 Barbara Wegiel
Barbara Wegiel Marta Vuerich3
Marta Vuerich3 Saeed Daneshmandi
Saeed Daneshmandi Pankaj Seth
Pankaj Seth- 1Department of Surgery, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 2Cancer Research Institute, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 3Division of Interdisciplinary Medicine, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 4Department of Medicine, Beth Israel Deaconess Medical Center, Boston, MA, United States
Tumor-induced immune tolerance permits growth and spread of malignant cells. Cancer cells have strong influence on surrounding cells and shape the hypoxic tumor microenvironment (TME) facilitating cancer progression. A dynamic change in glucose metabolism occurring in cancer cells and its influence on the TME are still poorly understood. Indeed, cancer and/or immune cells undergo rapid adaptation in metabolic pathways during cancer progression. Metabolic reprograming affects macrophages, T cells, and myeloid derived suppressor cells (MDSCs) among other immune cells. Their role in the TME depends on a nature and concentration of factors, such as cytokines, reactive oxygen species (ROS), growth factors, and most importantly, diffusible metabolites (i.e., lactate). Further, the amounts of available nutrients and oxygen as well as activity of microbiota may influence metabolic pathways in the TME. The roles of metabolites in regulating of the interaction between immune and cancer cell are highlighted in this review. Targeting metabolic reprogramming or signaling pathways controlling cell metabolism in the TME might be a potential strategy for anti-cancer therapy alone or in combination with current immunotherapies.
Introduction
Metabolic Switches in the TME During Cancer Progression and Responses to Therapy
Majority of cancers show complex metamorphoses in metabolism and composition of immune stroma during their progression (1, 2). This includes metabolic switch toward Warburg biology, which is associated with immune suppression. Therefore, understanding of the molecular mechanisms and signaling pathways regulating metabolism in the TME is crucial for developing novel therapies. Among current anti-cancer approaches, immunotherapy targets the immune cells in the TME and has been successfully applied for subset of patients with melanoma or lung cancer. However, only a small percentage of patients are fully responsive to this regimen. We and others propose that the therapeutic responses are partly dependent on the metabolic status of immune and cancer cells in TME (3–6). In early stages of the disease, immune cells suppress tumor growth, but as tumor becomes established, they favor cancer progression (7) and resistance to the therapy (8, 9). Here, we focus on describing metabolic switches in immune cells in the TME during cancer progression as the targets of future therapies.
Role of Metabolic Switch in Tumor-Associated Macrophages (TAMs) During Cancer Progression
TAMs are one of the main cells that define cancer development and progression (10, 11). TAMs are immunosuppressive and polarized toward an M2 phenotype in established tumors. M2-polarized TAMs promote angiogenesis and tissues remodeling. However, in some patients accumulation of TAMs in the TME was associated with better prognosis. Indeed, mice inoculated with tumors with high number of TAMs present in the niche at the time of initiation had smaller tumors (12). Further, Sica et al showed that recruitment of myeloid cells from the blood and their subsequent differentiation toward TAMs may lead to cancer regression (11). ROS and reactive nitrogen species (RNS) generated by TAMs can induce cancer cell death leading to tumor regression or genomic instability supporting malignant transformation. Moreover, M2-polarized TAMs suppress adoptive immunity as well as release of growth factors and matrix proteinases that block tumor progression (13). Cancer cells dictate differentiation and polarization of TAMs into an M2 phenotype via regulation of metabolic switches in the TME. Recent studies show the spatial organization of TAMs populations based on the hypoxia and lactate levels in the TME (14). Indeed, hypoxia promotes M2 like phenotype of TAMs characterized by low MHC-II (15). TAMs with high arginase-1 (Arg-1), and mannose receptor C type 1 (CD206) accumulate in the regions with lower blood and oxygen supply (Figure 1). In these regions TAMs express hypoxia responsive factor-1α (HIF-1α) and switch their metabolism to glycolytic fermentation. In the TME, TAMs are exposed to particularly high concentration of cancer cell-derived lactic acid. Intriguing, the lactic acid stabilizes HIF-1α in TAM under hypoxic as well as normoxic conditions. Lactic acid induces polarization toward M2-like TAMs, which are characterized by higher Arginase-1 (Arg-1) and VEGF levels (16) (Figure 1). Thus, cancer cells induce vicious cycle by generating lactate that further promotes acidic pH by activation of hypoxic responses in TAMs. Moreover, in response to lactic acid, TAMs produce high levels of anti-inflammatory cytokines (i.e., IL-10), which are immunosuppressive. We have recently shown that deletion of lactate dehydrogenase A (LDH-A) and subsequently lactate in myeloid cells leads to regression of lung cancer and is associated with stronger anti-cancer immune responses (6). These findings demonstrate extensive influence of TME metabolic reprogramming of TAMs away from M2 polarization.
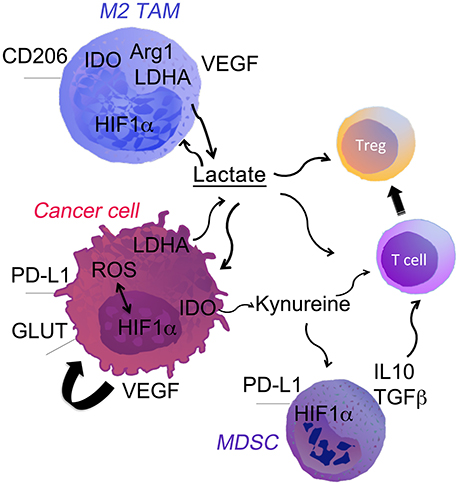
Figure 1. Cell fate in the hypoxic TME. Low oxygen levels, typical of the poorly vascularized tumor milieu, can affect both tumor and immune cells through the stabilization of HIF-1α. It directly promotes tumor growth through the upregulation of genes involved in glycolysis, LDH-A. HIF-1α up-regulation drives expression of PDL-1 and Arg-1 that potentiate the immune suppressive TME. Hypoxia-induced HIF1α and LDH-A-derived lactate strongly modulates the TME. Lactate acts directly on the cells or through the pH changes in the niche. The most notable effects are: (i) polarization of TAM into M2 macrophages; (ii) accumulation of myeloid-derived suppressor cells (MDSCs) and T regs; (iii) inhibition of T effector cells.
Multiple metabolic genes have been implicated in regulation of Mϕ polarization and control of TAMs phenotype (17). As an example, the tumor pyruvate kinase PKM2 inhibits LPS-induced IL-1β secretion, and thus limits the M1 phenotype (18). Further, O'Neill et al showed that PKM2 blockade suppressed PDL-1 expression on TAMs, dendritic cells, T cells and tumor cells (19). Such regulation occurs via direct binding of PKM2 and HIF1α to the PDL-1 promoter. Interestingly, we found that LDH-A/lactate similarly promoted PDL-1 expression in the TME (6). Although, the mechanism of lactate-induced PDL-1 expression is not known, it is possible that such regulation involves HIF1α (20) In addition to glycolytic switch, resting Mϕ or that present the TME are highly dependent on the glutamine-glutamate pathway (21). TAMs express high levels of the glutaminase, which is involved in the glutamine metabolism. Glutaminolysis is important for polarization of Mϕ to M2 phenotype by inducing high α-ketoglutarate/succinate ratio in the TME (22).
Metabolic Pathways Controlling Lymphocytes
T cells, B cells, and Natural Killer (NK) cells play a pivotal role in anti-cancer responses (23). In order to destroy cancer cells, effector T cells undergo activation followed by rapid proliferation. These cells are heavily dependent on the supply of oxygen and metabolites, which is limited in the TME. Hypoxia and low nutrient content change T cell metabolism and their anti-cancer responses. In such conditions T cells rapidly switch from oxidative phosphorylation to glycolysis (24). Simultaneously, these cells are exposed to extracellular lactic acid from the neighboring cells that strongly inhibits T cell expansion (25). Of note, naïve T cells depend on energy from fatty acid oxidation (FAO), oxidative phosphorylation, and glutamine metabolism; upon activation, T cells require more nutrients and rely on glucose and glutamine metabolism (26). Changes in lipid metabolism, are also implicated in modulation of CD8+ T-cell anti-tumor immunity. Inhibition of acetyl CoA acetyl transferase 1 (ACAT1) results in enhanced effector functions of CD8+ T cells. Recent publication by Patsoukis et al. highlights the role of PD-1 in T-cell development by promoting FAO via carnitine palmitoyltransferase (CPT1A) (26). Several compounds, such as inhibitor of lipogenic enzymes, have already been tested in preclinical models of cancer. This includes recent combinatorial studies with anti-PD-1 and ACAT1 inhibitor showing positive results in mouse melanoma (27, 28). Avasimibe, an ACAT1 inhibitor, is an established drug for atherosclerosis with a good human safety profile.
Myeloid Derived Suppressor Cells (MDSCs) Depend on Amino Acids and Fatty Acids Metabolism
MDSCs are part of the bone marrow derived cell population that accumulates under inflammatory conditions and in the TME (29). Human MDSCs are defined by expression of the alpha M-integrin CD11b and myeloid (CD14 and CD33) or granulocyte/neutrophil (CD15) markers. Murine MDSCs are characterized by simultaneous expression of Gr-1 and CD11b. MDSCs accumulate in the TME and strongly limit the anti-cancer immunity. MDSCs produce immunosuppressive cytokines such as IL-10 and TGF-β, which drive expansion of regulatory T cells and suppression of NK cells. The suppressive activity of MDSCs is strongly regulated by oxidative stress and amino acid metabolism (30). The depletion of important amino acids, like L-arginine, and L-tryptophan inhibit T cells proliferation and increase kynurenine. Kynurenine—a tryptophan metabolite—induces generation of T regulatory cells (24, 31). MDSCs rely on FAO, which is a major metabolic fuel for production of inhibitory cytokines. Thus, targeting of FAO is a possible strategy to limit MDSCs suppressive function. Raloxizine, a FDA approved FAO inhibitor blocked MSDCs expansion and functions in immunogenic preclinical models (32).
Metabolic Reprogramming of Cancer Cells as a Target of Anti-cancer Therapies
Warburg Effect and Glucose Metabolism in Cancer Cells
Proliferating tumor cells convert most of the glucose into lactate, even in highly oxygenated environments. This aerobic form of glycolysis is described as “Warburg effect” (33). We now know that the Warburg effect is a key feature of all hypoxic cells as well as highly proliferating cells, such as activated effector T cells (34). In contrary to the Warburg's suggestion, most of cancer cells do not have defects in mitochondria or oxidative phosphorylation (34, 35). Metabolic switch to fermentative glycolysis in cancer cells are triggered by various factors that favor tumor survival in hypoxic condition (Figure 1). Hypoxia is accompanied by increased in ROS. High-energy consuming processes including protein, DNA, and fatty acid synthesis are also accompanied by increase in ROS levels (36–39). Cancer cells produce high levels of ROS and are more susceptible to oxidative stress. Cytotoxic anticancer drugs that stimulate H2O2 production in cancer cells can induce tumor cell apoptosis (38). These treatments also select more aggressive cancer cell clones that have repressed mitochondrial activity (40) and higher ability to survive in hypoxic niche.
Glutamine Metabolism in Cancer Cells
Although not classified as an essential amino acid, under conditions of high energy demand, glutamine is a growth-limiting nutrient for cancer cells. A competition for glutamine between cancer cells and MDSC may take place as glutaminolysis supports MDSC maturation (41). The process of glutaminolysis, catalyzed by mitochondrial enzyme glutaminase, leads to conversion of glutamine into glutamate. Glutamate, is further converted into α-ketoglutarate which enters to the TCA, where it serves as a substrate for fatty acids, amino acids, and nucleotides syntheses. This process is controlled by growth factors and oncogenes (i.e., c-myc) (42). Additionally, glutamine can be converted into glutathione, which controls redox state balance. Glutaminolytic rate is considerably higher in cancer cells compare to normal non-malignant cells. Therefore, glutaminolysis has been identified as a potential vulnerability in cancer cells and is an important pharmaceutical target for anti-cancer drugs (43).
Lipid Metabolism in Cancer Cells
Metabolic reprogramming in cancer cells due to high energy demands affects lipid synthesis. Cancer cells upregulate monoacylglycerol lipase, acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), all which are involved in lipid metabolism. Increased expression of ATP citrate lyase (ACLY), which promotes cholesterol synthesis, is also a common feature of several tumors (44). Interestingly, neoplastic cells can acquire additional energy source from nearby adipocytes. Metastatic ovarian cancers cells convert triglycerides (TGs) from adipocytes into free fatty acids (FA) (45). Prostaglandin (PE) secretion is another common feature of neoplastic cells. PE supports MDSCs recruitment and activation as well as TAM polarization toward the M2 phenotype (46). Therefore, lipid metabolic reprogramming can be another important target for anti-cancer therapy.
Molecular Determinants of Metabolic Reprogramming: Role of Signaling Pathways in Driving Responses to Anti-cancer Therapies
Many cancer and immune cell-derived factors present in the tumor microenvironment modulate neighbor cell metabolism. We will discuss here examples of molecular signaling pathways and transcription factors leading to aberrant metabolism in cancer and immune cells in the TME (Figure 2). All these signaling pathways are targets of hypoxia or feed into the metabolic pathways associated with hypoxia and cell starvation.
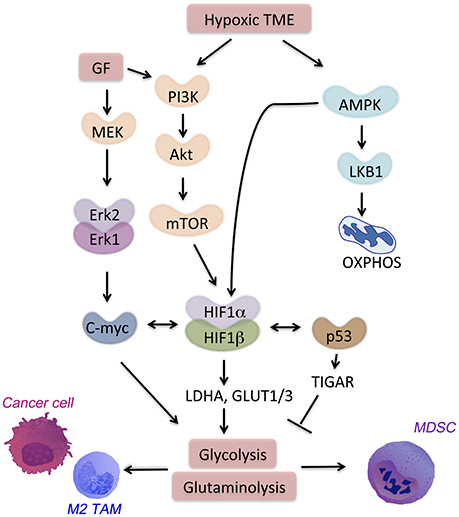
Figure 2. Signaling pathways in the hypoxic TME. HIF-1α is a major regulator of hypoxia-responsive genes in the TME. Multiple signaling pathways crosstalk with or activate HIF-1α. AMPK activation in the TME drives cell catabolism and is important for HIF-1α-regulated transcription. AMPK allows for metabolic adaptation in hypoxia with low ATP and lack of nutrient. Hypoxia also induces PI3K signaling through Akt-mTOR that promotes anti-apoptotic responses and metabolic shift toward glycolysis.
PI3K-AKT and mTOR Pathways
PI3K-AKT pathway regulates both pro-inflammatory and immunosuppressive responses, thus drives the outcome of the disease (47). The signaling through PI3K-AKT leads to the activation of mammalian target of rapamycin (mTOR) (48) (Figure 2). A metabolic regulator that couples nutrient sensing to metabolic stress, mTOR regulates important tumor-related processes. mTOR activates HIF-1α. Together with other transcription factors (i.e., c-myc and Oct1), HIF-1α promotes expression of glycolytic genes such as hexokinase 2 (HK2), LDH-A, glucose transporter 1 (GLUT1) and 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 (PFKFB3) as well as PD-L1 (49). In CD8+ T cells, mTOR sustains glycolysis, their effector functions as well as cytokine-driven differentiation into Th1, Th2, or Th17 subsets (50).
It is now recognized, that PI3K-δ and γ isoforms play a key role in recruiting the inflammatory cells into the TME, increased angiogenesis and tumor growth (51). Moreover, tumor-derived chemo attractants that activate the PI3K-γ isoform lead to enhanced expression of α4β1 integrin. The integrins allow myeloid cell adhesion and infiltration into the TME. These myeloid cells later become immunosuppressive TAMs. Moreover, small-molecule inhibitors targeting PI3K δ and γ suppress this process and the associated tumor growth.
AMPK Pathway
AMPK, a nutrient sensor, is a key regulator of oxidative phosphorylation as well as metabolic and oncogenic stress (52). AMPK suppresses anabolic pathways and activates cell catabolism. Activation of AMPK blocks cellular proliferation and tumor growth. However, in the absence of nutrients, loss of AMPK is not sufficient to promote cell proliferation. On the contrary, the loss of bioenergetics homeostasis leads to apoptosis of cancer cells that lack either AMPK or LKB1, a target of AMPK (Figure 2). AMPK regulates generation of CD8+ memory T cells, promotion of T regulatory cells, and suppression of T effector function (50, 53). The TME is characterized by enhanced hypoxia, nutrient deprivation and presence of anti-inflammatory cytokines (i.e., IL-10, IL-14, and TGF-β). In this niche, AMPK is activated in TAMs, MDSCs, and infiltrating T cells. AMPK activation leads to a metabolic switch toward oxidative phosphorylation and immunosuppression. Metformin, an activator of AMPK, showed a clinical efficacy on suppressing tumor growth (54). Interestingly, metformin supports expansion and survival of long-lived T memory cells by inhibition of glycolysis and promotion of oxidative phosphorylation in TME (55, 56). Targeting of AMPK in both immune cells and tumor cells might be a strategy to achieve a strong anti-tumor response in the hypoxic niche.
Transcription Factors: HIF-1α, c-myc, and p53
HIF-1α regulates several homeostatic functions of mammalian cells under hypoxia. The outcome of HIF-1α activation is different depends on the cell type and the downstream signaling. For instance, HIF-1α can directly promote tumor growth via binding to the promoters of the genes involved in glycolysis, such as glucose transporters (i.e., GLUT1, GLUT3) or enzymes (i.e., LDH-A) (57). Activation of HIF-1α blocks pyruvate dehydrogenase kinase (PDK) thereby suppresses TCA activity and facilitates glycolysis in cancer cells. HIF-1α can also drive inflammation, angiogenesis, and tissue remodeling in the TME. MDSCs under hypoxia suppress both antigen-specific and nonspecific T cells (58). Moreover, HIF-1α upregulation leads to increased expression of PD-L1 that amplifies MDSCs suppressive activity (20). The detailed role of hypoxia and HIF-1α will be discussed below.
Similarly to HIF-1α, c-myc modulates several metabolic pathways by regulating the expression of enzymes, such as LDH-A and pyruvate dehydrogenase kinase. However, c-myc in addition to driving glycolytic metabolism, and in contrast to HIF-1α activates mitochondria biogenesis. This is necessary for c-myc-driven generation of mitochondrial intermediates for synthesis of DNA, proteins and fatty acids. C-myc also regulates glutamine metabolism in cancer and stroma cells by inducing the expression of glutamine transporters (e.g., SLC5A1) and glutaminase 1 (GLS1, the initial enzyme of glutaminolysis) (59, 60). Fatty acid oxidation is another target of c-myc in subset of triple negative breast cancer tumors (61). In activated T cells, c-myc increases glycolysis, glutaminolysis, and polyamine synthesis (62). Importantly, alternative polarization of TAMs requires c-myc expression that directly regulates expression of CD209 among other genes associated with M2 polarization (63).
The tumor suppressor protein, p53, is mutated in most of cancer cells and affects glucose metabolism and hypoxic responses in the TME. In normal cells, p53 down-regulates GLUT1 and GLUT4 genes. In contrary, mutations in the DNA binding domain of p53 abrogate its inhibitory effect. This allows for high expression of glucose transporters and promotes glucose metabolism (64). Mutant p53 is linked to high expression of hexokinase 2 (HK2). HK2 is upregulated in many tumors and supports high rates of glucose catabolism (65). In normal cells, p53 suppresses the same genes by targeting of TP53-induced glycolysis and apoptosis regulator (TIGAR). TIGAR blocks glycolysis and blocks intracellular ROS levels (66). Moreover, p53 limits glutaminolysis. In response to DNA damage or oxidative stress, p53 promotes the expression of mitochondrial glutaminase (GLS2) (67). Interestingly, plasticity of TAMs is regulated by p53-regulated repression of c-myc expression (68), which leads to suppression of M2 polarization.
Hypoxia and Tumor Microenvironment
HIFs, a family of transcription factors, sense and respond to hypoxia. HIF-1α and HIF-2 α are the most widely studied family members (69). The activity of HIFs is regulated by post-transcriptional modifications, which involve hydroxylation of their proline and asparagine residues via prolyl hydroxylases (70). In normoxia, HIF-1α subunits are prolyl hydroxylated and tagged for proteasome degradation. During hypoxia, the HIF-1α protein translocates to the nucleus where it binds HIF-1β and regulates the transcription of multiple genes that harbor hypoxia response elements (HRE) in their promoters. Activities of HIF-1α and HIF-2α lead to upregulation of glucose transporters and multiple enzymes of the glycolytic pathway (71). Hypoxia blocks proryl-hydroxylase (PDH), via activation of PDK and decreases TCA cycle as well as the flow of pyruvate to mitochondria. HIF-1α mediates the metabolic switch from oxidative phosphorylation to aerobic glycolysis in cancer cells and immune cells in the TME (72). HIF-1α regulates the Th17/Treg balance in favor of the Th17 phenotype (73). Under hypoxic conditions, tumor cells have greater resistance to T cell-mediated cytotoxicity, due to HIF-1α-dependent transcriptional upregulation of PD-L1 (74). Similarly, PD-L1 expression in MDSCs in response to hypoxia further limits T cells cytolytic activity (20, 74). Therapeutic strategies targeting HIF pathway in cancer and immune cells might be beneficial for anti-tumor immune response (75).
Metabolic Interplay in the TME
There is evidence that targeting of aberrant metabolism in cancer cells inhibits tumor growth in animal models (76–81). Recent studies have also reported that targeting metabolic pathways in immune cells rather than directly in cancer cells may improve anti-cancer treatment (6, 82). The interactions between tumor and its microenvironment are driven in part by diffusible metabolites. This metabolic interplay in the TME, will be discussed in next paragraph with respect to nutrient competition and metabolic byproducts such as lactic acid. Changes in both metabolic substrates and products regulate immune and cancer cells in the TME.
Lactic Acid, as a Key Player in Tumor and Immune Metabolism
LDH-A, an enzyme responsible for the conversion of pyruvate to lactic acid, is significantly upregulated in cancer cells and is the main source of lactic acid in the tumor milieu. Extracellular lactic acid supports tumor expansion and immune evasion (6). Lactate strongly inhibits proliferation and anti-cancer function of human cytotoxic T lymphocytes (CTLs) (83, 84). We have recently reported that induction of global LDH-A deletion resulted in blockade of tumorigenesis (80). The effect of LDH-A deficiency in myeloid immune cells results in the same effect in KRAS-driven lung carcinoma model (6). It has been suggested, that acidosis induces hypoxia, HIF1α and immune checkpoint inhibitors such as PD-L1. We have demonstrated that LDH-A deletion in myeloid cells boosts anti-tumor T cell immunity through induction of IL-17-producing CD8+ T (Tc17) cells, likely via suppression of the lactate-driven PDL-1 expression (6). Furthermore, tumor-derived lactate supports the accumulation of myeloid-derived suppressor cells (MDSCs) while suppressing the function of natural killer (NK) cells and T lymphocytes (85, 86).
Although a major impact of lactate on the TME is acidification, lactate can be oxidized and provide energy in response to nutrient depletion. Recent studies have clarified that low pH in the presence of high lactate promotes M2 macrophage polarization (16, 87). Interestingly, treatment with proton pump inhibitors, which regulate the pH of TME, significantly limited tumor expansion (88). These treatments also restored functionality of tumor infiltrating lymphocytes and increased the therapeutic efficacy of adoptive immunotherapy in pre-clinical models (89). In another recent preclinical study, the effect of increasing alkalinity in TME in the context immunotherapy has been investigated (90). The combination of anti-PD-1 antibodies with bicarbonate administration led to improved antitumor responses in animals. These studies highlight how possibly reducing tumor acidity results in increased infiltration of cytotoxic T cell resulting in enhanced immune response.
Tryptophan and Glutamine Metabolism as a Target in the TME
Amino acid metabolism plays important roles in TME, similarly to glucose metabolism. L-Tryptophan is one of eight essential amino acids, which are required for protein synthesis and support important cellular activities, such as proliferation. It is, now established that the catabolism of the amino acid tryptophan (TRP), through the kynurenine signaling, maintains an immunosuppressive environment in cancer (91, 92). The breakdown of tryptophan is catalyzed by the rate limiting enzyme indoleamine 2,3-dioxygenase (IDO) and TRP-2,3-dioxygenase 2 (TDO). These enzymes play important roles in cancer immune escape (Figure 1). IDO is induced by hypoxia in dendritic cells or macrophages (93, 94). Inhibition of IDO results in rejection of allogenic fetuses (95), suggesting that tryptophan breakdown is necessary for maintaining immune tolerance. The enzymatic activity of indoleamine 2,3-dioxygenase (IDO) and TRP-2,3-dioxygenase 2 (TDO) results in depletion of TRP in the local microenvironment. This in turn induces an amino acid starvation response in T cells leading to cell cycle arrest and cell death (96, 97). Many preclinical studies have proposed and demonstrated enhance anti-immune response to a combination of anti PD-1 therapy with IDO inhibitors (98).
3-hydroxy-kynurenine (3-HK) (KYN) and kynurenic acid (KA) are regulatory metabolites by which IDO and TDO favor immune tolerance. These molecules accumulate in the TME and promote differentiation of T cells into forkhead box P3-positive (FOXP3+) Tregs (Figure 1). This process occurs through the activation of the transcription factor, aryl hydrocarbon receptor (AhR) (99, 100). Moreover, AhR signaling promotes effector T cell anergy. There is evidence that both enzymes are upregulated in several of the cancer types, electing the inhibition of their enzymatic activity as a new promising anti-cancer therapy.
The glutaminase gene, is characterized by two splice variants in human. Glutaminase coverts glutamine into glutamate and is a promising target for anti-cancer therapy (42, 43). The 6-diazo-5-oxo-L-norleucine (DON), a non-standard amino acid originally isolated from Streptomyces in a sample of Peruvian soil, was the first glutaminase inhibitor, as early as 1950. DON acts as a substrate analog for glutaminase, competing with glutamine for the binding of the active site of the enzyme and therefore reducing the glutamine catabolism. In 1954, Dion and colleagues hypothesized for the first time the use of DON as anti-tumor molecule (101). As of today, many inhibitors targeting glutamine metabolismhave been developed. This includes; CB-839, a selective orally bioavailable inhibitor of both glutaminase splice variants. CB-839 showed a potent anti-proliferative activity in a triple-negative breast cancer (TNBC) cell line (102) and has recently entered the Phase 1 clinical trial in combination with paclitaxel.
Gut Microbiota Influence Cancer Metabolism and Responses to Therapy
Recent studies suggest a role of gut microbiota in responses to the anti-cancer drugs (103, 104). The host immune response, and the drug toxicity are modulated by the intestinal flora. In part, these effects depend on the type of metabolites released by the different types of bacteria. For instance, β-glucuronidase, a carbolytic enzyme produced by gut-microbiota metabolites, controls the levels of the bioactive form of the chemotherapeutic drug Irinotecan and thus its side effects in the digestive tract (105). Furthermore, the metabolic activity of the gut microbiota induces ROS in the stroma. High level of ROS, prone epithelial cells to cytotoxic effect of oxaliplan, which is widely used for treatment of gastrointestinal malignancies (104, 106).
Further, gut microbiome modulates responses to immunotherapies. Microbial metabolites including the short-chain fatty acids (SCFAs) (107) such as butyrate, propionate, and acetate influence anti-cancer therapy. Butyrate, a histone deacetylase inhibitor (HDACi) regulates gene expression directly in the colon (108, 109). Furthermore, butyrate can be used as an energy source by colon cells. In 2014, Belcheva and colleagues demonstrated that butyrate supports growth of human adenomatous polyposis in mice models (110). In contrary, Donohoe and colleagues have reported that dietary fiber increase the butyrate levels in the colonic lumen and inhibit colorectal tumorigenesis in vivo (111). High butyrate–producing microbiota mitigate graft-versus host disease (GVHD) after allogenic bone marrow transplantation (112) suggesting its immunosuppressive effects. These observations highlight a possible role of the gut microbiome and their metabolites as pivotal players in the development of anti-cancer therapies.
Summary and Future Directions
A crosstalk between metabolism and immune regulation in the TME is the major focus of current studies in multiple laboratories. It is clear that the metabolic imbalance in cancer cells as well as stroma cells dictates survival of the tumor and immune cells, and controls resistance to anti-cancer therapy. The roles that the same metabolic enzymes play in tumor and immune cells warrant consideration of future therapies. Moreover, the nutrient composition and availability, acting directly on the cells in the TME or through the gut microbiota enzymes, can strongly drive the outcome of the treatment and disease progression. Based on current evidences in the literature, it is clear that metabolic modulation is considered as an innovative therapeutic approach. Moreover, metabolic targeting in combination with already approved checkpoint blockade treatments may provide novel ways to facilitate long-term remission or cure of cancer.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Our studies were partly supported by funding from: R01 DK104714, R21 CA169904 to BW, and R01CA169470, W81XWH-15-1-0686 to PS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Krstic J, Trivanovic D, Jaukovic A, Santibanez JF, Bugarski D. Metabolic plasticity of stem cells and macrophages in cancer. Front Immunol. (2017) 8:939. doi: 10.3389/fimmu.2017.00939
2. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
3. Norata GD, Caligiuri G, Chavakis T, Matarese G, Netea MG, Nicoletti A, et al. The cellular and molecular basis of translational immunometabolism. Immunity (2015) 43:421–34. doi: 10.1016/j.immuni.2015.08.023
4. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. (2016) 17:364–8. doi: 10.1038/ni.3415
5. Molon B, Cali B, Viola A. T cells and cancer: how metabolism shapes immunity. Front Immunol. (2016) 7:20. doi: 10.3389/fimmu.2016.00020
6. Seth P, Csizmadia E, Hedblom A, Vuerich M, Xie H, Li M, et al. Deletion of lactate dehydrogenase-a in myeloid cells triggers antitumor immunity. Cancer Res. (2017) 77:3632–43. doi: 10.1158/0008-5472.CAN-16-2938
7. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. (2015) 125:3365–76. doi: 10.1172/JCI80006
8. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
9. Kalinski P, Talmadge JE. Tumor immuno-environment in cancer progression and therapy. Adv Exp Med Biol. (2017) 1036:1–18. doi: 10.1007/978-3-319-67577-0_1
10. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. (2006) 25:315–22. doi: 10.1007/s10555-006-9001-7
11. Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer (2006) 42:717–27. doi: 10.1016/j.ejca.2006.01.003
12. Nemeth Z, Li M, Csizmadia E, Dome B, Johansson M, Persson JL, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget (2015) 6:33675–88. doi: 10.18632/oncotarget.5284
13. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. (2008) 267:204–15. doi: 10.1016/j.canlet.2008.03.028
14. Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci USA. (2017) 114:2934–9. doi: 10.1073/pnas.1700600114
15. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. (2014) 74:24–30. doi: 10.1158/0008-5472.CAN-13-1196
16. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513:559–63. doi: 10.1038/nature13490
17. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell (2008) 13:472–82. doi: 10.1016/j.ccr.2008.05.005
18. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LEMW, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
19. Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol. (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
20. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
21. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. (1986) 239:121–5. doi: 10.1042/bj2390121
22. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. doi: 10.1038/ni.3796
23. Biswas M, Terhorst C, Herzog RW. Treg: tolerance vs immunity. Oncotarget (2015) 6:19956–7. doi: 10.18632/oncotarget.4648
24. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
25. Mockler MB, Conroy MJ, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol. (2014) 4:107. doi: 10.3389/fonc.2014.00107
26. Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, et al. The role of metabolic reprogramming in T cell fate and function. Curr Trends Immunol. (2016) 17:1–12.
27. Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. (2010) 6:551–62. doi: 10.2217/fon.10.11
28. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531:651–5. doi: 10.1038/nature17412
29. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425. doi: 10.1158/0008-5472.CAN-06-3037
30. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
31. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
32. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
33. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
34. Maldonado EN, Lemasters JJ. Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J Pharmacol Exp Ther. (2012) 342:637–41. doi: 10.1124/jpet.112.192153
35. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
36. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. (1991) 51:794–8.
37. Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. (2000) 88:1880–9. doi: 10.1152/jappl.2000.88.5.1880
38. Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. (2005) 65:948–56.
39. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell (2006) 10:175–6. doi: 10.1016/j.ccr.2006.08.015
40. Sanchez-Arago M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis (2010) 31:567–76. doi: 10.1093/carcin/bgq012
41. Hammami I, Chen J, Bronte V, DeCrescenzo G, Jolicoeur M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochem Biophys Res Commun. (2012) 425:724–9. doi: 10.1016/j.bbrc.2012.07.139
42. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
43. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
44. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi: 10.1038/oncsis.2015.49
45. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
46. Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, et al. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J Immunol. (2011) 187:1157–65. doi: 10.4049/jimmunol.1100889
47. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. (2004) 30:193–204. doi: 10.1016/j.ctrv.2003.07.007
48. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
49. Noman MZ, Chouaib S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology (2014) 3:e954463. doi: 10.4161/21624011.2014.954463
50. Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. (2015) 36:257–64. doi: 10.1016/j.it.2015.02.007
51. Schmid MC, Franco I, Kang SW, Hirsch E, Quilliam LA, Varner JA. PI3-kinase gamma promotes Rap1a-mediated activation of myeloid cell integrin alpha4beta1, leading to tumor inflammation and growth. PLoS ONE (2013) 8:e60226. doi: 10.1371/journal.pone.0060226
52. Zadra G, Batista JL, Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol Cancer Res. (2015) 13:1059–72. doi: 10.1158/1541-7786.MCR-15-0068
53. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
54. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci USA. (2011) 108:18348–53. doi: 10.1073/pnas.1108856108
55. Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. (2014) 2:57. doi: 10.3978/j.issn.2305-5839.2014.06.01
56. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. (2015) 66:17–29. doi: 10.1146/annurev-med-062613-093128
57. Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, Rodriguez-Enriquez S, Moreno-Sanchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. (2009) 9:1084–101. doi: 10.2174/138955709788922610
58. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
59. Wise A. Transcriptional switches in the control of macronutrient metabolism. Nutr Rev. (2008) 66:321–5. doi: 10.1111/j.1753-4887.2008.00039.x
60. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458:762–5. doi: 10.1038/nature07823
61. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. (2016) 22:427–32. doi: 10.1038/nm.4055
62. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
63. Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood (2012) 119:411–21. doi: 10.1182/blood-2011-02-339911
64. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. (2004) 64:2627–33. doi: 10.1158/0008-5472.CAN-03-0846
65. Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem (1997) 272:22776–80. doi: 10.1074/jbc.272.36.22776
66. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
67. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. (2010) 107:7461–6. doi: 10.1073/pnas.1002459107
68. Li L, Ng DS, Mah WC, Almeida FF, Rahmat SA, Rao VK, et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ (2015) 22:1081–93. doi: 10.1038/cdd.2014.212
69. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
70. Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. (2013) 13:646–53. doi: 10.1016/j.coph.2013.04.009
71. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
72. Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. (2007) 39:231–4. doi: 10.1007/s10863-007-9081-2
73. Yu C, Wu G, Dang N, Zhang W, Zhang R, Yan W, et al. Inhibition of N-myc downstream-regulated gene 2 in prostatic carcinoma. Cancer Biol Ther. (2011) 12:304–13. doi: 10.4161/cbt.12.4.16382
74. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
75. Subtil FS, Wilhelm J, Bill V, Westholt N, Rudolph S, Fischer J, et al. Carbon ion radiotherapy of human lung cancer attenuates HIF-1 signaling and acts with considerably enhanced therapeutic efficiency. FASEB J. (2014) 28:1412–21. doi: 10.1096/fj.13-242230
76. Budihardjo II, Walker DL, Svingen PA, Buckwalter CA, Desnoyers S, Eckdahl S, et al. 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin Cancer Res. (1998) 4:117–30.
77. Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. (2009) 8:626–35. doi: 10.1158/1535-7163.MCT-08-1049
78. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13:227–32. doi: 10.1038/nrc3483
79. Currie E, Schulze A, Zechner R, Walther TC, Farese RVJr. Cellular fatty acid metabolism and cancer. Cell Metab. (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
80. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. (2014) 19:795–809. doi: 10.1016/j.cmet.2014.03.003
81. Lu W, Zhang Y, Zhou L, Wang X, Mu J, Jiang L, et al. miR-122 inhibits cancer cell malignancy by targeting PKM2 in gallbladder carcinoma. Tumour Biol. (2015) 37, 15615–1562. doi: 10.1007/s13277-015-4308-z
82. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
83. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
84. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer (2012) 131:633–40. doi: 10.1002/ijc.26410
85. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
86. Husain Z, Seth P, Sukhatme VP. Tumor-derived lactate and myeloid-derived suppressor cells: linking metabolism to cancer immunology. Oncoimmunology (2013) 2:e26383. doi: 10.4161/onci.26383
87. Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, et al. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer (2013) 133:1107–18. doi: 10.1002/ijc.28114
88. Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. (2004) 96:1702–13. doi: 10.1093/jnci/djh305
89. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. (2012) 72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272
90. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. (2016) 76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743
91. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
92. Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology (2002) 107:452–60. doi: 10.1046/j.1365-2567.2002.01526.x
93. Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. (2016) 76:818–30. doi: 10.1158/0008-5472.CAN-15-0977
94. Song X, Zhang Y, Zhang L, Song W, Shi L. Hypoxia enhances indoleamine 2,3-dioxygenase production in dendritic cells. Oncotarget (2018) 9:11572–80. doi: 10.18632/oncotarget.24098
95. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
96. Lob S, Konigsrainer A, Schafer R, Rammensee HG, Opelz G, Terness P. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood (2008) 111:2152–4. doi: 10.1182/blood-2007-10-116111
97. Platten MN, von Knebel D, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. (2014) 5:673. doi: 10.3389/fimmu.2014.00673
98. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer (2014) 2:3. doi: 10.1186/2051-1426-2-3
99. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65–71. doi: 10.1038/nature06880
100. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203. doi: 10.1038/nature10491
101. Dion H. 6-diazo-5-oxo-L-norleucine, A new tumor inhibitory substance. II: isolation and characterization antibiotics and chemotherapy. J Am Chem Soc. (1954) 78:3075–7.
102. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
103. Karin M, Jobin C, Balkwill F. Chemotherapy, immunity and microbiota–a new triumvirate? Nat Med. (2014) 20:126–7. doi: 10.1038/nm.3473
104. Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. (2015) 7:271ps271. doi: 10.1126/scitranslmed.3010473
105. Lin XB, Farhangfar A, Valcheva R, Sawyer MB, Dieleman L, Schieber A, et al. The role of intestinal microbiota in development of irinotecan toxicity and in toxicity reduction through dietary fibres in rats. PLoS ONE (2014) 9:e83644. doi: 10.1371/journal.pone.0083644
106. Hamer HM, Jonkers DM, Bast A, Vanhoutvin SA, Fischer MA, Kodde A, et al. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin Nutr. (2009) 28:88–93. doi: 10.1016/j.clnu.2008.11.002
107. Ganapathy V, Thangaraju M, Prasad PD, Martin PM, Singh N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol. (2013) 13:869–74. doi: 10.1016/j.coph.2013.08.006
108. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut (1980) 21:793–8. doi: 10.1136/gut.21.9.793
109. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr (2003) 133(7 Suppl):2485S–93S. doi: 10.1093/jn/133.7.2485S
110. Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell (2014) 158:288–99. doi: 10.1016/j.cell.2014.04.051
111. Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. (2014) 4:1387–97. doi: 10.1158/2159-8290.CD-14-0501
Keywords: tumor microenvironment (TME), metabolic reprogramming, immune stroma, HIF-1α, lactate
Citation: Wegiel B, Vuerich M, Daneshmandi S and Seth P (2018) Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 8:284. doi: 10.3389/fonc.2018.00284
Received: 27 February 2018; Accepted: 09 July 2018;
Published: 13 August 2018.
Edited by:
Suzie Chen, Rutgers University, The State University of New Jersey, United StatesReviewed by:
Vijay Pandey, Cancer Science Institute of Singapore, National University of Singapore, SingaporeFabrizio Martelli, Istituto Superiore di Sanità, Italy
Copyright © 2018 Wegiel, Vuerich, Daneshmandi and Seth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj Seth, cHNldGhAYmlkbWMuaGFydmFyZC5lZHU=
Barbara Wegiel, YndlZ2llbEBiaWRtYy5oYXJ2YXJkLmVkdQ==
†These authors have contributed equally to this work