that were supposed to complementary target the mRNA (76). A number of theoretical papers mathematically modeled this interaction (77, 78) and additional experimental data (79) showed this crosstalk is impossible because of the very low abundance of binding sites harbored by lncRNA or mRNA. The discovery of CDR1as with its repetitive structure and over 70 binding sites for the same miRNA relaunched the hypothesis. However, following in silico and experimental studies showed that no other circRNA has this inhibitory potential (25, 28). Therefore, a circRNA that usually is not abundant and has a limited number of interaction sites for a miRNA could not be a potent miRNA sponge and its function remains largely unknown. Moreover, the lack of a clear function of circRNAs reminds us of the fact that only a part of miRNAs are abundant enough in a tissue to truly exert a post-translational regulation of mRNA: miRNAs act more as buffers that maintain the translation in a state of equilibrium (80). Despite this, numerous papers present a dysregulated miRNA as the main mechanism of a pathogenic chain. Therefore, a better characterization of the function of circRNAs is necessary before truly demonstrating their implication in cancer. Moreover, it is important to realize that circRNAs, like miRNAs, are part of a complex molecular network. In order to systematically study their function it is necessary to assess their relationship not only with the linear mRNAs that arise from the common pre-mRNAs, but also the miRNAs and RBPs with which the circular transcript interacts and furthermore their downstream targets.
In conclusion, a basic understanding of the biology (i.e., biogenesis, function, localization, conservation) of circRNAs is necessary before trying to find a clinical application for these new molecules.
Author Contributions
Conception and design (MD); Provision of study materials (MD, GC); Collection and assembly of data (MD); Manuscript writing: (MD, GC); Figure Design (MD); Final approval of manuscript: (MD, GC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank Ana Carolina Paschoalini Mafra and Mr. Sergiu Dan Dragomir for their useful comments on the manuscript.
Funding
Work in Dr. Calin’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943-01 through the NIH Common Fund, Office of Strategic Coordination (OSC), the NIH/NCI grant 1 R01 CA182905-01, a U54 grant – UPR/MDACC Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Ladies Leukemia League grant, a CLL Moonshot Flagship project, a SINF 2017 grant, and the Estate of C. G. Johnson, Jr. The work of Mihnea Dragomir is supported by a POC grant, entitled “Clinical and economical impact of personalized targeted anti-microRNA therapies in reconverting lung cancer chemoresistance” – CANTEMIR, Competitivity Operational Program, 2014-2020, no. 35/01.09.2016, MySMIS 103375.
References
1. Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, et al. Scrambled exons. Cell (1991) 64(3):607–13. doi:10.1016/0092-8674(91)90244-S
2. Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, et al. Circular transcripts of the testis-determining gene Sry in adult-mouse testis. Cell (1993) 73(5):1019–30. doi:10.1016/0092-8674(93)90279-Y
3. Zaphiropoulos PG. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci U S A (1996) 93(13):6536–41. doi:10.1073/pnas.93.13.6536
4. Zaphiropoulos PG. Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol Cell Biol (1997) 17(6):2985–93. doi:10.1128/MCB.17.6.2985
5. Li XF, Lytton J. A circularized sodium-calcium exchanger exon 2 transcript. J Biol Chem (1999) 274(12):8153–60. doi:10.1074/jbc.274.12.8153
6. Houseley JM, Garcia-Casado Z, Pascual M, Paricio N, O’Dell KM, Monckton DG, et al. Noncanonical RNAs from transcripts of the Drosophila muscleblind gene. J Hered (2006) 97(3):253–60. doi:10.1093/jhered/esj037
7. Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One (2012) 7(2):e30733. doi:10.1371/journal.pone.0030733
8. Maass PG, Glazar P, Memczak S, Dittmar G, Hollfinger I, Schreyer L, et al. A map of human circular RNAs in clinically relevant tissues. J Mol Med (2017) 95(11):1179–89. doi:10.1007/s00109-017-1582-9
9. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu JZ, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA (2013) 19:141–57. doi:10.1261/rna.035667.112
10. Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell (2017) 66(1):22–37.e9. doi:10.1016/j.molcel.2017.02.017
11. Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of CircRNAs. Mol Cell (2017) 66(1):9–21.e7. doi:10.1016/j.molcel.2017.02.021
12. Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell (2015) 58(5):870–85. doi:10.1016/j.molcel.2015.03.027
13. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet (2013) 9(9):e1003777. doi:10.1371/journal.pgen.1003777
14. Kelly S, Greenman C, Cook PR, Papantonis A. Exon skipping is correlated with exon circularization. J Mol Biol (2015) 427(15):2414–7. doi:10.1016/j.jmb.2015.02.018
15. Pasman Z, Been MD, Garcia-Blanco MA. Exon circularization in mammalian nuclear extracts. RNA (1996) 2(6):603–10.
16. Dubin RA, Kazmi MA, Ostrer H. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene (1995) 167(1–2):245–8. doi:10.1016/0378-1119(95)00639-7
17. Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell (2014) 159(1):134–47. doi:10.1016/j.cell.2014.09.001
18. Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev (2014) 28(20):2233–47. doi:10.1101/gad.251926.114
19. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell (2015) 160(6):1125–34. doi:10.1016/j.cell.2015.02.014
20. Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell (2014) 56(1):55–66. doi:10.1016/j.molcel.2014.08.019
21. Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol (2004) 2(12):e391. doi:10.1371/journal.pbio.0020391
22. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol (2004) 22(8):1001–5. doi:10.1038/nbt996
23. Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep (2015) 10(2):170–7. doi:10.1016/j.celrep.2014.12.019
24. Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol (2017) 14(8):1035–45. doi:10.1080/15476286.2016.1271524
25. Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol (2014) 15(7):409. doi:10.1186/s13059-014-0409-z
26. Veno MT, Hansen TB, Veno ST, Clausen BH, Grebing M, Finsen B, et al. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol (2015) 16:245. doi:10.1186/s13059-015-0801-3
27. Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, et al. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep (2014) 9(5):1966–80. doi:10.1016/j.celrep.2014.10.062
28. You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci (2015) 18(4):603–10. doi:10.1038/nn.3975
29. Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, et al. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem (2015) 61(1):221–30. doi:10.1373/clinchem.2014.230433
30. Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PLoS One (2015) 10(10):e0141214. doi:10.1371/journal.pone.0141214
31. Li Y, Zheng QP, Bao CY, Li SY, Guo WJ, Zhao J, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res (2015) 25(8):981–4. doi:10.1038/cr.2015.82
32. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature (2013) 495(7441):333–8. doi:10.1038/nature11928
33. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature (2013) 495(7441):384–8. doi:10.1038/nature11993
34. Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. miRNA-dependent gene silencing involving ago2-mediated cleavage of a circular antisense RNA. EMBO J (2011) 30(21):4414–22. doi:10.1038/emboj.2011.359
35. Schneider T, Hung LH, Schreiner S, Starke S, Eckhof H, Rossbach O, et al. CircRNA-protein complexes: IMP3 protein component defines subfamily of circRNPs. Sci Rep (2016) 6:31313. doi:10.1038/srep31313
36. Hentze MW, Preiss T. Circular RNAs: splicing’s enigma variations. EMBO J (2013) 32(7):923–5. doi:10.1038/emboj.2013.53
37. Munker R, Calin GA. MicroRNA profiling in cancer. Clin Sci (Lond) (2011) 121(4):141–58. doi:10.1042/CS20110005
38. Kristensen LS, Hansen TB, Veno MT, Kjems J. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene (2018) 37(5):555–65. doi:10.1038/onc.2017.361
39. Bolha L, Ravnik-Glavac M, Glavac D. Circular RNAs: biogenesis, function, and a role as possible cancer biomarkers. Int J Genomics (2017) 2017:6218353. doi:10.1155/2017/6218353
40. Chen B, Huang S. Circular RNA: an emerging non-coding RNA as a regulator and biomarker in cancer. Cancer Lett (2018) 418:41–50. doi:10.1016/j.canlet.2018.01.011
41. Sand M, Bechara FG, Gambichler T, Sand D, Bromba M, Hahn SA, et al. Circular RNA expression in cutaneous squamous cell carcinoma. J Dermatol Sci (2016) 83(3):210–8. doi:10.1016/j.jdermsci.2016.05.012
42. Sand M, Bechara FG, Sand D, Gambichler T, Hahn SA, Bromba M, et al. Circular RNA expression in basal cell carcinoma. Epigenomics (2016) 8(5):619–32. doi:10.2217/epi-2015-0019
43. Sarkar D, Oghabian A, Bodiyabadu PK, Joseph WR, Leung EY, Finlay GJ, et al. Multiple isoforms of ANRIL in melanoma cells: structural complexity suggests variations in processing. Int J Mol Sci (2017) 18(7):1378. doi:10.3390/ijms18071378
44. Yu L, Gong X, Sun L, Zhou Q, Lu B, Zhu L. The circular RNA Cdr1as act as an oncogene in hepatocellular carcinoma through targeting miR-7 expression. PLoS One (2016) 11(7):e0158347. doi:10.1371/journal.pone.0158347
45. Xu L, Zhang M, Zheng X, Yi P, Lan C, Xu M. The circular RNA ciRS-7 (Cdr1as) acts as a risk factor of hepatic microvascular invasion in hepatocellular carcinoma. J Cancer Res Clin Oncol (2017) 143(1):17–27. doi:10.1007/s00432-016-2256-7
46. Tang W, Ji M, He G, Yang L, Niu Z, Jian M, et al. Silencing CDR1as inhibits colorectal cancer progression through regulating microRNA-7. Onco Targets Ther (2017) 10:2045–56. doi:10.2147/OTT.S131597
47. Weng W, Wei Q, Toden S, Yoshida K, Nagasaka T, Fujiwara T, et al. Circular RNA ciRS-7-A promising prognostic biomarker and a potential therapeutic target in colorectal cancer. Clin Cancer Res (2017) 23(14):3918–28. doi:10.1158/1078-0432.CCR-16-2541
48. Barbagallo D, Condorelli A, Ragusa M, Salito L, Sammito M, Banelli B, et al. Dysregulated miR-671-5p/CDR1-AS/CDR1/VSNL1 axis is involved in glioblastoma multiforme. Oncotarget (2016) 7(4):4746–59. doi:10.18632/oncotarget.6621
49. Li F, Zhang L, Li W, Deng J, Zheng J, An M, et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget (2015) 6(8):6001–13. doi:10.18632/oncotarget.3469
50. Huang G, Zhu H, Shi Y, Wu W, Cai H, Chen X. cir-ITCH plays an inhibitory role in colorectal cancer by regulating the Wnt/beta-catenin pathway. PLoS One (2015) 10(6):e0131225. doi:10.1371/journal.pone.0131225
51. Wan L, Zhang L, Fan K, Cheng ZX, Sun QC, Wang JJ. Circular RNA-ITCH suppresses lung cancer proliferation via inhibiting the Wnt/beta-catenin pathway. Biomed Res Int (2016) 2016:1579490. doi:10.1155/2016/1579490
52. Guo W, Zhang J, Zhang D, Cao S, Li G, Zhang S, et al. Polymorphisms and expression pattern of circular RNA circ-ITCH contributes to the carcinogenesis of hepatocellular carcinoma. Oncotarget (2017) 8(29):48169–77. doi:10.18632/oncotarget.18327
53. Yang C, Yuan W, Yang X, Li P, Wang J, Han J, et al. Circular RNA circ-ITCH inhibits bladder cancer progression by sponging miR-17/miR-224 and regulating p21, PTEN expression. Mol Cancer (2018) 17(1):19. doi:10.1186/s12943-018-0771-7
54. Yang W, Du WW, Li X, Yee AJ, Yang BB. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene (2016) 35(30):3919–31. doi:10.1038/onc.2015.460
55. Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res (2016) 44(6):2846–58. doi:10.1093/nar/gkw027
56. Du WW, Fang L, Yang W, Wu N, Awan FM, Yang Z, et al. Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ (2017) 24(2):357–70. doi:10.1038/cdd.2016.133
57. Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, et al. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark (2016) 16(1):161–9. doi:10.3233/CBM-150552
58. Li WH, Song YC, Zhang H, Zhou ZJ, Xie X, Zeng QN, et al. Decreased expression of Hsa_circ_00001649 in gastric cancer and its clinical significance. Dis Markers (2017) 2017:4587698. doi:10.1155/2017/4587698
59. Xu Y, Yao Y, Zhong X, Leng K, Qin W, Qu L, et al. Downregulated circular RNA hsa_circ_0001649 regulates proliferation, migration and invasion in cholangiocarcinoma cells. Biochem Biophys Res Commun (2018) 496(2):455–61. doi:10.1016/j.bbrc.2018.01.077
60. Ji W, Qiu C, Wang M, Mao N, Wu S, Dai Y. Hsa_circ_0001649: a circular RNA and potential novel biomarker for colorectal cancer. Biochem Biophys Res Commun (2018) 497(1):122–6. doi:10.1016/j.bbrc.2018.02.036
61. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun (2016) 7:11215. doi:10.1038/ncomms11215
62. Chen G, Shi Y, Liu M, Sun J. circHIPK3 regulates cell proliferation and migration by sponging miR-124 and regulating AQP3 expression in hepatocellular carcinoma. Cell Death Dis (2018) 9(2):175. doi:10.1038/s41419-017-0204-3
63. Li Y, Zheng F, Xiao X, Xie F, Tao D, Huang C, et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep (2017) 18(9):1646–59. doi:10.15252/embr.201643581
64. Okholm TLH, Nielsen MM, Hamilton MP, Christensen LL, Vang S, Hedegaard J, et al. Circular RNA expression is abundant and correlated to aggressiveness in early-stage bladder cancer. NPJ Genom Med (2017) 2:36. doi:10.1038/s41525-017-0038-z
65. Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y, et al. Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget (2016) 7(18):26680–91. doi:10.18632/oncotarget.8589
66. Jin H, Fang MF, Man ZR, Wang Y, Liu HC. Circular RNA 001569 acts as an oncogene and correlates with aggressive characteristics in hepatocellular carcinoma. Int J Clin Exp Patho (2017) 10(3):2997–3005.
67. Huang MG, Zhong ZY, Lv MX, Shu J, Tian Q, Chen JX. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget (2016) 7(30):47186–200. doi:10.18632/oncotarget.9706
68. Zhong ZY, Huang MG, Lv MX, He YF, Duan CZ, Zhang LY, et al. Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett (2017) 403:305–17. doi:10.1016/j.canlet.2017.06.027
69. Chen J, Li Y, Zheng QP, Bao CY, He J, Chen B, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett (2017) 388:208–19. doi:10.1016/j.canlet.2016.12.006
70. L'Abbate A, Tolomeo D, Cifola I, Severgnini M, Turchiano A, Augello B, et al. MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences. Leukemia (2018) doi:10.1038/s41375-018-0033-0
71. Verduci L, Ferraiuolo M, Sacconi A, Ganci F, Vitale J, Colombo T, et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biol (2017) 18:237. doi:10.1186/s13059-017-1368-y
72. Li TW, Shao YF, Fu LY, Xie Y, Zhu LW, Sun WL, et al. Plasma circular RNA profiling of patients with gastric cancer and their droplet digital RT-PCR detection. J Mol Med (2018) 96(1):85–96. doi:10.1007/s00109-017-1600-y
73. Zhang Y, Li J, Yu J, Liu H, Shen ZY, Ye GT, et al. Circular RNAs signature predicts the early recurrence of stage III gastric cancer after radical surgery. Oncotarget (2017) 8(14):22936–43. doi:10.18632/oncotarget.15288
74. Zhang Y, Liu H, Li WD, Yu J, Li J, Shen ZY, et al. CircRNA_100269 is downregulated in gastric cancer and suppresses tumor cell growth by targeting miR-630. Aging (Albany NY) (2017) 9(6):1585–94. doi:10.18632/aging.101254
75. Pritchard CC, Kroh E, Wood B, Arroyo JD, Dougherty KJ, Miyaji MM, et al. Blood cell origin of circulating microRNAs: a cautionary note for cancer biomarker studies. Cancer Prev Res (2012) 5(3):492–7. doi:10.1158/1940-6207.CAPR-11-0370
76. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell (2011) 146(3):353–8. doi:10.1016/j.cell.2011.07.014
77. Jens M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet (2015) 16(2):113–26. doi:10.1038/nrg3853
78. Vasilescu C, Tanase M, Dragomir M, Calin GA. From mobility to crosstalk. A model of intracellular miRNAs motion may explain the RNAs interaction mechanism on the basis of target subcellular localization. Math Biosci (2016) 280:50–61. doi:10.1016/j.mbs.2016.07.012
79. Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell (2014) 54(5):766–76. doi:10.1016/j.molcel.2014.03.045
Keywords: circular RNA, microRNA, non-coding RNA, cancer, biomarker
Citation: Dragomir M and Calin GA (2018) Circular RNAs in Cancer – Lessons Learned From microRNAs. Front. Oncol. 8:179. doi: 10.3389/fonc.2018.00179
Received: 28 March 2018; Accepted: 08 May 2018;
Published: 28 May 2018
Edited by:
Dario Palmieri, The Ohio State University, United StatesReviewed by:
David Langenberger, ecSeq Bioinformatics GmbH, GermanyYun Dai, Virginia Commonwealth University, United States
Copyright: © 2018 Dragomir and Calin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George A. Calin, Z2NhbGluQG1kYW5kZXJzb24ub3Jn