 Florinda Meléndez-Rodríguez
Florinda Meléndez-Rodríguez Olga Roche
Olga Roche Ricardo Sanchez-Prieto
Ricardo Sanchez-Prieto Julian Aragones
Julian Aragones- 1Research Unit, Hospital of Santa Cristina, Research Institute Princesa (IP), Autonomous University of Madrid, Madrid, Spain
- 2CIBER de Enfermedades Cardiovasculares, Carlos III Health Institute, Madrid, Spain
- 3Unidad Asociada de Biomedicina, Universidad de Castilla-La Mancha, Consejo Superior de Investigaciones Científicas (CSIC), Albacete, Spain
- 4Departamento de Ciencias Médicas, Facultad de Medicina, Universidad Castilla-La Mancha, Albacete, Spain
- 5Laboratorio de Oncología Molecular, Unidad de Medicina Molecular, Centro Regional de Investigaciones Biomédicas, Universidad de Castilla-La Mancha, Albacete, Spain
- 6Departamento de Biología del Cáncer, Instituto de investigaciones Biomedicas Alberto Sols, Universidad Autónoma de Madrid, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
The most common type of the renal cancers detected in humans is clear cell renal cell carcinomas (ccRCCs). These tumors are usually initiated by biallelic gene inactivation of the Von Hippel–Lindau (VHL) factor in the renal epithelium, which deregulates the hypoxia-inducible factors (HIFs) HIF1α and HIF2α, and provokes their constitutive activation irrespective of the cellular oxygen availability. While HIF1α can act as a ccRCC tumor suppressor, HIF2α has emerged as the key HIF isoform that is essential for ccRCC tumor progression. Indeed, preclinical and clinical data have shown that pharmacological inhibitors of HIF2α can efficiently combat ccRCC growth. In this review, we discuss the molecular basis underlying the oncogenic potential of HIF2α in ccRCC by focusing on those pathways primarily controlled by HIF2α that are thought to influence the progression of these tumors.
Introduction
Kidney cancer accounts for ~3% of all cancer diagnoses worldwide and most of them are classified as clear cell renal cell carcinomas (ccRCCs). The clear appearance of these tumor cells is the result of the intracellular lipid depositions (1–4). ccRCCs are initiated by biallelic gene inactivation of the Von Hippel–Lindau (VHL) factor in the renal epithelium. VHL is a component of the E3 ligase ubiquitin machinery essential for the regulation of hypoxia-inducible factors (HIFs). HIFs are transcription factors comprised of one HIFα subunit (HIF1α, HIF2α, or HIF3α) and a member of the HIFβ family also known as aryl hydrocarbon receptor nuclear translocator (ARNT). HIFβ subunits are stable but the stability of HIFα subunits is controlled by cellular oxygen availability through the prolyl 2-oxoglutarate-dependent Fe2+-dioxygenases PHD1, PHD2, and PHD3 (5, 6). In normoxic conditions, PHDs use oxygen to hydroxylate two conserved proline residues in the HIFα subunits. These hydroxylated prolyl residues can be recognized by the VHL/E3 ubiquitin ligase complex, leading to the proteasomal degradation of HIFα subunits (7, 8). However, in hypoxic conditions there is insufficient oxygen for PHDs to hydroxylate the HIFα subunits, precluding their recognition by VHL and leading to HIFα subunits stabilization and activation of an HIF-dependent transcriptional program (9–12). Thus, the loss of VHL in ccRCC leads to constitutive activity of HIFs in normoxic conditions and its transcriptional program, which are ultimately key determinants in the progression of ccRCC.
Contrasting Properties of HIF1α and HIF2α in ccRCC
Initial somatic inactivation of the VHL gene in precancerous renal tubule lesions leads to HIF1α activation (13), as well as a progressive gain in HIF1α and HIF2α expression in dysplastic and cystic lesions (13). Moreover, mouse models of ccRCC have also shown that renal epithelium-specific HIF1α or HIF2α gene inactivation impairs ccRCC formation (14–16), indicating that both HIF1α and HIF2α are involved in ccRCC initiation. However, HIF1α expression is lost in 30–40% of overt ccRCCs, since HIF1α acts as a tumor suppressor during further progression of ccRCC by attenuating autonomous VHL-deficient tumor cell proliferation (Figure 1). Conversely, HIF2α acts as an oncoprotein in ccRCC (17–19). Therefore, overt ccRCC can be subdivided into those cases where both HIF1α and HIF2α are expressed, and those that only show HIF2α expression characterized by enhanced ccRCC cell proliferation and adverse prognosis (Figure 1) (17, 20–22). Therefore, the oncoprotein potential of HIF2α in ccRCC has led to the development of the HIF2α antagonists PT2399 and PT2385 to combat the progression of these tumors (23, 24). These HIF2α antagonists show inhibitory effects in vivo than those of the tyrosine kinase inhibitor sunitinib, which is used as a standard first-line therapy for metastatic ccRCC. In addition, PT2385 also appears to improve disease control in a patient who had been administered prior with other pharmacological therapies for ccRCC (23). In this review, we will focus on the cell autonomous pathways primarily controlled by HIF2α that have been shown to contribute to ccRCC progression.
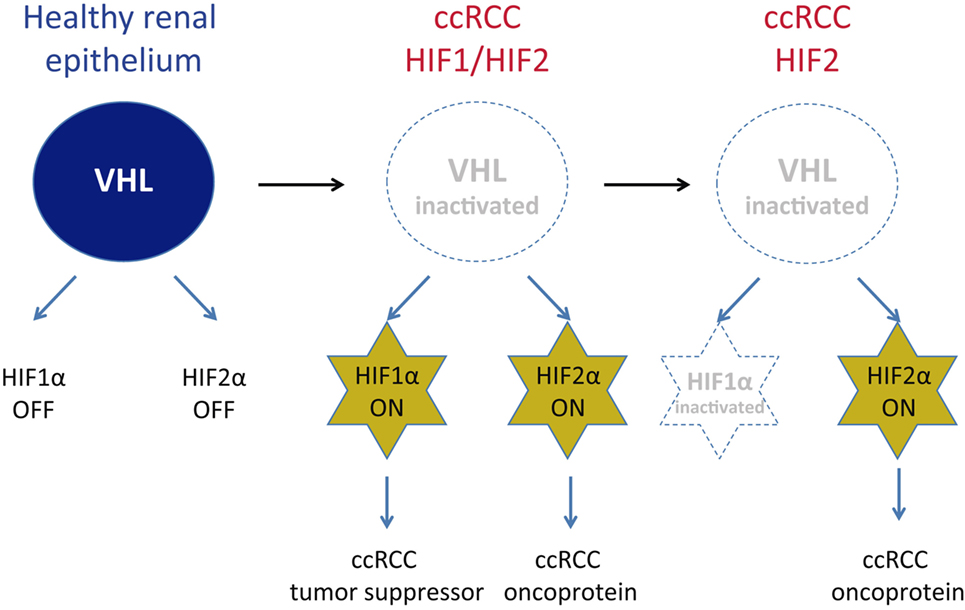
Figure 1. Expression of hypoxia-inducible factors (HIF)1α and HIF2α in Von Hippel–Lindau (VHL)-deficient clear cell renal cell carcinoma (ccRCC). The expression of VHL/E3 ubiquitin ligase complex leads to the proteasomal degradation of HIFα subunits, which assure that HIF1α and HIF2α inactivation in healthy renal epithelial cells. Upon VHL gene inactivation in ccRCC, HIF1α and HIF2α cannot be degraded and, therefore, are constitutively expressed in a large number of ccRCC. However, HIF1α acts as a ccRCC tumor suppressor and in this line, HIF1α locus is inactivated in some ccRCC while the expression of HIF2α—acting as a ccRCC oncoprotein—persists in some other ccRCC subtypes.
General Considerations About HIF2α-Dependent ccRCC Development
The protumoral potential of HIF2α in ccRCC have been studied extensively in VHL-deficient cell lines that express only HIF2α, such as the 786-O and A498 (24, 25) or those expressing both isoforms such as RCC4 cells (17, 26). Genetic or pharmacological inhibition of HIF2α usually in 786-O cells impairs their ability to form xenografts in nude mice and to generate colonies in soft agar conditions (18, 20, 25, 27, 28). However, such HIF2α inhibition does not alter the cell autonomous proliferation of these cells when they are grown in a petri dish under standard culture conditions (26–30). In RCC4 cells, inhibition of HIF2α can attenuate their normoxic in vitro cell proliferation under standard culture conditions (17, 26) although the extent of this effect is much less pronounced when compared with HIF2α inhibition in vivo in 786-O cells. Therefore, though the pro-proliferative properties of HIF2α in ccRCC can be appreciated in some cell culture conditions, they appear to be best observed when cells are subjected to experimental conditions that better mimic the three-dimensional solid tumor in vivo, such as xenografts in immunocompetent mice or colonies grown in soft agar.
A well-known target of HIF2α in ccRCC is vascular endothelial growth factor-a (VEGF-a), which drives ccRCC angiogenesis (23, 24, 31, 32). This HIF2/VEGF-a pathway does not seem to be required for in vitro 786-O cell proliferation but rather, it is essential for 786-O xenograft formation where intratumoral neoangiogenesis may be more critical (28). As such, this HIF2/VEGF-a pathway can explain the more pronounced impact of HIF2α on in vivo xenograft growth as opposed to in vitro cell proliferation (Figure 2). In this line, several pharmacological interventions to block ccRCC angiogenesis that target VEGF receptor (VEGFR) activity have shown clinical efficacy, such as sorafenib, pazopanib, or sunitinib (33).
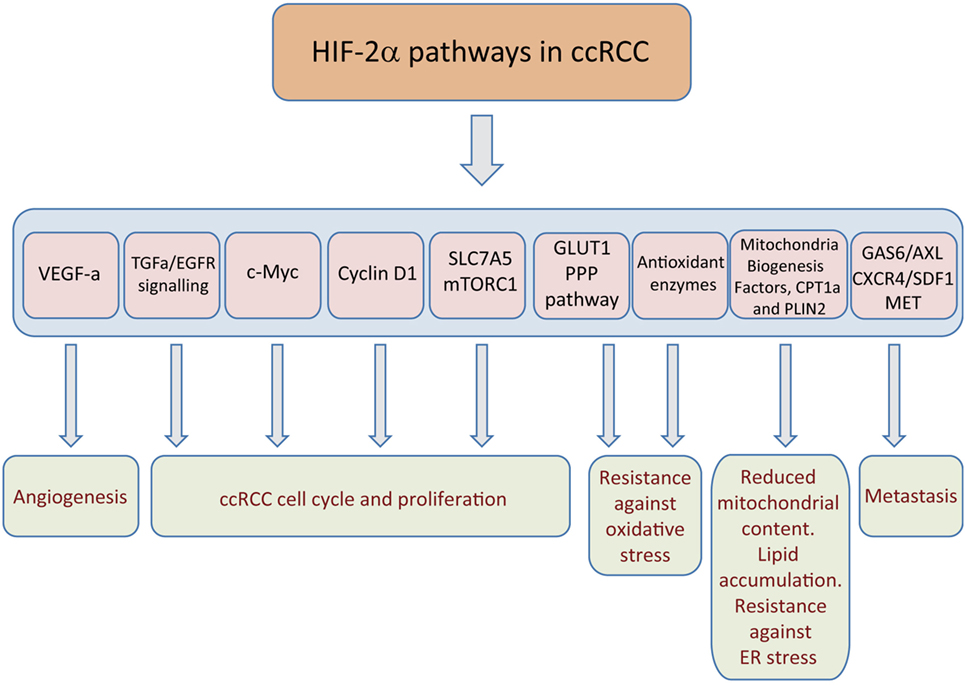
Figure 2. Hypoxia-inducible factor (HIF)2α-dependent pathways that sustain clear cell renal cell carcinoma (ccRCC) growth. The figure represents those target genes primarily controlled by HIF2α and those biological actions executed by these genes, such as tumor angiogenesis, cell autonomous proliferation, potentiation of glycolysis and pentose phosphate pathway (PPP), resistance to oxidative damage, endoplasmic reticulum (ER) stress, as well as metastasis.
HIF2α-Dependent Cell Autonomous Pathways in ccRCC Growth
Hypoxia-inducible factor 2α-driven angiogenesis is certainly relevant to the progress of ccRCC, affecting distant vascular cells in a non-cell autonomous manner. However, from here on we will focus on other key HIF2α dependent, cell autonomous mechanisms that are critical to not only explain the protumoral properties of HIF2α in ccRCC but also the more remarkable effect of HIF2α in in vivo settings that are relevant to investigate ccRCC progression.
HIF2α- and EGFR-Dependent Pathway
Regarding the effect of HIF2α in in vivo settings, it can be considered that cells in xenografts have less intratumoral access to nutrients or growth factors when compared with cells grown in standard monolayer cultures where nutrients and growth factors are more accessible. Indeed, the pro-proliferative potential of HIF2α on 786-O cells can best be observed when these cells are cultured in the low serum conditions (30). At the molecular level, HIF2α induces TGF-α expression, which in turns activates epidermal growth factor receptor (EGFR)-dependent cell signaling and proliferation (Figure 2) (30, 32). Moreover, it has been also shown that EGFR can be also transcriptionally regulated by HIF signaling (34) where HIF2α could also participate in its regulation in ccRCC. Interestingly, HIF2α can facilitate EGFR expression not only through transcriptional mechanisms but also by promoting EGFR mRNA translation. Indeed, the 3’ UTR region of EGFR mRNA contains an RNA binding site for HIF2α (rHRE) and the binding of HIF2α to this site accelerates EGFR mRNA translation. HIF2α forms a complex with the RNA-binding protein RBM4 and the cap-binding eIF4E2, an eIF4E homolog, which binds to the rHRE of EGRF mRNA and targets EGFR transcript to polysomes for active translation (35). Interestingly, this HIF2α–RBM4 complex has also been detected in ccRCC cells lines, irrespective of the oxygen tension (35), suggesting a possible role of this mechanism in ccRCC growth. Furthermore, HIF2α can also attenuate EGFR endocytosis, therefore, facilitating EGFR-dependent signaling in ccRCC cells (36). Moreover, HIF2α overexpression increases EGFR protein levels in 786-O cells (36). However, an independent study have shown that silencing endogenous HIF2α in 786-O does not alter EGFR protein levels in these cells (37).
Regarding the functional role of EGFR in ccRCC, silencing of EGFR signaling suppresses the ability of VHL-deficient 786-O cell lines to form xenografts. Along this line, it is also important to highlight that EGFR upregulation has been found in human ccRCC (34, 38, 39), which correlates with poor prognostic parameters (38). An additional study has also shown elevated EGFR expression in papillary RCC that is not characterized by HIF overactivation but in a lesser extent than in ccRCC (39). Furthermore, the expression of HCRP1, a repressor of EGFR signaling, is dampened significantly in ccRCC (40). In addition, it has also been found that the cell surface glycoprotein CUB domain-containing protein 1 (CDCP1) is induced through HIF2α—but not HIF1α—in response to hypoxia (41). EGF and CDCP1 cooperate to EGF-dependent intracellular signaling (42, 43), which suggest that this cooperation also occurs in ccRCC biology. In this line, CDCP1 is elevated in ccRCC and poor overall survival is found in patients with high CDCP1 expression (41).
In addition to EGFR function, HIF2α also potentiates the activity of the met proto-oncogene (MET) receptor (44–46). In this line, another tyrosin-kinase receptor AXL that is highly expressed in aggressive ccRCC tumors and associated with poor outcome has been shown to be a direct target gene of HIF2α but not HIF1α isoform (47). Indeed, GAS6/AXL signaling activates the met proto-oncogene (MET) receptor in an HGF-independent manner to optimize ccRCC migration and invasion without affecting primary tumor growth assessed using SN12L1 renal cancer cells (Figure 2) (47). Furthermore, HIF2α induces the expression of CXCR4 receptor, which facilitates the ability of its ligand—stromal cell-derived factor-1a (SDF-1a)—to promote ccRCC chemotaxis and influence patient survival (48–50).
Role of HIF2α in the Regulation of c-Myc, Cell Cycle Regulators and p53
A key event in the molecular biology of ccRCC is the activation of c-Myc, a pro-proliferative transcription factor (Figure 2) (51, 52). In addition, in ccRCC, HIF1α and HIF2α have opposite effects on some c-Myc target genes that are involved in the cell cycle, providing a molecular basis for the opposite properties of both these HIF isoforms during overt ccRCC progression. Indeed, HIF1α impairs the binding of c-Myc to the regulatory regions of genes involved in the cell cycle, reducing E2F and cyclin D2 expression, while augmenting the expression of the cell cycle blockers p21 and p27 (21, 51). In sharp contrast, the HIF2α isoform potentiates c-Myc activity on these genes, regulating gene expression to favor cell cycle progression of ccRCC cells (Figure 2) (21, 51). Furthermore, this HIF2α-c-Myc pathway controls the expression of key effectors of homologous recombination or spindle assembly checkpoint, which limit DNA damage during replication (21).
Regarding the role of HIF2α in regulating the cell cycle machinery, cyclin D1 is another HIF2α target found specifically in renal cancers (Figure 2) (19). Indeed, a genetic variant at chromosome 11q13.3 is associated with a predisposition to renal cancer by permitting HIF2α binding to a cyclin D1 enhancer (53). Cyclin D1 is a cell cycle regulator involved in cancer cell proliferation and development (54), and its relative contribution to ccRCC has recently been evaluated in cyclin D1-silenced 786-O cells. Silencing cyclin D1 in 786-O cells does not alter their in vitro cell growth, but it does markedly attenuate their ability to form xenografts (28). Hence, like other HIF2α-dependent genes described in this review, cyclin D1 appears to be specifically required to sustain ccRCC growth in vivo without having a major impact on in vitro cell culture conditions.
Finally, HIF2α also prevents the activity of the tumor suppressor p53 to favor ccRCC survival and protect ccRCC cells from radiation treatment (20). Indeed, HIF2α induces the expression of several antioxidant enzymes in ccRCC, which restrict the oxidative stress-dependent p53 activation (Figure 2) (20). Therefore, inhibition of HIF2α permits the accumulation of reactive oxygen species (ROS) and DNA damage, leading to apoptosis and reduced survival of ccRCC cells (20).
HIF2α and mTORC1 Activity in ccRCC
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that responds to amino acid availability, as well as energy status of the cell, playing a central role in cell growth and proliferation (55, 56). mTORC1 activation is required for ccRCC progression (57) and indeed, the allosteric inhibitors of mTORC1, everolimus, and temsirolimus, counteract ccRCC (58, 59). Such mTORC1 activation is also observed in mouse models of ccRCC in which Vhl and Pten, or Vhl and Pbrm1, are inactivated simultaneously in the epithelial cells of the genital tract (60, 61). This mTORC1 overactivation is a consequence of excess HIF2α activity in ccRCC (29). It should be considered first that mTORC1 responds to the availability of essential extracellular amino acids and second that amino acid supply presumably becomes compromised at the inner core of solid tumors (29, 62, 63). Along similar lines, HIF2α provides both an in vitro growth advantage to ccRCC cells as well as a potentiation of mTORC1 activity when cells are exposed to low amino acid supply, which to some extent could mimic the limited intratumoral amino acid availability (29).
At the molecular level, HIF2α induces the expression of the amino acid carrier SLC7A5 (LAT-1), which is essential to promote amino acid-dependent mTORC1 activation, and to sustain the potential of 786-O cells to form xenografts in nude mice (Figure 2) (29, 64). Enhanced SLC7A5 expression is not only found in 786-O VHL-deficient cells but also, in human ccRCC samples. Importantly, SLC7A5 protein is markedly elevated in VHL-deficient ccRCC samples—but not in non-clear renal cell carcinomas (ncRCC)—when compared with adjacent healthy kidney (29). An independent study has also confirmed this SLC7A5 elevation at RNA level in VHL-deficient ccRCC when compared with non-tumor samples (65). Importantly, increased SLC7A5 expression favors amino acid-dependent mTORC1 activity and cell proliferation, preferentially in low amino acid conditions (29, 66, 67). The relevance of SLC7A5 in low amino acid conditions can also explain the marked effect of HIF2α in in vivo ccRCC settings, when a limited intratumoral amino acid supply would be expected. Furthermore, the influence of HIF2α on SLC7A5 expression is not restricted to renal cell carcinoma and it has actually been found in other biological settings, such as cervical cancer cells (68) as well as in other non-tumoral scenarios (29, 69).
Paradoxically, HIF2α also increases the expression of REDD1, a well-recognized mTORC1 inhibitor (70). REDD1 is not capable of repressing mTORC1 in 786-O cells but this is not generalized, since it does not occur in other VHL-deficient cell lines (71). Therefore, in ccRCC a HIF2α-dependent mTORC1 activation pathway overrides the mTORC1 inhibitory potential of REDD1 (71). Indeed, it is likely that elevated REDD1 in ccRCC could limit full mTORC1 activation but not potent enough to prevent HIF2α-dependent mTORC1 activated pathways.
HIF2α and ccRCC Glucose and Lipid Metabolism
A central metabolic response to HIF activation is an anaerobic switch favoring glycolysis, while simultaneously repressing mitochondrial function (72–74). The HIF-dependent regulation of glycolysis is mainly executed by the HIF1α primarily inducing the vast majority of glycolytic enzymes (75–77). However, HIF2α is also a potent inducer of glucose transporter-1 (GLUT-1) (18, 19) and of enolase 2 (ENO2) (28) in ccRCC (Figure 2), which also anticipates an increased rate of glycolysis in cells expressing only HIF2α. Glycolytic intermediates corresponding to the first steps of the glycolytic pathway that are required to foster the pentose phosphate pathway are markedly elevated in human ccRCC, possibly contributing to counteract oxidative stress (Figure 2) (78–80). Regarding the relative contribution of GLUT-1 to ccRCC biology, VHL-deficient RCC4 cells are particularly sensitive to glucose deprivation and GLUT-1 silencing provokes apoptosis of these cells (81). Furthermore, pharmacological inhibition of GLUT-1 with STF-31 selectively kills VHL-deficient cells and attenuates the ability of 786-O cells to form xenografts (81). Surprisingly, an independent study has shown that silencing of GLUT-1 or ENO-2 does not prevent these 786-O cells from generating xenografts (28). The reasons for this discrepancy are unclear and possibly reflect differences between the pharmacological and genetic approaches to assure GLUT-1 inhibition in 786-O cells. Despite of this contrasting information, ccRCC are characterized by accumulation of glycolytic intermediates as mentioned above in line with GLUT-1 elevation. Furthermore, HIF2α not only increases GLUT-1 expression, probably to increase in the glycolytic rate but also, it simultaneously attenuates glucose oxidation in parallel with an increase in glutamine usage via the reductive carboxylation pathway (82). These metabolic actions seem to be executed even in the absence of HIF1α isoform, which has been more widely recognized to be essential to attenuate glucose oxidation and promote glutamine reductive carboxylation (83–85). Indeed, 786-O cells use reductive glutamine metabolism to generate the citrate that is essential to sustain lipogenic pathways that are required for cell proliferation (82).
In addition to this glucose and glutamine metabolism reprogramming, a key metabolic feature that defines ccRCC is the remarkable accumulation of intracellular lipid droplets (86). These lipid droplets are the sites where cells store neutral lipids, such as triglycerides, steryl esters, and retinyl esters that are surrounded by a phospholipid monolayer including associated lipid droplets surface proteins (87). HIF2α—together with HIF1α—participates in the lipid deposition in ccRCC. First, HIF2α drives lipid deposition in ccRCC by repressing fatty acid oxidation, specifically the rate-limiting component of mitochondrial fatty acid transport, carnitine palmitoyltransferase 1A (CPT1A) (88). Second, HIF2α can reduce mitochondrial content as well as key factors in mitochondrial biogenesis that also characterized VHL-deficient renal cell carcinoma cells (89–91) which can also explain not only reduced fatty acid oxidation but also glucose oxidation in ccRCC cells. Importantly, HIF2α, but not HIF1α, also controls the expression of Perilipin 2 (PLIN2) (Figure 2), a lipid droplet coat protein that regulates lipid storage and lipolysis (32). These droplets are functionally and physically associated with the endoplasmic reticulum (ER), and, therefore, this HIF2α-PLIN2-dependent lipid droplet formation not only favors the accumulation of excess fatty acids possibly as a consequence of reduced fatty acid oxidation but also avoids cytotoxic ER stress which facilitates the progression of ccRCC (Figure 2).
Conclusion
Hypoxia-inducible factor 2α drives numerous pathways that favor ccRCC proliferation and survival of these tumors. Importantly, these pathways seem to be specially relevant under in vivo conditions, where tumor mass can be characterized by compromised oxygen supply, nutrient availability (e.g., glucose or amino acids), possibly less access to growth factors, such as EGFR ligands, as well as stressful intratumoral conditions. In this line, HIF2α and their pathways seem less relevant when ccRCC cells are grown under in vitro monolayer conditions, where nutrient, oxygen supply and growth factors supply can be unlimited. In this line, HIF2α favors simultaneously the expression of GLUT-1 glucose transporter, SLC7A5 amino acid carrier, as well as VEGFa-dependent angiogenesis, which all together can favor ccRCC nutrient and oxygen supply in vivo. Moreover, HIF2α also potentiates EGFR signaling and promotes signals that alleviate oxidative and ER stress promoting ccRCC survival. Along this line, current therapies that focus on HIF2α inhibition have the potential to concomitantly affect all the pathways described here, without the need to focus on each individual pathway described here.
Author Contributions
JL wrote the manuscript and designs the general structure, sections, and topics to be discussed in this mini-review. FM-R, OR, and RSP wrote specific parts of this mini-review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from Ministerio de Educación y Ciencia (SAF2013-46058-R; SAF2016-76815), TV3 Marató (534/C/2016), and CIBERCV. OR has a contract for accessing the Spanish System of Science, Technology and Innovation (SECTI) funded by the University of Castilla La Mancha (UCLM).
References
1. Frew IJ, Moch H. A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu Rev Pathol (2015) 10:263–89. doi:10.1146/annurev-pathol-012414-040306
2. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer (2015) 15:55–64. doi:10.1038/nrc3844
3. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi:10.1038/nrdp.2017.9
4. Kaelin WG Jr. The VHL tumor suppressor gene: insights into oxygen sensing and cancer. Trans Am Clin Climatol Assoc (2017) 128:298–307.
5. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science (2001) 294:1337–40. doi:10.1126/science.1066373
6. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi:10.1016/S0092-8674(01)00507-4
7. Safran M, Kaelin WG Jr. HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest (2003) 111:779–83. doi:10.1172/JCI200318181
8. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–54. doi:10.1038/nrm1366
9. Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem (1996) 271:17771–8. doi:10.1074/jbc.271.30.17771
10. Ortiz-Barahona A, Villar D, Pescador N, Amigo J, del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res (2010) 38:2332–45. doi:10.1093/nar/gkp1205
11. Ratcliffe PJ, O’Rourke JF, Maxwell PH, Pugh CW. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol (1998) 201:1153–62.
12. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148:399–408. doi:10.1016/j.cell.2012.01.021
13. Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell (2002) 1:459–68. doi:10.1016/S1535-6108(02)00071-5
14. Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, et al. Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol Biol Cell (2009) 20:1089–101. doi:10.1091/mbc.E08-06-0566
15. Schonenberger D, Harlander S, Rajski M, Jacobs RA, Lundby AK, Adlesic M, et al. Formation of renal cysts and tumors in Vhl/Trp53-deficient mice requires HIF1alpha and HIF2alpha. Cancer Res (2016) 76:2025–36. doi:10.1158/0008-5472.CAN-15-1859
16. Schonenberger D, Rajski M, Harlander S, Frew IJ. Vhl deletion in renal epithelia causes HIF-1alpha-dependent, HIF-2alpha-independent angiogenesis and constitutive diuresis. Oncotarget (2016) 7:60971–85. doi:10.18632/oncotarget.11275
17. Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell (2007) 11:335–47. doi:10.1016/j.ccr.2007.02.006
18. Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell (2002) 1:237–46. doi:10.1016/S1535-6108(02)00043-0
19. Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol (2005) 25:5675–86. doi:10.1128/MCB.25.13.5675-5686.2005
20. Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, et al. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci U S A (2009) 106:14391–6. doi:10.1073/pnas.0907357106
21. Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell (2008) 14:435–46. doi:10.1016/j.ccr.2008.10.016
22. Salama R, Masson N, Simpson P, Sciesielski LK, Sun M, Tian YM, et al. Heterogeneous effects of direct hypoxia pathway activation in kidney cancer. PLoS One (2015) 10:e0134645. doi:10.1371/journal.pone.0134645
23. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539:112–7. doi:10.1038/nature19796
24. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature (2016) 539:107–11. doi:10.1038/nature19795
25. Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol (2003) 1:E83. doi:10.1371/journal.pbio.0000083
26. Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, et al. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov (2011) 1:222–35. doi:10.1158/2159-8290.CD-11-0098
27. Cho H, Kaelin WG. Targeting HIF2 in clear cell renal cell carcinoma. Cold Spring Harb Symp Quant Biol (2016) 81:113–21. doi:10.1101/sqb.2016.81.030833
28. Zhang T, Niu X, Liao L, Cho EA, Yang H. The contributions of HIF-target genes to tumor growth in RCC. PLoS One (2013) 8:e80544. doi:10.1371/journal.pone.0080544
29. Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F, Rodriguez-Vaello V, Marsboom G, de Carcer G, et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell (2012) 48:681–91. doi:10.1016/j.molcel.2012.09.017
30. Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S. Silencing of epidermal growth factor receptor suppresses hypoxia-inducible factor-2-driven VHL-/- renal cancer. Cancer Res (2005) 65:5221–30. doi:10.1158/0008-5472.CAN-05-0169
31. Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1alpha expression maintain vascular endothelial growth factor expression through HIF-2alpha. Carcinogenesis (2007) 28:529–36. doi:10.1093/carcin/bgl143
32. Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov (2015) 5:652–67. doi:10.1158/2159-8290.CD-14-1507
33. Coppin C, Kollmannsberger C, Le L, Porzsolt F, Wilt TJ. Targeted therapy for advanced renal cell cancer (RCC): a Cochrane systematic review of published randomised trials. BJU Int (2011) 108:1556–63. doi:10.1111/j.1464-410X.2011.10629.x
34. Roche O, Deguiz ML, Tiana M, Galiana-Ribote C, Martinez-Alcazar D, Rey-Serra C, et al. Identification of non-coding genetic variants in samples from hypoxemic respiratory disease patients that affect the transcriptional response to hypoxia. Nucleic Acids Res (2016) 44:9315–30. doi:10.1093/nar/gkw811
35. Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature (2012) 486:126–9. doi:10.1038/nature11055
36. Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med (2009) 15:319–24. doi:10.1038/nm.1922
37. Zhou L, Yang H. The von Hippel-Lindau tumor suppressor protein promotes c-Cbl-independent poly-ubiquitylation and degradation of the activated EGFR. PLoS One (2011) 6:e23936. doi:10.1371/journal.pone.0023936
38. Dordevic G, Matusan Ilijas K, Hadzisejdic I, Maricic A, Grahovac B, Jonjic N. EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J Biomed Sci (2012) 19:40. doi:10.1186/1423-0127-19-40
39. Thomasson M, Hedman H, Ljungberg B, Henriksson R. Gene expression pattern of the epidermal growth factor receptor family and LRIG1 in renal cell carcinoma. BMC Res Notes (2012) 5:216. doi:10.1186/1756-0500-5-216
40. Chen F, Deng J, Liu X, Li W, Zheng J. HCRP-1 regulates cell migration and invasion via EGFR-ERK mediated up-regulation of MMP-2 with prognostic significance in human renal cell carcinoma. Sci Rep (2015) 5:13470. doi:10.1038/srep13470
41. Emerling BM, Benes CH, Poulogiannis G, Bell EL, Courtney K, Liu H, et al. Identification of CDCP1 as a hypoxia-inducible factor 2alpha (HIF-2alpha) target gene that is associated with survival in clear cell renal cell carcinoma patients. Proc Natl Acad Sci U S A (2013) 110:3483–8. doi:10.1073/pnas.1222435110
42. Dong Y, He Y, de Boer L, Stack MS, Lumley JW, Clements JA, et al. The cell surface glycoprotein CUB domain-containing protein 1 (CDCP1) contributes to epidermal growth factor receptor-mediated cell migration. J Biol Chem (2012) 287:9792–803. doi:10.1074/jbc.M111.335448
43. Law ME, Ferreira RB, Davis BJ, Higgins PJ, Kim JS, Castellano RK, et al. CUB domain-containing protein 1 and the epidermal growth factor receptor cooperate to induce cell detachment. Breast Cancer Res (2016) 18:80. doi:10.1186/s13058-016-0741-1
44. Koochekpour S, Jeffers M, Wang PH, Gong C, Taylor GA, Roessler LM, et al. The von Hippel-Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol Cell Biol (1999) 19:5902–12. doi:10.1128/MCB.19.9.5902
45. Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, et al. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res (2006) 66:3699–705. doi:10.1158/0008-5472.CAN-05-0617
46. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell (2003) 3:347–61. doi:10.1016/S1535-6108(03)00085-0
47. Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A (2014) 111:13373–8. doi:10.1073/pnas.1404848111
48. Gassenmaier M, Chen D, Buchner A, Henkel L, Schiemann M, Mack B, et al. CXC chemokine receptor 4 is essential for maintenance of renal cell carcinoma-initiating cells and predicts metastasis. Stem Cells (2013) 31:1467–76. doi:10.1002/stem.1407
49. Micucci C, Matacchione G, Valli D, Orciari S, Catalano A. HIF2alpha is involved in the expansion of CXCR4-positive cancer stem-like cells in renal cell carcinoma. Br J Cancer (2015) 113:1178–85. doi:10.1038/bjc.2015.338
50. Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature (2003) 425:307–11. doi:10.1038/nature01874
51. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell (2007) 12:108–13. doi:10.1016/j.ccr.2007.07.006
52. Grampp S, Platt JL, Lauer V, Salama R, Kranz F, Neumann VK, et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat Commun (2016) 7:13183. doi:10.1038/ncomms13183
53. Schodel J, Bardella C, Sciesielski LK, Brown JM, Pugh CW, Buckle V, et al. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat Genet (2012) 44(420–5):S1–2. doi:10.1038/ng.2204
54. Pestell RG. New roles of cyclin D1. Am J Pathol (2013) 183:3–9. doi:10.1016/j.ajpath.2013.03.001
55. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell (2000) 103:253–62. doi:10.1016/S0092-8674(00)00117-3
56. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell (2009) 136:731–45. doi:10.1016/j.cell.2009.01.042
57. Battelli C, Cho DC. mTOR inhibitors in renal cell carcinoma. Therapy (2011) 8:359–67. doi:10.2217/thy.11.32
58. Barthelemy P, Hoch B, Chevreau C, Joly F, Laguerre B, Lokiec F, et al. mTOR inhibitors in advanced renal cell carcinomas: from biology to clinical practice. Crit Rev Oncol Hematol (2013) 88:42–56. doi:10.1016/j.critrevonc.2013.02.006
59. Kornakiewicz A, Solarek W, Bielecka ZF, Lian F, Szczylik C, Czarnecka AM. Mammalian target of rapamycin inhibitors resistance mechanisms in clear cell renal cell carcinoma. Curr Signal Transduct Ther (2014) 8:210–8. doi:10.2174/1574362409666140206222746
60. Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J (2008) 27:1747–57. doi:10.1038/emboj.2008.96
61. Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, et al. The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell renal cell carcinoma. Cell Rep (2017) 18:2893–906. doi:10.1016/j.celrep.2017.02.074
62. Marjon PL, Bobrovnikova-Marjon EV, Abcouwer SF. Expression of the pro-angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 by human breast carcinomas is responsive to nutrient deprivation and endoplasmic reticulum stress. Mol Cancer (2004) 3:4. doi:10.1186/1476-4598-3-4
63. Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell (2013) 50:200–11. doi:10.1016/j.molcel.2013.02.008
64. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi:10.1016/j.cell.2008.11.044
65. Betsunoh H, Fukuda T, Anzai N, Nishihara D, Mizuno T, Yuki H, et al. Increased expression of system large amino acid transporter (LAT)-1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer (2013) 13:509. doi:10.1186/1471-2407-13-509
66. Cormerais Y, Giuliano S, LeFloch R, Front B, Durivault J, Tambutte E, et al. Genetic disruption of the multifunctional CD98/LAT1 complex demonstrates the key role of essential amino acid transport in the control of mTORC1 and tumor growth. Cancer Res (2016) 76:4481–92. doi:10.1158/0008-5472.CAN-15-3376
67. Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep (2017) 21:3819–32. doi:10.1016/j.celrep.2017.12.002
68. Corbet C, Draoui N, Polet F, Pinto A, Drozak X, Riant O, et al. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res (2014) 74:5507–19. doi:10.1158/0008-5472.CAN-14-0705
69. Suhara T, Hishiki T, Kasahara M, Hayakawa N, Oyaizu T, Nakanishi T, et al. Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc Natl Acad Sci U S A (2015) 112:11642–7. doi:10.1073/pnas.1515872112
70. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev (2004) 18:2893–904. doi:10.1101/gad.1256804
71. Kucejova B, Pena-Llopis S, Yamasaki T, Sivanand S, Tran TA, Alexander S, et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res (2011) 9:1255–65. doi:10.1158/1541-7786.MCR-11-0302
72. Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab (2009) 9:11–22. doi:10.1016/j.cmet.2008.10.001
73. Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol (2011) 76:347–53. doi:10.1101/sqb.2011.76.010678
74. Xie H, Simon MC. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem (2017) 292:16825–32. doi:10.1074/jbc.R117.799973
75. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol (2003) 23:9361–74. doi:10.1128/MCB.23.24.9361-9374.2003
76. Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J (1998) 17:3005–15. doi:10.1093/emboj/17.11.3005
77. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269:23757–63.
78. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell (2016) 29:104–16. doi:10.1016/j.ccell.2015.12.004
79. Pinthus JH, Whelan KF, Gallino D, Lu JP, Rothschild N. Metabolic features of clear-cell renal cell carcinoma: mechanisms and clinical implications. Can Urol Assoc J (2011) 5:274–82. doi:10.5489/cuaj.10196
80. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499:43–9. doi:10.1038/nature12222
81. Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med (2011) 3:94ra70. doi:10.1126/scitranslmed.3002394
82. Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab (2013) 17:372–85. doi:10.1016/j.cmet.2013.02.002
83. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177–85. doi:10.1016/j.cmet.2006.02.002
84. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187–97. doi:10.1016/j.cmet.2006.01.012
85. Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A (2011) 108:19611–6. doi:10.1073/pnas.1117773108
86. Ericsson JL, Seljelid R, Orrenius S. Comparative light and electron microscopic observations of the cytoplasmic matrix in renal carcinomas. Virchows Arch Pathol Anat Physiol Klin Med (1966) 341:204–23. doi:10.1007/BF00961071
87. Thiam AR, Farese RV Jr, Walther TC. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol (2013) 14:775–86. doi:10.1038/nrm3699
88. Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun (2017) 8:1769. doi:10.1038/s41467-017-01965-8
89. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, et al. Suppression of PGC-1alpha is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep (2015) 12:116–27. doi:10.1016/j.celrep.2015.06.006
90. Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis (2002) 23:759–68. doi:10.1093/carcin/23.5.759
Keywords: Von Hippel–Lindau, hypoxia-inducible factors, hypoxia-inducible factor 2, kidney, renal cancer, clear cell renal cell carcinoma
Citation: Meléndez-Rodríguez F, Roche O, Sanchez-Prieto R and Aragones J (2018) Hypoxia-Inducible Factor 2-Dependent Pathways Driving Von Hippel–Lindau-Deficient Renal Cancer. Front. Oncol. 8:214. doi: 10.3389/fonc.2018.00214
Received: 05 February 2018; Accepted: 24 May 2018;
Published: 08 June 2018
Edited by:
Gabriel Leprivier, Heinrich Heine Universität Düsseldorf, GermanyReviewed by:
Kasper Rouschop, Maastricht University, NetherlandsBarak Rotblat, Ben-Gurion University of the Negev, Israel
Copyright: © 2018 Meléndez-Rodríguez, Roche, Sanchez-Prieto and Aragones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julian Aragones, amFyYWdvbmVzLmhscHJAc2FsdWQubWFkcmlkLm9yZw==