 Raghavendra Tejo Karthik Poluri1
Raghavendra Tejo Karthik Poluri1 Étienne Audet-Walsh1,2*
Étienne Audet-Walsh1,2*
- 1Department of Molecular Medicine, Axe Endocrinologie – Néphrologie du Centre de recherche du CHU de Québec, Université Laval, Québec, QC, Canada
- 2Centre de recherche sur le cancer de l’Université Laval, Québec, QC, Canada
The PTEN gene encodes for the phosphatase and tensin homolog; it is a tumor suppressor gene that is among the most frequently inactivated genes throughout the human cancer spectrum. The most recent sequencing approaches have allowed the identification of PTEN genomic alterations, including deletion, mutation, or rearrangement in about 50% of prostate cancer (PCa) cases. It appears that mechanisms leading to PTEN inactivation are cancer-specific, comprising gene mutations, small insertions/deletions, copy number alterations (CNAs), promoter hypermethylation, and RNA interference. The examination of publicly available results from deep-sequencing studies of various cancers showed that PCa appears to be the only cancer in which PTEN is lost mostly through CNA. Instead of inactivating mutations, which are seen in other cancers, deletion of the 10q23 locus is the most common form of PTEN inactivation in PCa. By investigating the minimal deleted region at 10q23, several other genes appear to be lost simultaneously with PTEN. Expression data indicate that, like PTEN, these genes are also downregulated upon loss of 10q23. These analyses raise the possibility that 10q23 is lost upon selective pressure not only to inactivate PTEN but also to impair the expression of surrounding genes. As such, several genes from this deleted region, which represents about 500 kb, may also act as tumor suppressors in PCa, requiring further studies on their respective functions in that context.
Introduction
The PTEN gene on chromosome 10q23 encodes for the phosphatase and tensin homolog, a tumor suppressor gene that is among the most frequently inactivated genes throughout the human cancer spectrum. Its lipid phosphatase activity allows PTEN to dephosphorylate phosphatidylinositol-triphosphate, therefore repressing the oncogenic PI3K/Akt/mTOR pathway. In prostate cancer (PCa), PTEN is frequently lost by deletion of the 10q23 region in tumors, which has been described several years ago (1–4), and prostate-specific deletion of Pten in mice leads to PCa development (5). The most recent sequencing approaches have allowed the identification of several types of genomic alterations of PTEN, including deletion, mutation, or rearrangement (including genomic inversions), and have further described PTEN alterations in about 50% of all PCa samples (6–14).
The inactivation rate of PTEN in PCa is similar to what has been described in other types of cancer, such as breast and endometrial cancers (15, 16). However, mechanisms leading to PTEN inactivation appear to be cancer-specific, comprising gene mutations, small insertions/deletions, copy number alterations (CNAs), promoter hypermethylation, and RNA interference (RNAi) (6–15). For example, endometrial cancer is characterized by microsatellite instability that is associated with frameshift mutations, which are the most frequent inactivating alterations in PTEN in that type of cancer (15, 17). In addition, patients with Cowden syndrome, who have a germline mutation in PTEN, also harbor a significantly higher risk of endometrial cancer (15, 18). Cowden syndrome is a rare autosomal-dominant condition that leads to an increased risk of breast, thyroid, and endometrial cancers (15, 19). However, patients with Cowden syndrome do not have increased risk of PCa, even though the loss of PTEN can be detected early in PCa patients (1, 6, 9, 13). It is reported that in PCa, between 2 and 15% of primary tumors harbor a PTEN mutation, while between 30 and 40% exhibit an important deletion on chromosome 10q23 (6, 10–13).
Importantly, many other genes are also present on the deleted region on 10q23, most of which have not been investigated in the context of PCa. Moreover, because PTEN is more frequently deleted in PCa through large genomic deletions and at a higher frequency compared with other types of cancer instead of the gene-specific mutations that occur in most malignancies, it raises the possibility that genes lost at the same times as PTEN in PCa also display important tumor suppressor functions. It is well accepted now that large genomic deletions can contain more than one important gene, but this concept was not investigated in the context of the loss of 10q23 in PCa. In this perspective article, we will discuss the genes that are lost along with PTEN upon deletion of the 10q23 locus that might well play a role in PCa development.
PTEN is More Frequently Altered through CNA Rather than Through a Specific Gene Mutation in PCa
The PTEN genomic status was first screened through the cancer spectrum across the different cohorts available on the cBio Cancer Genomics Portal from The Cancer Genome Atlas (TCGA) group (20, 21). Only cohorts with both mutation and CNA were kept for analysis. In most types of cancer, PTEN is often mutated (Figure 1, green color), with particularly high mutations rates in glioblastoma and uterine cancer, where the alterations rate is between 40 and 65% (Figure 1). Despite showing small CNA rates, gene mutations are the most common PTEN genomic alterations. Interestingly, the only type of cancer with high rates of PTEN genomic alterations particularly caused by CNA is PCa. In PCa, between 20 and 50% of all tumors exhibit PTEN alterations, with 60 and 90% of them being CNA instead of mutations (Figure 1). This suggests that CNA at the PTEN locus might affect more than PTEN itself and lead to the deletion of other tumor suppressor genes important to the etiology of PCa.
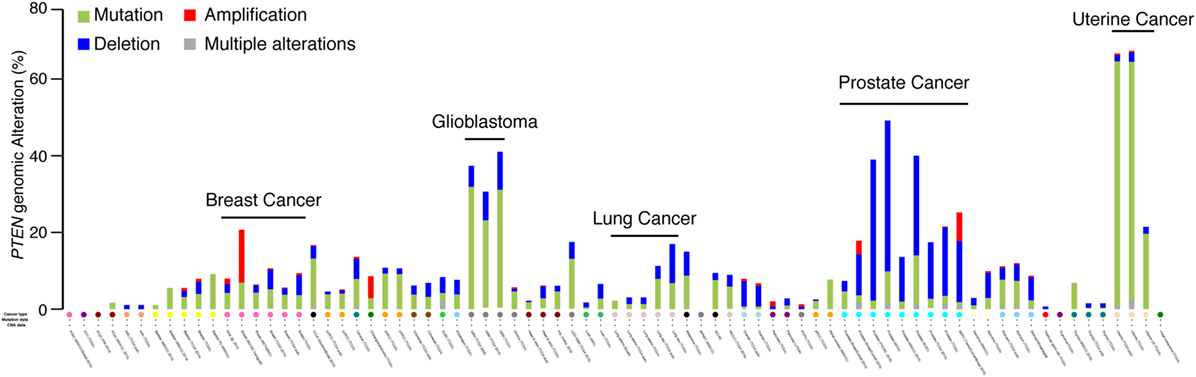
Figure 1. PTEN genomic alterations in the human cancer spectrum. Genomic alterations of the PTEN gene were visualized with the cBioPortal for Cancer Genomics (20, 21). Only cohorts with data on both mutation and copy number alterations are shown.
We further analyzed the PTEN genomic alteration status between the different PCa cohorts (Figure 2A), which included the Michigan, Stand Up To Cancer (SU2C), and Fred Hutchinson Cancer Research Center datasets, mostly comprising metastatic samples (12, 13, 22); the Trento/Cornell/Broad dataset, composed of metastatic neuroendocrine prostate cancer (23); the Broad/Cornell 2012, Broad/Cornell 2013, Memorial Sloan Kettering Cancer Center, and the two TCGA datasets, comprising mostly primary localized PCa (9, 11, 16, 24). In cohorts mostly composed of clinically localized tumors, PTEN genomic alterations, mostly CNA, ranged from 10 to 20%. This alteration frequency increased to 40–50% in cohorts of metastatic samples and castration-resistant PCa (CRPC) tumors, again mostly through CNA of PTEN. These results are consistent with previous reports linking PTEN loss to PCa aggressiveness as it is increased in more aggressive disease settings (25–28). Further investigation of PTEN alterations in the CRPC/metastatic cohorts confirmed higher rates of CNA in these tumors compared with the TCGA cohort, which is mostly composed of clinically localized tumors (Figure 2B). These data also confirmed that deletion of PTEN is the most frequent genomic alteration occurring at this locus in prostate tumors.
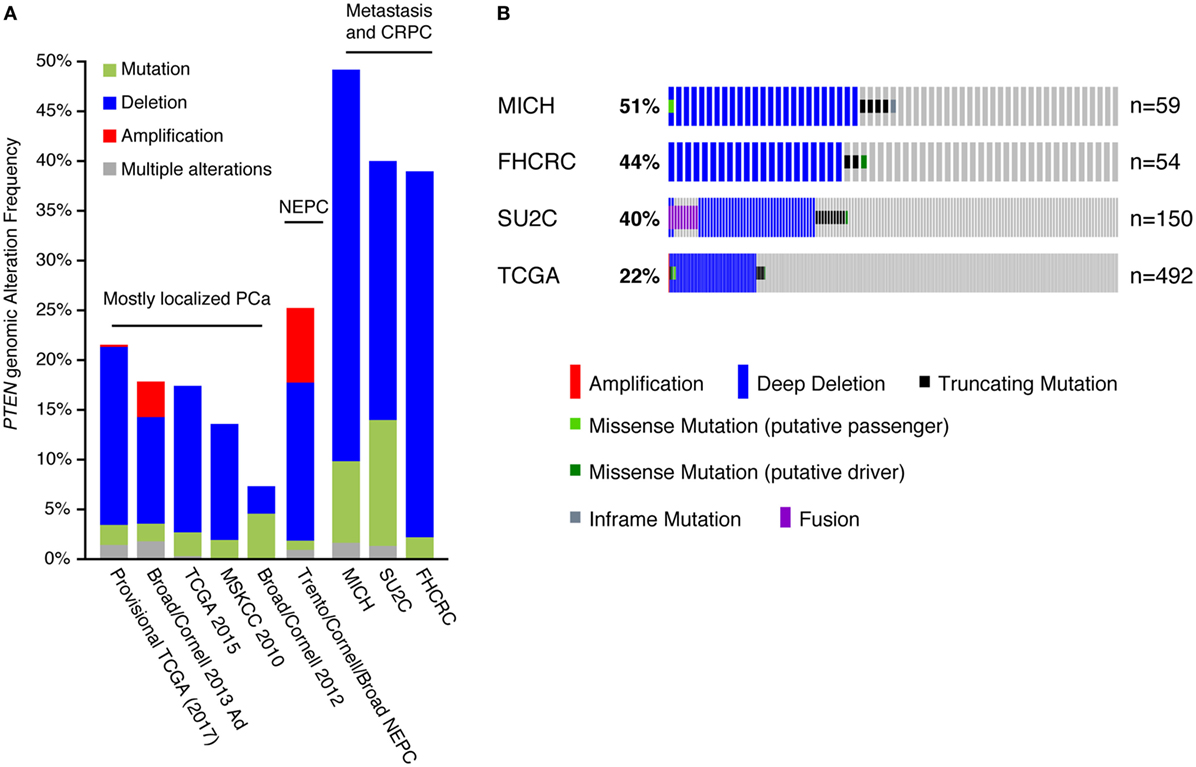
Figure 2. Copy number alteration (CNA) is the most frequent inactivation mechanism of PTEN in human prostate cancer (PCa). (A) Genomic alterations of the PTEN gene in PCa. Only cohorts with data on both mutation and CNAs are shown. Abbreviations: NEPC, neuroendocrine prostate cancer; CRPC, castration-resistant PCa. (B) Specific alterations of PTEN in the Michigan (MICH), Fred Hutchinson Cancer Research Center (FHCRC), Stand Up To Cancer (SU2C), and The Cancer Genome Atlas (TCGA) cohorts. Note that the proportion of alterations is slightly different than in (A): all tumors are shown in (A) while all patients are shown in (B) (some patients had more than one sample sequenced).
CNA at 10q23 Leads to Loss not only of PTEN but also of Several Additional Genes
Interestingly, visualization of CNA at the PTEN locus in the various cohorts available on the cBioportal indicated that deletion of PTEN often results in the loss of a large (>500 kb) genomic segment of chromosome 10q23 (Figure 3). Results from the metastatic cohort SU2C revealed that PTEN is commonly lost with other genes located at 10q23, including MINPP1, PAPSS2, KLLN, and ATAD1. Moreover, deletion at 10q23 frequently occurs in one of the RNLS introns. By also investigating other cohorts with high coverage at 10q23 CNA status, we observed a similar deletion pattern, notably in the localized PCa cohort from the provisional TCGA dataset (Figure 3, right). Again, the same similar minimal region seems to accompany the loss of PTEN, altering the same set of genes as in the more aggressive SU2C cohort, including the deletion breakpoint in the intronic region of RNLS. These results suggest that the loss of 10q23 in PCa cells does not solely inactivate the tumor suppressor PTEN, but that there is also a selective pressure to lose other gene(s) at this particular genomic region in this specific type of cancer. Accordingly, CNA at 10q23 significantly altered mRNA expression of PTEN, and deletions of PTEN resulted in decreased mRNA expression in both the SU2C and the TCGA cohorts (Figures 4A,B, respectively). Genes surrounding PTEN that are located in the minimal deleted regions (Figure 3) also have a similar pattern, with a significantly decreased expression with either shallow or deep deletions in the two cohorts (Figure 4). The only exception was PAPSS2, which was not significantly altered by deep or shallow deletion (Figure 4). As reported previously (13, 16), various inactivating mutations of PTEN were also detected in both cohorts, but at lower frequency than CNA.
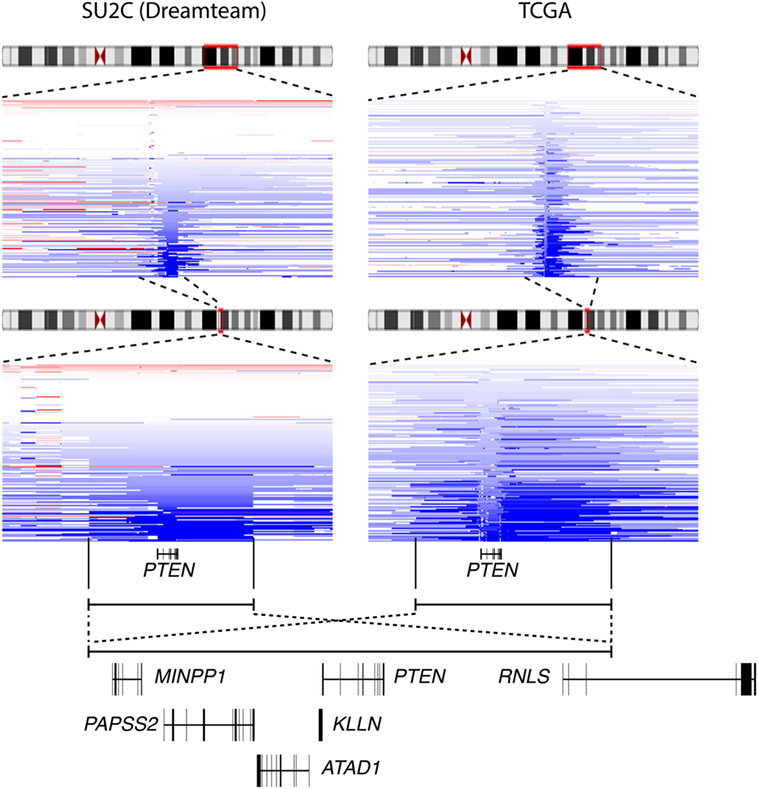
Figure 3. The minimal deletion at 10q23 in prostate cancer (PCa). Deletion status at the 10q23 locus in the Stand Up To Cancer (SU2C) (left) and the The Cancer Genome Atlas (TCGA) (right) PCa clinical datasets. Blue color indicates copy number loss at this locus. Genes present in that genomic regions are shown (bottom panel).
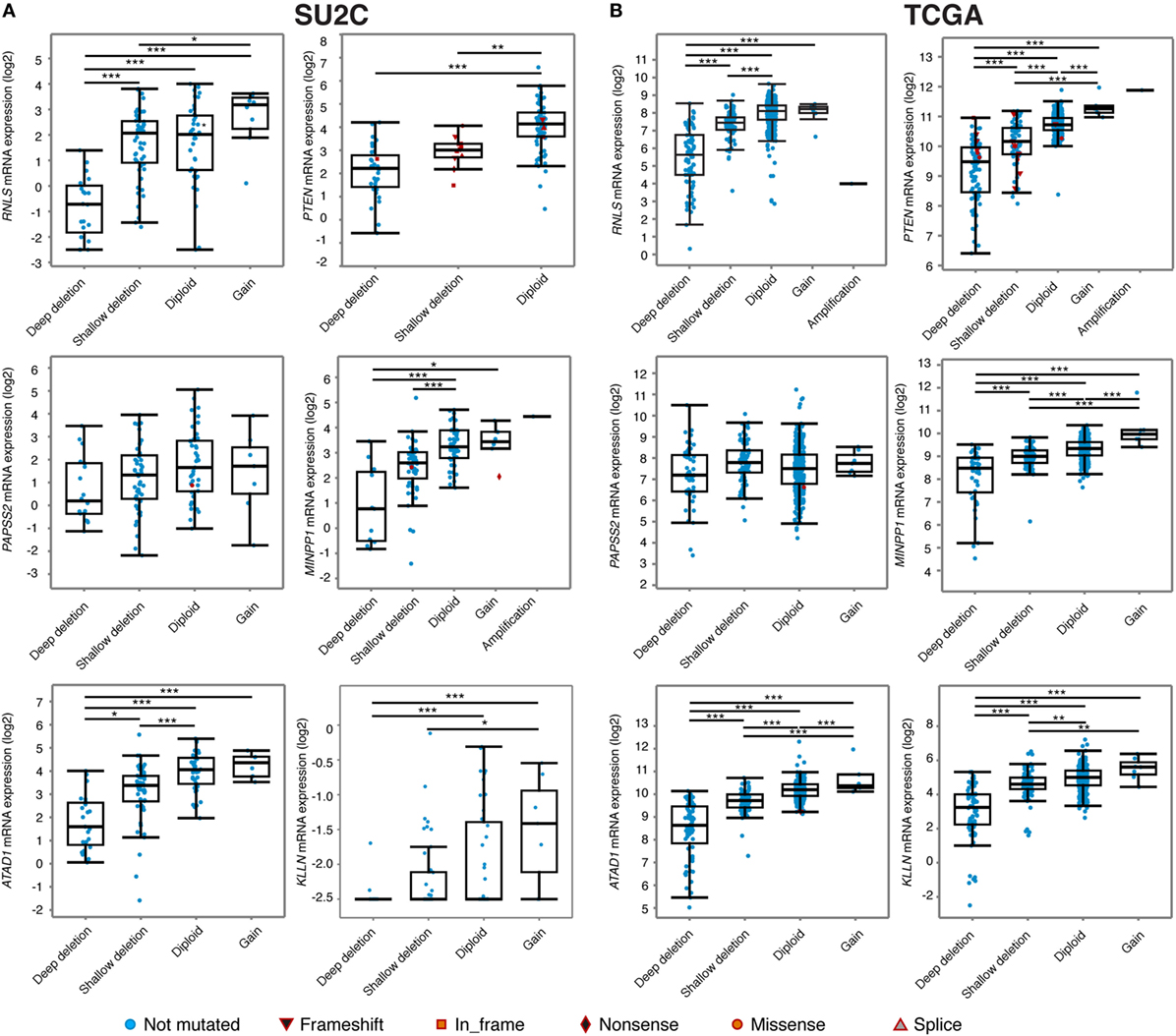
Figure 4. Relationship between genomic alterations at the 10q23 locus and genes encompassed within this region. Expression of six genes located at the minimal deleted regions at the 10q23 locus in the Stand Up To Cancer (SU2C) (A) and the The Cancer Genome Atlas (TCGA) (B) prostate cancer cohorts. ***p < 0.001; **p < 0.01; *p < 0.05 in ANOVA with post hoc Tukey’s Honest Significant Difference. Note that for RNLS and PTEN expression data in the TCGA cohort (B), the sample with an amplification at 10q23 was not included for statistics.
Future Direction in PCa Genomic Alteration Studies
Since the initial characterization of PTEN loss in PCa two decades ago (1–4), most studies on CNA at 10q23 have focused exclusively on PTEN as basically the only gene lost upon deletion at this specific locus, even in more recent deep-sequencing studies (6–14). Given its known role as a tumor suppressor in most types of cancer and because mutation of PTEN causes a hereditary syndrome with multiple cancer susceptibilities, PTEN is most certainly the main tumor suppressor gene lost with this deletion. However, other genes located in the minimal deleted region at 10q23 might play a significant role in PCa etiology because (1) CNA is the major genomic alteration in PCa, not a direct mutation of PTEN, which occurs in all other types of cancer; (2) a large region at 10q23 is deleted, comprising more than just PTEN and often including the loss of at least six other genes; and (3) some of these genes have already been associated with tumor suppressing functions in PCa or other cancers. The question remains as to what roles these genes play in prostate biology and PCa development.
KLLN encodes for the KILLIN protein, which has been identified as a P53 target required for S phase checkpoint control to eliminate precancerous cells (29). KLLN overexpression reduces PCa cell growth in vitro by decreasing the androgen receptor (AR) signaling, while its repression increases it; this is consistent with a tumor suppressor function of this gene (30). Interestingly, mutation in the promoter of KLLN is also associated with Cowden and Cowden-like syndromes, possibly by sharing its promoter with PTEN itself (31). ATAD1 depletion induces mitochondrial fragmentation and impairs respiration (32). It is notable that increased mitochondrial respiration is a key metabolic phenotype associated with PCa development and progression (33–37). RNLS encodes the renalase FAD-dependent metabolic enzyme (38), which has no currently known role in PCa. PAPSS2 encodes for PAPS synthase 2, which provides sulfate donors to sulfotransferase enzymes, including SULT2A1, which is a critical enzyme for dehydroepiandrosterone (DHEA) sulfation (39). DHEA and its sulfate form (DHEA-S) represent the major adrenal androgen precursors and therefore are important sources for intra-tumor androgen synthesis. This is particularly relevant during PCa progression and is a therapeutic target used in the clinic (adrenal androgens production is inhibited using abiraterone acetate) (40–42). The few patients identified with mutations in this gene are female, and heterozygous inactivation of PAPSS2 has been associated with polycystic ovary syndrome, premature puberty, hyperandrogenic anovulation, very low DHEA-S levels, and increased androgen levels (43). Even though PAPSS2 is not significantly decreased by CNA, intra-tumor loss of PAPSS2 could favor androgen excess and hyperactivation of AR, which is critical for tumor growth and cancer progression. MINPP1 encodes a phosphatase linked to inositol-3-phosphate metabolism, similar to PTEN functions (44). CFL1P1 is cofilin pseudogene 1 and has no known function. However, based on mRNA expression profiles across tissues from the data presented by Fagerberg et al. (45), it shows high expression specifically in the testis, possibly reflecting a function in the male reproductive system.
Further experiments are now required to characterize the role of these genes located near PTEN and lost along with this important tumor suppressor in PCa. Only a few in vitro models of human PCa exist, some that exhibit complete loss of PTEN, such as in PC3 cells; some that exhibit mutation of PTEN and partial loss at 10q23, such as LNCaP cells; and finally some that harbor wild-type PTEN, such as 22rv1 and DU145 cells (4, 9, 46). These wild-type PCa cell lines thus represent potential in vitro models to study the impact on PCa cell proliferation of PTEN inactivation, with and without inactivation of one or more of the other genes located within the minimal deleted region at 10q23 (KLLN, ATAD1, RNLS, PAPSS2, MINPP1, and CFLIP1). Because in most tumors there is a single copy lost at 10q23 in most tumors, and not a complete loss of both copies, repression of these genes with RNAi would mimic the gene expression decrease observed in tumor samples. The more recent genome editing technology using the CRISPR/Cas9 system would also allow the knockout of these genes along with PTEN to study their potential role as tumor suppressors of PCa in vitro (47).
Interestingly, the mouse genome exhibits a similar gene architecture around Pten, with at least the presence of Atad1, Rnls, Papss2, and Minpp1. The prostate-specific loss of Pten PCa mouse model (Ptenflox/flox;PB-Cre4+) is one of the most commonly used in vivo models to study PCa development and progression (5, 48). In this model, Pten is inactivated by deletion of exon 5 (49) and other genes surrounding Pten are not altered. The single knockout of these genes in the Ptenflox/flox;PB-Cre4+ would allow the study of their function as tumor suppressors in this PCa mouse model in vivo. In addition, the development of a new PCa mouse model by inducing the loss of 19qC1, the loci that contains Pten in the mouse genome, would mimic the human loss of 10q23 and would be highly informative on the biological functions as tumors suppressors of this region in comparison to the disruption of Pten only. This type of genetic engineering approach was successfully used to study oncogenic chromosomal rearrangements in mouse models of human cancers, such as the EML4-ALK oncogene in lung cancer (50). Indeed, induction of this gene fusion in adult mice promotes the development of non-small-cell lung cancers, clearly demonstrating the oncogenic properties of this chromosomal rearrangement. Inducing the loss of 19qC1 followed by rescue of Atad1, Rnls, Papss2, and Minpp1 would also be an interesting approach to study the role of all the genes lost along with PTEN in human and their impact on PCa development and progression.
Concluding Remarks
In summary, using publicly available results from deep-sequencing studies of various cancers, PCa appears to be the only cancer in which PTEN is inactivated mostly through CNA. Large genomic deletions often contain more than one important gene, and this is a concept that needs to be revisited in the context of PTEN loss in human PCa. Instead of inactivating mutations as seen in other cancers, deletion at 10q23 is the most common form of PTEN inactivation. Investigation of the minimal deleted region at 10q23 revealed that several other genes appear to be lost in addition to PTEN. Expression data indicate that, like PTEN, these genes are downregulated upon CNA, and, together with the CNA profile, suggest that these genes represent potential novel tumor suppressor genes in PCa. Their potential function as PCa tumor suppressors thus remained to be determined using state-of-the-art genetic engineering approaches in in vitro and in vivo models of PCa.
Author Notes
Data are publicly available through the TCGA cBioPortal web platform for cancer genomics. All ethics approvals and consents to publish were obtained for the original studies.
Author Contributions
EAW conceptualized the study and performed the analyses. RTKP and EAW analyzed the data and wrote the manuscript.
Conflict of Interest Statement
There are no competing financial interests to disclose. Data are publicly available through the TCGA cBioPortal web platform for cancer genomics. All ethics approvals and consents to publish were obtained for the original studies. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
EAW is funded by the Fondation du CHU de Québec and the Axe endocrinologie – néphrologie du Centre de recherche du CHU de Québec – Université Laval.
References
1. Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res (1997) 57(22):4997–5000.
2. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275(5308):1943–7. doi:10.1126/science.275.5308.1943
3. Teng DH, Hu R, Lin H, Davis T, Iliev D, Frye C, et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res (1997) 57(23):5221–5.
4. Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res (1998) 58(13):2720–3.
5. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell (2003) 4(3):209–21. doi:10.1016/S1535-6108(03)00215-0
6. Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, et al. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res (2007) 67(18):8504–10. doi:10.1158/0008-5472.CAN-07-0673
7. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463(7283):899–905. doi:10.1038/nature08822
8. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature (2010) 466(7308):869–73. doi:10.1038/nature09208
9. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
10. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature (2011) 470(7333):214–20. doi:10.1038/nature09744
11. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet (2012) 44(6):685–9. doi:10.1038/ng.2279
12. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi:10.1038/nature11125
13. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161(5):1215–28. doi:10.1016/j.cell.2015.05.001
14. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature (2017) 541(7637):359–64. doi:10.1038/nature20788
15. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer (2011) 11(4):289–301. doi:10.1038/nrc3037
16. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell (2015) 163(4):1011–25. doi:10.1016/j.cell.2015.10.025
17. Zhou XP, Kuismanen S, Nystrom-Lahti M, Peltomaki P, Eng C. Distinct PTEN mutational spectra in hereditary non-polyposis colon cancer syndrome-related endometrial carcinomas compared to sporadic microsatellite unstable tumors. Hum Mol Genet (2002) 11(4):445–50. doi:10.1093/hmg/11.4.445
18. Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol (2007) 34(5):428–34. doi:10.1053/j.seminoncol.2007.07.009
19. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst (2013) 105(21):1607–16. doi:10.1093/jnci/djt277
20. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi:10.1158/2159-8290.CD-12-0095
21. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):l1. doi:10.1126/scisignal.2004088
22. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med (2016) 22(4):369–78. doi:10.1038/nm.4053
23. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22(3):298–305. doi:10.1038/nm.4045
24. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell (2013) 153(3):666–77. doi:10.1016/j.cell.2013.03.021
25. Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, Soares FA, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer (2007) 97(5):678–85. doi:10.1038/sj.bjc.6603924
26. Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res (2011) 17(20):6563–73. doi:10.1158/1078-0432.CCR-11-1244
27. Phin S, Moore MW, Cotter PD. Genomic rearrangements of PTEN in prostate cancer. Front Oncol (2013) 3:240. doi:10.3389/fonc.2013.00240
28. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer (2015) 15(12):701–11. doi:10.1038/nrc4016
29. Cho YJ, Liang P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc Natl Acad Sci U S A (2008) 105(14):5396–401. doi:10.1073/pnas.0705410105
30. Wang Y, Radhakrishnan D, He X, Peehl DM, Eng C. Transcription factor KLLN inhibits tumor growth by AR suppression, induces apoptosis by TP53/TP73 stimulation in prostate carcinomas, and correlates with cellular differentiation. J Clin Endocrinol Metab (2013) 98(3):E586–94. doi:10.1210/jc.2012-3490
31. Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA (2010) 304(24):2724–31. doi:10.1001/jama.2010.1877
32. Chen YC, Umanah GK, Dephoure N, Andrabi SA, Gygi SP, Dawson TM, et al. Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail-anchored proteins. EMBO J (2014) 33(14):1548–64. doi:10.15252/embj.201487943
33. Costello LC, Franklin RB. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch Biochem Biophys (2016) 611:100–12. doi:10.1016/j.abb.2016.04.014
34. Audet-Walsh É, Dufour CR, Yee T, Zouanat FZ, Yan M, Kalloghlian G, et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev (2017) 31(12):1228–42. doi:10.1101/gad.299958.117
35. Audet-Walsh E, Vernier M, Yee T, Laflamme CE, Li S, Chen Y, et al. SREBF1 activity is regulated by an AR/mTOR nuclear axis in prostate cancer. Mol Cancer Res (2018). doi:10.1158/1541-7786.MCR-17-0410
36. Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, et al. Androgen-dependent repression of ERRgamma reprograms metabolism in prostate cancer. Cancer Res (2017) 77(2):378–89. doi:10.1158/0008-5472.CAN-16-1204
37. Audet-Walsh E, Yee T, Tam IS, Giguère V. Inverse regulation of DHT synthesis enzymes 5alpha-reductase types 1 and 2 by the androgen receptor in prostate cancer. Endocrinology (2017) 158(4):1015–21. doi:10.1210/en.2016-1926
38. Moran GR, Hoag MR. The enzyme: renalase. Arch Biochem Biophys (2017) 632:66–76. doi:10.1016/j.abb.2017.05.015
39. Oostdijk W, Idkowiak J, Mueller JW, House PJ, Taylor AE, O’Reilly MW, et al. PAPSS2 deficiency causes androgen excess via impaired DHEA sulfation – in vitro and in vivo studies in a family harboring two novel PAPSS2 mutations. J Clin Endocrinol Metab (2015) 100(4):E672–80. doi:10.1210/jc.2014-3556
40. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med (2011) 364(21):1995–2005. doi:10.1056/NEJMoa1014618
41. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med (2013) 368(2):138–48. doi:10.1056/NEJMoa1209096
42. Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med (2017) 377(4):352–60. doi:10.1056/NEJMoa1704174
43. Noordam C, Dhir V, McNelis JC, Schlereth F, Hanley NA, Krone N, et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N Engl J Med (2009) 360(22):2310–8. doi:10.1056/NEJMoa0810489
44. Chi H, Yang X, Kingsley PD, O’Keefe RJ, Puzas JE, Rosier RN, et al. Targeted deletion of Minpp1 provides new insight into the activity of multiple inositol polyphosphate phosphatase in vivo. Mol Cell Biol (2000) 20(17):6496–507. doi:10.1128/MCB.20.17.6496-6507.2000
45. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics (2014) 13(2):397–406. doi:10.1074/mcp.M113.035600
46. Squillace RM, Miller D, Wardwell SD, Wang F, Clackson T, Rivera VM. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. Int J Oncol (2012) 41(2):425–32. doi:10.3892/ijo.2012.1487
47. Li X, Wu R, Ventura A. The present and future of genome editing in cancer research. Hum Genet (2016) 135(9):1083–92. doi:10.1007/s00439-016-1713-3
48. Wu X, Gong S, Roy-Burman P, Lee P, Culig Z. Current mouse and cell models in prostate cancer research. Endocr Relat Cancer (2013) 20(4):R155–70. doi:10.1530/ERC-12-0285
49. Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis (2002) 32(2):148–9. doi:10.1002/gene.10036
Keywords: steroid, androgen, castration-resistant, androgen receptor, neuroendocrine, androgen deprivation therapy
Citation: Poluri RTK and Audet-Walsh É (2018) Genomic Deletion at 10q23 in Prostate Cancer: More Than PTEN Loss? Front. Oncol. 8:246. doi: 10.3389/fonc.2018.00246
Received: 03 April 2018; Accepted: 18 June 2018;
Published: 29 June 2018
Edited by:
Massimo Bonora, Albert Einstein College of Medicine, United StatesReviewed by:
Andrea Lunardi, University of Trento, ItalyAndrew M. Chan, The Chinese University of Hong Kong, Hong Kong
Copyright: © 2018 Poluri and Audet-Walsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Étienne Audet-Walsh, RXRpZW5uZS5BdWRldC1XYWxzaEBjcmNodWRlcXVlYmVjLnVsYXZhbC5jYQ==