 Isabel Fabregat1,2,3*
Isabel Fabregat1,2,3* Daniel Caballero-Díaz1,3*
Daniel Caballero-Díaz1,3*- 1TGF-β and Cancer Group, Oncobell Program, Bellvitge Biomedical Research Institute, Barcelona, Spain
- 2Department of Physiological Sciences, School of Medicine, University of Barcelona, Barcelona, Spain
- 3Oncology Program, CIBEREHD, National Biomedical Research Institute on Liver and Gastrointestinal Diseases, Instituto de Salud Carlos III, Barcelona, Spain
The Transforming Growth Factor-beta (TGF-β) family plays relevant roles in the regulation of different cellular processes that are essential for tissue and organ homeostasis. In the case of the liver, TGF-β signaling participates in different stages of disease progression, from initial liver injury toward fibrosis, cirrhosis and cancer. When a chronic injury takes place, mobilization of lymphocytes and other inflammatory cells occur, thus setting the stage for persistence of an inflammatory response. Macrophages produce profibrotic mediators, among them, TGF-β, which is responsible for activation -transdifferentiation- of quiescent hepatic stellate cells (HSC) to a myofibroblast (MFB) phenotype. MFBs are the principal source of extracellular matrix protein (ECM) accumulation and prominent mediators of fibrogenesis. TGF-β also mediates an epithelial-mesenchymal transition (EMT) process in hepatocytes that may contribute, directly or indirectly, to increase the MFB population. In hepatocarcinogenesis, TGF-β plays a dual role, behaving as a suppressor factor at early stages, but contributing to later tumor progression, once cells escape from its cytostatic effects. As part of its potential pro-tumorigenic actions, TGF-β induces EMT in liver tumor cells, which increases its pro-migratory and invasive potential. In parallel, TGF-β also induces changes in tumor cell plasticity, conferring properties of a migratory tumor initiating cell (TIC). The main aim of this review is to shed light about the pleiotropic actions of TGF-β that explain its effects on the different liver cell populations. The cross-talk with other signaling pathways that contribute to TGF-β effects, in particular the Epidermal Growth Factor Receptor (EGFR), will be presented. Finally, we will discuss the rationale for targeting the TGF-β pathway in liver pathologies.
Introduction
The liver shows an unique regenerative response to injuries produced by physical or toxic treatments, which induce tissue damage (1–3). Liver injuries can be classify depending on their persistence or duration and can develop acute and chronic liver diseases. Acute liver injuries can be completely restored, without any evidence of the injury, only withdrawing the damaging agent in a short period of time. In these cases, the liver architecture and function remain stable. However, long-time exposure with the damaging agent generates progressive liver damage, parenchyma alterations and vascular architectural distortion, which eventually results in liver fibrosis, cirrhosis, and ultimately, hepatocellular carcinoma (HCC), which is the end-stage of most chronic liver diseases (4, 5).
Chronic liver diseases are characterized by a parenchyma damage with a continued wound healing response, tissue remodeling, inflammatory environment and an altered molecular signaling pathways. Strong evidences point out the relevant role of the Transforming Growth Factor beta (TGF-β) signaling during all phases of the development of liver fibrosis and hepatocarcinogenesis. Perturbation of signaling by TGF-β family members is often seen in different diseases, including malignancies, inflammatory and fibrotic conditions (6). Under physiological conditions, TGF-β has a cytostatic and pro-apoptotic role in adult hepatocytes, which is critical for the control of liver mass. Loss of these functions may result in hyperproliferative disorders and cancer (7–9). Indeed, in early-stage carcinomas, TGF-β exerts tumor-suppressing activities, inducing cell cycle arrest and apoptosis. However, in late-stage carcinomas, once cells acquire resistance to its suppressive effects, TGF-β actions switch to pro-oncogenic, conferring cell survival, inducing cell migration and invasion, mediating immune alterations and microenvironment modifications (10, 11).
Recent evidences suggest that many of the pathological TGF-β effects could be related with its capacity to regulate cell plasticity, contributing to modifications in the phenotype of different liver cell populations. Cell plasticity refers to the interconversion of different stem cell pools, activation of facultative stem cells, and dedifferentiation, transdifferentiation or phenotypic transition of differentiated cells within a tissue (12) and is related with the ability of cells to reversibly change their phenotype and to take on characteristics of other cell types (13). The most studied and classic event related with cell plasticity is the epithelial-mesenchymal transition (EMT) and the opposite mesenchymal-epithelial transition (MET) (14). After specific stimuli, the cells suffer genetic and epigenetic changes, as well as cytoskeleton remodeling, which alter their phenotype and functions. TGF-β induces EMT in hepatocytes (15) and it is responsible for activation of hepatic stellate cells (HSC) to myofibroblasts (MFB) (16), both effects contributing to liver fibrosis. Moreover, during hepatocarcinogenesis TGF-β could also mediate an EMT process in liver tumor cells. This review will update recent evidences indicating the relevance of TGF-β signaling pathway in the regulation of the cell plasticity during the progression and pathogenesis of liver chronic diseases, as well as the molecular mechanisms involved. Finally, we will discuss the rationale for targeting the TGF-β pathway in liver pathologies.
TGF-β Signaling Pathways
In humans, the pleiotropic TGF-β cytokine superfamily includes different members, such as bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs) and TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3). TGF-β signaling pathways regulates different cellular processes playing essential roles in proliferation, migration, differentiation, or cell death. These processes are essential for the homeostasis of tissues and organs and TGF-β signaling deregulation contributes to human disease. TGF-β1 (TGF-β from now on) has essential roles in liver physiology and pathology and contribute to all stages of disease progression: from liver injury through inflammation, fibrosis, cirrhosis and HCC (7, 8).
Most of the functions of the cells involved in the fibrotic tissue and in the tumor microenvironment are under the control of TGF-β: promotes MFB differentiation, the recruitment of immune cells, affects epithelial and endothelial cell differentiation and inhibits the anti-tumor immune responses (17, 18). Besides TGF-β responses could be different depending on the cell type, its receptors are expressed on most of the cells and its signaling pathway is very similar in all of them (6). All TGF-β isoforms are synthesized within the cell as pro-peptide precursors containing a pro-domain, named Latency-Associated peptide (LAP), and the mature domain. This latent form is secreted to the extracellular matrix (ECM) and stored as a fast and available pool of TGF-β, without a novo synthesis (19). By different mechanisms, TGF-β is cleaved and the bioactive form signals via binding to its specific kinase receptor at the cell surface of target cells. Stored TGF-β could be activated by the cell contractile force, which is transmitted by integrins (20, 21). Specific integrins and matrix protein interactions could be required for activation of the latent form of TGF-β. Integrins αv are the major regulators of the local activation of latent TGF-β and in this activation it is required the RGD (Arg-Gly-Asp) sequence (21). Integrin αv deletion in HSC protected mice from CCl4-induced hepatic fibrosis (22). A recent review summarized the crosstalk between TGF-β and tissue extracellular matrix components (23).
TGF-β binds to its receptors triggering the formation of a heterotetrameric complex of type I and type II serine/threonine kinase receptors, in which the constitutively active type II receptor phosphorylates and activates the type I receptor. There are several types of both type I and type II receptors, but TGF-β preferentially signals through activin receptor-like kinase 5 (ALK5) type I receptor (TβRI) and the TGF-β type II receptor (TβRII). In addition, endoglin and betaglican (TβRIII), also called accessory receptors, bind TGF-β with low affinity and present it to the TβRI and TβRII. Activated receptor complexes mediate canonical TGF-β signaling through phosphorylation of the Receptor Associated SMADs (R-SMADs) at their carboxy-terminal. Humans express eight SMAD proteins that can be classified into three groups: R-SMADs, Cooperating SMADs (Co-SMADs) and Inhibitory SMADs (I-SMADs: SMAD6 and SMAD7). Among the R-SMADs, SMAD2 and 3 mediate the TGF-β1 branch of signaling (6, 8). After phosphorylation, R-SMADs form a trimeric complex with SMAD4, which translocates to the nucleus and associates with other transcription factors in order to regulate gene expression (7, 8). In addition to the canonical SMAD pathway, TGF-β is able to use non-SMAD effectors to mediate some of its biological responses, including non-receptor tyrosine kinases proteins such as Src and FAK, mediators of cell survival (e.g., NF-kB, PI3K/Akt pathways), MAPK (ERK1/2, p38 MAPK, and JNK among others), and Rho GTPases like Ras, RhoA, Cdc42, and Rac1. Interestingly, these pathways can also regulate the canonical SMAD pathway and are involved in TGF-β-mediated biological responses (Figure 1) (8, 24–26).
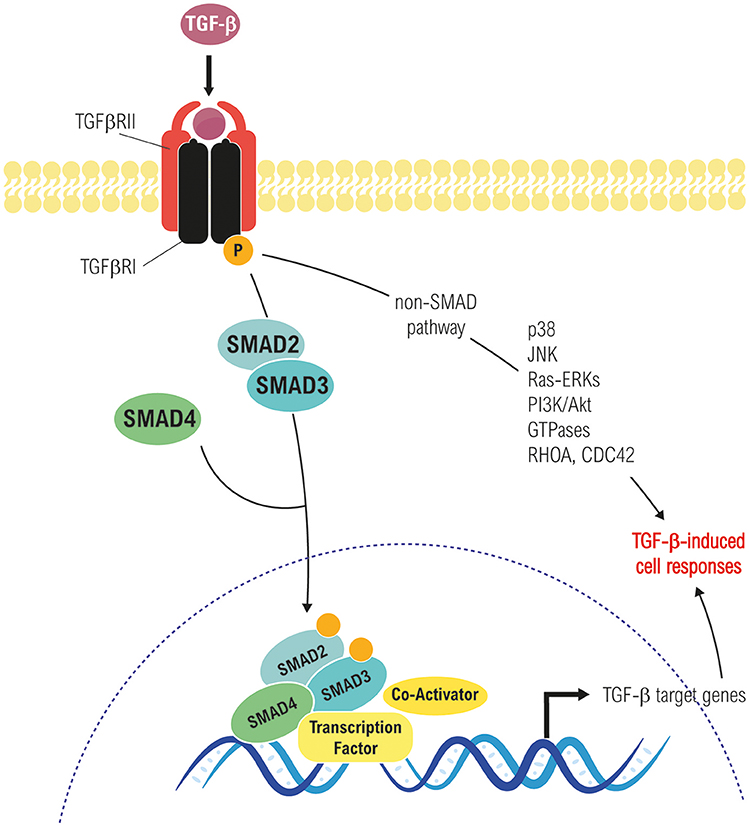
Figure 1. Canonical (Smad-dependent) and non-canonical (Smad-independent) TGF-β signaling pathways. Both converge in transcriptional-dependent and independent effects on cell proliferation, differentiation, apoptosis/survival, migration, etc., in a cell and context-dependent manner.
Liver Fibrosis
Liver fibrosis is a common pathological chronic liver disease, consequence of a continued injury with a huge accumulation of extracellular matrix proteins, mainly enriched in fibrillar collagens, due to a multiple reparative and regenerative processes (5, 27, 28). After liver damage, reparative mechanisms are triggered to replace necrotic and apoptotic hepatocytes, generating wound healing and inflammatory responses that are essential for liver regeneration (5). However, if the damage persists over a long time, the excessive accumulation of extracellular matrix proteins (collagens I, II, and III, undulin, fibronectin, laminin, elastin, proteoglycans and hyaluronan) could replace parenchymal areas leading fibrosis to a cirrhotic state. In advanced stages, it develops an abnormal liver architecture, altered vascularization and fibrotic septa surroundings with regenerative nodules. Liver systemic failure, portal hypertension, high susceptibility to infection and high risk to develop HCC are the main clinical consequences of cirrhosis (28, 29). Interestingly, multiple clinical reports have reported that liver insult eradication can regret liver fibrosis in huge number of patients, mostly during the first stages (29–32). In the development of liver fibrosis, TGF-β plays crucial roles regulating the different stages of the disease, among them, the control of cell plasticity of different liver cell populations, which is summarize in the Figure 2 and we detail in the next chapters.
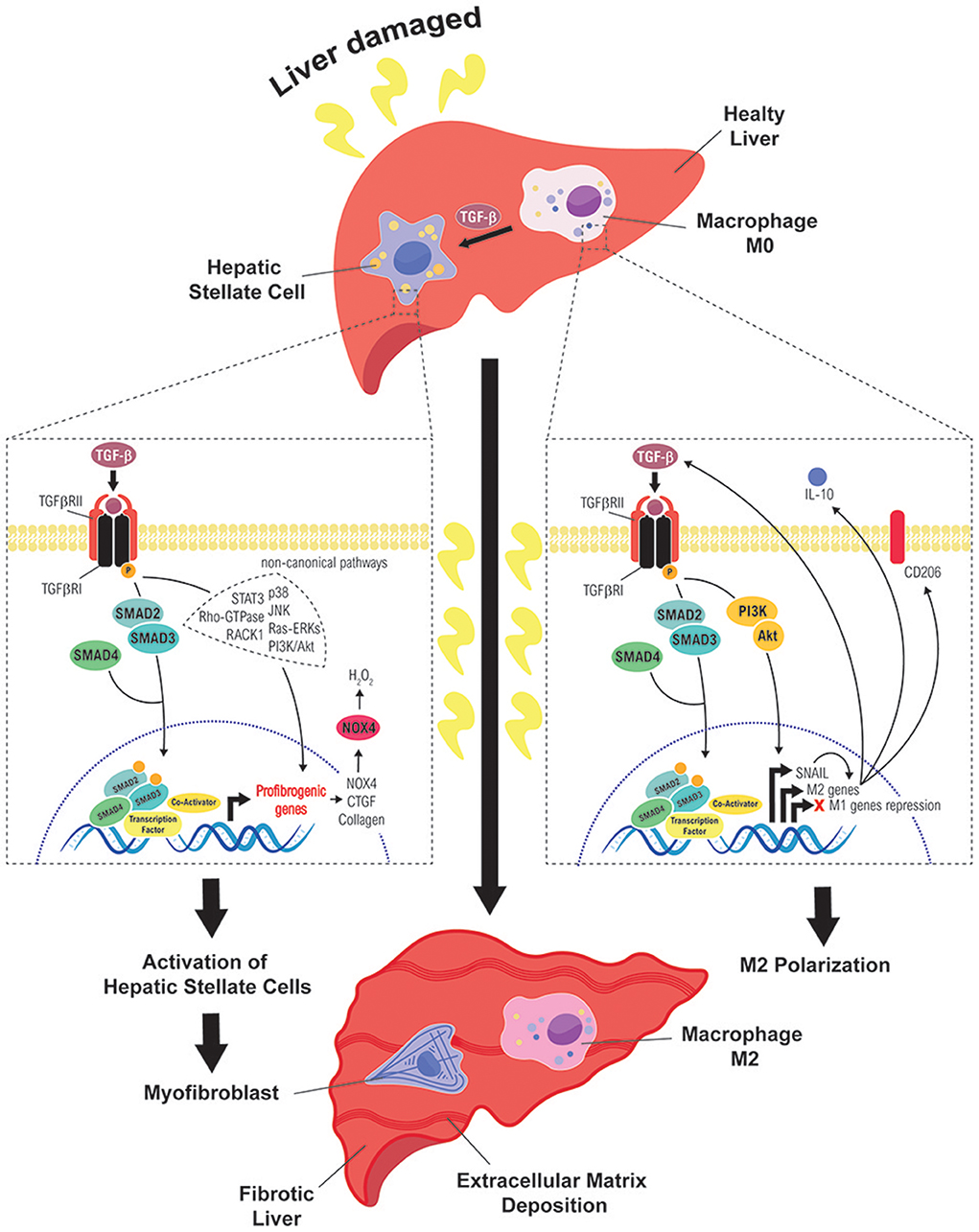
Figure 2. Role of TGF-β in the cell plasticity of hepatic stellate cells and macrophages during liver fibrosis. Different routes followed by TGF-β signals to mediate activation of HSC into MFB (left) or polarization of macrophages to a M2 state (right), which contribute to sustain a fibrotic and immunosuppressive environment, favorable to the initiation of a hepatocarcinogenic process.
TGF-β Regulates Macrophage Plasticity During Liver Fibrosis
Inflammation plays a key role in liver fibrosis development. After injury takes place, infiltration of immune system cells -macrophages, lymphocytes, eosinophils, and plasma cells- arises to the damaged place. Lymphocytes produce cytokines and chemokines, which activate macrophages. Activated macrophages stimulate inflammatory cells such as lymphocyte, among others, over-activating and maintaining the inflammatory environment (33). During fibrosis, macrophages produce pro-fibrotic factors such as TGF-β and platelet derived growth factor (PDGF), control ECM turnover by regulating the balance of various matrix metalloproteases and tissue inhibitors of metalloproteinases (TIMPs) (27, 34, 35) and are found very close to collagen-producing MFB (36–38) suggesting the macrophages relevance in the activation of MFB. In this sense, hepatic macrophages have been described as a potential targets against fibrosis (39, 40).
Macrophages represent a heterogeneous cell population with a huge cell plasticity, where diverse microenvironment stimuli polarize them into different phenotypes (41). There are mainly two sources of hepatic macrophages: liver resident macrophages, also called Kupffer cells (42), and circulating monocytes (inflammatory recruited macrophages) (43). Besides the origin, both could play significant roles in the development of fibrosis. In vitro and in vivo studies described that both Kupffer cells and monocyte-derived macrophages can activate HSC and induce their transdifferentiation by paracrine mechanisms, including TGF-β (44–47). Resident hepatic macrophages secrete the chemokine CCL2 (a potent chemoattractant) in order to recruit monocytes which could increase and promote fibrosis. Although, it was described that the pro-fibrotic functions of these resident macrophages remain functional even when recruited macrophages are pharmacologically inhibited using CCL2 antagonists (48). Transgenic rats that express a mutated form of the CCL2 (acting as a negative-mutant), and tail vein injection of adenovirus that overexpress a truncated form of TGF-β receptor II (acting as a negative-receptor mutant) attenuate liver fibrosis in a DEN-induced fibrosis model in rats (49), suggesting the relevance of inflammation and TGF-β pathway during this disease.
In the early stages, activated macrophages secrete pro-inflammatory cytokines and produce reactive oxygen species (ROS), while in late stages macrophages have been associated with release of anti-inflammatory factors, attenuating inflammation and promoting tissue regeneration (43, 50). Macrophages are classify into M1, also known as classical or pro-inflammatory; and M2, also known as alternative or anti-inflammatory macrophages (51, 52). It is not easy to strictly separate both liver macrophage populations, since they could show common gene expression, and even more M2 macrophages are classify also in different subclasses. For that reason, it has been proposed that could be more adequate to separate them according to their functions: defensive, restorative and regulatory macrophages (53). In the classical classification, M1 macrophages are associated with inflammatory diseases due to microbicidal activity (through their capacity to produce ROS and their phagocytic functions), antigen presentation and antitumor activity. M1 macrophages prevail during the onset of injury (54) and are related with the release of metalloproteinases that degrade ECM and promote EMT/Endothelial-to-mesenchymal transition (EndMT). On the other hand, M2 macrophages secrete anti-inflammatory factors such as IL-10, arginase, TGF-β, and HO-1. Their polarization is promoted by IL-4 and IL-13, and are characterized for the expression of Arg1, Ym1, and Fizz, secretion of angiogenic factors such as IL-8, VEGF, and EGF4 and increased mannose receptor (CD206), with lower ROS production (47, 50). M2 macrophages stimulate an anti-inflammatory environment and promote regeneration and wound healing. However, if injury becomes chronic, M2 macrophages take up a pro-fibrotic role secreting pro-fibrotic factors such as TGF-β, PDGF, among others (47).
Nowadays it is clear that macrophages are essential players in the regulation of liver fibrosis and they are an important source of TGF-β but, could this cytokine regulate the phenotype between M1 and M2 macrophages and their functions? Recent data described that TGF-β could induce M2-like macrophage polarization via SNAIL (55). SNAIL-overexpression in human THP-1 macrophages promotes M2 markers (such as CD206), induces the expression of the anti-inflammatory IL-10 and inhibits pro-inflammatory M1-related cytokines (TNF-α and IL-12). By contrast, SNAIL knockdown by siRNA technology abolishes TGF-β-M2-induced phenotype and partly restores M1 polarization through up-regulation of pro-inflammatory cytokines. The canonical SMAD2/3 and the non-canonical PI3K/AKT signaling pathways are crucial for TGF-β-induced SNAIL overexpression in THP-1 cells. The blockade of TGF-β/SNAIL signaling restores the production of pro-inflammatory cytokines. Likewise, TGF-β also stimulates murine BMDM macrophages to display an M2-like phenotype characterized by high levels of IL-10 and low levels of IL-12p70, and M1-specific markers. Macrophages isolated from fibrotic mouse livers show higher balance of M2/M1 macrophages in comparison to control mice (56). In other fibrotic animal models, such as lung fibrosis, TGF-β could modulate M2 responses (57); and in kidney, TGF-β/Smad3-dependt pathway could transdifferentiate M2-macrophages to myofibroblast favoring kidney fibrosis (58). Moreover, TβRII–/– mice show a defective polarization to M2-macrophages (59). Fibrosis-induced model in rats by thioacetamide show that both M1 and M2-macrophagues polarizations occur during development of the disease (60). Overall results show up that M2-activation/polarization has a relevant role in the development of fibrosis in mice and patients with liver fibrosis (61). However, due to the heterogenicity and higher plasticity of macrophages and the complexity of their study in vivo models in liver, further works are needed in order to clarify the molecular mechanisms whereby TGF-β pathway promote the polarization and the pro-fibrotic functions of macrophages in vivo models. Current data seems to indicate that both hepatic and recruited macrophages play relevant roles in the progression and reversion of liver fibrosis. Targeting both and the reorientation of their phenotypes are arising as attractive therapies (62).
TGF-β Regulates Liver Epithelial Cells Plasticity During Liver Fibrosis
“Activated” fibroblast or MFB are the main producing cells of fibrogenesis mediators and ECM components, participating actively in their accumulation (63). In the liver, the most fibrogenic MFB are endogenous and their origin is controversial and still unclear, but nowadays there are accepted different sources (63–65), among them, portal and resident fibroblasts (66), activation and differentiation of HSC (more details in the next section) (16, 67), bone marrow-derived fibrocytes (68), liver epithelial cells (hepatocytes and cholangiocytes) that undergo EMT (69–71), endothelial cells that undergo EndMT (66, 72), vascular smooth muscle cells and pericytes (73).
EMT-clear example of cellular plasticity- is a process that drives a de-differentiation of epithelial cells to a mesenchymal-like phenotype increasing their migratory and invasive properties (13, 14, 74, 75). The reverse process is called as MET and allows cells to differentiate into different organs and tissues. In a tumorigenic context, mesenchymal migratory tumor cells undergo MET to metastasize (76). The EMT process includes loss of epithelial genes, such as E-cadherin and cytokeratins (8, 18 and 19), and up-regulation of mesenchymal genes, such as N-cadherin, alfa-Smooth Muscle Actin (α-SMA, ACTA2 gene) which correlate with the expression of EMT transcription factors (EMT-TFs) Snail (SNA1 gene), Slug (SNA2 gene), Twist and ZEB (74, 75). Intermediates states are also found between EMT and MET (77). Partial EMT is described for cells that co-express both epithelial and mesenchymal markers. Even more, EMT is classified into different subclasses related with the biological context (78): Type 1 EMT is involved in development stages; type 2 EMT concerns regenerative process and organ fibrosis; and type 3 EMT is related with metastatic process.
During liver fibrosis, type 2 EMT plays a relevant role in the appearance of a pro-fibrotic fibroblast phenotype. Bipotent adult hepatic progenitor cells, which possess the cell plasticity to differentiate into hepatocytes and cholangiocytes after different stimuli (79), are able to undergo EMT in response to liver injury during cholestatic liver fibrosis. Hepatocyte plasticity could play relevant roles during the progression of chronic liver diseases. Mouse hepatocytes that survive to the apoptotic effects of TGF-β, could regulate -in a TGF-β dependent manner- the expression of fibrosis-related genes, such as Connective Tissue Growth Factor (CTGF) or fibronectin, and EMT-related genes, such as Snail, and β-catenin (15, 71, 80), with downregulation of epithelial markers (81). Even more, primary adult hepatocytes could transdifferentiate to a more fibroblastic-like phenotype with loss of cell–cell contacts and polarity, after TGF-β treatment (80). Indeed, hepatocyte-derived fibroblasts are an additional and significant lineage of mesenchymal cells that contribute to progression of liver fibrosis. Zeisberg and collaborators elegantly demonstrate that adult hepatocytes can undergo an EMT process after TGF-β stimuli, contributing to the in vivo pool of fibroblast during liver fibrosis (82). The role of hepatocytes during liver fibrosis in vivo related with TGF-β was also previously described in a transgenic animal model which overexpress SMAD7 (inhibitor of the pathway) specifically in hepatocytes. These transgenic animals have attenuated the TGF-β signaling and EMT, with less ECM depositions and improved CCl4-dependent liver damage and fibrosis (83). Bone morphogenetic protein-7 (BMP-7), a member of the TGF-β family which plays opposite roles to TGF-β, induces MET. Primary rat hepatocytes treated with TGF-β upregulate the expression levels of fibrotic markers, whereas BMP-7 treatment upregulated E-cadherin and decreases SMAD2/3 phosphorylation levels. Even more, in CCl4-treated rats treated with TGF-β, which show advanced fibrosis with higher expression of α-SMA and lower E-cadherin, the fibrotic situation was rescued after BMP-7 treatment (84). Moreover, cholangiocytes -another epithelial cell population- activate, proliferate and change into a more fibroblastic phenotype, increasing the expression of pro-fibrotic cytokines and factors such as TGF-β, PDGF and CTGF (85). These results open new ideas about how epithelial liver cells, through an EMT process, could generate mesenchymal/fibroblastic cells, which could be relevant in the progression of the fibrotic diseases.
Relevance of TGF-β and HSC Differentiation During Liver Fibrogenesis
In the normal liver, HSC (around 5–8% of the cells in the liver) are in a quiescent phenotype hosted in the space of Disse between hepatic epithelial and the sinusoidal endothelial cells (86). HSC are characterized by the store of vitamin A, lipid droplets and the expression of a large number of adipogenic genes and neural markers. After liver insults, different paracrine and autocrine signals are triggered promoting the HSC activation -transdifferentiation- from a quiescent state to an activated myofibroblastic phenotype. MFB are characterized by the expression of α-SMA, loss of retinoids and lipid droplets and de novo expression of receptors for mitogenic, fibrogenic and chemotactic factors, leading an increase in proliferation and survival, enhanced synthesis of matrix proteins (predominantly fibrillar collagens) and inhibitors of matrix degradation TIMPs, and secretion of pro-inflammatory cytokines and chemokines. This provokes the progressive scar formation and the development of liver fibrosis (29, 32).
HSC activation is one of the most important steps during liver fibrosis and is mediated by different signals, such as growth factors (PDGF and CTGF, among others), lipidic mediators, as well as ROS and cytokines produced by hepatocytes, cholangiocytes, endothelial cells, macrophages (Kupffer cells) and immune cells (67, 86). Among these cytokines, TGF-β plays a master role in the activation of the HSC to MFB (16). In fact, some of the previous factors stimulate the expression, production and activation of TGF-β, which at the end is responsible for the activation of HSC (87). Furthermore, MFB demonstrate a growth stimulatory effect in response to TGF-β (88), which also contributes to the maintenance of their myofibroblastic phenotype (89). SMAD3 has been identified as the main mediator of the TGF-β-induced fibrogenic transcriptional program, particularly the up-regulation of collagen expression (7, 8, 30, 31). HSC isolated from SMAD3 knock-out mice showed lower expression of Collagen1A1 mRNA mediated by p38 MAPK (30). Interestingly, it has been proposed that TGF-β activates the p38 MAPK pathway, further leading to SMAD3 phosphorylation at the linker region in the cultured MFBs, which promoted hetero-complex formation and nuclear translocation of SMAD3 and SMAD4 (31). These results would indicate that non-canonical activation of the SMAD3/SMAD4 transcriptional activity accounts for SMAD3-dependent extracellular matrix production in MFBs.
During liver fibrogenesis, activated HSC express CTGF, which acts downstream of TGF-β modulating the ECM production. CTGF mRNA expression is under the control of the canonical TGF-β/SMAD3 and non-canonical ERKs, JNK, p38, and STAT3 pathways (90, 91). Moreover, receptor for activated C-kinase 1 (RACK1), a scaffold protein involved in numerous cellular processes and signaling pathways, is another TGF-β downstream target involved in the HSC activation. RACK1 is able to induce pro-fibrogenic pathways in a TGF-β-dependent manner, contributing to differentiation, proliferation, and migration of HSC (92, 93). Indeed, in this migratory phenotype and in remodeling of the cytoskeleton in TGF-β-activated HSC is also involved the role of Rho guanosine triphosphatase (Rho GTPase) signaling (94). TGF-β also regulates the expression of TRPM7 (transient receptor potential melastatin 7) in a SMAD3-depend manner, which inhibition attenuates TGF-β-induced expression of MFB markers (95).
Mild to moderate liver fibrosis may be reversible. The reversion process is related with the elimination of the damaging stimuli. During liver fibrosis reversion, activated HSC (or myofibroblast) reverted to an inactivated phenotype. In this state, inactivated HSC decrease the expression of fibrogenic genes (including COL1A1 and ACTA2) and up-regulate the expression of some quiescence-associated genes like PPARγ (96, 97). Furthermore, inactivating some of the TGF-β downstream signals, such as ROS production, allows the reversion of the myofibroblast phenotype (89).
Role of ROS During HSC Activation by TGF-β
Oxidative stress plays a relevant role in the sequence of events following TGF-β activation of HSC. Actually, antioxidants can inhibits HSC transdifferentiation into MFB (98, 99). In both normal physiological and pathological conditions, ROS are critical intermediates. Oxidative stress plays a role during both initial inflammatory phase and its progression to fibrosis (100). Oxidative stress markers have been detected in experimental liver fibrosis/cirrhosis animals and in the biopsy and serum samples from liver cirrhotic patients (101). It is well known that ROS may act upstream and downstream of the TGF-β pathway. Upstream, ROS, through LAP activation and subsequent TGF-β release, promote fibrosis activating latent TGF-β (102) and/or via matrix metalloproteinases activation (103). Indeed, LAP/TGF-β complex has been suggested to function as an oxidative stress sensor (104). Furthermore, in many cell types such as HSC and hepatocytes, ROS may up-regulate the expression and secretion of TGF-β in a positive feedback loop (105, 106). ROS may be generated in the liver by multiple sources, including the cytochrome p450 family members, peroxisomes, mitochondrial respiratory chain, xanthine oxidase, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. Worthy to note, accumulating evidence indicates that NADPH oxidases (NOX)-mediated ROS have a critical role in HSC activation and hepatic fibrogenesis (101) mediating TGF-β actions.
NOX enzyme family generate ROS, either hydrogen peroxide or superoxide as the primary species, during oxygen catalytic metabolism for arrange of signaling functions and host defense. There are described seven NOX isoforms in mammalian cells (NOX1-5, DUOX1, and DUOX2). Liver cells (either parenchymal and non-parenchymal) express different members of the NOX family. Hepatocytes and HSC express NOX1, NOX2, NOX4, DUOX1, and DUOX2; endothelial cells express mainly NOX1, NOX2, and NOX4; and Kupffer cells express the phagocytic NOX2 (101, 107). NOXs proteins could be playing relevant roles during liver fibrosis development (101, 108). Both NOX1- and NOX2-deficient HSC had decreased ROS generation and failed to upregulate collagen and TGF-β in response to angiotensin II (109). Of relevance, NOXes mediate TGF-β activation of HSC to MFB process (89, 110). In different organs, such as heart, the main mediator for the activation of MFB is NOX4, downstream from TGF-β (111), lung (112) and kidney (113). In in vivo models of liver fibrosis, the levels of NOX4 are up-regulated, as well as in patients with chronic hepatitis C virus derived infection, increasing along the fibrosis degree. HSC respond to TGF-β inducing NOX4-derived ROS (105), which play a key role in hepatic MFB in both in vivo and in vitro (89, 114). In NOX4 knock-out animals and in NOX4 downregulated cells the TGF-β-transactivation of HSC is attenuated (89, 114), and even more, the myofibroblastic state could also be reversible (89). During liver fibrosis, NOX4 is required for both HSC activation and maintenance of the activated phenotype in MFB in a TGF-β-dependent manner and mediates the TGF-β pro-apoptotic response in hepatocytes, which might be relevant to blunt regeneration and create a pro-fibrogenic microenvironment. In this sense, apoptotic hepatocytes after liver injury generate apoptotic bodies which were described to promote HSC survival (115). HSC can engulf and clear apoptotic hepatocytes bodies inducing their activation in JAK1/STAT3-dependent pathway and a NOX-dependent PI3K/Akt/NF-κB induction pathway, concomitant with a production of ECM components (115). Recent evidences show up the dual role of NOX1/NOX4 pharmacological inhibitors in decreasing both the apparition of fibrogenic markers and hepatocyte apoptosis in vivo (114, 116), highlighting the relevance of NOX1 and NOX4 in liver fibrosis and opening new perspectives for its treatment. Actually, NOX1 and NOX4 signaling mediates hepatic fibrosis through activation of HSC (114, 117). Indeed, it was recently described that NOX4, downstream TGF-β, would play a role in the acquisition and maintenance of the MFB phenotype (89). Deficiency of NOX1 and NOX4 attenuates liver fibrosis in mice after CCl4 treatment. Activated HSC and ROS generation are also attenuated in HSC lacking NOX1 and NOX4, suggesting NOX1 and NOX4 play important roles in liver fibrosis and injury through regulating inflammation, proliferation and fibrogenesis in HSC (117). Therefore, targeting NOX1/4 is emerging as a new and attractive therapy for liver fibrosis in order to impair the pathological effects of TGF-β over this disease.
Human Hepatocellular Carcinoma (HCC)
HCC is a major public health problem worldwide with almost 800,000 new cases each year and its incidence is increasing in Europe and worldwide. In 2015 reports from World Health Organization, liver cancer is the second leading cause of cancer-related deaths, following lung cancer (118). HCC is the most common primary liver malignancy in adults. Intriguingly, there are significant differences on the incidence when considering the gender, being the male to female ratio estimated to be 2.4. This difference is mainly attributed to the different exposition to risk factors, as well as the influence of androgens and oestrogens on HCC progression (119). Exposition to risk factors also determines the incidence of liver cancer regarding age or ethnicity and the highest incidence of HCC is found in Asia and sub-Saharian Africa (120–122). In most cases, HCC develops within an established background of chronic liver disease. Progressive hepatic fibrosis frequently evolves to cirrhosis, which is the largest risk factor for developing liver cancer. Up to 90% of cases of HCC arise in the setting of advanced fibrosis or cirrhosis regardless of etiology (121, 123–125).
During hepatocarcinogenesis, a complex multistep process, many signaling cascades are altered as a result of genetic and epigenetic changes that contribute to a heterogeneous molecular profile. Furthermore, cellular plasticity increases the complexity of the cellular heterogenicity. Indeed, tumor heterogeneity in HCC is impressive: it can be observed between patients, between nodules in the same patient (i.e., second primary tumors after curative treatment or synchronous multifocal tumors of different clonality) and even within a single tumor nodule (126). Many molecular mechanisms are known to be clearly involved in HCC (127). Signaling pathways are related mainly with cell proliferation, angiogenesis, invasion, and metastasis. IGF-1, Epidermal Growth Factor (EGF), PDGF, Hepatocyte Growth Factor (HGF), VEGF, as well as TGF-β, are the most frequent growth factors and cytokines involved in HCC development. (128). The role of the microenvironment in tumor initiation and progression in HCC is critical and HCC cells could acquire an abnormal phenotype due to tissue remodeling altering their biological behavior (129, 130).
Role of TGF-β During Hepatocarcinogenesis
As it was mentioned before, TGF-β signaling -in the liver- participates in all stages of disease progression, from initial liver injury through inflammation and fibrosis, to cirrhosis and cancer (7, 8). During early stages of tumorigenesis TGF-β acts as a tumor suppressor, while in late stages it acts with a pro-tumorigenic role, promoting invasiveness and metastasis once cells become resistant to its suppressor effects (8, 131). In non-transformed hepatocytes and HSC, the cytostatic effects of TGF-β are often dominant over the opposing mitogenic signals; however, carcinoma-derived cells are usually refractory to growth inhibition by this cytokine. Activation of the TGF-β pathway induces antiproliferative responses due to the regulation of the cell cycle at G1-S by inhibiting c-MYC and cyclin-dependent kinase complex (CDK)-1-6 and 7 and regulates cycling inhibitors such as p21 and p15 (132–134). Smad4 –/– mice could develop head and neck cancer demonstrating the role of the TGF-β pathway as cytostatic regulator (135). Other proteins involved in the regulation of the pathway, such as β-II spectrin, which plays a role as scaffold for SMAD3 and SMAD4 and their subsequent activation after TGF-β. Sptbn2 heterozygote mutants develop HCC indicating that TGF-β signaling and β-II spectrin suppress hepatocarcinogenesis, potentially through cyclin D1 deregulation (136). TGF-β pathway also activates the NRF2 transcription factor which is involved in the expression of many cytoprotective genes which are relevant in the protection of the cells against toxic insults, and its depletion increases tumorigenic process (137). These data show up the relevance of the cytoprotective and suppressor role of TGF-β pathway, which could be altered during the carcinogenesis process. Malignant cells surpass the suppressive effects of TGF-β either through inactivation of core components of the pathway (such as TGF-β receptors and/or SMADs) or by downstream alterations repressing the tumor-suppressive arm. In late stages, liver cancer cells take advantages from the TGF-β-dependent pathways to acquire capabilities that contribute to tumor progression, such as production of autocrine mitogens, release of pro-metastatic cytokines and chemokines and up-regulation of receptors that mediate the response to them (10, 138–140). In this sense, different evidences suggest that TGF-β plays a dual role in hepatocarcinogenesis. On one side, as commented above, TGF-β inhibits proliferation and induces apoptosis in hepatocytes and liver tumor cells (141, 142), but simultaneously, it activates survival pathways, such as Akt, and induces an increase in the expression of anti-apoptotic BCL-2-related proteins (143–145), a process that is related to the capacity of TGF-β to transactivate c-Src and EGFR pathways, among others (146). Interestingly, the inhibition of the EGFR increases the apoptotic response to TGF-β (146, 147). In fact, in hepatocytes, TGF-β-induced apoptosis could be counteracted by EGF (an important survival signal) (141, 148) a process that requires activation of the PI3K/Akt axis to counteract TGF-β-induced upregulation of the NOX4, oxidative stress and mitochondrial-dependent apoptosis (149, 150). Another member of the NOX family, NOX1, is involved in this anti-apoptotic role. TGF-β-mediated activation of NOX1 promotes autocrine growth of liver tumor cells through the activation of the EGFR pathway, via upregulation of EGFR ligands expression through a c-Src (151) and NF-κB (152) dependent mechanism. The autocrine loop of EGFR activated by TGF-β in non-transformed hepatocytes and liver cancer cells requires the activity of the metalloprotease TACE/ADAM17 (142, 146) in a Caveolin-1/Src/NOX1 dependent manner (153, 154). This proliferation can be impaired by the addition of the NOX inhibitor VAS2870 (155). Moreover, TGF-β is able to mediate the production of EGFR ligands, which eventually confers resistance to its pro-apoptotic effects in hepatocytes (149, 152) and HCC cells (156). Importantly, the capacity of hepatocytes to survive to TGF-β is also dependent on their differentiation status (157). Thus, rat hepatoma cells respond to TGF-β inducing survival signals, whereas adult hepatocytes do not (142). In the same way, different features of HCC cell lines, like the activation of the EGFR or MEK/ERK pathways, may provoke different outcomes after TGF-β exposure (156, 158).
Once cells overcome the cytostatic and apoptotic effects of TGF-β, this cytokine regulates cell plasticity, a fact that has been elegantly evidenced in a study by Coulouarn and col., where they proposed different liver TGF-β gene signatures, defining a cohort of genes related to its tumor suppressor capacity and another cohort of genes related with its tumor promoting effects: the early and the late TGF-β-signatures. The “early” TGF-β signature is associated to genes involved in growth inhibition and apoptosis, whereas the termed “late” TGF-β signature is associated to EMT, migration and invasion (159). Of relevance, this study also discriminated HCC cell lines by degree of invasiveness. Interestingly, the early response pattern is associated with longer, and the late response pattern with shorter, survival in human HCC patients. In addition, tumors expressing the late TGF-β-responsive genes displayed invasive phenotype, increased tumor recurrence and accurately predicted liver metastasis. In the development of liver hepatocarcinogenesis, TGF-β plays crucial roles regulating the different stages of the disease, some of these roles are summarize in the Figure 3 and we detail in the next chapters.
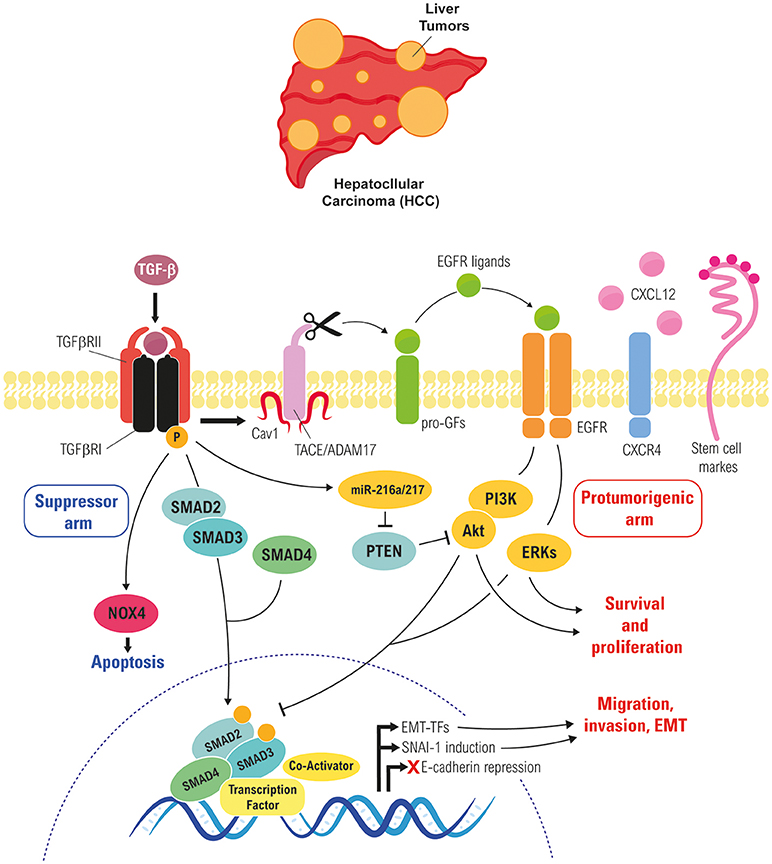
Figure 3. Role of TGF-β in regulating EMT of liver tumor cells. Cross-talk between the TGF-β and the EGFR pathways in liver tumor cells, which rescues cells from TGF-β-induced apoptosis and allows them to respond to it undergoing a partial or full EMT, which increase their migratory/invasive and stemness properties.
TGF-β Promotes EMT in HCC
Tumor cells that overcome the suppressor effects of TGF-β become ready to respond to this cytokine inducing other effects, such as EMT, processes that contribute to either fibrosis and/or tumor dissemination (160). Neoplastic transformation of hepatocytes and progenitor cells, which both are epithelial-like, to a mesenchymal-like phenotype boost heterogeneity in HCC (75).
TGF-β is one of the strongest inducers of EMT under both physiological and pathological context (161), regulating the expression and activity of EMT-TFs (14). Different in vitro studies support the idea that TGF-β induces EMT in non-tumorigenic epithelial cells, transforming them into a fibroblast-like phenotype. For example, alveolar epithelia cells via FoxM1/Snail1 can undergo EMT after TGF-β exposure (162, 163); mammary epithelial cells undergo EMT in a TGF-β/PI3K/mTOR pathway (164, 165). After liver insult, non-transformed hepatocytes can undergo EMT as an adaptative response to move and scape from damaged, inflammatory, hypoxic and redox-activated microenvironment allowing them to find better conditions (166). Moreover, TGF-β induces anti-apoptotic signals in transformed hepatocytes, through the activation of the EGFR pathway (143, 146), and liver cells that overcome TGF-β pro-apoptotic effects undergo EMT in a Snail1-dependent manner conferring resistance to apoptosis (15, 71, 142, 156). Besides apoptotic resistance, mesenchymal-like phenotype increases migratory properties in HCC cells through activation of the CXCR4/CXCL12 axis in TGF-β-dependent manner (139), a mechanism that would contribute to tumor progression in HCC patients (167). Interestingly, CXCR4 is localized in the migratory front of the tumors and is coincident with TGF-β signaling overactivation, suggesting this pathway as a future prognostic factor to predict patient response to TGF-β therapies. MicroRNAs (miRNA) are also involved in the regulation of EMT and in the progression and development of HCC. MiR-181, which is regulated by TGF-β, is overexpressed in HCC samples and is associated with and EMT phenotype (168–170). In hepatocytes, miR-181 induces an EMT-like response and mimics TGF-β-effects, upregulating MMP2, α-SMA and vimentin, downregulating E-cadherin and inducing morphological changes.
Upon HCC development, the excessive growth of transformed cells also generates hostile nodules for cancer cells due to oxygen depletion in internal areas (hypoxic environment) (171) as compared to tumor stroma borders, which induces tumor cell necrosis. Malignant hepatocytes or progenitor cells could undergo EMT as an option to escape from these places and to move toward a cytokine/chemokine better and enriched microenvironment, as well as a resistance mechanisms of survival to cell death stimuli (172, 173). In addition, hypoxic factors, such as HIF-1α, could stimulate EMT in hepatocytes in a TGF-β-dependent manner due to hypoxic hepatocytes secrete enzymes that activate latent TGF-β (174).
TGF-β Regulates Cancer Stem Cell Plasticity
The variability in the prognosis of HCC patients suggests that it may comprise several distinct biological phenotypes, but individuals with HCC who shared a gene expression pattern with fetal hepatoblasts showed to have a poor prognosis (175). Several evidences provide insight into the role of TGF-β in regulating the cancer stem cell niche (176), much less is known about the potential crosstalk between TGF-β-induced EMT in the HCC cells and the acquisition of stem cell properties (74, 177). It is proved that liver epithelial cells that undergo a TGF-β-dependent EMT process show a less differentiated phenotype. Chronic TGF-β treatment in rat and human fetal hepatocytes, as well as in human HCC cells, promotes a mesenchymal-like phenotype concomitant with decreased expression of specific hepatic genes and the appearance of stem cell features, reminiscent of a progenitor-like phenotype (156, 177, 178). Cancer stem cells (CSC) or tumor-initiating cells (TICs) in the liver could derive from hepatic progenitor cells exposed to chronic TGF-β-exposure during hepatocarcinogenesis (179). TGF-β is involved in the neoplastic transformation of liver progenitor cells, through a miR-216a/PTEN/Akt-dependent pathway (179), concomitant with FOXO3a nuclear exportation. FOXO transcription factors are implicated in a huge cellular events and are also related with the neoplastic phenotypes linked to PI3K/Akt activation (180).
It is suggested that the expression of stem-related genes could also be mediating the acquisition of an EMT phenotype. In this sense, the stem-related CD44 or CD133 are not only involved in the acquisition of stem properties, but also in the switch to a more mesenchymal, migratory phenotype (181, 182). TGF-β-mediated mesenchymal-like phenotype is regulated by CD44, and its overexpression provokes down-regulation in E-cadherin expression and up-regulation of vimentin, which correlate with higher phospho-SMAD2-positive nuclei and poor prognosis in HCC patients (181, 183). Moreover, intermediate EMT states have recently been identified as crucial drivers of organ fibrosis and tumor progression (14). During partial-EMT stage, both epithelial and mesenchymal stem genes can be expressed. In this sense, it is worthy to mention that in certain HCC cell lines, TGF-β-treatment induces the expression of mesenchymal genes, such as VIM (vimentin), and the mesenchymal-related stem-related genes CD44 and CD90, but simultaneously, they express E-cadherin and the epithelial-related stem genes EPCAM or CD133 (177). Interestingly, this partial EMT phenotype confers to the HCC cells the highest stemness stage concomitant with an increased migratory/invasive capacity (177).
CSCs could contribute to the failure of therapies to abolish malignant tumors. In pre-clinical assays, cancer stem-like spheres from de-differentiated HCC-derived cell lines show increased expression of stemness markers (CD44), and higher resistance to anticancer drugs (184). Furthermore, the acquisition of some mesenchymal properties and the expression of CD44 impair the HCC cell response to sorafenib-induced apoptosis (183). For this, novel strategies are focused to target CSC development. It is interesting to mention that treatment with inhibitors of the TGF-β pathway, such as Galunisertib (LY2157299–a selective ATP-mimetic inhibitor of TGF-β receptor I) decreased the stemness related genes of mesenchymal HCC cells and their ability to form colonies, liver spheroids and invasive growth (185). Resminostat, a novel orally histone deacetylases inhibitor, has been demonstrated as a good therapy in the SHELTER study (a phase I/II clinical study) in mono and combination therapy with sorafenib (186). The combination therapy revealed an advantage in terms of overall survival and time to progression. In HCC cells with a mesenchymal phenotype, caused by autocrine expression of TGF-β, resminostat sensitizes them to the apoptotic response induced by sorafenib (187). Mesenchymal-related gene expression was decreased in resminostat-treated HCC cells. This event is concomitant with an epithelial-related gene expression increase, more organized tight junctions and lower invasive growth. Indeed, resminostat down-regulated CD44 expression is coincident with a decrease in the stemness properties. These results reinforce the strong impact of the TGF-β-induced mesenchymal/stemness phenotype on HCC drug resistance.
Liver Cancer Stroma Cell Plasticity and TGF-β
TGF-β display multiple effects on the microenvironment (188), which plays a relevant role in HCC development and progression. In the stroma, TGF-β induces microenvironment changes, including generation of cancer-associated fibroblasts (CAFs) (23) that play a relevant role in facilitating the production of growth factors and cytokines, which contribute to cell proliferation, invasion and neoangiogenesis, being related with poor prognosis (189–192). Different origins are described, but in the liver CAFs could be originated from epithelial cells–hepatocytes, cholangiocytes- and HSC that undergo an EMT process. To promote their tumoral functions, CAFs need the expression of EMT-TFs. Indeed, Snail expression in CAFs is necessary for their response to TGF-β, increased production of fibronectin and stiffness of the ECM (23). CAFs are also a potent source of TGF-β and are described to promote the migration and invasion of HCC cells in vitro and facilitate the HCC metastasis to the bone, brain and lung in NOD/SCID mice (193).
In HCC microenvironment anti-tumor response is impaired due to various immune suppressive elements, such as Tumor Associated Macrophages (TAMs) and regulatory T cells (Treg) (194–196). Similarly to that occurs with macrophages in fibrosis, during HCC progression, TAMs are mainly polarized toward an M2 phenotype, due to the higher levels of TGF-β (among others factors) (197). M2-like macrophages are major players in the connection between inflammation and cancer, involved in functions such as: promotion of tumor cell proliferation, ECM turnover, inhibition of adaptive immunity, among others (198, 199), TAMs are correlated with angiogenesis, metastasis, and poor prognosis (200, 201). Moreover, TAMs can promote cancer stem cell properties in a TGF-β-dependent manner (202). CAFs can also educate Natural Killer (NK) cells (203), dendritic cells (204) and upregulate the production of Treg in a TGF-β-dependent manner (205). CAFs promote Treg cell induction, through upregulation and activation of the human B7 homolog 1(B7-H1)/programmed death 1 (PD-1) signaling, which are involved in immunosuppressive functions in a mTOR/Akt dependent-manner (206). Accumulating evidence indicates that the immune system microenvironment plays key roles in the development of HCC (207, 208). CD4+ naïve T cells show an enormous cell plasticity and under TGF-β-treatment could differentiate into Treg cells (209). Poor prognosis in HCC patients is associated with infiltration liver tissue prognosis in HCC patients. Treg cells -Foxp3-positive cells- are involved in immune homeostasis, peripheral tolerance and prevention of autoimmunity (210, 211). TGF-β induces the expression of the transcriptional factor Foxp3 involved in the conversion of naïve CD4+CD25 T cells to CD4+CD25+ Treg cells with potent immunosuppressive potential (212). Blocking TGF-β signaling with SM-16 (TGF-β inhibitor) significant decreases the percentage of Treg cells in liver tissue concomitant with an attenuation of the hepatocarcinogenesis process in a DEN-model (213). Interestingly, addition of exogenous TGF-β restores the expression level of Foxp3 and the presence of Treg cells. Exogenous addition of TGF-β normalizes Treg cell numbers and promoted their cell differentiation. Moreover, In HCC patient samples, the expression of both genes, TGFB1 and FOXP3, correlate positively and are involved in tumoral progression. (213). On summary, TGF-β promotes tumor immune escape and survival by maintaining natural Treg levels, inducing Treg cell differentiation and TAMs polarization into M2-phenotype.
New Therapies to Inhibit the TGF-β Pathway
Developing an effective therapy to target the TGF-β pathway in liver pathologies requires a better understanding of its complex role in this organ, considering its pleiotropic effects on cell proliferation, death and differentiation of different liver cell types, its ability to induce EMT in epithelial cells or EndEMT in the endothelial ones, as well as its capacity to act as an immune modulator. In spite of this, targeting TGF-β was proposed a good approach to delay the progression of liver diseases and, in particular, of HCC (9, 214, 215). Indeed, first experiments indicated that inhibiting the TGF-β pathway in HCC cell lines blocked migration and invasion of HCC cells by up-regulating E-cadherin and down-regulating CXCR4 (167, 188), as well as inhibiting the upregulated levels of CTGF induced by TGF-β, reducing the stromal component of the microtumoural environment and slowing the HCC growth in vivo (216). These data suggested that there could be a mechanistic use for targeting TGF-β in HCC clinical trials.
Several different strategies have been proposed to inhibit the TGF-β pathway in liver pathologies, including the use of chimeric proteins, monoclonal antibodies, peptide inhibitors, small molecules that inhibit the receptors' kinase activity and antisense oligonucleotides (217). First studies demonstrated the efficiency of potential peptide inhibitors of TGF-β1 (derived from TGF-β1 and from its type III receptor) in vitro and in vivo in reducing liver fibrosis (218, 219). These peptide inhibitors were proved to be also useful in enhancing the efficacy of antitumor immunotherapy (220). To increase the delivery efficiency, in a recent study, one of these peptides (P-17) was loaded separately into folic acid-functionalized nano-carriers made of bovine serum albumin. Cellular studies demonstrated the targeting efficiency of the hybrid carriers (221).
In the last years the interest has been focused on the TβRI kinase inhibitor Galunisertib, developed by Lilly (LY2157299, a selective ATP-mimetic inhibitor of TβRI) that has proved more efficient than neutralizing humanized antibodies, such as D10 against TβRII, in blocking the canonical TGF-β signaling in HCC cells, experiments that supported the use of this drug in preclinical and clinical trials (9, 222). Despite limited antiproliferative effects, Galunisertib yielded potent anti-invasive properties in HCC models and in ex vivo tumor tissue samples from patients (223). Worthy to note, in combination, Galunisertib potentiated the effect of sorafenib efficiently by inhibiting proliferation and increasing apoptosis. Galunisertib also reduced the expression of stemness-related genes, such as CD44 and THY1, in vitro and in ex vivo human HCC specimens, overcoming stemness-derived aggressiveness (185). Furthermore, it also showed antitumor activity through the activation of CD8+ T-cell antitumor responses (224). Recent studies have also suggested the potential efficiency of Galunisertib as antifibrotic drug. In ex vivo studies, using both healthy and cirrhotic human precision-cut liver slices, Galunisertib reduced fibrosis-related transcription, which correlated with a significant inhibition in the production and maturation of collagens (225). Furthermore, in an experimental preclinical model (Abcb4ko mice) the treatment with Galunisertib decreased the expression of several fibrogenic genes, such as collagens (Col1a1 and Col1a2), Tgfb1 and Timp1, and reduced the ECM/stromal components, fibronectin and laminin-332, as well as the carcinogenic β-catenin pathway (226).
A phase II clinical trial using Galunisertib in patients with advanced HCC to test safety, time to progression and overall survival (OS) is ongoing (NCT01246986 http://clinicaltrials.gov). Preliminary data show that patients with higher levels of circulating TGF-β1 are more likely to respond to therapy with Galunisertib. TGF-β1 reduction in response to the treatment is related to improvement in OS when compared to patients with non-TGF-β1 reduction. Some efforts are being made in optimizing the delivery of Galunisertib in form of novel polymeric nano-micelles, to avoid acidic pH of gastrointestinal tract, colon alkaline pH and anti-immune recognition (227).
Once it is proven the safety and the benefit of using Galunisertib in HCC, biomarkers will be extremely useful in the proper selection of patients who might benefit from receiving the drug. In this sense, high TGFB1 expression in HCC patients, concomitant with high expression of the genes that mediate its invasive effects, such as PDGF, CXCR4, or CD44, (167, 177, 228) would anticipate a benefit for the use of Galunisertib. Furthermore, a recent study has proposed SKIL and PMEPA1 as strongly downregulated by Galunisertib, correlating with endogenous TGFβ-1 (185, 229). These target genes identified may also serve as biomarkers for the stratification of HCC patients undergoing treatment with Galunisertib. Since biopsy is not frequent in HCC patients, new areas of research must be focused on the improvement of liquid biopsies in these patients to develop the possibility that this kind of analysis may be done in tumor circulating cells.
Finally, new approaches to interfere not only the TGF-β canonical, but also the non-canonical pathways must be developed in the next future, as previously mentioned, the switch from tumor-suppressive to pro-oncogenic TGF-β actions could be directed by its crosstalk with Receptor Tyrosine Kinases, in particular, EGFR. So, interference with EGFR signaling, by employing approved targeted drugs, in TGF-β/SMAD-positive HCC patients might be effective in improving the effectiveness of Galunisertib.
Concluding Remarks
TGF-β plays unique actions in modulating cell plasticity, and the liver reveals as a tissue where these actions would be very relevant during the response to injuries that cause chronic diseases. In general terms, TGF-β-induced changes in cell plasticity may converge in transdifferentiation toward a different phenotype, such as the case of activation of HSC to MFB or the dedifferentiation/acquisition of stem cell properties in hepatocytes and liver tumor cells. But it may also proportionate to the cells new capabilities, such as cell survival or increase in migratory/invasive properties that the liver tumor cells acquire when they undergo EMT in response to TGF-β. And, worthy to note the essential role that TGF-β plays inducing Treg cell differentiation and TAMs polarization into M2-phenotype, which promotes tumor immune escape and survival. Despite all current knowledge, there are still many gaps that need to be clarify. However, these evidences point toward the use of tools that target the TGF-β signaling pathway to counteract liver disease progression.
Authors Contributions
Both authors equally contribute to the writing and revision of this work. Figures were designed and elaborated by DC-D and supervised by IF.
Funding
Grants: Ministry of Science, Innovation and Universities, Spain (cofounded by FEDER funds/Development Fund—a way to build Europe): SAF2015-64149-R. DC-D was recipient of a fellowship from the FPI program, Ministry of Economy, Industry and Competitiveness, Spain: BES-2016 0077564. The CIBEREHD, National Biomedical Research Institute on Liver and Gastrointestinal Diseases, is funded by the Instituto de Salud Carlos III, Spain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to give thanks to Esther Bertran and Judit López-Luque for all their support and advices.
References
1. Rozga J. Hepatocyte proliferation in health and in liver failure. Med Sci Monit. (2002) 8:RA32–8.
2. Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology (2012) 56:769–75. doi: 10.1002/hep.25670
3. Michalopoulos GK. Advances in liver regeneration. Expert Rev Gastroenterol Hepatol. (2014) 8:897–907. doi: 10.1586/17474124.2014.934358
4. Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology (2008) 134:1641–54. doi: 10.1053/j.gastro.2008.03.002
5. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. (2014) 14:181–94. doi: 10.1038/nri3623
6. Heldin C-H, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. (2016) 8:a022053. doi: 10.1101/cshperspect.a022053
7. Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. (2012) 347:245–56. doi: 10.1007/s00441-011-1246-y
8. Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G., et al. TGF-β signalling and liver disease. FEBS J. (2016) 283:2219–32. doi: 10.1111/febs.13665
9. Giannelli G, Mikulits W, Dooley S, Fabregat I, Moustakas A, ten Dijke, P., et al. The rationale for targeting TGF-β in chronic liver diseases. Eur J Clin Invest. (2016) 46:349–61. doi: 10.1111/eci.12596
11. Giannelli G, Rani B, Dituri F, Cao Y, Palasciano G. Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut (2014) 63:1668–76. doi: 10.1136/gutjnl-2014-307323
12. Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nature Cell Biol. (2017) 19:1133–41. doi: 10.1038/ncb3611
13. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science (2013) 342:1234850. doi: 10.1126/science.1234850
14. Nieto MA, Huang RYJ, Jackson RA, Thiery JP. EMT: 2016. Cell (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
15. Franco DL, Mainez J, Vega S, Sancho P, Murillo MM, de Frutos CA, et al. Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J Cell Sci. (2010) 123:3467–77. doi: 10.1242/jcs.068692
16. Dewidar B, Soukupova J, Fabregat I, Dooley S. TGF-β in hepatic stellate cell activation and liver fibrogenesis: updated. Curr Pathobiol Rep. (2015) 3:291–305. doi: 10.1007/s40139-015-0089-8
17. Kim KK, Sheppard D, Chapman HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. (2017) 10:a022293. doi: 10.1101/cshperspect.a02229.
18. Pickup MW, Owens P, Moses HL. TGF-β, Bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harb Perspect Biol. (2017) 9:a022285. doi: 10.1101/cshperspect.a022285
19. Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol. (2016) 8:a021907. doi: 10.1101/cshperspect.a021907
20. Thomas GJ, Hart IR, Speight PM, Marshall JF. Binding of TGF-beta1 latency-associated peptide (LAP) to alpha(v)beta6 integrin modulates behaviour of squamous carcinoma cells. Br J Cancer (2002) 87:859–67. doi: 10.1038/sj.bjc.6600545
21. Khan Z, Marshall JF. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. (2016) 365:657–73. doi: 10.1007/s00441-016-2474-y
22. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. (2013) 19:1617–24. doi: 10.1038/nm.3282
23. Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, et al. TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mole Sci. (2018) 19:1294. doi: 10.3390/ijms19051294
24. Drabsch Y, ten Dijke P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. (2012) 31:553–68. doi: 10.1007/s10555-012-9375-7
25. Heldin C-H, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. (2012) 586:1959–70. doi: 10.1016/j.febslet.2012.02.037
26. Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. (2013) 341:30–40. doi: 10.1016/j.canlet.2013.02.048
28. Schuppan D. Liver fibrosis: common mechanisms and antifibrotic therapies. Clin Res Hepatol Gastroenterol. (2015) 39 (Suppl. 1):S51–59. doi: 10.1016/j.clinre.2015.05.005
29. Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. (2013) 305:C789–99. doi: 10.1152/ajpcell.00230.2013
30. Cao Q, Mak KM, Lieber CS. DLPC decreases TGF-β1-induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am J Physiol Gastrointestinal Liver Physiol. (2002) 283:G1051–61. doi: 10.1152/ajpgi.00128.2002
31. Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology (2003) 38:879–89. doi: 10.1002/hep.1840380414
32. Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. (2013) 304:C216–25. doi: 10.1152/ajpcell.00328.2012
33. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30:245–57. doi: 10.1055/s-0030-1255354
34. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. (1989) 170:727–37. doi: 10.1084/jem.170.3.727
35. Koslowski R, Seidel D, Kuhlisch E, Knoch KP. Evidence for the involvement of TGF-beta and PDGF in the regulation of prolyl 4-hydroxylase and lysyloxidase in cultured rat lung fibroblasts. Exp Toxicol Pathol. (2003) 55:257–64. doi: 10.1078/0940-2993-00323
36. Leicester KL, Olynyk JK, Brunt EM, Britton RS, Bacon BR. CD14-positive hepatic monocytes/macrophages increase in hereditary hemochromatosis. Liver Int. (2004) 24:446–51. doi: 10.1111/j.1478-3231.2004.0943.x
37. Li H, You H, Fan X, Jia J. Hepatic macrophages in liver fibrosis: pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. (2016) 3:e000079. doi: 10.1136/bmjgast-2016-000079
38. Ramm GA, Nair VG, Bridle KR, Shepherd RW, Crawford DH. Contribution of hepatic parenchymal and nonparenchymal cells to hepatic fibrogenesis in biliary atresia. Am J Pathol. (1998) 153:527–35. doi: 10.1016/S0002-9440(10)65595-2
39. Heymann F, Trautwein C, Tacke F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm Allergy Drug Targets (2009) 8:307–18. doi: 10.2174/187152809789352230
40. Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. (2013) 123:1887–901. doi: 10.1172/JCI66028
41. Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. (2015) 5:683. doi: 10.3389/fimmu.2014.00683
42. Naito M, Hasegawa G, Ebe Y, Yamamoto T. Differentiation and function of Kupffer cells. Med Electron Microsc (2004) 37:16–28. doi: 10.1007/s00795-003-0228-x
43. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
44. De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology (2007) 132:1937–46. doi: 10.1053/j.gastro.2007.02.033
45. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology (2009) 50:261–74. doi: 10.1002/hep.22950
46. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58:1461–73. doi: 10.1002/hep.26429
47. Sun YY, Li XF, Meng XM, Huang C, Zhang L, Li J. Macrophage phenotype in liver injury and repair. Scand J Immunol. (2017) 85:166–74. doi: 10.1111/sji.12468
48. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
49. Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology (2005) 128:138–46. doi: 10.1053/j.gastro.2004.10.005
50. Braga TT, Agudelo JSH, Camara NOS. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol. (2015) 6:602. doi: 10.3389/fimmu.2015.00602
51. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
52. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
53. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
54. Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med. (2011) 13:e23. doi: 10.1017/S1462399411001943
55. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
56. Bai L, Liu X, Zheng Q, Kong M, Zhang X, Hu R, et al. M2-like macrophages in the fibrotic liver protect mice against lethal insults through conferring apoptosis resistance to hepatocytes. Sci Rep. (2017) 7:10518. doi: 10.1038/s41598-017-11303-z
57. Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. (2011) 43:154–62. doi: 10.1016/j.biocel.2010.10.013
58. Wang S, Meng X-M, Ng Y-Y, Ma FY, Zhou S, Zhang Y, et al. TGF-β/Smad3 signalling regulates the transition of bone marrowderived macrophages into myofibroblasts during tissue fibrosis. Oncotarget (2015) 7:8809–22. doi: 10.18632/oncotarget.6604
59. Gong D, Shi W, Yi S, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. (2012) 13:31. doi: 10.1186/1471-2172-13-31
60. Wijesundera KK, Izawa T, Tennakoon AH, Murakami H, Golbar HM, Katou-Ichikawa C, et al. M1- and M2-macrophage polarization in rat liver cirrhosis induced by thioacetamide (TAA), focusing on Iba1 and galectin-3. Exp Mol Pathol. (2014) 96:382–92. doi: 10.1016/j.yexmp.2014.04.003
61. Bility MT, Nio K, Li F, McGivern DR, Lemon SM, Feeney ER, et al. Chronic hepatitis C infection-induced liver fibrogenesis is associated with M2 macrophage activation. Sci Rep. (2016) 6:39520. doi: 10.1038/srep39520
62. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
63. Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepato-Biliary-Pancreat Sci. (2015) 22:512–8. doi: 10.1002/jhbp.245
64. Elpek GÖ. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J Gastroenterol. (2014) 20:7260–76. doi: 10.3748/wjg.v20.i23.7260
65. Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U. S. A. (2014) 111:E3297–3305. doi: 10.1073/pnas.1400062111
66. Wells RG. Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis. (2008) 12:759–68. doi: 10.1016/j.cld.2008.07.008
67. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
68. Russo FP, Alison MR, Bigger BW, Amofah E, Florou A, Amin F, et al. The bone marrow functionally contributes to liver fibrosis. Gastroenterology (2006) 130:1807–21. doi: 10.1053/j.gastro.2006.01.036
69. Del Castillo G, Murillo MM, Alvarez-Barrientos A, Bertran E, Fernández M, Sánchez A, et al. Autocrine production of TGF-beta confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: role of EGF receptor ligands. Exp Cell Res. (2006) 312:2860–71. doi: 10.1016/j.yexcr.2006.05.017
70. Kaimori A, Potter J, Kaimori J-Y, Wang C, Mezey E, Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem. (2007) 282:22089–101. doi: 10.1074/jbc.M700998200
71. Valdés F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernández M, et al. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol Cancer Res. (2002) 1:68–78.
72. Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest. (1990) 86:1641–8. doi: 10.1172/JCI114886
73. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
74. Fabregat I, Malfettone A, Soukupova J. New insights into the crossroads between EMT and stemness in the context of cancer. J Clin Med. (2016) 5:37. doi: 10.3390/jcm5030037
75. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. (2016) 65:798–808. doi: 10.1016/j.jhep.2016.05.007
76. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer (2002) 2:442–54. doi: 10.1038/nrc822
77. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
78. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. (2009) 119:1429–37. doi: 10.1172/JCI36183
79. Walkup MH, Gerber DA. Hepatic stem cells: in search of. Stem Cells (2006) 24:1833–40. doi: 10.1634/stemcells.2006-0063
80. Weng HL, Ciuclan L, Liu Y, Hamzavi J, Godoy P, Gaitantzi H, et al. Profibrogenic transforming growth factor-beta/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology (2007) 46:1257–70. doi: 10.1002/hep.21806
81. Xie G, Diehl AM. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G881–90. doi: 10.1152/ajpgi.00289.2013
82. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. (2007) 282:23337–47. doi: 10.1074/jbc.M700194200
83. Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology (2008) 135:642–59. doi: 10.1053/j.gastro.2008.04.038
84. Bi WR, Xu GT, Lv LX, Yang CQ. The ratio of transforming growth factor-β1/bone morphogenetic protein-7 in the progression of the epithelial-mesenchymal transition contributes to rat liver fibrosis. Genet Mol Res. (2014) 13:1005–14. doi: 10.4238/2014.February.20.2
85. Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology (2004) 127:1565–77. doi: 10.1053/j.gastro.2004.08.006
86. Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. (2013) 123:1902–10. doi: 10.1172/JCI66369
87. Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. (2011) 79:944–56. doi: 10.1038/ki.2010.516
88. Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology (2000) 31:1094–106. doi: 10.1053/he.2000.6126
89. Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernández-Rodriguez CM, Pinedo F, et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE (2012) 7:e45285. doi: 10.1371/journal.pone.0045285
90. Ding Z, Jin G, Liang H, Wang W, Chen W, Datta PK, et al. Transforming growth factor β induces expression of connective tissue growth factor in hepatic progenitor cells through Smad independent signaling. Cell Signal. (2013) 25:1981–92. doi: 10.1016/j.cellsig.2013.05.027
91. Liu Y, Liu H, Meyer C, Li J, Nadalin S, Königsrainer A, et al. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J Biol Chem. (2013) 288:30708–19. doi: 10.1074/jbc.M113.478685
92. Jia D, Duan F, Peng P, Sun L, Liu X, Wang L, et al. Up-regulation of RACK1 by TGF-β1 promotes hepatic fibrosis in mice. PLoS ONE (2013) 8:e60115. doi: 10.1371/journal.pone.0060115
93. Liu M, Peng P, Wang J, Wang L, Duan F, Jia D, et al. RACK1-mediated translation control promotes liver fibrogenesis. Biochem Biophys Res Commun. (2015) 463:255–61. doi: 10.1016/j.bbrc.2015.05.040
94. Li L, Wang JY, Yang CQ, Jiang W. Effect of RhoA on transforming growth factor β1-induced rat hepatic stellate cell migration. Liver Int. (2012) 32:1093–102. doi: 10.1111/j.1478-3231.2012.02809.x
95. Fang L, Huang C, Meng X, Wu B, Ma T, Liu X, et al. TGF-β1-elevated TRPM7 channel regulates collagen expression in hepatic stellate cells via TGF-β1/Smad pathway. Toxicol Appl Pharmacol. (2014) 280:335–44. doi: 10.1016/j.taap.2014.08.006
96. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U. S. A. (2012) 109:9448–53. doi: 10.1073/pnas.1201840109
97. Liu X, Xu J, Brenner DA, Kisseleva T. Reversibility of liver fibrosis and inactivation of fibrogenic myofibroblasts. Curr Pathobiol Rep. (2013) 1:209–14. doi: 10.1007/s40139-013-0018-7
98. Foo N-P, Lin S-H, Lee Y-H, Wu M-J, Wang Y-J. α-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-β. Toxicology (2011) 282:39–46. doi: 10.1016/j.tox.2011.01.009
99. Abhilash PA, Harikrishnan R, Indira M. Ascorbic acid supplementation down-regulates the alcohol induced oxidative stress, hepatic stellate cell activation, cytotoxicity and mRNA levels of selected fibrotic genes in guinea pigs. Free Radic Res. (2012) 46:204–13. doi: 10.3109/10715762.2011.647691
100. Torok NJ. Dysregulation of redox pathways in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G667–74. doi: 10.1152/ajpgi.00050.2016
101. Crosas-Molist E, Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. (2015) 6:106–11. doi: 10.1016/j.redox.2015.07.005
102. Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab Invest. (2004) 84:1013–23. doi: 10.1038/labinvest.3700109
103. Wang L, Clutter S, Benincosa J, Fortney J, Gibson LF. Activation of transforming growth factor-beta1/p38/Smad3 signaling in stromal cells requires reactive oxygen species-mediated MMP-2 activity during bone marrow damage. Stem Cells (2005) 23:1122–34. doi: 10.1634/stemcells.2004-0354
104. Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res. (2006) 166:839–48. doi: 10.1667/RR0695.1
105. Proell V, Carmona-Cuenca I, Murillo MM, Huber H, Fabregat I, Mikulits W. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp Hepatol. (2007) 6:1. doi: 10.1186/1476-5926-6-1
106. Boudreau HE, Emerson SU, Korzeniowska A, Jendrysik MA, Leto TL. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: a new contributor to HCV-induced oxidative stress. J Virol. (2009) 83:12934–46. doi: 10.1128/JVI.01059-09
107. Liang S, Kisseleva T, Brenner DA. The Role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol. (2016) 7:17. doi: 10.3389/fphys.2016.00017
108. Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal. (2014) 20:2854–72. doi: 10.1089/ars.2013.5619
109. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology (2011) 53:1730–41. doi: 10.1002/hep.24281
110. Crosas-Molist E, Bertran E, Fabregat I. Cross-Talk Between TGF-β and NADPH Oxidases During Liver Fibrosis and Hepatocarcinogenesis. Curr Pharm Des. (2015) 21:5964–76. doi: 10.2174/1381612821666151029112126
111. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. (2005) 97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D
112. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. (2009) 15:1077–81. doi: 10.1038/nm.2005
113. Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, et al. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. (2010) 21:93–102. doi: 10.1681/ASN.2009020146
114. Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in v0ivo. Free Radic Biol Med. (2012) 53:289–96. doi: 10.1016/j.freeradbiomed.2012.05.007
115. Jiang JX, Mikami K, Venugopal S, Li Y, Török NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. (2009) 51:139–48. doi: 10.1016/j.jhep.2009.03.024
116. Aoyama T, Paik Y-H, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology (2012) 56:2316–27. doi: 10.1002/hep.25938
117. Lan T, Kisseleva T, Brenner DA. Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS ONE (2015) 10:e0129743. doi: 10.1371/journal.pone.0129743
118. European Association For The Study Of The Liver and European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. (2012) 56:908–43. doi: 10.1016/j.jhep.2011.12.001
119. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science (2007) 317:121–4. doi: 10.1126/science.1140485
120. Tabrizian P, Roayaie S, Schwartz ME. Current management of hepatocellular carcinoma. World J Gastroenterol. (2014) 20:10223–37. doi: 10.3748/wjg.v20.i30.10223
121. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut (2014) 63:844–55. doi: 10.1136/gutjnl-2013-306627
122. Balogh J, Victor D, Asham EH, Burroughs SG, Boktour M, Saharia A, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma (2016) 3:41–53. doi: 10.2147/JHC.S61146
123. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet (2012) 379:1245–55. doi: 10.1016/S0140-6736(11)61347-0
124. Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology (2016) 150:835–53. doi: 10.1053/j.gastro.2015.12.041
125. Wallace MC, Friedman SL. Hepatic fibrosis and the microenvironment: fertile soil for hepatocellular carcinoma development. Gene Expr. (2014) 16:77–84. doi: 10.3727/105221614X13919976902057
126. Lu L-C, Hsu C-H, Hsu C, Cheng A-L. Tumor heterogeneity in hepatocellular carcinoma: facing the challenges. Liver Cancer (2016) 5:128–38. doi: 10.1159/000367754
127. Blagotinsek K, Rozman D. Targeting signalling pathways in hepatocellular carcinoma. Curr Pharm Des. (2017) 23:170–5. doi: 10.2174/1381612822666161006160005
128. Moeini A, Cornellà H, Villanueva A. Emerging signaling pathways in hepatocellular carcinoma. Liver Cancer (2012) 1:83–93. doi: 10.1159/000342405
129. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology (2013) 144:512–27. doi: 10.1053/j.gastro.2013.01.002
130. Rani B, Cao Y, Malfettone A, Tomuleasa C, Fabregat I, Giannelli G. Role of the tissue microenvironment as a therapeutic target in hepatocellular carcinoma. World J Gastroenterol. (2014) 20:4128–40. doi: 10.3748/wjg.v20.i15.4128
131. Achyut BR, Yang L. Transforming growth factor-β in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology (2011) 141:1167–78. doi: 10.1053/j.gastro.2011.07.048
132. Sanchez A, Alvarez AM, Benito M, Fabregat I. Transforming growth factor beta modulates growth and differentiation of fetal hepatocytes in primary culture. J Cell Physiol. (1995) 165:398–405. doi: 10.1002/jcp.1041650221
133. Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell (2000) 103:295–309. doi: 10.1016/S0092-8674(00)00121-5
134. Majumdar A, Curley SA, Wu X, Brown P, Hwang JP, Shetty K, et al. Hepatic stem cells and transforming growth factor β in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2012) 9:530–8. doi: 10.1038/nrgastro.2012.114
135. Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. (2009) 119:3408–19. doi: 10.1172/JCI38854
136. Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene (2007) 26:7103–10. doi: 10.1038/sj.onc.1210513
137. Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res. (2008) 1:187–91. doi: 10.1158/1940-6207.CAPR-08-0028
138. Seoane J. Escaping from the TGFbeta anti-proliferative control. Carcinogenesis (2006) 27:2148–56. doi: 10.1093/carcin/bgl068
139. Bertran E, Caja L, Navarro E, Sancho P, Mainez J, Murillo MM, et al. Role of CXCR4/SDF-1 alpha in the migratory phenotype of hepatoma cells that have undergone epithelial-mesenchymal transition in response to the transforming growth factor-beta. Cell Signal. (2009) 21:1595–606. doi: 10.1016/j.cellsig.2009.06.006
140. Cepeda EB, Dediulia T, Fernando J, Bertran E, Egea G, Navarro E, et al. Mechanisms regulating cell membrane localization of the chemokine receptor CXCR4 in human hepatocarcinoma cells. Biochim Biophys Acta (2015) 1853:1205–18. doi: 10.1016/j.bbamcr.2015.02.012
141. Fabregat I, Herrera B, Fernández M, Alvarez AM, Sánchez A, Roncero C, et al. Epidermal growth factor impairs the cytochrome C/caspase-3 apoptotic pathway induced by transforming growth factor beta in rat fetal hepatocytes via a phosphoinositide 3-kinase-dependent pathway. Hepatology (2000) 32:528–35. doi: 10.1053/jhep.2000.9774
142. Caja L, Ortiz C, Bertran E, Murillo MM, Miró-Obradors MJ, Palacios E, et al. Differential intracellular signalling induced by TGF-beta in rat adult hepatocytes and hepatoma cells: implications in liver carcinogenesis. Cell Signal. (2007) 19:683–94. doi: 10.1016/j.cellsig.2006.09.002
143. Valdés F, Murillo MM, Valverde AM, Herrera B, Sánchez A, Benito M, et al. Transforming growth factor-beta activates both pro-apoptotic and survival signals in fetal rat hepatocytes. Exp Cell Res. (2004) 292:209–18. doi: 10.1016/j.yexcr.2003.08.015
144. Wilkes MC, Mitchell H, Penheiter SG, Doré JJ, Suzuki K, Edens M., et al. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. (2005) 65, 10431–10440. doi: 10.1158/0008-5472.CAN-05-1522
145. Song K, Wang H, Krebs TL, Danielpour D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. (2006) 25:58–69. doi: 10.1038/sj.emboj.7600917
146. Murillo MM, del Castillo G, Sánchez A, Fernández M, Fabregat I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene (2005) 24:4580–87. doi: 10.1038/sj.onc.1208664
147. Park SS, Eom YW, Kim EH, Lee JH, Min DS, Kim S, et al. Involvement of c-Src kinase in the regulation of TGF-beta1-induced apoptosis. Oncogene (2004) 23:6272–81. doi: 10.1038/sj.onc.1207856
148. Fabregat I, Sánchez A, Alvarez AM, Nakamura T, Benito M. Epidermal growth factor, but not hepatocyte growth factor, suppresses the apoptosis induced by transforming growth factor-beta in fetal hepatocytes in primary culture. FEBS Lett. (1996) 384:14–8. doi: 10.1016/0014-5793(96)00266-9
149. Carmona-Cuenca I, Herrera B, Ventura J-J, Roncero C, Fernández M, Fabregat I. EGF blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J Cell Physiol. (2006) 207:322–30. doi: 10.1002/jcp.20568
150. Carmona-Cuenca I, Roncero C, Sancho P, Caja L, Fausto N, Fernández M, et al. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. (2008) 49:965–76. doi: 10.1016/j.jhep.2008.07.021
151. Sancho P, Fabregat I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J Biol Chem. (2010) 285:24815–24. doi: 10.1074/jbc.M110.114280
152. Murillo MM, Carmona-Cuenca I, Del Castillo G, Ortiz C, Roncero C, Sánchez A, et al. Activation of NADPH oxidase by transforming growth factor-beta in hepatocytes mediates up-regulation of epidermal growth factor receptor ligands through a nuclear factor-kappaB-dependent mechanism. Biochem J. (2007) 405:251–9. doi: 10.1042/BJ20061846
153. Moreno-Càceres J, Mainez J, Mayoral R, Martín-Sanz P, Egea G, Fabregat I. Caveolin-1-dependent activation of the metalloprotease TACE/ADAM17 by TGF-β in hepatocytes requires activation of Src and the NADPH oxidase NOX1. FEBS J. (2016) 283:1300–10. doi: 10.1111/febs.13669
154. Moreno-Càceres J, Caballero-Díaz D, Chike Nwosu Z, Meyer C, López-Luque J, Malfettone A, et al. The level of caveolin-1 expression determines response to TGF-β as a tumour suppressor in hepatocellular carcinoma cells. Cell Death Dis. (2017) 8:e3098. doi: 10.1038/cddis.2017.469
155. Sancho P, Fabregat I. The NADPH oxidase inhibitor VAS2870 impairs cell growth and enhances TGF-β-induced apoptosis of liver tumor cells. Biochem Pharmacol. (2011) 81:917–24. doi: 10.1016/j.bcp.2011.01.007
156. Caja L, Bertran E, Campbell J, Fausto N, Fabregat I. The transforming growth factor-beta (TGF-β) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J Cell Physiol. (2011) 226:1214–23. doi: 10.1002/jcp.22439
157. Sánchez A, Alvarez AM, López Pedrosa JM, Roncero C, Benito M, Fabregat I. Apoptotic response to TGF-beta in fetal hepatocytes depends upon their state of differentiation. Exp Cell Res. (1999) 252:281–91. doi: 10.1006/excr.1999.4624
158. Caja L, Sancho P, Bertran E, Iglesias-Serret D, Gil J, Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. (2009) 69:7595–602. doi: 10.1158/0008-5472.CAN-09-1482
159. Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology (2008) 47:2059–67. doi: 10.1002/hep.22283
160. Moustakas A, Heldin CH. Induction of epithelial-mesenchymal transition by transforming growth factor β. Semin Cancer Biol. (2012) 22:446–54. doi: 10.1016/j.semcancer.2012.04.002
161. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal (2014) 7:re8. doi: 10.1126/scisignal.2005189
162. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM., et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. (2005) 166:1321–32. doi: 10.1016/S0002-9440(10)62351-6
163. Balli D, Ustiyan V, Zhang Y, Wang I-C, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. (2013) 32:231–44. doi: 10.1038/emboj.2012.336
164. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. (2000) 275:36803–10. doi: 10.1074/jbc.M005912200
165. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. (2007) 178:437–51. doi: 10.1083/jcb.200611146
166. Pinzani M. Epithelial-mesenchymal transition in chronic liver disease: fibrogenesis or escape from death? J Hepatol. (2011) 55:459–65. doi: 10.1016/j.jhep.2011.02.001
167. Bertran E, Crosas-Molist E, Sancho P, Caja L, Lopez-Luque J, Navarro E, et al. Overactivation of the TGF-β pathway confers a mesenchymal-like phenotype and CXCR4-dependent migratory properties to liver tumor cells. Hepatology (2013) 58:2032–44. doi: 10.1002/hep.26597
168. Wang B, Hsu S-H, Majumder S, Kutay H, Huang W, Jacob ST, et al. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene (2010) 29:1787–97. doi: 10.1038/onc.2009.468
169. Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. (2013) 123:150–63. doi: 10.1172/JCI64946
170. Li L, Xu QH, Dong YH, Li GX, Yang L, Wang LW, et al. MiR-181a upregulation is associated with epithelial-to-mesenchymal transition (EMT) and multidrug resistance (MDR) of ovarian cancer cells. Eur Rev Med Pharmacol Sci. (2016) 20:2004–10.
171. Chen C, Lou T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget (2017) 8:46691–703. doi: 10.18632/oncotarget.17358
172. Jou J, Diehl AM. Epithelial-mesenchymal transitions and hepatocarcinogenesis. J Clin Invest. (2010) 120:1031–4. doi: 10.1172/JCI42615
173. Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev. (2010) 29:351–78. doi: 10.1007/s10555-010-9225-4
174. Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. (2010) 30:669–82. doi: 10.1111/j.1478-3231.2010.02205.x
175. Lee J-S, Heo J, Libbrecht L, Chu I-S, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. (2006) 12:410–6. doi: 10.1038/nm1377
176. Rao S, Zaidi S, Banerjee J, Jogunoori W, Sebastian R, Mishra B, et al. Transforming growth factor-β in liver cancer stem cells and regeneration. Hepatol Commun. (2017) 1:477–93. doi: 10.1002/hep4.1062
177. Malfettone A, Soukupova J, Bertran E, Crosas-Molist E, Lastra R, Fernando J, et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. (2017) 392:39–50. doi: 10.1016/j.canlet.2017.01.037
178. del Castillo G, Alvarez-Barrientos A, Carmona-Cuenca I, Fernández M, Sánchez A, Fabregat I. Isolation and characterization of a putative liver progenitor population after treatment of fetal rat hepatocytes with TGF-beta. J Cell Physiol. (2008) 215:846–55. doi: 10.1002/jcp.21370
179. Wu K, Ding J, Chen C, Sun W, Ning BF, Wen W., et al. (2012). Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 56, 2255–2267. doi: 10.1002/hep.26007
180. Paik J-H, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell (2007) 128:309–23. doi: 10.1016/j.cell.2006.12.029
181. Mima K, Okabe H, Ishimoto T, Hayashi H, Nakagawa S, Kuroki H, et al. CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. (2012) 72:3414–23. doi: 10.1158/0008-5472.CAN-12-0299
182. Nomura A, Banerjee S, Chugh R, Dudeja V, Yamamoto M, Vickers SM, et al. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget (2015) 6:8313–22. doi: 10.18632/oncotarget.3228
183. Fernando J, Malfettone A, Cepeda EB, Vilarrasa-Blasi R, Bertran E, Raimondi G, et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int J Cancer (2015) 136:E161–172. doi: 10.1002/ijc.29097
184. Hashimoto N, Tsunedomi R, Yoshimura K, Watanabe Y, Hazama S, Oka M. Cancer stem-like sphere cells induced from de-differentiated hepatocellular carcinoma-derived cell lines possess the resistance to anti-cancer drugs. BMC Cancer (2014) 14:722. doi: 10.1186/1471-2407-14-722
185. Rani B, Malfettone A, Dituri F, Soukupova J, Lupo L, Mancarella S, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. (2018) 9:373. doi: 10.1038/s41419-018-0384-5
186. Bitzer M, Horger M, Giannini EG, Ganten TM, Wörns MA, Siveke JT, et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma - The SHELTER study. J Hepatol. (2016) 65:280–8. doi: 10.1016/j.jhep.2016.02.043
187. Soukupova J, Bertran E, Peñuelas-Haro I, Urdiroz-Urricelqui U, Borgman M, Kohlhof H, et al. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget (2017) 8:110367–79. doi: 10.18632/oncotarget.22775
188. Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting transforming growth factor (TGF)-betaRI inhibits activation of beta1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology (2009) 49:839–50. doi: 10.1002/hep.22731
189. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432:332–7. doi: 10.1038/nature03096
190. Eiró N, Vizoso FJ. Importance of tumor/stroma interactions in prognosis of hepatocellular carcinoma. Hepatobiliary Surg Nutr. (2014) 3:98–101. doi: 10.3978/j.issn.2304-3881.2014.02.12
191. Kubo N, Araki K, Kuwano H, Shirabe K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. (2016) 22:6841–50. doi: 10.3748/wjg.v22.i30.6841
192. Lau EYT, Lo J, Cheng BYL, Ma MKF, Lee JMF, Ng JKY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. (2016) 15:1175–89. doi: 10.1016/j.celrep.2016.04.019
193. Liu J, Chen S, Wang W, Ning B-F, Chen F, Shen W, et al. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. (2016) 379:49–59. doi: 10.1016/j.canlet.2016.05.022
194. Kuang D-M, Zhao Q, Peng C, Xu J, Zhang J-P, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. (2009) 206:1327–37. doi: 10.1084/jem.20082173
195. Zhang HH, Mei MH, Fei R, Liu F, Wang JH, Liao WJ, et al. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J Viral Hepat. (2010) 17 (Suppl. 1):34–43. doi: 10.1111/j.1365-2893.2010.01269.x
196. Sprinzl MF, Galle PR. Immune control in hepatocellular carcinoma development and progression: role of stromal cells. Semin Liver Dis. (2014) 34:376–88. doi: 10.1055/s-0034-1394138
197. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. (2009) 86:1065–73. doi: 10.1189/jlb.0609385
198. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8.
199. Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
200. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. (2006) 66:605–12. doi: 10.1158/0008-5472.CAN-05-4005
201. Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today (2012) 42:1–7. doi: 10.1007/s00595-011-0058-8
202. Fan Q-M, Jing Y-Y, Yu G-F, Kou X-R, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. (2014) 352:160–8. doi: 10.1016/j.canlet.2014.05.008
203. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. (2012) 318:154–61. doi: 10.1016/j.canlet.2011.12.020
204. Cheng J-T, Deng Y-N, Yi H-M, Wang G-Y, Fu B-S, Chen W-J, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis (2016) 5:e198. doi: 10.1038/oncsis.2016.7
205. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. (2010) 31:220–7. doi: 10.1016/j.it.2010.04.002
206. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
207. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
208. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
209. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. (2010) 10:554–67. doi: 10.1038/nri2808
210. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
211. Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
212. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
213. Shen Y, Wei Y, Wang Z, Jing Y, He H, Yuan J, et al. TGF-β regulates hepatocellular carcinoma progression by inducing treg cell polarization. CPB (2015) 35:1623–32. doi: 10.1159/000373976
214. Giannelli G, Mazzocca A, Fransvea E, Lahn M, Antonaci S. Inhibiting TGF-β signaling in hepatocellular carcinoma. Biochim Biophys Acta (2011) 1815:214–23. doi: 10.1016/j.bbcan.2010.11.004
215. Zhang S, Sun W-Y, Wu J-J, Wei W. TGF-β signaling pathway as a pharmacological target in liver diseases. Pharmacol. Res. (2014) 85:15–22. doi: 10.1016/j.phrs.2014.05.005
216. Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology (2010) 51:523–34. doi: 10.1002/hep.23285
217. Fabregat I, Fernando J, Mainez J, Sancho P. TGF-beta signaling in cancer treatment. Curr Pharm Des. (2014) 20:2934–47. doi: 10.2174/13816128113199990591
218. Ezquerro IJ, Lasarte JJ, Dotor J, Castilla-Cortázar I, Bustos M, Peñuelas I., et al. A synthetic peptide from transforming growth factor beta type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine (2003) 22:12–20. doi: 10.1016/S1043-4666(03)00101-7
219. Dotor J, López-Vázquez AB, Lasarte JJ, Sarobe P, García-Granero M, Riezu-Boj JI., et al. Identification of peptide inhibitors of transforming growth factor beta 1 using a phage-displayed peptide library. Cytokine (2007) 39:106–15. doi: 10.1016/j.cyto.2007.06.004
220. Llopiz D, Dotor J, Casares N, Bezunartea J, Díaz-Valdés N, Ruiz M, et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int J Cancer (2009) 125:2614–23. doi: 10.1002/ijc.24656
221. Hanafy NAN, Quarta A, Di Corato R, Dini L, Nobile C, Tasco V, et al. Hybrid polymeric-protein nano-carriers (HPPNC) for targeted delivery of TGFβ inhibitors to hepatocellular carcinoma cells. J Mater Sci Mater Med. (2017) 28:120. doi: 10.1007/s10856-017-5930-7
222. Dituri F, Mazzocca A, Fernando J, Peidrò FJ, Papappicco P, Fabregat I., et al. Differential inhibition of the TGF-β signaling pathway in HCC Cells Using the Small Molecule Inhibitor LY2157299 and the D10 Monoclonal Antibody against TGF-β Receptor Type II. PLoS ONE (2013) 8:e67109. doi: 10.1371/annotation/c943a596-3965-4a5b-a27c-55c16685ea32
223. Serova M, Tijeras-Raballand A, Dos Santos C, Albuquerque M, Paradis V, Neuzillet C, et al. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget (2015) 6:21614–27. doi: 10.18632/oncotarget.4308
224. Liang S, Hu J, Xie Y, Zhou Q, Zhu Y, Yang X. A polyethylenimine-modified carboxyl-poly(styrene/acrylamide) copolymer nanosphere for co-delivering of CpG and TGF-β receptor I inhibitor with remarkable additive tumor regression effect against liver cancer in mice. Int J Nanomedicine (2016b) 11:6753–62. doi: 10.2147/IJN.S122047
225. Luangmonkong T, Suriguga S, Bigaeva E, Boersema M, Oosterhuis D, de Jong KP, et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br J Pharmacol. (2017) 174:3107–17. doi: 10.1111/bph.13945
226. Hammad S, Cavalcanti E, Werle J, Caruso ML, Dropmann A, Ignazzi A, et al. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch Toxicol. (2018) 92:2297–309. doi: 10.1007/s00204-018-2231-y
227. Hanafy NAN, Quarta A, Ferraro MM, Dini L, Nobile C, De Giorgi ML, et al. Polymeric Nano-micelles as novel cargo-carriers for LY2157299 liver cancer cells delivery. Int J Mol Sci (2018) 19: E748. doi: 10.3390/ijms19030748
228. Fischer ANM, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene (2007) 26:3395–405. doi: 10.1038/sj.onc.1210121
Keywords: TGF-β, plasticity, liver, cancer biology, fibrosis, HCC, EMT, hepatic stellate cell
Citation: Fabregat I and Caballero-Díaz D (2018) Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 8:357. doi: 10.3389/fonc.2018.00357
Received: 22 June 2018; Accepted: 13 August 2018;
Published: 10 September 2018.
Edited by:
Ubaldo Emilio Martinez-Outschoorn, Thomas Jefferson University, United StatesReviewed by:
Przemyslaw Blyszczuk, Universität Zürich, SwitzerlandAnna Laurenzana, Università degli Studi di Firenze, Italy
Copyright © 2018 Fabregat and Caballero-Díaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Fabregat, aWZhYnJlZ2F0QGlkaWJlbGwuY2F0
Daniel Caballero-Díaz, ZGNhYmFsbGVyb0BpZGliZWxsLmNhdA==