 Emily E. Lederman1
Emily E. Lederman1 Michael R. King
Michael R. King- 1Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, United States
- 2Department of Biomedical Engineering, Vanderbilt University, Nashville, TN, United States
Background: Metastasis through the bloodstream contributes to poor prognosis in many types of cancer. A unique approach to target and kill colon, prostate, and other epithelial-type cancer cells in the blood has been recently developed that uses circulating leukocytes to present the cancer-specific, liposome-bound Tumor Necrosis Factor (TNF)-related apoptosis inducing ligand (TRAIL) on their surface along with E − selectin adhesion receptors. This approach, demonstrated both in vitro with human blood and in mice, mimics the cytotoxic activity of natural killer cells. The resulting liposomal TRAIL-coated leukocytes hold promise as an effective means to neutralize circulating tumor cells that enter the bloodstream with the potential to form new metastases.
Methods: The computational biology study reported here examines the mechanism of this effective signal delivery, by considering the kinetics of the coupled reaction cascade, from TRAIL binding death receptor to eventual apoptosis. In this study, a collision of bound TRAIL with circulating tumor cells (CTCs) is considered and compared to a prolonged exposure of CTCs to soluble TRAIL. An existing computational model of soluble TRAIL treatment was modified to represent the kinetics from a diffusion-limited 3D reference frame into a 2D collision frame with advection and adhesion to mimic the E − selectin and membrane bound TRAIL treatment. Thus, the current model recreates the new approach of targeting cancer cells within the blood. The model was found to faithfully reproduce representative observations from experiments of liposomal TRAIL treatment under shear.
Results: The model predicts apoptosis of CTCs within 2 h when treated with membrane bound TRAIL, while apoptosis in CTCs treated with soluble TRAIL proceeds much more slowly over the course of 10 h, consistent with previous experiments. Given the clearance rate of soluble TRAIL in vivo, this model predicts that the soluble TRAIL method would be rendered ineffective, as found in previous experiments.
Conclusion: This study therefore indicates that the kinetics of the coupled reaction cascade of liposomal E − selectin and membrane bound TRAIL colliding with CTCs can explain why this new approach to target and kill cancer cells in blood is much more effective than its soluble counterpart.
Background
Cancer metastasis accounts for more than 90% of cancer-related deaths (1). In many types of cancer, circulating tumor cells are shed from the primary tumor site into peripheral circulation where they then can extravasate into extravascular space to form metastatic tumors (2–4). Recent studies have shown that CTCs from primary tumors express sialyated carbohydrate ligands which interact with selectins on the surface of the endothelium (5, 6). These selectins can begin to tether to the sialyated carbohydrate ligands, in a fashion similar to leukocyte interaction with endothelium. These rapid force-dependent binding interactions can trigger rolling adhesion and eventually firm adhesion to the endothelium, facilitating survival and formation of micrometastases (7–9). Surgery and radiation, while proven effective in treating primary tumors, pose challenges due to the limited detectability of distant micrometastases.
New methods to target CTCs have been developed in vivo and hold promise in reducing the metastatic load and the formation of new tumors. One recent technology uses leukocytes as a drug delivery mechanism. Leukocytes and CTCs are similar in size and rigidity, causing both to migrate to the near wall region of blood vessels. For every CTC, there are ~1 × 106 leukocytes circulating, which effectively surround the CTC, making leukocytes an attractive carrier for cancer drug delivery (10–13).
It has been shown that functionalizing leukocytes with liposomes decorated with TRAIL (memTRAIL) and E − selectin (ES), an adhesion molecule, is an effective way of treating circulating cancer cells in flowing human blood in vitro, and in the peripheral circulation of mice in vivo (12, 14). This method of treatment is more effective than soluble TRAIL (sTRAIL); however, the mechanism of this enhanced apoptosis response has not yet been fully elucidated. Beyond concentrating memTRAIL in the close vicinity of CTCs, there are two other key reasons why this method of treatment is believed to be so effective. First, the shearing caused by blood can help to promote the collision of memTRAIL with CTC, effectively increasing the on-rate of binding. Previous studies have shown that increased shear has a direct correlation with the sensitivity of cancer cells to TRAIL (15). Secondly, it is possible that E − selectin briefly tethers the liposome to the CTC after collision, effectively reducing the slip velocity after collision and lowering the off-rate of TRAIL binding DRs.
Several models have been built to gain a better quantitative understanding of the reaction cascade pathway that takes place when tumor cells exposed to sTRAIL undergo apoptosis, but previous models have not considered the memTRAIL interacting with CTCs in a tethered 2-D frame of reference. Some important considerations which must be captured in such a model are 2-D binding reaction kinetics, the effects of a slip velocity, and the effects of cell adhesion. This, in turn, will better represent the case of leukocytes functioned with memTRAIL, in a shearing blood flow, with E − selectin temporarily tethering CTCs to the treated leukocytes. Our model builds off and significantly extends Albeck et al.'s model which captures the coupled reactions of TRAIL-induced apoptosis by modeling the downstream reaction pathways initiated by TRAIL's binding to death receptors 4 and 5, through the use of numerically integrating reaction rate laws via MATLAB's ordinary differential equation (ODE) solver (16). During apoptosis, the potent effector caspase 3 (C3) is activated by extracellular stimuli such as TRAIL. C3 degrades the proteome and activates DNAses, which dismantle chromosomes of cells committed to die (17). Caspase activation represents an irreversible change in cell fate regulated by the assembly of complexes on death receptors, binding of pro- and anti-apoptotic members of the Bcl-2 family to each other in cytosolic and mitochondrial compartments, mitochondria-to-cytosol translocation of Smac and cytochrome c (CyC), and the direct repression of caspases by inhibitor apoptosis proteins (IAPs) (18–24). In the ODE-based model of C3 regulation, the mass action kinetics of a typical CTC undergoing apoptosis are captured to better understand this memTRAIL model by examining the interplay of each reagent's concentration within the reaction cascade as a function of time. From this, new insights are revealed to explain why the sheared memTRAIL model is notably more effective in inducing apoptosis in CTCs.
Results
Specific Reactant Concentration Profiles
Specific values of TRAIL, DR, k+ and k− were used for the following four cases of TRAIL binding to DRs: sTRAIL, memTRAIL without shear, memTRAIL with shear but without adhesion, and memTRAIL with both shear and adhesion (Table 1). These four cases were chosen to mimic experiments carried out by Mitchell et al., which showed that TRAIL was most potent when TRAIL and E − selectin were tethered to the surface of a liposome, and sheared during treatment of tumor cells (14). It has been suggested that E − selectin helps promote the binding of TRAIL and DR by causing the liposomes to adhere to CTCs (14). This effect of adhesion on memTRAIL binding DR was included in the model.
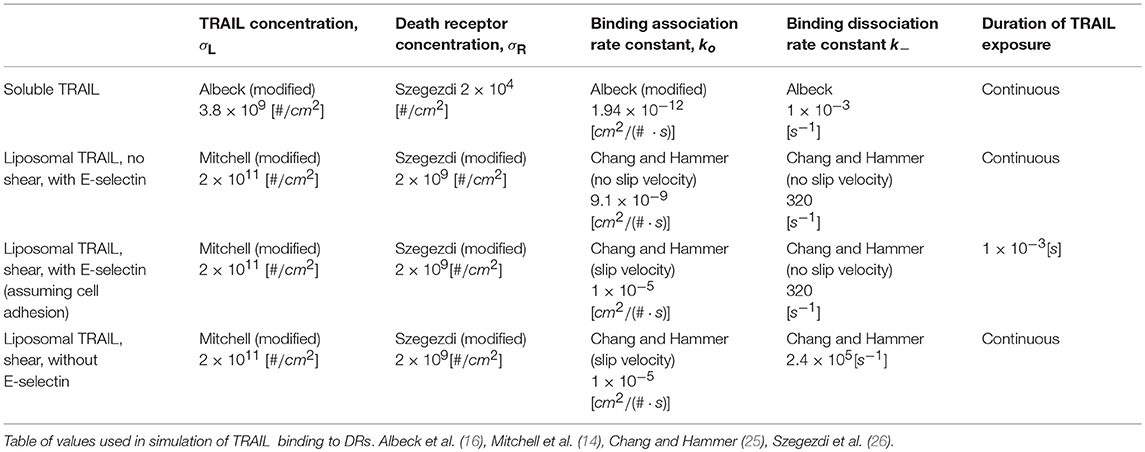
Table 1. lists the parameter values used in the different simulation conditions.
cPARP Instantaneous Concentrations
Each concentration profile was normalized with its maximum concentration, to better examine the relative time progression of the reaction pathway rather than the relative concentrations within the reaction pathway. When focusing on cleavage of PARP, memTRAIL under shear with adhesion induced apoptosis the fastest of all of the treatment methods, at Td = 1.8 h (Figure 1). This value is very close to that found experimentally by Mitchel et al, where there was a 98% reduction in reported CTCs, after allowing the ES/TRAIL treatment to circulate in mice for 2.5 h, when compared with mice treated with ES alone (14). Following that, the second fastest treatment method was memTRAIL without adhesion in shear at Td = 3.5 h. memTRAIL without shear and sTRAIL had much longer times to apoptosis at Td = 12.5 h and Td = 10.5 h, respectively.
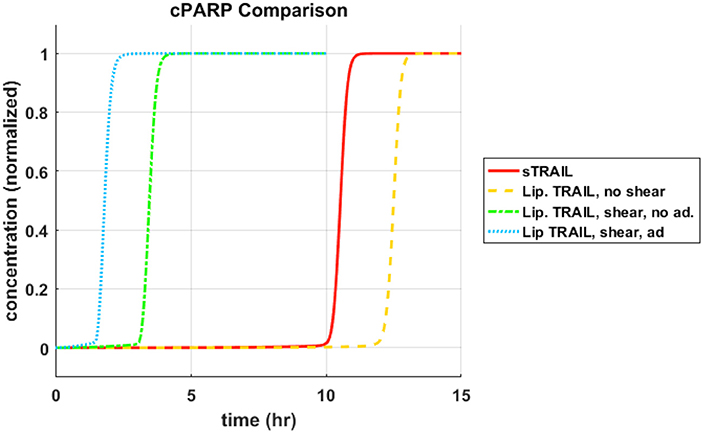
Figure 1. cPARP concentration profile as function of time for sTRAIL and memTRAIL with shear and adhesion.
To reveal the underlying mechanism at work here, the pathway was divided into two sections, pre-mitochondrial pathway and post-mitochondrial pathway. Within these two sections, the proteins participating in the specific reactions, or the essential reactants' concentrations were plotted as a function of time to identify why sheared memTRAIL delivery is a more efficient method of drug delivery both in simulations and experimentally.
Pre-mitochondrial Species Concentration Profiles
It was observed that the pre-mitochondrial pathway transition (marked by a sudden graded response in the reactant concentrations) coincided with the cleavage of PARP for higher values of Td (Figures 2A–C); however, this happened before the transition of cPARP for smaller values of Td (Figure 2D). It was also observed that an initial change in reagent concentration was present for the memTRAIL case with shear and adhesion (Figure 2D) but less prominent for the other treatment methods. These observations suggest that the pre-mitochondrial pathway is activated much faster for memTRAIL with adhesion and shear than for the other treatment methods.
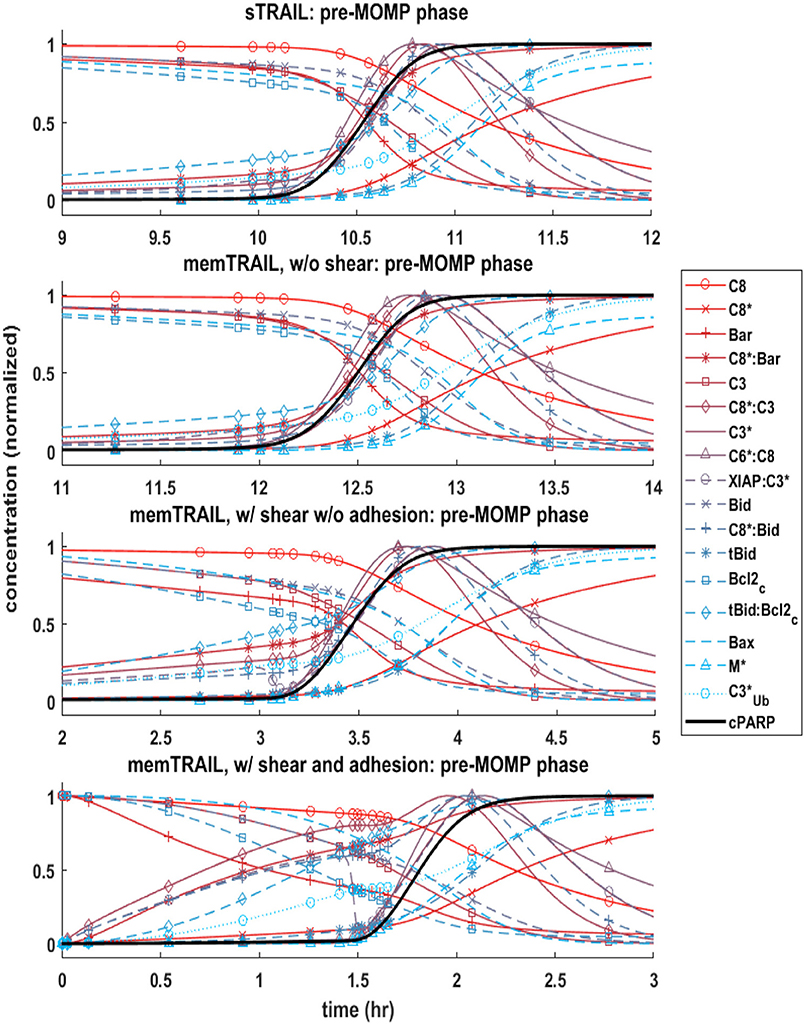
Figure 2. Concentration profiles of cPARP and other important proteins/protein complexes as a function of time for 4 different cases of TRAIL binding to DRs.
Post-mitochondrial Species Concentration Profiles
Next, the post mitochondrial pathway, which transitions during and after the permeabilization of the mitochondrial outer membrane, was considered. It was observed that the reagents underwent a sharper transition as Td decreased for different treatment methods, indicating that this pathway is less inhibited by upstream reagents for memTRAIL delivery with shear and adhesion (Figure 3).
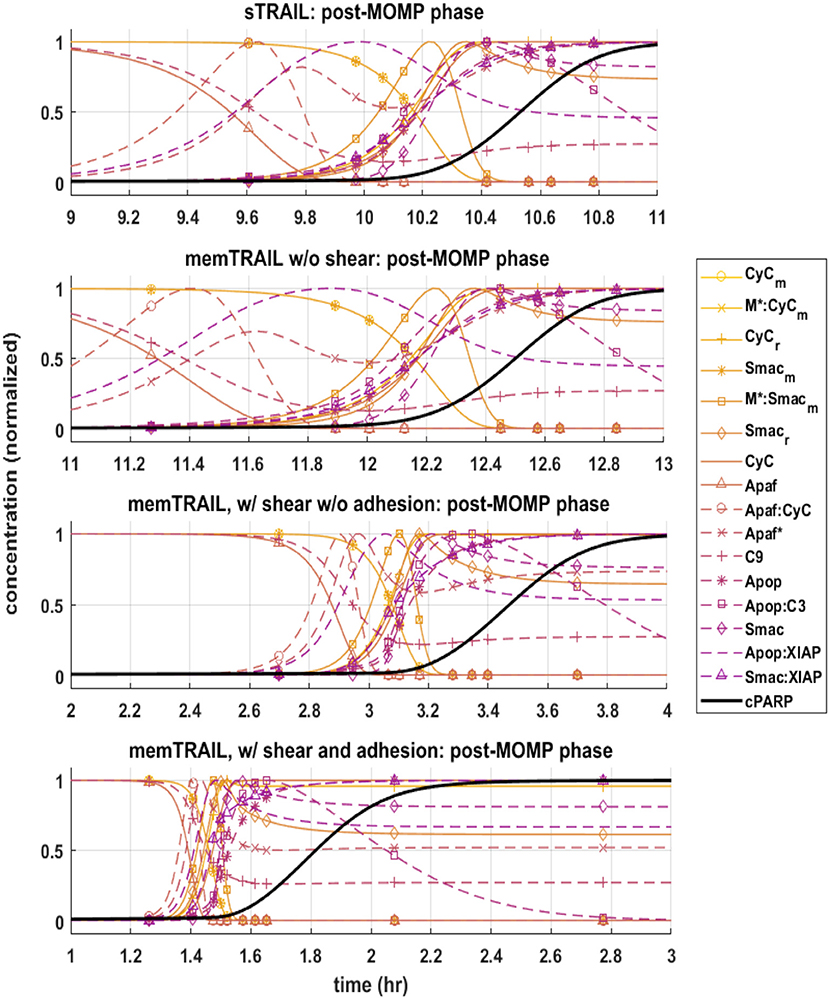
Figure 3. Pre-mitochondrial pathway species concentration profiles as a function of time for four different cases of TRAIL binding to DRs: sTRAIL, memTRAIL without shear, memTRAIL with shear and without adhesion, and memTRAIL with shear and adhesion.
C3 and XIAP Concentration Profiles
Given these observations regarding the pre and post mitochondrial membrane reaction pathways, we next considered where the two pathways meet. This sheds light on the specific mechanism that allows for faster apoptosis in CTCs exposed to memTRAIL with shear and adhesion. Two species are of greatest importance in this analysis: C3 and XIAP. To simplify the comparison, only two critical cases were considered – sTRAIL and memTRAIL with shear and cell adhesion. Given that the time until apoptosis had already been quantified and that we were most interested in how each reactant profile emerges with respect to cPARP's transition, Td was subtracted from the time vector to re-center each image around the time of cell death. The curves were normalized to the maximum concentration of reagent for sTRAIL pathway. In cases where the reagent concentration is relatively higher for reactants within the memTRAIL pathway, then the profile displays values greater than 1, and if relatively less, then values less than 1.
It is evident that a sudden increase emerges in concentration of species C8*:C3, XIAP:C3⋆, and for the liposomal TRAIL method (Figure 4B). One notable difference in the relative quantities of reagents. C8*:C3,Apop:C3, C3*, XIAP:C3*, and Apop:XIAP comparing the two cases is that all had lower maximum concentrations than their sTRAIL pathway counterparts, while C3*:PARP and XIAP:C3* peaked at higher values. Another notable difference between the two conditions is the order in which reagents emerge. Apop:XIAP and XIAP transitioned before other reagents for the sTRAIL pathway, while C8*:C3, XIAP:C3* and had already completed 40–75% of their respective transitions by the time Apop:XIAP and XIAP began to transition for the memTRAIL pathway (Figure 4).
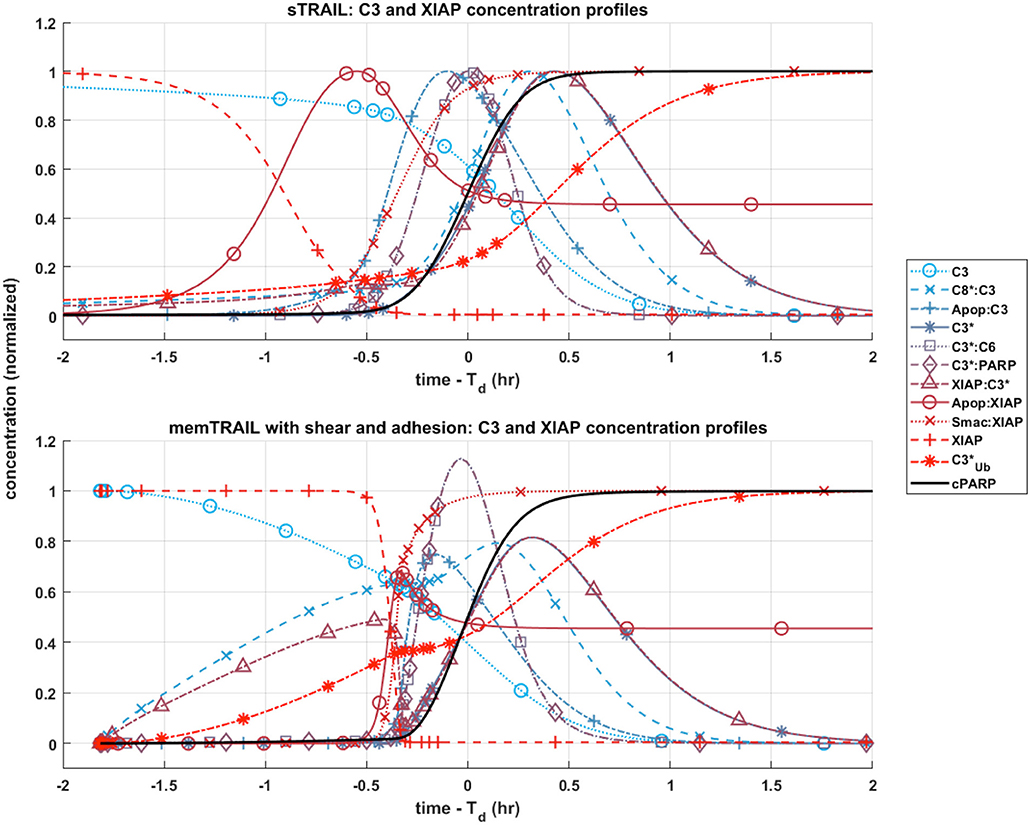
Figure 4. Post-mitochondrial pathway species concentration profiles as a function of time for four different cases of TRAIL bindng to DRs: sTRAIL, memTRAIL without shear, memTRAIL with shear without adhesion, and memTRAIL with shear and adhesion.
Mapping Time Until Apoptosis
A sensitivity study was conducted to determine why liposome bound TRAIL (memTRAIL) acts as a more potent drug delivery mechanism. In this study, apoptosis was quantified by the time it takes for the variable PARP to cleave and form cPARP, which indicates the end of the reaction pathway from TRAIL binding to DR to eventual cell death. This point was defined as the time when cPARP was halfway through its transition time, 0.5 × cPARPmax, Td. In order to gain insights about general trends, mappings of Td were created as a function of different combinations of the following parameters: Death Receptor (DR) concentration, TRAIL concentration, forward binding association rate constant of TRAIL binding DR, k+, and backwards binding dissociation rate constant, k−. Reasonable values of each parameter were determined, as specified in the Methods section.
Varying: TRAIL and DR Concentration
The first analysis considered a range of DR and TRAIL concentrations (Figure 5A). As DR concentration was increased, it was noted that Td decreased as expected. Interestingly, a bimodal dependence was observed, with Td starting high for lower concentrations of sTRAIL, reaching a minimum at some intermediate value, and then trending upwards again at very high concentrations of sTRAIL and low concentrations of DR. For high concentrations of DR an increase in Td was not observed for increasing concentrations of memTRAIL. However, at lower DR concentrations the increase in Td with increasing memTRAIL was observed.
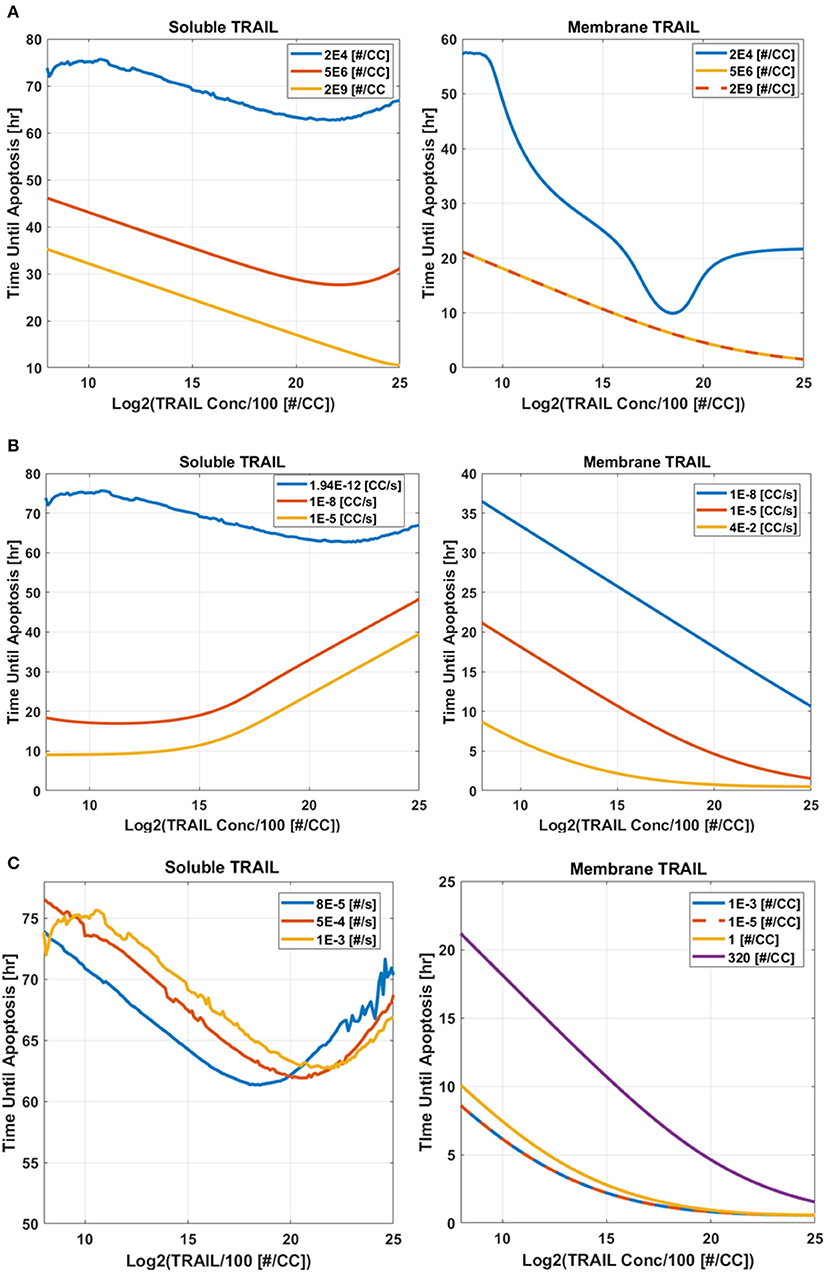
Figure 5. Time until apoptosis occurs as a function of sTRAIL and memTRAIL concentration, for different values of (A) DR concentration, (B) k+, (C) k−.
Varying: TRAIL Concentration and k+
TRAIL concentration and k+ were also varied (Figure 5B). As k+ was increased, time until apoptosis decreased, and a minimum was found as a function of sTRAIL concentration at low values of k+. However, for memTRAIL a consistent decrease in time until apoptosis was found.
Varying: TRAIL Concentration and k−
Td was calculated as a function of TRAIL concentration and k− (Figure 5C). As k− was increased, Td increased; at very high values of k−, a characteristic decrease, and then increase in Td as a function of increasing sTRAIL concentration was observed. For memTRAIL, only a decrease in Td was observed with increasing memTRAIL concentration. As k− was increased, Td increased for memTRAIL similar to sTRAIL.
Taken together, these findings suggest that there is an optimum concentration of sTRAIL that minimizes the time necessary for a cell to experience apoptosis, whereas memTRAIL maintains its potency despite the increasing concentrations, independent of k− and k+. Low DR concentration does, however, effect memTRAIL potency.
Discussion
Previous experimental work has shown that leukocyte-tethered TRAIL (memTRAIL) is much more effective than soluble TRAIL (sTRAIL) at inducing apoptosis in CTCs (14). The model presented here offers an explanation. Given that both leukocytes and CTCs travel along similar streamlines in the blood flow and the high ratio of ~1 × 106 leukocytes per CTC, each CTC is expected to come into frequent contact with leukocytes throughout the vascular network (10). With this higher effective concentration of memTRAIL present in the vicinity of the CTCs, an elevated on-rate caused by shearing, and a reduced off-rate caused by E − selectin (ES) induced adhesion it was shown that memTRAIL induces apoptosis in under 2 h while sTRAIL takes far longer at 10 + h. These findings are consistent with those of Mitchell et al., and support the observation that treatment is much more effective when tethering the ES/TRAIL liposomes to leukocytes rather than relying on sTRAIL and its rapid clearance rate in the circulation (12, 14).
Given these findings, a deeper understanding was sought as to why the complete reaction proceeded more quickly, by varying the dynamics of TRAIL binding to DRs. First, four key parameters, DR concentration, TRAIL concentration, k+, and k− were varied to determine the effect on the time until apoptosis, Td, after initial binding of TRAIL to DR. These simulations showed that given certain conditions, higher concentrations of TRAIL slow the overall reaction pathway. This suggests that the kinetics of TRAIL binding to DRs does not completely regulate how rapidly the overall apoptosis reaction proceeds, but rather affects how downstream reagents proceed in initiating cell death.
Four specific cases of binding were considered in greater detail: sTRAIL, memTRAIL without shear, memTRAIL with shear but without adhesion, and finally memTRAIL with shear and adhesion. For each of these cases, we looked at how key reagents unfold with respect to one another. For memTRAIL, pre-mitochondrial reagents started to transition immediately and completed their transition before cPARP transitioned. On the other hand, in considering the sTRAIL pathway to cell death, most of the pre-mitochondrial pathway reagents transitioned simultaneously with cPARP, which indicates that the pre-mitochondrial pathway is the limiting pathway for the sTRAIL case, but not for the memTRAIL with shear and adhesion case.
The relative concentrations of several key species between the two pathways were considered to focus in on the mechanism causing rapid apoptosis in CTCs treated in shear with ES/TRAIL liposomes. Due to the new configuration of memTRAIL conjugated to the surface of the liposome and the higher binding rate constants induced by shearing, the initial memTRAIL binding DR reaction occurred much faster, promoting the availability of its downstream reactants. The surge in concentration of one of these reactants, C8*, pushed the reaction of C8*binding C3 forward rapidly, thus activating C3 to form C3*. XIAP quickly engaged the available C3*, as shown by the rapid increase in XIAP:C3* concentration but lack of increase in C3*alone. This increase leads to a critical difference between the two pathways. As XIAP:C3* concentration elevates, C3* is converted to in an irreversible reaction, which decreases the amount of usable C3* until a critical point where XIAP:C3* is driven in the reverse direction to favor the unbound C3* and XIAP complex. This is marked by the sudden but temporary decrease in XIAP:C3* complex concentration. This new equilibrium point liberates more C3* for participating in the memTRAIL pathway than for the sTRAIL pathway, which promotes a steep and rapid uptake by PARP and C6 to form C3*:PARP and C3*:C6 respectively. Since C3*:PARP peaks at a higher value, PARP is cleaved faster and apoptosis occurs more rapidly.
This sequence of events could also explain why extremely high values of sTRAIL lead to higher Td and memTRAIL is more resistant to increasing Td. If the initial binding pathway proceeded too quickly, then would consume too much of C3* from the reaction pathway before C3* was able to bind to PARP, retarding the reaction of C3* cleavage of PARP. Thus, the efficiency of ES/TRAIL liposomes can be attributed to two effects. First, as the initial TRAIL binding DR pathway occurs more rapidly, the downstream reactions begins sooner, leading to earlier apoptosis. The second effect leading to ES/TRAIL liposome efficiency is the intricate balance of , resetting the bound and unbound equilibrium of C3* and XIAP. While it is initially faster to have C3* and XIAP bind quickly to allow downstream reactions to begin, this increased rate can reach a point of diminishing returns, where too much C3* is irreversibly converted to , reducing the available C3* for downstream reactions which ultimately result in the cleavage of PARP.
Conclusions
These results faithfully recapitulate the experimental findings of Mitchell et al. (14), where sTRAIL and tethered ES/TRAIL liposomes were both tested in mice to neutralize intravenously injected CTCs Given renal clearance mechanisms, our model suggests that although sTRAIL will eventually become effective if exposure could be sustained sufficiently long, it is unlikely to have sufficient time to act on the CTCs in vivo to induce apoptosis before being cleared from the circulation. This was the case in the previous experimental study of Mitchell et al. (14), as sTRAIL was ineffective at treating the mice whereas ES/TRAIL functionalized leukocytes showed 98.5% efficiency at clearing injected cancer cells after just 2 h of circulation. Given a new effective concentration of memTRAIL, an elevated k+ of memTRAIL binding DR caused by the slip velocity between the cell surfaces in shear flow, and a lowered off rate caused by ES adhesion, our simulation reproduces the behavior observed experimentally by Mitchell et. al, (14) and shed light on the enhanced efficacy of TRAIL/ES liposome therapy.
Methods
2D Binding: Initial Conditions
Bound TRAIL
The model of Albeck et al. was modified, along with the provided initial conditions and rate constants, to capture the sheared liposomal TRAIL treatment of CTCs (16). First, it was necessary to determine the concentration of TRAIL bound to the surface of a liposome, to provide appropriate initial conditions for the model (16). Mitchell et al. estimated that there were ~65 TRAIL molecules on average bound to the surface of each liposome of diameter ~100 nm (14). For spherical liposomes, this corresponds to a surface density of liposomal TRAIL of . The 2D density of death receptor on the surface of CTC was estimated, by assuming that all death receptors were located on the surface of the CTC. Given that there are 1 × 104 DR4/cellular volume and the volume of the cell to is 1 × 10−9cm3, for a roughly spherical cell the surface density can be calculated to be .
Soluble TRAIL
An effective spatial availability of sTRAIL was determined that corresponds to conditions in the previous experiments of Mitchell et al. (14). Plasma concentration of sTRAIL was estimated to be 1 μg/mL. For a cellular volume of 1 × 10−9cm2, it was approximated that the concentration of sTRAIL available for surface reaction is 1.85 × 104 TRAIL/cellular volume, or more appropriately, 3.8 × 109 TRAIL/cm2.
2D Binding: Reaction Rate Constants
Binding Association Rate Constant
Chang and hammer approach: shear
Next, the original 3D model was modified to more appropriately represent the binding kinetics of liposomal TRAIL in shear, via the binding association and dissociation rate constants.
An analysis was carried out employing Chang and Hammer's approach for determining binding association and dissociation rates for a 2D surface binding to a 2D surface with a relative slip velocity between the surfaces (25). The first step in this analysis required a determination of the slip velocity between liposomal TRAIL attached to leukocytes and CTCs in shear. It was assumed that the centroid of a TRAIL functionalized leukocyte and the centroid of an interacting CTC were ~10 μm apart (the sum of their radii), d, when they convect past each other. Given a uniform shear rate, S, of 1000 s−1 as in typical blood flow (27), it was determined that the relative velocity of the centers was S × d = V = 1 cm/s. This slip velocity, the sum of the lateral diffusivities of TRAIL and death receptor, and the reactive radius of our reagents were used to determine the Peclet number, Pe = V · a/D, which is the dimensionless ratio of bulk flow (advection) to diffusive flow of reagent. A diffusivity, D, was used of order 1 × 10−9cm2/s (28) and a reactive radius, a, of 5 × 10−6cm (29) to yield a Pe of 5000. In Chang and Hammer's analysis, since Pe ≫ 1, the Nusselt number, another measure of bulk flow to diffusive flow, can be approximated as Nu = 2Pe/π, yielding a Nu of 3183. From this, the enhanced forward association rate constant was determined to be .
Chang and hammer approach: unsheared (diffusion limit)
It also was necessary to determine the forward association rate constant, again in 2D binding, for the unsheared case to apply the model to the static control conditions in the experiments of Mitchell et al. This simulation condition involved no slip velocity, and thus Pe = 0. When Pe = 0, , where b 1/2 the mean distance between ligand and receptor and a is the reactive radius. b was estimated to be of order of magnitude b = 10 × 10−6 cm, yielding Nu = 2.9 and .
Bell approach: unsheared (diffusion limit)
As a check on the assumptions made, Bell's approach, which does not take into account a relative slip velocity, was also considered (30). Bell's approach proposes that ko = 2πD, giving us . This value is of the same order of magnitude for the unsheared case using Chang and Hammer's approach.
Modification of albeck approach
Albeck, and others have measured a forward association rate constant of sTRAIL binding death receptor with value equal to 2.4 × 105M−1s−1. To be consistent with the dimensionality of the molecular participants described above in 2D Binding: Initial Conditions, this value was converted to 1.94 × 10−12cm2/s, which directly follows from a CTC volume of 1 × 10−9cm2 and surface area of 4.8 × 10−6cm2.
Binding Dissociation Rate Constant
Chang and hammer approach: sheared
To determine the off rate for the sheared case, the average duration of encounter, τ, which for Pe ≫ 1, can be approximated as τ ~ 8a/(3|V|π), was estimated. Again, a is the reactive radius and |V| is the slip velocity, yielding τ ~ 4.2 × 10−6s. Next, the dimensionless duration time, , and the dimensionless Damköhler number, , were determined, where kin is the intrinsic forward reaction rate, equal to 1 × 109s−1 (29). Given these two parameters, the probability of binding was expressed as . From this, the overall forward rate of reaction was found to be . Given that kf = kokin/(kin + k−), k− was found to be .
Chang and hammer approach: unsheared (diffusion limit)
The binding dissociation rate constant was estimated for the unsheared case. Here, τ is given as , yielding τ = 3.1 × 10−3 s, Λ = 0.125, δ = 2.5 × 107, P ~ 1 for a and a .
Bell approach: unsheared (diffusion limit)
To check the assumptions made, Bell's approach was referenced, which does not take into account a relative slip velocity. Bells approach states that , yielding . This value is within one order of magnitude for the unsheared case obtained separately via Chang and Hammer's approach.
E-Selectin Effects on Binding Adhesion
Mitchell et al. demonstrated E-selectin as an adhesive targeting protein that simultaneously promotes the establishment of TRAIL-functionalized leukocytes, as well as close surface interactions with CTCs. To represent this adhesive interactions between TRAIL-coated leukocytes and colliding CTCs, the duration of time that E-selectin was expected to be adhered to CTCs before separating in shear flow was estimated to be of the order 1 × 10−3s (31). During this time, E-selectin tethering was assumed to effectively reduce the slip velocity between the two cells to zero, and thus set the binding dissociation rate to the diffusion limited case (unsheared). This was implemented within the simulation by executing the numerical integration for 1 × 10−3s with σL as the initial condition. Following this interval, the numerical integration using the last concentration from the first interval as the initial condition for all reagents except TRAIL which was then set to zero.
Biochemical Mathematical Modeling
The TRAIL-induced apoptosis biochemical reactions were modeled using one of the following mass-action paradigm equations as referenced in Albeck et al. (16):
Where E is an enzyme or other protein, S is the substrate or binding partner of E, and P is the product of the specific reaction i. K+i, k+i, and k−i are the catalytic, forward, and backward reaction rates, respectively.
For molecules that translocate from the mitochondria to the cytoplasm, the molecules in each compartment are assumed to be well mixed. When the mitochondria is perturbed, allowing for transport, the number of molecules x vary with time, t. Therefore, the concentration of x is defined by the following ODE as also seen in Albeck et al. (16).
Simulation and Visualization
The MATLAB mass matrix solver ODE15s was used to numerically solve the ODE system. Graphs were prepared using MATLAB.
Author Contributions
EL designed, implemented and evaluated the algorithmic methods and suggested improvements of the model. JH performed additional computational analysis and prepared some figures and interpretation of results. MK conceived the model and provided input to the algorithms' development. EL wrote the manuscript, and JH revised the manuscript. All authors edited the manuscript, read and approved the final manuscript.
Funding
This work was funded by the National Institutes of Health, Grant No. R01CA203991 to MK.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chaffer CL, Weinberg R. A perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi: 10.1126/science.1203543
2. Riethdorf S, Wikman H, Pantel K. Review: biological relevance of disseminated tumor cells in cancer patients. Int J Cancer (2008) 123:1991–2006. doi: 10.1002/ijc.23825
3. Maheswaran S, Haber DA. Circulating tumor cells a window into cancer biology and.pdf. Howard Hughes Med Inst. (2010) 20:96–9. doi: 10.1016/j.gde.2009.12.002
5. McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int J Cancer (2009) 125:1298–305. doi: 10.1002/ijc.24409
6. Van Ginhoven TM, Van Den Berg JW, Dik WA, Ijzermans JNM, De Bruin RWF. Preoperative dietary restriction reduces hepatic tumor load by reduced E-selectin-mediated adhesion in mice. J Surg Oncol. (2010) 102:348–53. doi: 10.1002/jso.21649
7. Gassmann P, Kang ML, Mees ST, Haier J. In vivo tumor cell adhesion in the pulmonary microvasculature is exclusively mediated by tumor cell–endothelial cell interaction. BMC Cancer (2010) 10:177. doi: 10.1186/1471-2407-10-177
8. Köhler S, Ullrich S, Richter U, Schumacher U. E-/P-selectins and colon carcinoma metastasis: first In vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br J Cancer (2010) 102:602–9. doi: 10.1038/sj.bjc.6605492
9. Rahn JJ, Chow JW, Horne GJ, Mah BK, Emerman JT, Hoffman P, et al. MUC1 mediates transendothelial migration in vitro by ligating endothelial cell ICAM-1. Clin Exp Metastasis (2005) 22:475–83. doi: 10.1007/s10585-005-3098-x
10. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. (2011) 192:373–82. doi: 10.1083/jcb.201010021
11. Mitchell MJ, King MR. Leukocytes as carriers for targeted cancer drug delivery. Expert Opin Drug Deliv. (2015) 12:375–92. doi: 10.1517/17425247.2015.966684
12. Wayne EC, Chandrasekaran S, Mitchell MJ, Chan MF, Lee RE, Schaffer CB, et al. TRAIL-coated leukocytes that prevent the bloodborne metastasis of prostate cancer. J Control Release (2016) 223:215–23. doi: 10.1016/j.jconrel.2015.12.048
13. Mitchell MJ, King MR. Unnatural killer cells to prevent bloodborne metastasis: inspiration from biology and engineering Unnatural killer cells to prevent bloodborne metastasis: inspiration from biology and engineering. Expert Rev Anticancer Ther. (2014) 14:641–4. doi: 10.1586/14737140.2014.916619
14. Mitchell MJ, Wayne EC, Rana K, Schaffer CB, King MR. TRAIL-coated leukocytes that kill cancer in the circulation. Proc Natl Acad Sci USA. (2014) 111:930–5. doi: 10.1073/pnas.1316312111
15. Mitchell MJ, King MR. Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via trimeric death receptors. New J Phys. (2013) 15:015008. doi: 10.1088/1367-2630/15/1/015008
16. Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK. Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol. (2008) 6:e299. doi: 10.1371/journal.pbio.0060299
17. Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. (2003) 10:76–100. doi: 10.1038/sj.cdd.4401160
18. Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. (1995) 14:5579–88.
19. Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJD, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. (2006) 8:1348–58. doi: 10.1038/ncb1499
20. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell (2002) 2:183–92. doi: 10.1016/S1535-6108(02)00127-7
21. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell (2000) 102:43–53. doi: 10.1016/S0092-8674(00)00009-X
22. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102:33–42. doi: 10.1016/S0092-8674(00)00008-8
23. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell (1996) 86:147–57.
24. Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. (1998) 17:2215–23.
25. Chang K, Hammer D. The forward rate of binding of surface-tethered reactants: effect of relative motion between two surfaces. Biophys J. (1999) 76:1280–92.
26. Szegezdi E, van der Sloot AM, Mahalingam D, O'Leary L, Cool RH, Muñoz IG, et al. Kinetics in signal transduction pathways involving promiscuous oligomerizing receptors can be determined by receptor specificity: apoptosis induction by TRAIL. Mol Cell Proteomics (2012) 11:M111.013730. doi: 10.1074/mcp.M111.013730
27. Turitto VT, Baumgartner HR. Platelet interaction with subendothelium in flowing rabbit blood: effect of blood shear rate. Microvasc Res. (1979) 17:38–54.
28. Varadhachary AS, Edidin M, Hanlon AM, Peter ME, Krammer PH, Salgame P. Phosphatidylinositol 3'-kinase blocks CD95 aggregation and caspase-8 cleavage at the death-inducing signaling complex by modulating lateral diffusion of CD95. J Immunol. (2001) 166:6564–9. doi: 10.4049/jimmunol.166.11.6564
29. Hammer DA, Lauffenburger DA. A dynamical model for receptor-mediated cell adhesion to surfaces in viscous shear flow. Cell Biophys. (1989) 14:139–73.
Keywords: cancer, apoptosis, TRAIL, modeling, circulating tumor cells (CTC)
Citation: Lederman EE, Hope JM and King MR (2018) Mass Action Kinetic Model of Apoptosis by TRAIL-Functionalized Leukocytes. Front. Oncol. 8:410. doi: 10.3389/fonc.2018.00410
Received: 21 June 2018; Accepted: 06 September 2018;
Published: 29 October 2018.
Edited by:
Owen McCarty, Oregon Health & Science University, United StatesReviewed by:
Monica M. Burdick, Ohio University, United StatesPaul Kenneth Newton, University of Southern California, United States
Copyright © 2018 Lederman, Hope and King. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael R. King, bWlrZS5raW5nQHZhbmRlcmJpbHQuZWR1