 Georg Gruenbacher
Georg Gruenbacher Martin Thurnher
Martin Thurnher- Immunotherapy Research Unit, Department of Urology, Medical University of Innsbruck, Innsbruck, Austria
Mevalonate metabolism provides cancer and immune cells with diverse products to ensure cell functionality. Similar metabolic reprogramming that raises mevalonate metabolism to higher levels appears to drive both, epithelial mesenchymal transition (EMT) of cancer cells, a reverse differentiation program that generates cancer cells with stem cell properties, and immune cell training for increased responsiveness to secondary stimulation. In this review, we address how mevalonate metabolism supports cancer development and stemness on the one hand, and on the other promotes immune responsiveness. In view of this dual nature of mevalonate metabolism, strategies to manipulate this metabolic pathway as part of anti-cancer therapies require careful analysis of risks versus benefits.
Introduction
The metabolic reprogramming that drives both, the malignant process and immunity, results in increased glycolysis and glutaminolysis, which fuel the mitochondrial citrate cycle to generate ATP (Figure 1). Such a metabolic constellation also facilitates the partial export of mitochondrial citrate into the cytosol. After hydrolysis by ATP citrate lyase (ACLY), acetyl-CoA becomes available for mevalonate generation and metabolism as well as for fatty acid synthesis (lipogenesis), a process that is driven by signaling through AKT and mTOR (mechanistic target of rapamycin). While AKT and mTOR generally promote glycolysis-driven lipogenesis (1), AKT has been reported to specifically control ACLY (2, 3). These lipogenic pathways provide cholesterol and fatty acids for membrane biogenesis and are therefore essential for cancer cell growth and proliferation (4–6). The expression of mevalonate pathway genes is governed by the sterol regulatory element-binding protein (SREBP) transcription factors. SREBPs have been shown to stimulate gene expression in the lipogenic pathways (7), including ACLY and HMGCR (HMG-CoA reductase) (Figure 1).
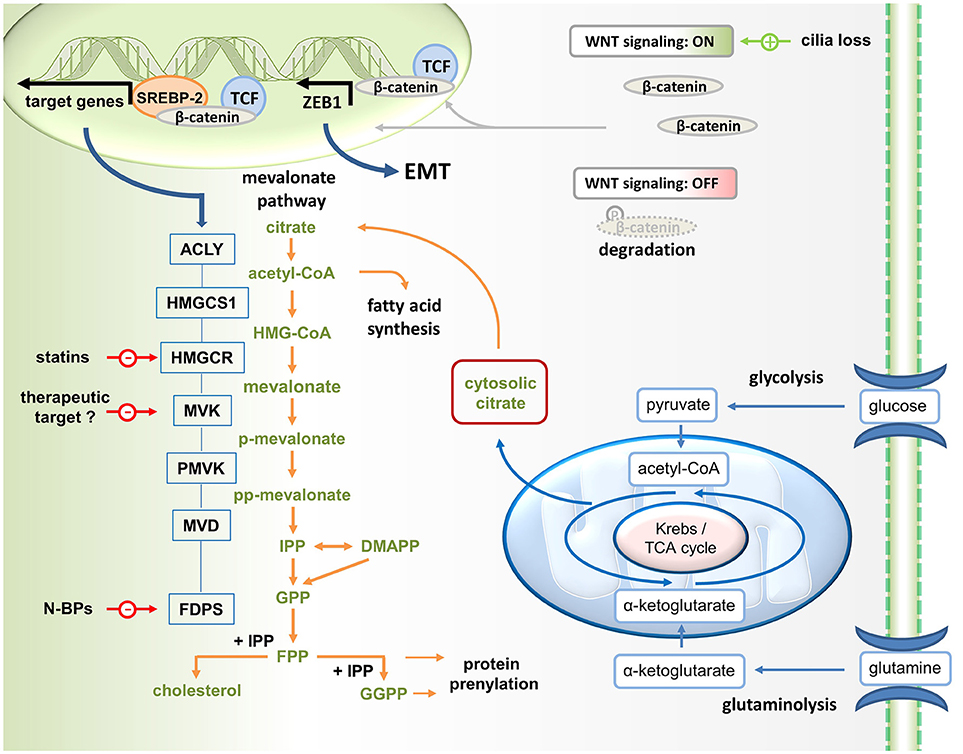
Figure 1. Wnt/ß-catenin driven mevalonate metabolism promotes EMT. Mevalonate metabolism is fueled by glycolysis. Glucose-derived pyruvate enters the TCA cycle. Since some citrate is exported into the cytosol for lipogenesis (i.e., mevalonate metabolism and fatty acid synthesis), glutaminolysis generates α-ketoglutarate to replenish TCA cycle intermediates. Canonical Wnt signaling, which can be activated for instance in response to cilia loss, promotes the translocation of β-catenin to the nucleus, where it acts as coactivator of TCF factors in the transcription of target genes. In addition, ß-catenin may stimulate mevalonate metabolism by forming a complex with SREBP2 on the promoters of mevalonate pathway genes, including HMGCR (HMG-CoA reductase, the target of statins). In addition, β-catenin/TCF4 binds directly to the promoter of ZEB1, a key inducer of EMT, and activates its transcription. N-BPs, nitrogen-containing bisphosphonates.
Mevalonate Metabolism Promotes EMT and Tumorigenesis
Metabolic reprogramming toward increased mevalonate pathway activity may also be associated with EMT (8–11). EMT, which describes the ability of epithelial cells to transdifferentiate into mesenchymal cells, generates cells with stem cell features and migratory capacity (9). In malignant cells, EMT produces cancer stem cells with an invasive phenotype and thus promotes metastasis formation (12).
A phenotypic hallmark of EMT is the loss of E-cadherin expression. EMT-inducing transcription factors such as Snail1, Snail2 (Slug), ZEB1, ZEB2, and TCF3 as well as KLF8 (Kruppel-like factor 8) can directly suppress E-cadherin expression. Growth factor signaling (TGFß, FGF, EGF, HGF) as well as Wnt/ß-catenin signaling, Notch signaling and hypoxia can induce EMT. CD44 expression has also been correlated with EMT, thereby promoting tumor invasion, metastasis, chemoresistance and recurrence (13, 14). As a cell surface protein CD44 interacts with various receptor tyrosine kinases to modulate cellular signaling (15). Oncogenic Ras signaling has been shown to promote CD44 splicing (16) leading to the expression of distinct CD44 isoforms in proliferating cells and tumors. Importantly, a switch in alternative splicing of CD44 has been reported to be essential for EMT and especially CD44v isoforms have been identified as cancer stem cell surface markers (17).
A study of the epithelial membrane lipidome revealed that epithelial cells undergoing EMT increase cholesterol levels (18), indicative of enhanced mevalonate pathway activity. As a consequence, the resulting mesenchymal cells were more sensitive to statin treatment, which also reduced membrane fluidity and migratory capacity as well as metastatic potential (19), confirming the importance of mevalonate metabolism in EMT.
In addition to cholesterol, mevalonate metabolism provides farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP), which are the activated forms of farnesyl and geranylgeranyl serving as substrates of the respective transferases in post-translational protein prenylation (Figure 1). Farnesylation or geranylgeranylation is required for the biological activity of many Ras superfamily members.
The role of the RAS superfamily of GTPases in tumorigenesis is well established (20). Uncontrolled flux through the mevalonate pathway promotes sustained Ras protein activation via constitutive prenylation. The tumor suppressor p53 controls mevalonate metabolism. However, gain-of-function p53 mutations may cause p53-mediated enhancement of mevalonate metabolism and subsequently also of protein prenylation, facilitating malignant transformation (21, 22). Sustained accumulation of mutant p53 mediated by geranylgeranylated RhoA (23) generates a self-perpetuating dynamic that may further promote malignancy.
A recent study by Deng et al. reported that targeted disruption of cilium biogenesis (ciliogenesis) sensitizes normal cells (pancreatic cells (HPDE6C7) and NIH3T3 cells) to oncogene (K-Ras)-driven malignant transformation through activating the mevalonate pathway (24). The authors observed disrupted ciliogenesis in mouse (breast and liver) as well as in human (lung, ovary, and pancreas) cancer cell lines (24). Moreover, cilia loss was detected in a mouse model of a precursor of pancreatic ductal adenocarcinoma. These observations were consistent with previous reports of ciliogenesis defects in human glioma, breast, and kidney cancer (25, 26).
Deng et al. found that cilia loss significantly increased the expression of ACLY, which determines the availability of cytosolic acetyl-CoA, the precursor to mevalonate (Figure 1). In addition, cilia loss promotes expression of several mevalonate pathway genes including HMGCR encoding MHG-CoA reductase, which catalyzes mevalonate formation. Statin-mediated MHG-CoA reductase inhibition could effectively reverse the oncogenic phenotype induced by cilia disruption. Cells with disrupted ciliogenesis appeared to be more dependent on mevalonate metabolism for survival and, thus, were more sensitive to statin treatment. This is in line with another recent study reporting that sensitization to statin treatment is related to H-Ras-induced EMT through activation of ZEB1 (27), which is a key inducer of epithelial transdifferentiation.
In the study by Deng et al., the reprogramming in response to cilia loss included activation of the Wnt–β-catenin signaling pathway (24), which also induces EMT. In the canonical Wnt pathway, stimulation promotes the translocation of β-catenin to the nucleus, where it acts as coactivator of T-cell and lymphoid enhancer (TCF–LEF) factors in the transcriptional activation of target genes (28). The potentially oncogenic Wnt–β-catenin signaling pathway triggers an EMT via ZEB1 (29). Deng et al. reported that several genes encoding mevalonate pathway enzymes, including HMGCR which encodes the pathway-initiating enzyme HMG-CoA reductase, turned out to be direct target genes of β-catenin–TCF signaling (Figure 1). Mechanistically, Wnt–β-catenin signaling appeared to stimulate mevalonate metabolism by complex formation between β-catenin and SREBP2 on the promoters of the genes involved in the mevalonate pathway.
Taken together, these observations suggested that oncogenic signaling, for instance via K-Ras, can suppress ciliogenesis resulting in a cellular reprogramming that is characterized by β-catenin–TCF driven mevalonate metabolism and EMT-like differentiation. In accordance with such a view, deficiency of primary cilia in kidney epithelial cells has recently been shown to induce EMT (30).
Mevalonate Metabolism Promotes Cancer Stemness
The protooncogene MYC is well known to induce metabolic reprogramming, including stimulation of lipogenesis (31). Myc-driven mevalonate metabolism has recently been shown to maintain brain tumor-initiating cells (BTICs) (32). Such stem-like cancer cells share signaling and metabolic preferences with cancer cells upon EMT (10). In BTIC models numerous mevalonate pathway genes were activated. Consistent with the importance of mevalonate metabolism for stemness, mevalonate pathway genes were suppressed when differentiation was induced in BTICs. Likewise, inactivation of HMG-CoA reductase, the mevalonate-generating enzyme that initiates the pathway, either by attenuation of gene expression or by statin-mediated enzyme inhibition reduced self-renewal capacity and tumorigenicity of these cells. In addition, statin treatment also reduced Myc expression in BTICs (32).
Myc is a main driver of stemness in different types of cancer (33). Myc promotes the formation of tumor spheres (34), which are considered a surrogate marker of self-renewal capacity (35). Sphere formation initially requires detachment from a tissue and its extracellular matrix (ECM), which is mediated by the downregulation of genes encoding distinct integrins involved in cell-to-cell contact as well as by the upregulation of genes encoding matrix metallopetidases, thus promoting ECM degradation. This process of spinning off and establishing an independent spherical colony through anchorage-independent growth is reminiscent of the EMT-driven initiation of metastasis. Importantly, Myc-driven sphere formation has been shown to be associated with increased expression of sterol regulatory element binding factors and transcriptional activation of cholesterol synthesis in the mevalonate pathway (34). Collectively, these findings indicate that mevalonate metabolism drives EMT-induced stemness.
Mevalonate Metabolism Enhances Innate Immunity
A metabolic reprogramming toward enhanced flux through the mevalonate pathway similar to that observed in cancer cells and cancer stem cells has been observed in innate immune cells, which as a consequence exhibit increased responsiveness. As a metabolic pathway that supports cell growth and proliferation, mevalonate metabolism is already required during macrophage development. M-CSF, a main driver of myelopoiesis, promotes glycolysis, which in turn stimulates Myc-driven activation of lipogenic pathways (36). Such M-CSF induced metabolic reprogramming included the transcription of multiple genes involved in mevalonate generation and in cholesterol biosynthesis. Conversely, attenuation of mevalonate metabolism impaired myelopoiesis (36).
In addition to its role in macrophage development, mevalonate metabolism is crucial for the effector function of M1 macrophages, which exhibit strong tumoricidal activity in response to classical activation by the Th1 cytokine interferon-γ (IFN-γ) plus lipopolysaccharide (LPS) (37). In M1 macrophages, GM-CSF increases glycolytic capacity. GM-CSF thus primes M1 macrophages for a particularly strong glycolytic response upon LPS stimulation, also resulting in significantly higher levels of pro-inflammatory cytokines. Since glycolysis fuels mevalonate metabolism, it was not surprising that GM-CSF primed macrophages also contained higher levels of acetyl-CoA, the precursor to mevalonate, and displayed increased expression of HMG-CoA reductase, the mevalonate-generating enzyme, which is inhibited by statins. The ability of statins to inhibit cytokine responses of M1 macrophages and to prevent the priming effect of GM-CSF highlights the role of mevalonate metabolism in M1 macrophage function.
Both, M-CSF and GM-CSF have also been implicated in the induction of EMT. For instance, M-CSF can promote EMT of lung cancer cells via canonical Wnt signaling (38). An EMT-inducing potential of GM-CSF has been reported in colon cancer. GM-CSF-trained cancer cells displayed mesenchymal features, including motility and chemoresistance, and ZEB1 as well as MAPK/ERK signaling were identified as critical factors of GM-CSF-driven EMT (39).
Although memory has long been considered a specific feature of adaptive immune cells, i.e. B and T cells, recent work has demonstrated that innate immune cells can also develop a form of memory after microbial stimulation during infections or vaccinations (40). Similar to antigen priming of lymphocytes, the repetitive stimulation of monocytes with Bacillus Calmette–Guérin (BCG) or β-glucan via toll-like receptors facilitated the increased production of inflammatory cytokines and reactive oxygen intermediates in response to subsequent challenges. Akt–mTOR-driven metabolic reprogramming toward enhanced flux through the mevalonate pathway was identified as the underlying mechanism that drives improved innate immunity.
Although initially developed as a tuberculosis vaccine, BCG treatment is also among the most effective cancer immunotherapies, and in high-risk, non-muscle-invasive bladder cancer, the repetitive intravesical instillation of BCG is still the standard adjuvant treatment according to the European Association of Urology (EAU) guidelines (41, 42). The relevance of mevalonate metabolism in BCG-induced immune training was demonstrated when statins were shown to impair both, the memory of monocytes in the in vitro model (40) and the clinical outcome of BCG treatment for bladder cancer (43).
A recent study performed in mice but with important clinical implications demonstrated that access of BCG to the bone marrow enhances myelopoiesis and educates hematopoietic stem cells to generate trained monocytes, which are highly protective against M. tuberculosis infection (44). Likewise, administration of ß-glucan to mice was shown to also enhance myelopoiesis in the bone marrow via local production of IL-1ß and GM-CSF (45). In expanding progenitors, several mevalonate pathway genes were significantly upregulated, including HMGCR, encoding HMG-CoA reductase. Intriguingly, ß-glucan-enhanced myelopoiesis also turned out to be protective to chemotherapy-induced myeloablation.
Statins are HMG-CoA reductase inhibitors and thus block the production of mevalonate suggesting that lack of the precursor prevents the generation of immunity-promoting products such as cholesterol or the protein prenylation substrates FPP and GGPP. However, mevalonate itself was recently identified as a key mediator of innate immune training. Mevalonate was found to improve the functionality of the insulin-like growth factor 1 (IGF1) receptor, leading to the activation of mTOR and subsequent histone modifications in inflammatory pathways (46). Importantly, IGF1 receptor signaling also induces EMT and contributes to metastasis and drug resistance in different types of tumors (47), indicating parallels between EMT and trained innate immunity.
Once mevalonate is formed by HMG-CoA reductase, the pathway continues by mevalonate phosphorylation through mevalonate kinase (MVK). In humans, MVK mutation may strongly attenuate enzyme activity, leading to hyper IgD syndrome (HIDS). This block in mevalonate metabolism is well known to result in sterile inflammation including periodic fever attacks. A novel aspect, however, is that accumulation of mevalonate induces a trained immunity phenotype in monocytes from these patients via improved IGF1 receptor functionality similar to BCG or ß-glucan-induced immune training (46). Transcriptional profiling of trained monocytes from HIDS patients revealed upregulation of the EMT-inducing transcription factor ZEB1 and of the EMT-associated cancer stem cell promoting chemokine IL8 (46, 48, 49).
Another phenotypic marker of EMT is the increased expression of the neural cell adhesion molecule (NCAM) (8, 50). NCAM is expressed by neurons and glia as well as skeletal muscle cells. The same protein, however, is also expressed in the hematopoietic system, where it is referred to as CD56. Among blood cells, CD56 is mainly associated with NK cells (51), subsets of γδ T cells (52) and CD8+ T cells (53) as well as on a subset of dendritic cells (52, 54, 55). Intriguingly, in all cases increased CD56 (NCAM) expression was associated with enhanced cellular functionality.
Collectively, these observations suggest that an EMT-like reprogramming toward enhanced flux through the mevalonate pathway similar to that observed in cancer cells may also promote innate immunity.
Enhanced Mevalonate Metabolism in Cancer Cells Alerts γδ T Cells
As a growth and proliferation-promoting pathway mevalonate metabolism also contributes to tumorigenesis (21, 22) and immune surveillance of this potentially oncogenic pathway is therefore desirable. A subset of human γδ T cells, which are innate-like T cells with strong antitumor activity, is capable of recognizing and killing cancer cells with a hyperactive mevalonate pathway (56). Vγ9Vδ2 T cells, which represent the main γδ T cell population in human peripheral blood, recognize small isoprenoid phosphoantigens (57, 58). The mevalonate pathway intermediate isopentenyl diphosphate (IPP) is a strong agonist of γδ T cells expressing the Vγ9Vδ2 T cell receptor and IPP-activated Vγ9Vδ2 T cells can proliferate, produce effector cytokines such as interferon (IFN)-γ and display natural killer cell-like cytotoxicity, especially in the presence of interleukin-2 (IL-2). Nitrogen-containing bisphosphonates (N-BPs) are another class of mevalonate pathway manipulating drugs, which inhibit FPP synthase (Figure 1). Although N-BPs have been developed for the treatment of osteoporosis, N-BPs also have a long history in cancer treatment (59). N-BPs may exhibit direct antitumor effects but may also generate therapeutic effects through immune activation. Treatment of cancer cells or antigen-presenting cells with N-BPs causes IPP accumulation in these cells (60). As a consequence these cells acquire the capacity to activate Vγ9Vδ2 T cells. In addition to IPP accumulation, N-BP-mediated FPP synthase inhibition also causes downstream depletion of FPP and GGPP, resulting in an inflammatory response that depends on caspase-1 activated IL-1ß and IL-18 (54, 55). These cytokines can costimulate the IPP-driven activation of Vγ9Vδ2 T cells (52) and may facilitate concomitant NK cell activation (55). Similar to N-BP treatment, ectopic expression of HMGCR (Figure 1) in cancer cells increases the flux through the mevalonate pathway and also renders these cells targets of Vγ9Vδ2 T cells (60). Collectively, these and other findings indicate that Vγ9Vδ2 T cells participate in immune surveillance of the potentially oncogenic mevalonate pathway.
Mevalonate Metabolism May Also Enhance Adaptive Immunity
The metabolic reprogramming toward increased glycolysis during T cell activation is controlled by Myc (61) and ensures the availability of building blocks required for cell growth, proliferation and effector function. Myc-driven glycolysis fuels lipogenesis. At the transcriptional level, T cell receptor triggering results in the SREBP2-dependent activation of genes encoding enzymes involved in mevalonate generation and metabolism (7). In contrast to activated T cells, which preferentially engage glycolysis, memory T cells appear to rely on fatty acid oxidation (FAO) (62). As resting cells, memory T cells use FAO-derived ATP for survival. The role of the mevalonate pathway in this particular metabolic constellation is unclear. Fatty acid breakdown generates acetyl-CoA, which can be oxidized in the mitochondria or serve as a substrate in protein acetylation and, in principle, also as a precursor in mevalonate synthesis. Further work is needed to clarify the role of mevalonate metabolism in memory T cells.
The transcription factor GATA-3 has been shown to control Myc expression and to be critical for peripheral T cell maintenance through IL-7 signaling (63). The peripheral maintenance of GATA-3 deficient CD8 T cells was impaired with reduced IL-7 receptor (IL-7R) expression. These findings indicated that GATA-3 is required for IL-7R expression in CD62LhiCD44hi central memory CD8 T cells. Both, CD44 and IL-7R (CD127) have been described as markers of EMT (13, 64). Moreover, CD44 expression has been reported to function as a sensitive reporter of tonic mTOR-S6 kinase signaling (65), a signaling pathway well known to promote mevalonate metabolism (66). GATA-3 robustly upregulates Myc (63), which drives the metabolic reprogramming associated with EMT. Canonical Wnt signaling, which promotes EMT and mevalonate metabolism, also enhances GATA-3 expression (67), altogether suggesting that mevalonate metabolism may also play a role in memory T cells.
Concluding Remarks
It is meanwhile well accepted that glycolysis-driven mevalonate metabolism governs the functions of activated cells, including cancer and immune cells. To target one population without impairing the other represents a great therapeutic challenge. Among mevalonate pathway inhibitors (20), statins inhibit the pathway early on and actually prevent mevalonate generation. The increase of mevalonate pathway activity that occurs during EMT sensitizes cancer cells to the antitumor effects of statins (19). While statins may thus have direct antitumor effects, also by reducing cancer stemness (32), they will concomitantly also suppress innate immunity and, in particular, prevent the phenomenon of trained immunity in monocytes that critically depends on mevalonate (46). In addition, statins preclude accumulation of IPP and, as a consequence, they prevent Vγ9Vδ2 T cells from exercising their cancer surveillance function (60). The recent observation that HIDS patients, which lack MVK, have a trained immunity phenotype (46), suggests that MVK might be an attractive target. Pharmacological inhibition of MVK causes mevalonate accumulation and thus immune training of monocytes. At the same time, MVK inhibition would deplete downstream metabolites such as FPP and GGPP in cancer cells. This effect will attenuate protein prenylation of multiple Ras family members and thus antagonize cancer stemness. However, just like the statins, MVK inhibition would prevent of IPP accumulation and γδ T cell activation. Still left to be evaluated are the N-BPs, which inhibit protein prenylation (20), generate immune training effects (54) and expand Vγ9Vδ2 T cells (68). Appropriate administration and combination, for instance with cytokines, may therefore lead to regimen that exploit the full potential of N-BPs.
Author Contributions
MT gathered information and wrote the first draft of the manuscript. GG participated in writing and editing of the final manuscript and prepared the figure. Both authors read and approved the submitted version.
Funding
This work was supported by the Austrian Science Fund (FWF; P 28923-B28).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors MT and GG.
References
1. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
2. Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, et al. ATP citrate lyase (ACLY): a promising target for cancer prevention and treatment. Curr Drug Targets (2015) 16:156–63. doi: 10.2174/1389450115666141224125117
3. Osinalde N, Mitxelena J, Sanchez-Quiles V, Akimov V, Aloria K, Arizmendi JM, et al. Nuclear Phosphoproteomic Screen Uncovers ACLY as Mediator of IL-2-induced Proliferation of CD4+ T lymphocytes. Mol Cell Proteomics (2016) 15:2076–92. doi: 10.1074/mcp.M115.057158
4. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer (2016) 16:718–31. doi: 10.1038/nrc.2016.76
5. Gruenbacher G, Thurnher M. Mevalonate Metabolism in Immuno-Oncology. Front Immunol. (2017) 8:1714. doi: 10.3389/fimmu.2017.01714
6. Gruenbacher G, Thurnher M. Mevalonate metabolism governs cancer immune surveillance. Oncoimmunology (2017) 6:e1342917. doi: 10.1080/2162402X.2017.1342917
7. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell (2008) 134:97–111. doi: 10.1016/j.cell.2008.04.052
8. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
9. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
10. Morandi A, Taddei ML, Chiarugi P, Giannoni E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front Oncol. (2017) 7:40. doi: 10.3389/fonc.2017.00040
11. Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. (2017) 284:3132–44. doi: 10.1111/febs.14090
12. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer (2005) 5:744–9. doi: 10.1038/nrc1694
13. Deng J, Wang L, Chen H, Hao J, Ni J, Chang L, et al. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget (2016) 7:55771–88. doi: 10.18632/oncotarget.9908
14. Dhar D, Antonucci L, Nakagawa H, Kim JY, Glitzner E, Caruso S, et al. Liver cancer initiation requires p53 inhibition by CD44-enhanced growth factor signaling. Cancer Cell (2018) 33:1061–77 e6. doi: 10.1016/j.ccell.2018.05.003
15. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. (2003) 4:33–45. doi: 10.1038/nrm1004
16. Cheng C, Yaffe MB, Sharp PA. A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. (2006) 20:1715–20. doi: 10.1101/gad.1430906
17. Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. (2011) 121:1064–74. doi: 10.1172/JCI44540
18. Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci USA. (2011) 108:1903–7. doi: 10.1073/pnas.1019267108
19. Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep. (2014) 4:7593. doi: 10.1038/srep07593
20. Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. (2007) 6:541–55. doi: 10.1038/nrd2221
21. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA. (2010) 107:15051–6. doi: 10.1073/pnas.0910258107
22. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. doi: 10.1016/j.cell.2011.12.017
23. Ingallina E, Sorrentino G, Bertolio R, Lisek K, Zannini A, Azzolin L, et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat cell biol. (2018) 20:28–35. doi: 10.1038/s41556-017-0009-8
24. Deng YZ, Cai Z, Shi S, Jiang H, Shang YR, Ma N, et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J Exp Med. (2018) 215:177–95. doi: 10.1084/jem.20170399
25. Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. (2009) 15:1062–5. doi: 10.1038/nm.2020
26. Seeger-Nukpezah T, Little JL, Serzhanova V, Golemis EA. Cilia and cilia-associated proteins in cancer. Drug Discov Today Dis Mech. (2013) 10:e135–e42. doi: 10.1016/j.ddmec.2013.03.004
27. Yu R, Longo J, van Leeuwen JE, Mullen PJ, Ba-Alawi W, Haibe-Kains B, et al. Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Cancer Res. (2018) 78:1347–57. doi: 10.1158/0008-5472.CAN-17-1231
28. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell (2006) 127:469–80. doi: 10.1016/j.cell.2006.10.018
29. Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA. (2011) 108:19204–9. doi: 10.1073/pnas.1108977108
30. Han SJ, Jung JK, Im SS, Lee SR, Jang BC, Park KM, et al. Deficiency of primary cilia in kidney epithelial cells induces epithelial to mesenchymal transition. Biochem Biophys Res Commun. (2018) 496:450–4. doi: 10.1016/j.bbrc.2018.01.079
31. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. (2013) 3:a014217. doi: 10.1101/cshperspect.a014217
32. Wang X, Huang Z, Wu Q, Prager BC, Mack SC, Yang K, et al. MYC-Regulated mevalonate metabolism maintains brain tumor-initiating cells. Cancer Res. (2017) 77:4947–60. doi: 10.1158/0008-5472.CAN-17-0114
33. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. (2014) 4:a014241. doi: 10.1101/cshperspect.a014241
34. Liu M, Xia Y, Ding J, Ye B, Zhao E, Choi JH, et al. Transcriptional profiling reveals a common metabolic program in high-risk human neuroblastoma and mouse neuroblastoma sphere-forming cells. Cell Rep. (2016) 17:609–23. doi: 10.1016/j.celrep.2016.09.021
35. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. (2003) 17:1253–70. doi: 10.1101/gad.1061803
36. Karmaus PWF, Herrada AA, Guy C, Neale G, Dhungana Y, Long L, et al. Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. J Exp Med. (2017) 214:2629–47. doi: 10.1084/jem.20161855
37. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
38. Yu YX, Wu HJ, Tan BX, Qiu C, Liu HZ. CSF-1R regulates non-small cell lung cancer cells dissemination through Wnt3a signaling. Am J Cancer Res. (2017) 7:2144–56.
39. Chen Y, Zhao Z, Chen Y, Lv Z, Ding X, Wang R, et al. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci Rep. (2017) 7:8265. doi: 10.1038/s41598-017-08047-1
40. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
41. Babjuk M, Burger M, Zigeuner R, Shariat SF, van Rhijn BW, Comperat E, et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2013. Eur Urol. (2013) 64:639–53. doi: 10.1016/j.eururo.2013.06.003
42. Pichler R, Gruenbacher G, Culig Z, Brunner A, Fuchs D, Fritz J, et al. Intratumoral Th2 predisposition combines with an increased Th1 functional phenotype in clinical response to intravesical BCG in bladder cancer. Cancer Immunol Immunother. (2017) 66:427–40. doi: 10.1007/s00262-016-1945-z
43. Hoffmann P, Roumeguere T, Schulman C, van Velthoven R. Use of statins and outcome of BCG treatment for bladder cancer. N Engl J Med. (2006) 355:2705–7. doi: 10.1056/NEJMc062714
44. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG Educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell (2018) 172:176–90 e19. doi: 10.1016/j.cell.2017.12.031
45. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell (2018) 172:147–61 e12. doi: 10.1016/j.cell.2017.11.034
46. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell (2018) 172:135-46 e9. doi: 10.1016/j.cell.2017.11.025
47. Li H, Batth IS, Qu X, Xu L, Song N, Wang R, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer (2017) 16:6. doi: 10.1186/s12943-016-0576-5
48. Dominguez C, David JM, Palena C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin Cancer Biol. (2017) 47:177–84. doi: 10.1016/j.semcancer.2017.08.002
49. Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. (2011) 71:5296–306. doi: 10.1158/0008-5472.CAN-11-0156
50. Lehembre F, Yilmaz M, Wicki A, Schomber T, Strittmatter K, Ziegler D, et al. NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J. (2008) 27:2603–15. doi: 10.1038/emboj.2008.178
51. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
52. Gruenbacher G, Gander H, Rahm A, Nussbaumer W, Romani N, Thurnher M. CD56+ human blood dendritic cells effectively promote TH1-type gammadelta T-cell responses. Blood (2009) 114:4422–31. doi: 10.1182/blood-2009-06-227256
53. Pittet MJ, Speiser DE, Valmori D, Cerottini JC, Romero P. Cutting edge: cytolytic effector function in human circulating CD8+ T cells closely correlates with CD56 surface expression. J Immunol. (2000) 164:1148–52. doi: 10.4049/jimmunol.164.3.1148
54. Nussbaumer O, Gruenbacher G, Gander H, Thurnher M. DC-like cell-dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by gammadelta T lymphocytes. Blood (2011) 118:2743–51. doi: 10.1182/blood-2011-01-328526
55. Gruenbacher G, Gander H, Nussbaumer O, Nussbaumer W, Rahm A, Thurnher M. IL-2 costimulation enables statin-mediated activation of human NK cells, preferentially through a mechanism involving CD56+ dendritic cells. Cancer Res. (2010) 70:9611–20. doi: 10.1158/0008-5472.CAN-10-1968
56. Simoes AE, Di Lorenzo B, Silva-Santos B. Molecular Determinants of Target Cell Recognition by Human gammadelta T Cells. Front Immunol. (2018) 9:929. doi: 10.3389/fimmu.2018.00929
57. Gruenbacher G, Nussbaumer O, Gander H, Steiner B, Leonhartsberger N, Thurnher M. Stress-related and homeostatic cytokines regulate Vgamma9Vdelta2 T-cell surveillance of mevalonate metabolism. Oncoimmunology (2014) 3:e953410. doi: 10.4161/21624011.2014.953410
58. Gruenbacher G, Gander H, Rahm A, Idzko M, Nussbaumer O, Thurnher M. Ecto-ATPase CD39 inactivates isoprenoid-derived Vgamma9Vdelta2 T cell phosphoantigens. Cell Rep. (2016) 16:444–56. doi: 10.1016/j.celrep.2016.06.009
59. Berenson JR, Hillner BE, Kyle RA, Anderson K, Lipton A, Yee GC, et al. American Society of Clinical Oncology clinical practice guidelines: the roleof bisphosphonates in multiple myeloma. J Clin Oncol. (2002) 20:3719–36. doi: 10.1200/JCO.2002.06.037
60. Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. (2003) 197:163–8. doi: 10.1084/jem.20021500
61. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
62. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
63. Wang Y, Misumi I, Gu AD, Curtis TA, Su L, Whitmire JK, et al. GATA-3 controls the maintenance and proliferation of T cells downstream of TCR and cytokine signaling. Nat Immunol. (2013) 14:714–22. doi: 10.1038/ni.2623
64. Qu H, Zou Z, Pan Z, Zhang T, Deng N, Chen G, et al. IL-7/IL-7 receptor axis stimulates prostate cancer cell invasion and migration via AKT/NF-kappaB pathway. Int Immunopharmacol. (2016) 40:203–10. doi: 10.1016/j.intimp.2016.08.017
65. Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios(+) T cells and autoantibodies. Elife (2013) 2:e01020. doi: 10.7554/eLife.01020
66. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. (2012) 12:325–38. doi: 10.1038/nri3198
67. Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. (2009) 10:992–9. doi: 10.1038/ni.1762
Keywords: epithelial mesenchymal transition (EMT), mevalonate metabolism, stemness, trained immunity, Wnt signaling
Citation: Gruenbacher G and Thurnher M (2018) Mevalonate Metabolism in Cancer Stemness and Trained Immunity. Front. Oncol. 8:394. doi: 10.3389/fonc.2018.00394
Received: 09 July 2018; Accepted: 31 August 2018;
Published: 20 September 2018.
Edited by:
Andreas Pircher, Innsbruck Medical University, AustriaReviewed by:
Anna Rita Cantelmo, Université Lille Nord de France, FranceSimona Pisanti, Università degli Studi di Salerno, Italy
Copyright © 2018 Gruenbacher and Thurnher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Thurnher, bWFydGluLnRodXJuaGVyQGktbWVkLmFjLmF0