 Alexandra Vander Ark
Alexandra Vander Ark Jingchen Cao
Jingchen Cao Xiaohong Li
Xiaohong Li- Program for Skeletal Disease and Tumor Microenvironment, Center for Cancer and Cell Biology, Van Andel Research Institute, Grand Rapids, MI, United States
Enzalutamide, a second-generation small-molecule inhibitor of the androgen receptor (AR), has been approved for patients who failed with androgen deprivation therapy and have developed castration-resistant prostate cancer. More than 80% of these patients develop bone metastases. The binding of enzalutamide to the AR prevents the nuclear translocation of the receptor, thus inactivating androgen signaling. However, prostate cancer cells eventually develop resistance to enzalutamide treatment. Studies have found resistance both in patients and in laboratory models. The mechanisms of and approaches to overcoming such resistance are significant issues that need to be addressed. In this review, we focus on the major mechanisms of acquired enzalutamide resistance, including genetic mutations and splice variants of the AR, signaling pathways that bypass androgen signaling, intratumoral androgen biosynthesis by prostate tumor cells, lineage plasticity, and contributions from the tumor microenvironment. Approaches for overcoming these mechanisms to enzalutamide resistance along with the associated problems and solutions are discussed. Emerging questions, concerns, and new opportunities in studying enzalutamide resistance will be addressed as well.
Introduction
Of the estimated 26,000 prostate cancer (PCa) deaths in 2016 in the United States, over 80% involved bone metastases (1–3). Second-line hormonal therapies such as enzalutamide (also known as MDV-3100) improve overall patient survival only by several months in about 50% of the patients, and almost all patients develop drug resistance (4–8). There is an urgent need to determine the mechanisms of drug resistance, identify new approaches for overcoming such resistance, and from that knowledge, develop better treatments for PCa bone metastasis.
The first-line treatment option for men with PCa is hormonal therapy. Huggins and Hodges discovered in 1941 that removing androgens or providing estrogens could inhibit the progression of PCa (9). Despite the initial response of most patients to androgen deprivation therapy (ADT), the disease typically progresses to a castration-resistant state within 18–24 months (10, 11). Castration-resistant prostate cancer (CRPC) is defined by disease progression despite hormonal therapies, which is often indicated by an increase of prostate-specific antigen (PSA), a target of androgen signaling activation. As the disease progresses, the CRPC ultimately metastasizes to bone and later to other organs. Patients with metastatic CRPC (mCRPC) have a poor prognosis and a predicted survival of fewer than 2 years from the initial time of progression; such patients account for a large portion of the PCa deaths per year (2, 3, 12, 13). In 2012, enzalutamide, which significantly improved patient survival, was approved. The time line of FDA-approved treatments and their respective effects are shown in Figure 1 (5, 6, 14, 15). However, the survival benefits of enzalutamide were achieved in only up to 50% of PCa patients treated, and patients who initially responded eventually developed resistance (4). Studies of the mechanisms of intrinsic and acquired resistance led to approaches to overcome it.
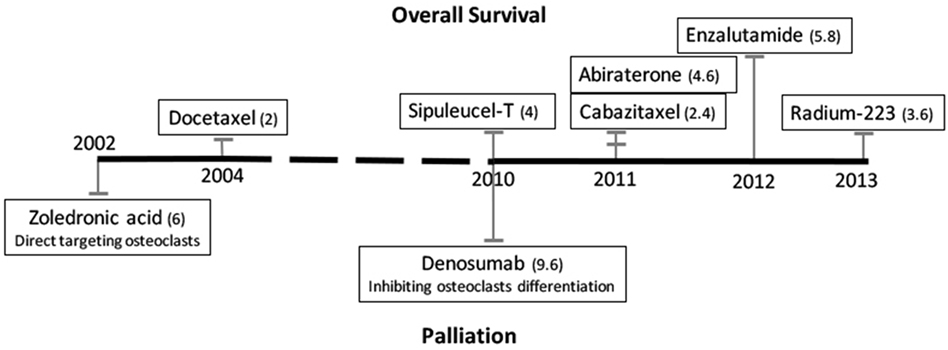
Figure 1. Time line of FDA-approved treatments for patients with castration-resistant prostate cancer bone metastases. Treatments that improve patients’ overall survival are listed above the black line, and palliation treatments are listed below. The numbers in parentheses are median months of overall increased survival or median months delayed to first detection of skeletal-related events, respectively.
Enzalutamide Function at the Molecular Level
The androgen receptor (AR) is a transcription factor and a member of the nuclear receptor superfamily. It consists of an N-terminal transactivation domain (exon 1), a central DNA-binding domain (DBD; exons 2–3), a C-terminal ligand-binding domain (LBD; exons 4–8), and a hinge region between the DBD and LBD that is involved in nuclear localization and degradation (16). The N terminus has a unique LxxLL-like motif, which binds to a hydrophobic cleft of the C terminus that is generated by ligand binding to the receptor. This binding also stabilizes the ligand binding-caused N terminal–C terminal physical interaction of the receptor. The N–C interaction is initially intramolecular in the cytoplasm and is necessary for nuclear localization, but the interaction changes to intermolecular in the nucleus (17–22).
Androgen receptor antagonists all bind to the LBD (23). Binding of the first-generation antagonists (such as flutamide or bicalutamide) does not prevent the intra- or intermolecular N–C interactions (19). Thus, after binding of these antagonists, the AR still translocates to the nucleus, binds to chromatin, and acts as an agonist. This antagonist to agonist switch is partially due to the highly conserved overall configurations of LBD binding with ligand (testosterone or dihydrotestosterone), bicalutamide, or flutamide (24). Enzalutamide binds to the AR with an eightfold higher affinity than bicalutamide (25). Furthermore, no intramolecular N–C interaction was found upon binding of enzalutamide, which blocks the translocation of the receptor to the nucleus and therefore blocks subsequent signaling activation (20).
Enzalutamide Resistance in PCa Cells and Approaches to Overcoming IT
Because enzalutamide binds to the AR LBD, the presence of splicing variants that lack the LBD and of constitutively activated AR in PCa cells apparently confer the intrinsic resistance to enzalutamide. AR-V7 in circulating tumor cells (CTC) has been associated with enzalutamide resistance in PCa patients, although larger-scale studies are needed to conclude that AR-v7 is the cause of resistance (26–30). On the other hand, the overwhelming majority of enzalutamide-treated patients who have robust declines in PSA eventually develop resistance, with increasing PSA and/or progression of bone lesions that suggest acquired resistances. We discuss several major mechanisms of acquired resistance in the following sections, based on the original research publication and recent reviews (21, 31–33).
AR Mutations and Splice Variants (AR-V)
One of the AR mutant F876L (substitution of phenylalanine for leucine at the 876 position), which confers enzalutamide resistance in vitro and in vivo (34, 35), spontaneously emerged among enzalutamide-resistant clones. These clones appeared after weeks of continuous enzalutamide treatments of PCa cell lines with AR either in culture or xenografted in immune comprised mice. The F876L mutation is in the LBD and is adjacent to the homozygous T877A mutation in LNCaP cells. The F876L mutation is heterozygous, with a mutant allelic mRNA frequency of approximately 40% in patient samples. Mechanistically, a positive correlation of increased AR activation and E2F1 activation was found in these resistant clones, correspondently, cyclin-dependent kinase (CDK) 4/6 inhibitor has been tested as an approach to overcoming F876L-based resistance. However, the specificity of the inhibitor is a concern, because of the key role of CDK4/6 in cell cycle and because one of the CDK4/6 inhibitors, LEE011, was shown to significantly reduce the viability of both parental and enzalutamide-resistant cells (34).
Other studies have focused on developing novel small-molecule inhibitors targeting the AR mutants. One example is darolutamide (ODM-201) (36), an AR antagonist. Darolutamide significantly inhibited the growth of an enzalutamide-resistant PCa clone such as the LNCaP-derived MR49F cell line that has the F877L mutation in vitro and in vivo. Mechanistically, darolutamide was able to inhibit the transcriptional activity of three AR mutants that confer enzalutamide resistance, such as F877L, F877L/T878A, and H875Y/T878A. Darolutamide has a different chemical structure than enzalutamide, and in silico simulation of the drug’s mode of action showed a distinct structure of darolutamide binding with the AR.
Another development is an AR degradation enhancer, ASC-J9 [5-hydroxy-1,7-bis(3,4-dimethoxyphenyl)-1,4,6-heptatrien-3-one] (37–39). ASC-J9 suppressed enzalutamide-resistant PCa progression (40), and it degrades not only wild-type AR but also AR-F876L and splice variants of such as AR-v7 (41–45). ASC-J9 degrades the AR by enhancing its association with the murine double minute protein 2 (MDM2) (41), an E3 ubiquitin ligase that drives AR clearance via proteasome-mediated degradation. However, this has not been confirmed by rescuing the AR degradation using proteasome inhibitors. It is not known whether the same mechanism applies for the downregulation of AR-F877L by ASC-J9. This is possible, because one amino acid mutation may completely change the AR structure and endow it with enzalutamide resistance but still allow it to interact with MDM2. ASC-J9 was also shown to have additional effects that contribute to the inhibitions of PCa progression and metastasis. For example, in PCa cells, ASC-J9 was able to suppress expression fatty acid synthase that stimulates PCa cell growth and invasion (46) or to inhibit the phosphorylation of STAT3 that promotes PCa stem/progenitor cells invasion (39); in the cells of the tumor microenvironment, ACS-J9 could suppress the CD4+ T cell migration that contribute to the progression of prostatitis (47), or the macrophage infiltration that stimulated PCa cell invasion (38). Together, these studies suggested that ACS-J9 was very promising in battling PCa. However, its broader effects on other signaling pathways and on cells from the microenvironment need to be explored further before being translated to clinical trials.
Furthermore, BET [bromodomain (BRD) and extraterminal] inhibitors, such as JQ1 and OTX015, have been shown to overcome enzalutamide resistance of CRPC conferred by AR-V (48, 49). BET inhibitors target the amino-terminus of BRD proteins, BRD4, and exhibit proliferation inhibition in a wide range of cancers, including CRPC. The underlying mechanistic studies suggested that JQ1 inhibited expression of full-length AR and AR-V, thus overcoming enzalutamide resistance (50). However, BET inhibitors as single agents may not succeed in the clinic because of a recent study showing acquired resistance to BET inhibitors in CRPC cells (51). It is not known whether the combination of BET inhibitors with enzalutamide induce resistance, or whether combination of BET inhibitors with CDK9 and/or PARP (poly ADP ribose polymerase) inhibitors overcomes enzalutamide resistance.
In summary, the above approaches target the outcomes of enzalutamide resistance. We believe targeting the causes of drug resistance will be most effective, which will require answers to pivotal questions such as: How does enzalutamide treatment induce mutations of AR? and What types of PCa cells will develop enzalutamide resistance?
Bypassing Signaling Pathways
The bypassing of AR signaling through increasing glucocorticoid receptor (GR) expression at both the mRNA and protein levels has been identified in enzalutamide-resistant PCa cells, and increases of GR have been confirmed in clinical samples (52, 53). The activation of the GR by dexamethasone is sufficient to confer enzalutamide resistance, and a novel GR antagonist, arylpyrazole compound 15, can restore sensitivity to the drug (52–54). However, blocking glucocorticoid signaling is neither practical nor effective, because glucocorticoid is essential for life, and also because GR antagonists activate AR target genes (52). Therefore, researchers have focused on how enzalutamide increases glucocorticoid signaling. One recent study (55) found that enzalutamide treatment sustained the level of cortisol, the active ligand for the GR, thus elevating glucocorticoid signaling and producing a loss of 11β-hydroxysteroid dehydrogenase-2 (11β-HSD2), the enzyme that converts cortisol to the inactive form, cortisone. It was further shown that 11β-HSD2 loss was mediated by an increase of AMFR, an ubiquitin E3-ligase autocrine mobility factor receptor. Approaches such as overexpression of 11β-HSD2 or knock-down of AMFR were effective in reversing enzalutamide resistance. Future research will need to address how to translate these approaches to therapies for patients.
Androgen signaling not only activates but also suppresses various downstream target genes, some of which are oncogenic, such as c-Met (56), c-Myb (57), or enhancer of zeste homolog 2 (EZH2) (58). Blocking either any of these proteins or their respective downstream targets was shown to inhibit castration-resistant PCa cell proliferation (in vitro) or growth (in vivo). Logically, since enzalutamide treatment induces c-Met, c-Myb, and EZH2 or activates the target genes, the combination of enzalutamide with inhibition of each of these targets may have better efficacy. However, further preclinical and clinical testing are needed for confirmation and validation.
On the other hand, enzalutamide treatment was shown to activate PI3K/AKT signaling by reducing FK506 binding protein 5 (a chaperone for the PHLPP) that destabilizes PHLPP (PH domain and leucine rich repeat protein phosphatases), the AKT phosphatase (59). Combining enzalutamide with BEZ235 (a dual PI3K and mTORC1/2 inhibitor) or AKT1/2 inhibitor led to significant inhibition of PCa tumor growth in mice (59). Another study showed increased noncanonical Wnt signaling, independent of increased GR, in CTC obtained from enzalutamide-resistant PCa patients compared with enzalutamide-naïve PCa patients by analyzing the RNA sequences of the single CTC (60). Furthermore, increased Wnt5A was confirmed to represent the noncanonical Wnt signaling in CTC, as well as in enzalutamide-treated or enzalutamide-resistant LNCaP PCa cells (60). However, the effectiveness of inhibiting PCa growth by combining enzalutamide with medicinal approaches for blocking noncanonical Wnt signaling or Wnt5A has not been tested. This avenue to overcoming enzalutamide resistance is practical since the Wnt5a antagonist has been tested in clinical trials in breast and colorectal cancers (61). Enzalutamide treatment has also been shown to increase autophagy, in which the AMPK pathway was upregulated (62). Combinations of enzalutamide with autophagy inhibitors enhanced the therapeutic response, particularly when using clomipramine or metformin, both of which are FDA approved as antidepressant and antidiabetic drugs, respectively. However, increasing autophagy is a general cause by blocking AR signaling, because the same effect was also observed in bicalutamide-resistant cells (62).
Other bypass pathways have also been reported. The associations of β-catenin, miRNA16, and oncostatin M with enzalutamide resistance were identified by gene profiling of parental cells versus enzalutamide-resistant cells (31). Correlations between each of these and CRPC cells had been reported in various studies (63–67). However, the effectiveness of overcoming enzalutamide resistance by blocking each target individually or in combination needs to be tested.
Intratumoral Androgen Biosynthesis
Intratumoral androgen biosynthesis has been recognized as one of the mechanisms of resistance to ADT and the development of CRPC (68–71). Elevated androgen levels were recently found in the bone marrow of patients treated with enzalutamide (72). An increase of intratumoral androgen biosynthesis is reported to be involved in enzalutamide resistance (73). Mechanistic studies revealed that several genes involved in the androgen synthesis pathway were significantly increased in enzalutamide-resistant PCa cells relative to the parental cells, including AKR1C3 (aldo-keto reductase family 1 member C3) (73).
AKR1C3 is an enzyme that participates in the conversion of weak androgens such as androstenediones to more-active androgens such as testosterone or dihydrotestosterone. Knock-down of the AKR1C3 gene or blocking of the AKR1C3 enzyme using a drug such as indomethacin can overcome enzalutamide resistance. Furthermore, overexpression of AKR1C3 can cause enzalutamide resistance in PCa cells (73). While this was a thorough study both in vitro and in vivo, validation from patient samples is needed to justify more effort in translating this work.
Lineage Switching
When lung cancer is exposed to targeted therapies, such as epidermal growth factor receptor (EGFR) inhibitor, adenocarcinoma cells can switch from EGFR dependent to EGFR independent with expression of neuroendocrine lineage markers, or to cells that lack all EGFR expression (74, 75). Researchers of Dr. Swayers’ group applied this idea to determine whether lineage plasticity is a novel mechanism of enzalutamide resistance in PCa. They found that combined TP53 and RB1 alterations were significantly higher in disease progressed tumors from men under treatment with enzalutamide or abiraterone (an androgen synthesis inhibitor) (76). The combined knock-down of TP53 and RB1 in human PCa LNCaP/AR and CWR22Pc-EP cells did confer enzalutamide resistance in vitro. Further characterization of the cells revealed increased expression of neuroendocrine markers but decreased luminal cell markers in both of the double knock-down cells compared with their respective parental LNCaP/AR and CWR22Pc-EP luminal PCa cells, and the cells switch from AR dependent to AR independent. In PCa, lineage switching from adenocarcinoma to neuroendocrine PCa cells has been reported, but without complete understanding of the mechanism that drives the plasticity (77). A back-to-back publication showed that loss of RB1 and TP53 genes conferred the PCa lineage plasticity, metastasis, and enzalutamide resistance by developing a genetically engineered mouse model crossed with a Pten knockout mouse (78). Together, these data suggest lineage plasticity under selective pressure such as enzalutamide treatment. Finally, pluripotency transcription factor, SOX2, was identified to be required for the lineage plasticity and enzalutamide resistance by the loss of TP53 and RB1. Based on these significant advances, the future directions are to investigate how to target SOX2, discover the mechanisms of enzalutamide-induced loss of TP53 and RB1, and to determine whether these mechanisms are targetable.
Contributions from the Tumor Microenvironment
Cancer is a systemic disease, and studies showed that paracrine factors such as hepatocyte growth factor, Wnts, basic fibroblast growth factor (or FGF2), and some cytokines from the microenvironment contribute to PCa initiation, progression, and metastasis (64, 79–83). Therefore, it is possible that paracrine factors influence the efficacy of enzalutamide treatment. The most studied and most easily targeted factor from the microenvironment is interleukin 6 (IL6) (84–88), which signals through the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway. One of the functions of IL6/JAK/STAT3 in PCa cells is stimulating the expression of stemness/self-renewal genes. IL6/JAK/STAT3 also induces the expression of suppressor of cytokine signaling 3 (SOCS3), which in turn suppresses the transcriptional activity of IL6/JAK/STAT3.
Culig led a study (84) to determine the effect of long-term enzalutamide treatment in an inflammatory environment, studying PCa cells treated with enzalutamide plus IL6 for 3 weeks. They found this combination treatment suppressed self-renewal genes and AR target genes in PCa cells that had SOCS3 overexpression, but the AR target genes were induced in PCa cells with SOCS3 knocked out, suggesting that uncontrolled IL6/JAK/STAT3 transactivation of androgen signaling conferred enzalutamide resistance. This study showed that an inflammatory microenvironment could control the efficacy of enzalutamide by fine-tuning signaling of the inflammatory cytokine IL6 (84). Others reported that bicalutamide or enzalutamide could promote macrophage infiltration that subsequently enhances PCa invasion. The mechanistic studies showed that blocking AR signaling in macrophages increased the expression of CCL2 through inhibiting the expression of PIAS3 (protein inhibitor of STAT3) and subsequent increase of STAT3 phosphorylation (38). To overcome this effect, ASC-J9, the AR degrader, could directly inhibit the STAT3 phosphorylation and activation (38).
In PCa bone metastases, because the bone matrix is a reservoir rich in cytokines, we suspect that there are other cytokines that could also contribute to enzalutamide resistance, such as IL8, stromal cell-derived factor 1 (also known CXCL12), and granulocyte colony-stimulating factor, which have been identified in cancer drug resistance (89). On the other hand, the PCa bone metastatic lesions are results of dysregulated activities of osteoclasts and osteoblasts that were hijacked by cancer cells. However, we do not know whether and how osteoclasts or osteoblasts affect enzalutamide resistance. Targeting osteoclasts that resorb bones using zoledronic acid or denosumab is effective in palliation (5, 6). Metastasized PCa cells adhere in the osteoblast niche, developing into overt metastatic lesions (90–95). Will combining enzalutamide with approaches that target the proliferation or differentiation of osteoblasts or osteoclasts be better therapies? Will the effects of enzalutamide on osteoblasts or osteoclasts contribute to the drug resistance? We believe combination therapies using enzalutamide plus approaches that target the bone microenvironment (either paracrine factors or the bone cells themselves) might be able to prevent and/or overcome the enzalutamide resistant in mCRPC.
Summary and Conclusion
Currently, enzalutamide is the best therapeutic drug for improving the survival of mCRPC patients. Due to their heterogeneity and plasticity, cancer cells have various mechanisms of drug resistance that we have begun to learn about, knowledge that will lead to targeted approaches to improving the efficacy of enzalutamide. The current mechanisms of enzalutamide resistance, the approaches for overcoming resistance, and the problems and solutions associated with these mechanisms are summarized in Table 1. The ultimate solution to drug resistance will be found only through future studies on the effects and mechanisms of enzalutamide action at the cellular and molecular levels.
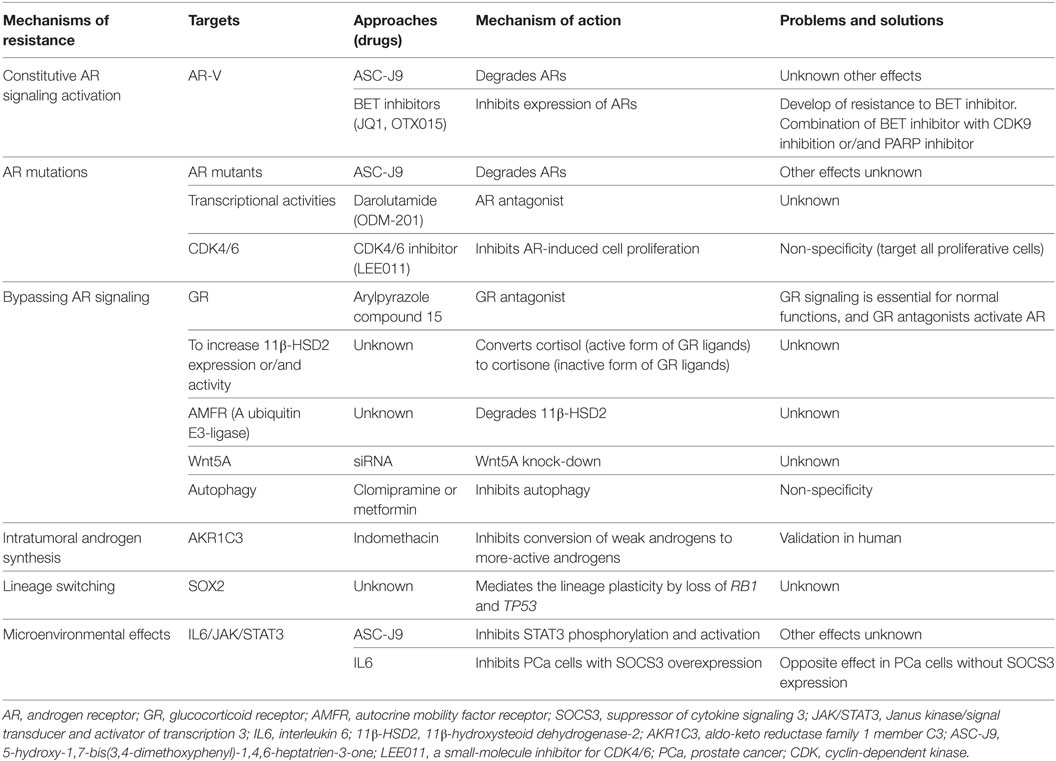
Table 1. Summary of mechanisms and approaches of overcoming the enzalutamide resistance in preclinical PCa model.
Author Contributions
Designed, wrote, and finalized the manuscript: XL. Wrote parts of the manuscript: AA and JC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by DOD PCRP (W81XWH-12-1-0271 to XL) and Van Andel Research Institute start-up funding (53010A to XL). We thank David Nadziejka for his technical editing of this manuscript. The content is solely the responsibility of the authors.
References
1. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res (2006) 12:6243s–9s. doi:10.1158/1078-0432.CCR-06-0931
2. Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin (2016) 66:271–89. doi:10.3322/caac.21349
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin (2016) 66:7–30. doi:10.3322/caac.21332
4. Schalken J, Fitzpatrick JM. Enzalutamide: targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int (2016) 117:215–25. doi:10.1111/bju.13123
5. El-Amm J, Freeman A, Patel N, Aragon-Ching JB. Bone-targeted therapies in metastatic castration-resistant prostate cancer: evolving paradigms. Prostate Cancer (2013) 2013:210686. doi:10.1155/2013/210686
6. El-Amm J, Aragon-Ching JB. The changing landscape in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Med Oncol (2013) 5:25–40. doi:10.1177/1758834012458137
7. McCain J. Drugs that offer a survival advantage for men with bone metastases resulting from castration-resistant prostate cancer: new and emerging treatment options. P T (2014) 39:130–43.
8. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med (2012) 367:1187–97. doi:10.1056/NEJMoa1207506
9. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol (2002) 168:9–12. doi:10.1016/S0022-5347(05)64820-3
10. Antonarakis ES, Blackford AL, Garrett-Mayer E, Eisenberger MA. Survival in men with nonmetastatic prostate cancer treated with hormone therapy: a quantitative systematic review. J Clin Oncol (2007) 25:4998–5008. doi:10.1200/JCO.2007.11.1559
11. Armstrong AJ, Garrett-Mayer E, de Wit R, Tannock I, Eisenberger M. Prediction of survival following first-line chemotherapy in men with castration-resistant metastatic prostate cancer. Clin Cancer Res (2010) 16:203–11. doi:10.1158/1078-0432.CCR-09-2514
12. Pond GR, Sonpavde G, de Wit R, Eisenberger MA, Tannock IF, Armstrong AJ. The prognostic importance of metastatic site in men with metastatic castration-resistant prostate cancer. Eur Urol (2014) 65:3–6. doi:10.1016/j.eururo.2013.09.024
13. Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, et al. Meta-analysis evaluating the impact of site of metastasis on overall survival in men with castration-resistant prostate cancer. J Clin Oncol (2016) 34:1652–9. doi:10.1200/JCO.2015.65.7270
14. Body JJ, Casimiro S, Costa L. Targeting bone metastases in prostate cancer: improving clinical outcome. Nat Rev Urol (2015) 12:340–56. doi:10.1038/nrurol.2015.90
15. Vander Ark AW, Woodford E, Li X. Molecular mechanisms of therapies for prostate cancer with bone metastasis. J Explor Res Pharmacol (2016) 1:35–9. doi:10.14218/JERP.2016.00023
16. Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog (2011) 10:20. doi:10.4103/1477-3163.83937
17. Choong CS, Kemppainen JA, Wilson EM. Evolution of the primate androgen receptor: a structural basis for disease. J Mol Evol (1998) 47:334–42. doi:10.1007/PL00006391
18. Langley E, Kemppainen JA, Wilson EM. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J Biol Chem (1998) 273:92–101. doi:10.1074/jbc.273.1.92
19. Schaufele F, Carbonell X, Guerbadot M, Borngraeber S, Chapman MS, Ma AA, et al. The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc Natl Acad Sci U S A (2005) 102:9802–7. doi:10.1073/pnas.0408819102
20. Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene (2014) 33:2815–25. doi:10.1038/onc.2013.235
21. Boudadi K, Antonarakis ES. Resistance to novel antiandrogen therapies in metastatic castration-resistant prostate cancer. Clin Med Insights Oncol (2016) 10:1–9. doi:10.4137/CMO.S34534
22. Culig Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr Mol Biol Rep (2017) 3:230–5. doi:10.1007/s40610-017-0079-1
23. Monaghan AE, McEwan IJ. A sting in the tail: the N-terminal domain of the androgen receptor as a drug target. Asian J Androl (2016) 18:687–94. doi:10.4103/1008-682X.181081
24. Lallous N, Dalal K, Cherkasov A, Rennie PS. Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int J Mol Sci (2013) 14:12496–519. doi:10.3390/ijms140612496
25. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (2009) 324:787–90. doi:10.1126/science.1168175
26. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol (2017) 35:2149–56. doi:10.1200/JCO.2016.70.1961
27. Lokhandwala PM, Riel SL, Haley L, Lu C, Chen Y, Silberstein J, et al. Analytical validation of androgen receptor splice variant 7 detection in a clinical laboratory improvement amendments (CLIA) laboratory setting. J Mol Diagn (2017) 19:115–25. doi:10.1016/j.jmoldx.2016.08.003
28. Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol (2016) 2:1441–9. doi:10.1001/jamaoncol.2016.1828
29. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res (2013) 73:483–9. doi:10.1158/0008-5472.CAN-12-3630
30. Hoefer J, Akbor M, Handle F, Ofer P, Puhr M, Parson W, et al. Critical role of androgen receptor level in prostate cancer cell resistance to new generation antiandrogen enzalutamide. Oncotarget (2016) 7:59781–94. doi:10.18632/oncotarget.10926
31. Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget (2016) 7:26259–74. doi:10.18632/oncotarget.8456
32. Armstrong CM, Gao AC. Adaptive pathways and emerging strategies overcoming treatment resistance in castration resistant prostate cancer. Asian J Urol (2016) 3:185–94. doi:10.1016/j.ajur.2016.08.001
33. Chism DD, De Silva D, Whang YE. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev Anticancer Ther (2014) 14:1369–78. doi:10.1586/14737140.2014.928594
34. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov (2013) 3:1030–43. doi:10.1158/2159-8290.CD-13-0142
35. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov (2013) 3:1020–9. doi:10.1158/2159-8290.CD-13-0226
36. Borgmann H, Lallous N, Ozistanbullu D, Beraldi E, Paul N, Dalal K, et al. Moving towards precision urologic oncology: targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201). Eur Urol (2018) 73:4–8. doi:10.1016/j.eururo.2017.08.012
37. Lin TH, Lee SO, Niu Y, Xu D, Liang L, Li L, et al. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/enzalutamide versus anti-androgen receptor ASC-J9(R) lead to promotion versus suppression of prostate cancer metastasis. J Biol Chem (2013) 288:19359–69. doi:10.1074/jbc.M113.477216
38. Lin TH, Izumi K, Lee SO, Lin WJ, Yeh S, Chang C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (enzalutamide) or casodex (bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis (2013) 4:e764. doi:10.1038/cddis.2013.270
39. Wen S, Tian J, Niu Y, Li L, Yeh S, Chang C. ASC-J9((R)), and not casodex or enzalutamide, suppresses prostate cancer stem/progenitor cell invasion via altering the EZH2-STAT3 signals. Cancer Lett (2016) 376:377–86. doi:10.1016/j.canlet.2016.01.057
40. Wang R, Sun Y, Li L, Niu Y, Lin W, Lin C, et al. Preclinical study using Malat1 small interfering RNA or androgen receptor splicing variant 7 degradation enhancer ASC-J9((R)) to suppress enzalutamide-resistant prostate cancer progression. Eur Urol (2017) 72:835–44. doi:10.1016/j.eururo.2017.04.005
41. Lai KP, Huang CK, Chang YJ, Chung CY, Yamashita S, Li L, et al. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol (2013) 182:460–73. doi:10.1016/j.ajpath.2012.10.029
42. Izumi K, Chang C. Targeting inflammatory cytokines-androgen receptor (AR) signaling with ASC-J9((R)) to better battle prostate cancer progression. Oncoimmunology (2013) 2:e26853. doi:10.4161/onci.26853
43. Wang R, Lin W, Lin C, Li L, Sun Y, Chang C. ASC-J9((R)) suppresses castration resistant prostate cancer progression via degrading the enzalutamide-induced androgen receptor mutant AR-F876L. Cancer Lett (2016) 379:154–60. doi:10.1016/j.canlet.2016.05.018
44. Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med (2007) 13:348–53. doi:10.1038/nm1547
45. Yamashita S, Lai KP, Chuang KL, Xu D, Miyamoto H, Tochigi T, et al. ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia (2012) 14:74–83. doi:10.1593/neo.111436
46. Wen S, Niu Y, Lee SO, Yeh S, Shang Z, Gao H, et al. Targeting fatty acid synthase with ASC-J9 suppresses proliferation and invasion of prostate cancer cells. Mol Carcinog (2016) 55:2278–90. doi:10.1002/mc.22468
47. Lin SJ, Chou FJ, Lin CY, Chang HC, Yeh S, Chang C. New therapy with ASC-J9(R) to suppress the prostatitis via altering the cytokine CCL2 signals. Oncotarget (2016) 7:66769–75. doi:10.18632/oncotarget.11484
48. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature (2014) 510:278–82. doi:10.1038/nature13229
49. Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res (2016) 14:324–31. doi:10.1158/1541-7786.MCR-15-0472
50. Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res (2015) 43:5880–97. doi:10.1093/nar/gkv262
51. Pawar A, Gollavilli PN, Wang S, Asangani IA. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep (2018) 22:2236–45. doi:10.1016/j.celrep.2018.02.011
52. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell (2013) 155:1309–22. doi:10.1016/j.cell.2013.11.012
53. Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer (2014) 5:72–89. doi:10.1007/s12672-014-0173-2
54. Wang JC, Shah N, Pantoja C, Meijsing SH, Ho JD, Scanlan TS, et al. Novel arylpyrazole compounds selectively modulate glucocorticoid receptor regulatory activity. Genes Dev (2006) 20:689–99. doi:10.1101/gad.1400506
55. Li J, Alyamani M, Zhang A, Chang KH, Berk M, Li Z, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife (2017) 6:1–17. doi:10.7554/eLife.20183
56. Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res (2007) 67:967–75. doi:10.1158/0008-5472.CAN-06-3552
57. Li L, Chang W, Yang G, Ren C, Park S, Karantanos T, et al. Targeting poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage response pathway in castration-resistant prostate cancer. Sci Signal (2014) 7:ra47. doi:10.1126/scisignal.2005070
58. Bohrer LR, Chen S, Hallstrom TC, Huang H. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology (2010) 151:5136–45. doi:10.1210/en.2010-0436
59. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell (2011) 19:575–86. doi:10.1016/j.ccr.2011.04.008
60. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science (2015) 349:1351–6. doi:10.1126/science.aab0917
61. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene (2017) 36:1461–73. doi:10.1038/onc.2016.304
62. Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr, et al. Targeting autophagy overcomes enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene (2014) 33:4521–30. doi:10.1038/onc.2014.25
63. Truica CI, Byers S, Gelmann EP. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res (2000) 60:4709–13.
64. Placencio VR, Sharif-Afshar AR, Li X, Huang H, Uwamariya C, Neilson EG, et al. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res (2008) 68:4709–18. doi:10.1158/0008-5472.CAN-07-6289
65. Schweizer L, Rizzo CA, Spires TE, Platero JS, Wu Q, Lin TA, et al. The androgen receptor can signal through Wnt/beta-catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol (2008) 9:4. doi:10.1186/1471-2121-9-4
66. Yan X, Liang H, Deng T, Zhu K, Zhang S, Wang N, et al. The identification of novel targets of miR-16 and characterization of their biological functions in cancer cells. Mol Cancer (2013) 12:92. doi:10.1186/1476-4598-12-92
67. Godoy-Tundidor S, Cavarretta IT, Fuchs D, Fiechtl M, Steiner H, Friedbichler K, et al. Interleukin-6 and oncostatin M stimulation of proliferation of prostate cancer 22Rv1 cells through the signaling pathways of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Prostate (2005) 64:209–16. doi:10.1002/pros.20235
68. Penning TM. Androgen biosynthesis in castration-resistant prostate cancer. Endocr Relat Cancer (2014) 21:T67–78. doi:10.1530/ERC-14-0109
69. Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell (2013) 154:1074–84. doi:10.1016/j.cell.2013.07.029
70. Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. Br J Cancer (2014) 111:1249–54. doi:10.1038/bjc.2014.268
71. Hara N, Nishiyama T. Androgen metabolic pathway involved in current and emerging treatment for men with castration resistant prostate cancer: intraprostatic androgens as therapeutic targets and endocrinological biomarkers. Curr Drug Targets (2014) 15:1215–24. doi:10.2174/1389450115666141024114736
72. Efstathiou E, Titus M, Wen S, Hoang A, Karlou M, Ashe R, et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol (2015) 67:53–60. doi:10.1016/j.eururo.2014.05.005
73. Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res (2015) 75:1413–22. doi:10.1158/0008-5472.CAN-14-3080
74. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun (2015) 6:6377. doi:10.1038/ncomms7377
75. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med (2011) 3:75ra26. doi:10.1126/scitranslmed.3002003
76. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (2017) 355:84–8. doi:10.1126/science.aah4307
77. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22:298–305. doi:10.1038/nm.4045
78. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (2017) 355:78–83. doi:10.1126/science.aah4199
79. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science (2004) 303:848–51. doi:10.1126/science.1090922
80. Li X, Martinez-Ferrer M, Botta V, Uwamariya C, Banerjee J, Bhowmick NA. Epithelial Hic-5/ARA55 expression contributes to prostate tumorigenesis and castrate responsiveness. Oncogene (2011) 30:167–77. doi:10.1038/onc.2010.400
81. Li X, Placencio V, Iturregui JM, Uwamariya C, Sharif-Afshar AR, Koyama T, et al. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene (2008) 27:7118–30. doi:10.1038/onc.2008.293
82. Li X, Sterling JA, Fan KH, Vessella RL, Shyr Y, Hayward SW, et al. Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol Cancer Res (2012) 10:494–503. doi:10.1158/1541-7786.MCR-11-0506
83. Meng X, Vander Ark A, Daft P, Woodford E, Wang J, Madaj Z, et al. Loss of TGF-beta signaling in osteoblasts increases basic-FGF and promotes prostate cancer bone metastasis. Cancer Lett (2018) 418:109–18. doi:10.1016/j.canlet.2018.01.018
84. Handle F, Erb HH, Luef B, Hoefer J, Dietrich D, Parson W, et al. SOCS3 modulates the response to enzalutamide and is regulated by androgen receptor signaling and CpG methylation in prostate cancer cells. Mol Cancer Res (2016) 14:574–85. doi:10.1158/1541-7786.MCR-15-0495
85. Culig Z, Puhr M. Interleukin-6 and prostate cancer: current developments and unsolved questions. Mol Cell Endocrinol (2017) 462(Pt A):25–30. doi:10.1016/j.mce.2017.03.012
86. Culig Z, Puhr M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol (2012) 360:52–8. doi:10.1016/j.mce.2011.05.033
87. Malinowska K, Neuwirt H, Cavarretta IT, Bektic J, Steiner H, Dietrich H, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer (2009) 16:155–69. doi:10.1677/ERC-08-0174
88. Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer (2012) 48:85–93. doi:10.1016/j.ejca.2011.10.014
89. Jones VS, Huang RY, Chen LP, Chen ZS, Fu L, Huang RP. Cytokines in cancer drug resistance: cues to new therapeutic strategies. Biochim Biophys Acta (2016) 1865:255–65. doi:10.1016/j.bbcan.2016.03.005
90. Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem (2008) 105:370–80. doi:10.1002/jcb.21835
91. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest (2011) 121:1298–312. doi:10.1172/JCI43414
92. Pedersen EA, Shiozawa Y, Pienta KJ, Taichman RS. The prostate cancer bone marrow niche: more than just ‘fertile soil’. Asian J Androl (2012) 14:423–7. doi:10.1038/aja.2011.164
93. Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer (2005) 5:21–8. doi:10.1038/nrc1528
94. Tai HC, Chang AC, Yu HJ, Huang CY, Tsai YC, Lai YW, et al. Osteoblast-derived WNT-induced secreted protein 1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget (2014) 5:7589–98. doi:10.18632/oncotarget.2280
Keywords: enzalutamide resistance, castration-resistant prostate cancer, androgen receptor mutants, androgen receptor splice variants, bypass, intratumoral androgen biosynthesis, lineage plasticity, tumor microenvironment
Citation: Vander Ark A, Cao J and Li X (2018) Mechanisms and Approaches for Overcoming Enzalutamide Resistance in Prostate Cancer. Front. Oncol. 8:180. doi: 10.3389/fonc.2018.00180
Received: 21 March 2018; Accepted: 08 May 2018;
Published: 24 May 2018
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Tristan M. Sissung, National Institutes of Health (NIH), United StatesDhiraj Kumar, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2018 Vander Ark, Cao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohong Li, eGlhb2hvbmcubGlAdmFpLm9yZw==