 Mayur Tanna
Mayur Tanna Rami I. Aqeilan
Rami I. Aqeilan- 1Faculty of Medicine, The Lautenberg Center for Immunology and Cancer Research, Institute for Medical Research, Israel-Canada (IMRIC), Hebrew University of Jerusalem, Jerusalem, Israel
- 2Department of Cancer Biology & Genetics, Ohio State University Wexner Medical Center, Columbus, OH, United States
The WW domain–containing oxidoreductase (WWOX) gene encompasses a common fragile sites (CFS) known as FRA16D, and is implicated in cancer. WWOX encodes a 46kDa adaptor protein, which contains two N-terminal WW–domains and a catalytic domain at its C–terminus homologous to short–chain dehydrogenase/reductase (SDR) family proteins. A high sequence conservation of WWOX orthologues from insects to rodents and ultimately humans suggest its significant role in physiology and homeostasis. Indeed, data obtained from several animal models including flies, fish, and rodents demonstrate WWOX in vivo requirement and that its deregulation results in severe pathological consequences including growth retardation, post–natal lethality, neuropathy, metabolic disorders, and tumorigenesis. Altogether, these findings set WWOX as an essential protein that is necessary to maintain normal cellular/physiological homeostasis. Here, we review and discuss lessons and outcomes learned from modeling loss of WWOX expression in vivo.
Introduction
As a result of perturbed DNA synthesis, chromosomal fragile sites display genomic perturbations in the form of gaps, breaks, constrictions and rearrangements at specific DNA loci on metaphase chromosomes (1–6). The first human chromosomal fragile site was presented in 1965 by Dekaban that was found in an irradiated female patient who received X–rays for the treatment of eczematous dermatitis. These sites harbored two types of chromosomal abnormalities; (a) frequent breaks in the long arm of the chromosome number 9 and (b) moderately increased rate of chromosome–type aberrations occurring at random in all chromosomes (7). Later, in 1970, the name “fragile site” was used to explain recurrent chromosomal breaks on chromosome 16, which followed Mendelian segregation within a large family in a dominant fashion showing linkage of the heptoglobin alpha locus (8). Subsequently, chromosomal fragile sites associated with X–linked mental retardation within different families were reported (9–12). These preliminary reports led to the classification of chromosomal fragile sites as common fragile sites (CFSs) and rare fragile sites, depending on their frequency in the population as well as their inheritance pattern and further subdivided these sites based on the type of inducing chemical (5, 9–14). Ultimately, these early cytogenetic findings intrigued the geneticist community to further explore fragile sites and study their relevance in human diseases.
CFSs are the largest class of chromosomal fragile sites, which represent a component of normal chromosome structure that are observed in all individuals. CFSs are late replicating regions and are known as preferential hotspots for metaphase chromosome breaks and chromosome rearrangements due to partial DNA replication stress (4, 5). In addition, CFSs are reported to be targets of replication stress in preneoplasia as well as sensors for DNA damage (15–18). Normally, CFSs are stable in cell culture settings but are highly susceptible to form chromosome breaks when DNA synthesis is impeded by using low doses of aphidicolin (APH) or hydroxurea (HU) (2, 4). As a consequence, chromosome breaks at 20 CFSs were revealed in lymphocytes treated with low doses of APH causing over 80% of the cytogenetic lesions. FRA16D, a genomic region where the WWOX gene resides, was noted as one of the most highly expressed CFSs amongst the 20 CFSs identified in human lymphocytes (4). Today, we know that there are about 90 CFSs that are tissue–specific and are induced at a stressor-specific manner [reviewed in (19)]. Based on these findings and others, CFSs were proposed to be involved in chromosome rearrangements observed in cancer (20–23).
WWOX, encompassing FRA16D at 16q23.2, was cloned and mapped in early 2000 by two research groups simultaneously explaining the significance of this archetypal fragile gene in human cancer (24, 25). The WWOX gene spans over 1 Mb of the genomic locus with nine exons separated by large introns encoding 2.2kb mRNA and 1.245kb ORF, which then translates into a 414–amino acid protein product (26). Following these studies, murine Wwox (also known as Wox1) was cloned suggesting the presence of WWOX orthologues in rodents (27). The WWOX protein, as the name suggests, contains two N-terminal WW domains and catalytic domain at its C-terminus homologous to short chain dehydrogenase/reductase (SDR) family proteins. Through its WW1 domain, WWOX interacts with partner proteins harboring proline–tyrosine (PY) motifs and acts as an adaptor protein regulating their localization transactivation, and stability (28, 29). Interestingly, the region of homozygous deletions reported in various malignancies coincides with FRA16D where intron eight spans ~750 kb of the WWOX gene (25, 30, 31).
Loss of WWOX expression has been described in various tumors including breast, colon, esophageal, gastric, lung, ovarian, and prostate cancers [reviewed in (32)]. Besides its location in a fragile site, WWOX altered expression was also attributed to hypermethylation of its promoter, regulation by microRNAs and posttranslational modifications [reviewed in (26, 33)]. As a result of its dyregulation, several lines of evidence revealed that WWOX affects DNA damage response (DNA repair and apoptosis) (6, 27, 34, 35), cellular metabolism (36–42) and numerous signaling pathways (26, 29, 38, 43–46). Altogether, these observations have led to the generation of animal models mimicking WWOX loss of function in vivo to further investigate its physiological significance.
Phylogenetic analysis of the amino acid sequences of WWOX protein reveals that WWOX is an evolutionary conserved protein as a discrete orthologue between insects to humans. The conserved sequences of WW domains and SDR domain in WWOX imply its tight regulation across species. All species exhibit distinct sequence relationships of WWOX orthologues through evolution except the closest organism Caenorhabditis elegans, which lacks the two N–terminal WW domains but have sequence homology to WWOX SDR domain suggesting common substrate of this putative enzymatic domain independent of its WW domain interactions (47). Indeed, there is a remarkable conservation in sequence, for example, between human FRA16D/WWOX and mouse FRA8E1/Wwox both at exon as well as intron levels exhibiting 94% identity along with 96% similarity at protein level (27, 48). Additionally, FRA16D/WWOX shares 49% identity and 66% similarity with Drosophila melanogaster homologous protein CG7221/DmWWOX (49).
Several lessons were learned from modeling WWOX perturbations in vivo using different animal models facilitating dissecting its significance in physiology and disease states. For example, after a thorough scrutiny of Wwox mutant mice, it was proposed that WWOX behaves as a tumor suppressor gene (50–52). Apart from its role in cancer, many reports revealed direct involvement of WWOX in neurological as well as metabolic disorders. Homozygous mutations in WWOX are reported in patients with epilepsy and mental retardation (53, 54). Many reports also indicate the role of WWOX in different metabolic disorders including lipid metabolism (55, 56), obesity (57), and type 2 diabetes (58). Increasing lines of evidence are, therefore, suggesting the significant contribution of WWOX in human maladies and requiring deeper understanding of WWOX function both at the molecular and cellular levels. In this review article, we describe the knowledge and conclusions learned by modeling loss of WWOX expression in vivo using various animal models including mice, rats, Drosophila and zebrafish.
Animal Models of Wwox Ablation
Mice
Considering the notion that WWOX is frequently lost in human cancers led to generation and characterization of mouse models that depict this ablation to further deepen our understanding of WWOX function in physiology and in pathophysiology. Several mouse models were established and studied.
Phenotypic Analysis of Wwox Null Mice
The first knockout (KO) mouse model for Wwox was established by Aqeilan et al. (50). In this article, the authors altered ~6 kb of DNA sequences within the genomic locus of murine Wwox using homologous recombination. This was achieved by replacing exons 2–4 of Wwox with a targeting cassette thus generating a Wwox mutant allele and later homozygous Wwox KO (Wwox −/−) mice. These mice were monitored very carefully and phenotyped. While wild type (WT), heterozygous (Wwox +/−), and Wwox −/− mice appeared normal in size and were indistinguishable at birth, Wwox −/− pups showed retarded post–natal growth from day 3 and eventually 100% of mice died at 3–4 weeks of age. These KO pups did not exhibit any histological lesions in the main organs but displayed severe metabolic defects and analysis of blood serum chemistry revealed altered levels of carbohydrates, proteins, and lipids hence associating WWOX with cellular metabolism (59). For example, Wwox −/− mice display reduced serum lipid levels (50, 60) as well as impaired expression of steroidogenic enzymes (61).
Bone phenotype of Wwox null mice
Severe bone metabolic disease is one of the major and prominent phenotypes observed in Wwox −/− mice. Although no abnormalities in the skeletal patterning was observed, Wwox −/− mice experienced impaired bone metabolism as there was a decrease in the density of trabeculae bone along with thinning of the inner cortex from postnatal day 7 leading to eventual development of smaller limbs in proportion to the body weight of KO pups. This delay in bone formation was the result of cell–autonomous defect in osteoblast differentiation beginning at the mineralisation stage. Reduced trabecular member connectivity and bone surface area in both Wwox +/− and Wwox −/− pups was seen at day 15 in three–dimensional μCT images but the mineral content of trabecular bone in Wwox +/− mice was not significantly changed in comparison to WT. Serum calcium levels were 50% reduced and serum phosphate levels were increased by 20% in Wwox −/− pups indicating a metabolic bone disease. In contrast, Wwox +/− pups did show any altered levels of serum calcium and phosphorous (59)
A physical, functional and molecular link between WWOX and RUNX2, the master regulator of bone differentiation, was later reported. WWOX suppresses transactivation of RUNX2 resulting in repression of many RUNX2 targets. This intimate relationship between WWOX and RUNX2 was also documented in cancer (62–65). In addition to impaired osteoblast differentiation, Wwox −/− mice also exhibited enhanced osteoclast activity (59), though the molecular mechanism of the later is poorly studied. Therefore, it is plausible to conclude that WWOX loss contributes to an osteopenic phenotype and bone metabolic disease. Although this phenotype is very prominent in mice, supporting evidence linking WWOX with osteopenia or osteoporosis in human patients is still lacking. This could be due to the fact that WWOX loss of function due to germline mutations in human patients is postnatal lethal or that patients were not carefully assessed for bone density.
Steroidogenesis defect in Wwox null mice
Detailed analysis of the phenotypic abnormalities of Wwox −/− mice also revealed that these pups are born with gonadal abnormalities displaying impaired spermatogenesis (61). A comprehensive study focusing on the defects in the reproductive system of both male and female Wwox−/− mice revealed an altered expression of several steroidogenesis related enzymes responsible for this phenotype. Lack of Leydig cells and remarkable reduction of serum testosterone was noted in Wwox−/− male mice likely led to the observed testicular hypoplasia. Likewise, Wwox−/− female mice have also displayed abnormalities in their reproductive system manifested by impaired theca cells proliferation and small primary follicles resulting in reduced ovary size. Additionally, expression of numerous genes encoding steroid biosynthesis enzymes, such as Hsd3b6 and Cyp11a1, is downregulated in the reproductive organs of Wwox −/− pups when compared with WT and Wwox +/− littermates (61). Altogether, these observations suggest that WWOX has an important role in steroidogenesis and its loss could be associated with infertility though future studies are required to dissect this interesting phenotype.
Lessons Learnt on WWOX Tumor Suppressor Function From Studying Wwox Mutant Mice
Wwox null mice
As introduced, altered WWOX expression has been reported in several human malignancies (32). Hence, Wwox ablation in mice has been predicted to associate with either tumor initiation or progression. The first in vivo evidence associating WWOX with tumor suppression was revealed when juvenile Wwox −/− mice developed femoral neoplastic focal lesions resembling early osteosarcoma (50, 63). These findings were based on histology sectioning through paraffin–embedded bones as well as screening by micro (μ)–CT imaging of intact limbs. These observations paved the way to hypothesize that WWOX deficiency can contribute to osteosarcoma development. Indeed, several reports have shown that WWOX deletion or altered expression is a frequent event in osteosarcoma (63, 64, 66, 67). Since Wwox −/− mice die prematurely, osteosarcoma development could not be followed in adult mice. Nevertheless, a more recent report revealed that somatic ablation of Wwox in committed osteoblasts accelerate osteoblastic osteosarcoma formation in Trp53f/f mice (68) (also see below).
Wwox–heterozygous (Wwox+/−) mice
The fact that Wwox ablation in mice leads to early postnatal lethality (50) prompted studying WWOX tumor suppressor function using Wwox–heterozygous (Wwox+/−) mice. The incidence of spontaneous tumor formation in Wwox+/− mice was significantly higher than that of WT littermates. Tumors formed in Wwox+/− mice included development of lung papillary carcinomas and lymphoblastic leukemia, but not osteosarcoma (50). Interestingly, several reports have documented altered WWOX expression in lung cancer (33, 69–71) and lymphomas (72–76) suggesting WWOX as a potential tumor suppressor in these malignancies.
As introduced earlier, perturbations in WWOX is commonly observed in various tumor kinds, other than lung cancer and lymphoma (32). One of the prominent cancer types that display absent or reduced WWOX expression is breast cancer (77–82). Monitoring of Wwox+/− mice for mammary tumor development in B6/129 mixed genetic background revealed very low incidence (52). These observations led the Aqeilan group to transfer the Wwox+/− allele onto C3H, a mammary tumor susceptible genetic background. Heterozygous WwoxC3H+/− mice were generated by backcrossing the Wwox+/− allele onto C3H pure genetic background mice, for at least 4 generations (51). Monitoring of these mice demonstrated that inactivation of a single Wwox allele is associated with increased incidence of mammary tumors. These tumors very much resemble those observed in human basal-like breast cancer (BLBC) with WWOX alteration (83). Poorly differentiated invasive ductal carcinomas and squamous cell carcinomas were observed. Immunohistochemistry staining of hormone receptors: estrogen receptor (ER) and progesterone receptor (PR) were absent/reduced in WwoxC3H+/− mammary tumors. These results, therefore, suggest that heterozygous Wwox loss could be sufficient for developing mammary tumors of BLBC-like nature (51). These results were further confirmed in a subsequent recent study by Abdeen and colleagues using a tissue-specific knockout mouse model of Wwox (84) (also see below).
Chemical–induced tumorigenesis in Wwox–heterozygous (Wwox+/−) mice
To further investigate WWOX tumor suppressor function, a number of studies tested susceptibility of Wwox+/− mice to chemical–induced tumorigenesis. In one study, Wwox+/− mice were treated with ethyl nitrosourea (ENU)–a mutagen that is commonly used to study tumor spectrum in a given mouse strain. As expected, the incidence of tumor formation, especially lung cancer and lymphoblastic lymphoma, was significantly higher in ENU–treated Wwox+/− mice in comparison with ENU–treated WT mice (50). In addition, various types of epithelial tumors like liver hemangiomas, chondrosarcomas, fibroadenoma, and squamous cell carcinomas were observed in ENU–treated Wwox+/− mice. It was also noted that multiplicity of tumors is significantly higher in ENU–treated Wwox+/−mice in comparison with ENU–treated WT mice. Of note, signs of lymphoma aggressiveness were also higher in ENU-treated Wwox+/− mice suggesting that WWOX loss could lead to more advanced tumors (50). These results could suggest that Wwox+/− mice might be a good model system to study the effects of carcinogens and chemoprevention studies.
In a subsequent study, the susceptibility of loss of one Wwox allele on N – nitrosomethylbenzylamine (NMBA)–induced forestomach tumors (modeling human esophageal cancer) was assessed. Cohorts of Wwox+/− and WT mice were treated with NMBA, an environmental carcinogen that is extensively utilized to induce esophageal and forestomach tumors in rodents, and monitored for tumor formation. Consistent with WWOX tumor suppressor function, it was observed that tumor incidence and multiplicity were higher in NMBA–treated Wwox+/− mice in comparison to NMBA–treated WT littermates (52). These results confirm that perturbations in WWOX could be associated with esophegeal tumor formation (85) and further suggest its important tumor suppressor function.
Wwox hypomorphic mice
This oncosuppressor function of WWOX was again strengthened by the phenotypes observed in a mouse model generated by the Aldaz group. In late 2007, Ludes–Meyers and co-workers reported the generation of a hypomorphic mouse strain containing mutated Wwox alleles using a gene trap approach (86). This mutagenesis resulted in the expression of a Wwox splice variant containing intact WW domains but no SDR domain, thus, generating homozygous Wwox mutant mouse named as gene trap alleles: Wwoxgt/gt. In these mice, WWOX expression was undetectable in most organs but low levels were found in a minority of tissues, hence, concluding that Wwoxgt/gt mice are WWOX hypomorphs. Unlike Wwox −/− mice (50), Wwoxgt/gt mice were viable for up to two years but had decreased survival rate when compared to WT mice. This increased viability phenotype was presumed to be a result of low expression of WWOX in some tissues, which might have been sufficient for postnatal survival of Wwoxgt/gt mice (86). Consistent with the Aqeilan Wwox −/− mouse model (50), higher incidence of tumor formation was noted in Wwoxgt/gt mice. Wwoxgt/gt female mice developed spontaneous B–cell lymphomas that was noted to be invasive in nature with a high degree of infiltration to lymph nodes, pancreas, kidneys, and liver. On the contrary, Wwoxgt/gt male mice developed lung adenomas as a characteristic of its genetic background. In addition, poorer reproductive capabilities were observed in both young and old Wwoxgt/gt male mice. The reduced fertility phenotype was predicted to be a consequence of severe degeneration of numerous seminiferous tubules causing premature testicular degeneration, therefore, indicating the important role of WWOX in the development and function of the reproductive system (86). These reports ultimately explained that loss of WWOX expression strongly correlates with tumor formation and aggression, impaired metabolism and defective steroidogenesis, as reported in (50).
Conditional Wwox–Knockout Models
Wwox-null mice suffer from post–natal lethality, hence restricting studying WWOX functions in adult mice (50). This certainly limits our understanding of WWOX comprehensive function in physiology and homeostasis as well as tumorigenesis. To overcome this limitation, the Aldaz group was the first to generate mice carrying a conditional mutant allele of Wwox by targeting exon 1 with LoxP–recombination sites (87). Wwoxlox/lox mice were then bred with a general deleter (EIIA–Cre) line to develop total Wwox KO mice. In agreement with conventional Wwox −/− mice results (50), these conditional Wwox KO mice displayed growth retardation and eventually post–natal lethality in comparison to HET and WT mice. The relative weights of multiple organs were significantly reduced whereas brain weight was increased in conditional Wwox KO pups consistent with previous reports on Wwox–null mice (59). Histology studies of spleen in these conditional Wwox KO mice revealed signs of splenic atrophy showing reduced cellularity of red pulp and lymphoid aggregates of white pulp. These mice also displayed signs of leukopenia with lower white blood cell count suggesting impaired hematopoiesis. Interestingly, conditional Wwox KO mice had significantly reduced bicarbonate levels in blood, a condition similar to metabolic acidosis. Blood chemistry results of these mice also displayed hypoglycaemia, hypocalcemia as well as had 2–fold higher blood urea nitrogen (BUN) causing kidney failure (87).
In parallel, the Aqeilan group also generated a conditional Wwox knockout model (named Wwoxf/f) by flanking exon 1 of Wwox with loxp sites (88). Wwoxf/f mice were also crossed with EIIA–Cre general deleter mice to generate Wwox KO (WwoxΔ/Δ) mice in all tissues. As expected, WwoxΔ/Δ mice displayed significant growth impairments and 100% of mice died by 3–4 weeks of age as a consequence of hypoglycemia and a severe metabolic disorder. Femurs of these mice displayed osteopenic phenotype, defected bone mineralisation process and presence of osteosarcoma–like cells, as observed in previously generated Wwox −/− mice (59). Altogether, both conditional mouse models (87, 88) were very much alike and recapitulated the null phenotype [Wwox −/− mice] (50). Nevertheless, there have been also some inconsistencies in phenotyping these models that shall be further investigated in future studies. Importantly, these models will be beneficial in investigating WWOX function in adult stages (see below).
Conditional Wwox ablation in mammary tissue
As mentioned earlier, perturbations in WWOX is a common event in breast tumors (32). Therefore, there has been tremendous efforts to uncover whether WWOX somatic deletion leads to mammary tumor formation. The Aldaz group generated a mammary-specific knockout of Wwox in a mixed 129SV/C57B1/6 genetic background using BK5–Cre and MMTV–Cre transgenic strains. Unlike the MMTV–Cre model which had no effect on mice survival, WWOX homozygous deletion using BK5–Cre mice resulted in premature death for unidentified reasons, as reported by the authors (89). Interestingly, both models (BK5 and MMTV) showed aberrant mammary branching morphogenesis displaying defects in branching development, impairment in ductal invasion, and expansion as well as abnormal mammary epithelium development with few ducts having no or reduced number of branching (89). Similar results on mammary development were observed in WwoxMGE−/− mice in B6/129 mixed genetic background, generated by the Aqeilan group using their Wwoxf/f mice upon breeding with MMTV–Cre transgenic mice (90). Surprisingly, none of these models developed mammary tumors (89, 90) hence questioning initiation role of tumor suppressor WWOX. This observation also led to propose that WWOX is a non–classical tumor suppressor, however this still remains to be proven as it depends on various factors, among which is dependency on cell of origin, combination with other genetic changes or even genetic background of the mouse model utilized as shown previously (51).
Recently, the Aqeilan group reported the first mouse model of somatic deletion of Wwox in mammary epithelium with mammary tumors resembling basal–like breast cancer (BLBC) (84). The authors back–crossed WwoxMGE−/− mice onto a mammary tumor susceptible background, C3H/HeJ mice, for seven rounds to generate WwoxΔMMTV. These mice were monitored for the incidence of tumor formation and indeed the majority (~76%) developed mammary tumors with median latency of 270 days whereas no tumors were obtained in WT control littermates. Histological and pathological characterization of these tumors revealed their Grade III invasive ductal carcinoma nature with occasional lung metastasis. The expression of ER and PR was negative in these tumors but ~60% were positive for CK14 hence resembling human BLBC. Further molecular analysis of WwoxΔMMTV tumors revealed that somatic loss of WWOX in mammary epithelium results in reduced expression and activity of p53. Interestingly when comparing WwoxΔMMTV tumors to those of Trp53ΔMMTV they cluster very closely indicating that WWOX and p53 cooperate to antagonize breast cancer development. Furthermore, perturbations in WWOX and TP53 co-occur and are correlated with poor survival of breast cancer patients. Altogether, these findings reveal WWOX as an important breast cancer tumor suppressor that regulates p53 function and activity (84)
Conditional ablation of Wwox in hepatocytes
Since Wwox −/− mice displayed reduced levels of serum lipids (50) and low expression of steroidogenic enzymes (61), a detailed analysis regarding role of WWOX in lipid metabolism was performed. Microarray analysis of total Wwox null mice (Wwox −/−) (50) and liver tissue–specific Wwox KO (Wwox hep−/−) mice (60) revealed altered levels of key regulators of high density lipoprotein (HDL) metabolism. Additionally, reduced levels of crucial biomolecules, ApoA−1 and AbcA1, necessary to reverse cholesterol transport and generate nascent HDL particles, were observed in both Wwox null and Wwox hep−/− mice. Interestingly, Wwox hep−/− male mice showed a small decrease in mRNA levels of AbcA1 but a significant reduction at protein levels. Conversely, unchanged mRNA and protein levels of ABCA1 were observed in Wwox hep−/− female mice. This discrepancy of ABCA1 expression at transcriptional as well as translational levels in both the sexes of Wwox hep−/− mice was considered to be a gender specific regulation of HDL metabolism. On the contrary, protein levels of ApoA–I were reduced in both Wwox hep−/− mice genders. These results were consistent with the observations reported in human disorders of HDL biogenesis suggesting a key role of WWOX in regulating HDL physiology (55). Additionally, older Wwox hep−/− female mice exhibited an increase in triglyceride (TG) levels and in very low–density lipoprotein (VLDL)–TG content within serum lipoproteins confirming previously reported association of Wwox and TG levels (50). Irrespective of the significant and prominent deregulation of lipid metabolism observed in Wwox hep−/− mice, microarray analysis also identified differential expression of several lipid–related canonical pathways specifically revealing the genes involved in cholesterol homeostasis, hydrolysis and biosynthesis of TG and fatty acid between WT and Wwox hep−/− mice. Further, network analysis of the microarray data demonstrated decreased HDL metabolism as well as upregulation of Angptl4, Fasn, Pltp, Gpam, Lipg, and downregulation of ApoA-I, Lpl, Insig2; the key genes involved in several lipid metabolic pathways, hence shedding light on the global and important effects of Wwox ablation in liver. Thus, this comprehensive examination of WWOX loss in hepatocytes demonstrated overall deregulation of lipid metabolic pathways along with gender specific regulation of HDL and TG metabolism indicating vital implications of WWOX in atherosclerosis and cardiovascular diseases (60).
In a more recent report, the effect of Wwox deletion in hepatocytes was assessed on development of liver cancer and liver regeneration (39). Given that WWOX is frequently altered in liver cancer, Abu–Remaileh and colleagues generated a mouse model with specific targeted deletion of murine Wwox alleles in hepatocytes (WwoxΔHep) and studied consequences on liver biology and development of hepatocellular carcinoma (HCC). Interestingly, WwoxΔHep mice exhibited more potent liver regeneration upon partial hepatectomy. This effect was accompanied with elevated Ki67 staining, a marker of proliferation, and higher levels of c–Myc transcripts, one of the major regulators of liver regeneration. Monitoring WwoxΔHep mice for 2–years didn't reveal increase in spontaneous liver cancer incidence in B6–129 background. Nevertheless, combined ablation of WWOX in hepatocytes and treatment with diethylnitrosamine (DEN), a known liver carcinogen, increased HCC tumor burden and load (39). This outcome was associated with increased levels of HIF1α, WWOX partner that drives aerobic glycolysis (36). WWOX deficiency also resulted in upregulation of HIF1α glycolytic target genes feeding enhanced proliferation. Inhibition of HIF1α activity in DEN–treated WwoxΔHep mice through systemic treatment with digoxin significantly attenuated tumor development suggesting that accelerated HCC development is indeed mediated by increased HIF1α activity. It has been also shown that feeding DEN–treated WwoxΔHep mice with high fat diet synergize to result in enhanced HCC formation. Altogether, these findings indicate that perturbations in WWOX could increase the risk of HCC development likely, but not only, through alteration in glucose metabolism (39).
Conditional ablation of Wwox in osteoblasts
Osteosarcoma is a highly aggressive and metastatic form of bone cancer that is frequent in adolescents and young adults. As mentioned before, WWOX loss has been associated with human osteosarcoma (63, 64, 66) and some animal models provide support for murine osteosarcoma development (50, 62, 88). Furthermore, WWOX protein is inversely associated with expression and function of RUNX2 (62, 63, 91), which is highly expressed in osteosarcoma and various metastatic cancers (64, 92, 93). To better clarify the role of WWOX in osteosarcoma, the Aqeilan group generated two osteoblast-specific knockout mouse models in which WWOX is ablated in either pre–osteoblasts (WwoxΔOsx1) or in fully mature osteoblasts (WwoxΔOc) (68). Analysis of these mice revealed that WwoxΔOsx1 mice exhibit a severe defect in osteogenesis, which was associated with induction of p53. Deletion of Trp53 in WwoxΔOsx1 mice rescued the osteogenic defect and resulted in the development of high penetrance of poorly differentiated osteosarcomas. The murine phenotype very much resembled human osteosarcoma in different aspects including histology, gene expression resistance to chemotherapy and metastatic behavior (68). Strikingly, it was demonstrated that WWOX might undergo loss of heterozygosity and that it promotes p53 loss of heterozygosity supporting a the emerging role of WWOX in DNA damage response and genomic stability (35, 94) ultimately confirming an intimate relationship between the two tumor suppressors. Importantly, co–occurrence of WWOX and p53 inactivation has been demonstrated as a common event in osteosarcoma (68). Altogether these findings provided the first in vivo evidence that WWOX suppresses osteosarcomagenesis through regulating p53 activity.
Significance of WWOX in Neurological Disorders
The high levels of WWOX in the different parts of the brain (95, 96), suggests an indispensable role of WWOX in central nervous system (CNS) homeostasis. Initially, low WWOX expression was reported in the hippocampus of Alzheimer's disease (AD) patients (97) suggesting a plausible role of WWOX in the biology of AD. The Chang group and collaborators have subsequently generated a Wwox null mouse by targeting exons 1, 2, 3, and 4 to evaluate physiological significance of WWOX in this neurodegenerative disease. In agreement with previously published data (50), KO mice survived only for about a month (98). Interestingly, it was revealed that homozygous loss of Wwox leads to the aggregation of Tau and TPC6AΔ (known to be involved in AD), in brain cortex of juvenile Wwox null mice, prior to their death (98). This suggests that WWOX plays a crucial role in inhibiting the aggregation of these plaque forming proteins which cause neurodegenerative diseases (98). Whether specific deletion of Wwox in hippocampal or cortical neurons would be associated with or contribute to AD in mice is still unknown.
Numerous subsequent reports documented a number of WWOX mutations in different neurological disorders. Indeed, WWOX germline mutations were found associated with developmental retardation, ataxia, early onset of epilepsy and intellectual deficiency (53, 54, 99–103). WWOX-mutant patients display a wide range of neurological behaviors ranging from progressive microcephaly, global developmental delay, seizure disorders, bilateral optic atrophy, and spastic quadriplegia in very young infants (~1.5 months) (known as WOREE phenotype, for WWOX–related epileptic encephalopathy), to non–progressive microcephaly and less severe phenotype in adolescence–adult with later onset at 9–12 months (associated with spinocerebellar ataxia type 12 (SCAR12)) (101). This wide range of phenotypic abnormalities could be due to the nature of these mutations. Relatively milder phenotypes seem to originate from missense point mutations (P47T and P47R, G372R), whereas severe manifestations were observed in nonsense mutations (R54*, K297*, and W335*) or partial/complete deletions [reviewed in (38)]. Therefore, WWOX genotypes might correlate with the reported phenotypes, although analysis of more patients would be required to further support this relationship.
WWOX involvement in epilepsy was also documented in animal models. Indeed, Wwox gene mutations were first associated with epilepsy and ataxia in mice. Mallaret et al. showed that the short–lived Wwox KO mouse displays spontaneous and audiogenic seizures (53). This phenotype was also observed in a spontaneous homozygous rat mutation of Wwox, (lethal dwarfism, ataxia, and epilepsy) presenting similar phenotype as the Wwox KO mice and symptoms similar to mutant WWOX patients (104). The molecular function and importance of WWOX in epilepsy and ataxia is largely unknown and is currently under investigation by several labs.
Rats
The role of WWOX in biology was also documented in rats as an animal model. The phenotype of spontaneously mutated–Lethal dwarfism with epilepsy (lde) in rats display severe dwarfism, early post–natal lethality, a high incidence of epileptic seizures, male hypogonadism and the presence of numerous extracellular vacuoles of different sizes in the hippocampus and amygdala of mutant brain (105). To define the genes responsible for the lde phenotype, the authors backcrossed lde/lde rats with Brown Norway rats to generate lde/lde rats with an altered genetic background displaying all the pleotropic phenotypes. Mapping of 1.5–Mbp region of chromosome 19 within lde locus revealed a 13 base pair deletion in exon 9 of Wwox transcript that caused a frame–shift mutation resulting in the translation of aberrant amino acid sequences at WWOX C–terminus. Considering epileptogenesis as congenital in lde/lde rats, these rats were exposed to different sound stimulus resulting in audiogenic seizures that had a pattern beginning with wild running followed by tonic–clonic convolusions. There was no correlation between the age of rat and the incidence of audiogenic seizures. Overall, these results further suggest an important role for WWOX in the CNS as WWOX loss leads to neuronal excitation (104).
WWOX was also described to be a putative target to alleviate symptoms for patients who experience a neuronal injury (106). Here, the authors studied the functional significance of rat Wwox and other transcription factors necessary to decide injured neuronal cell fate. Nuclear WWOX levels were rapidly increased in the neurons of sciatic nerve transection model in rats. This induction was accompanied with activation of other known genes involved in neuronal injury such as NF–κB activation and phosphorylation of CREB and JNK1. Eventually, the authors concluded that the elevation of WWOX levels signals the injured neurons to undergo apoptosis through activation of pro-apoptotic pathways (106).
Altogether, Wwox manipulation in the rat is associated with important phenotypes highlighting WWOX physiological functions, nevertheless the early death of these rats precludes analysis of adult phenotypes. It is of note that some of the phenotypes reported in the mouse models were not reported or observed in the rat model. This could be related to the approach used in genetic manipulation and/or genetic background.
Drosophila Melanogaster
A high amino acid sequence conservation along with distinct phylogenetic relationship of WWOX across species proposes retention of the major biological functions governed by the WWOX gene. The Drosophila orthologue of WWOX (DmWwox) was investigated with an aim to study its involvement in WWOX biological functions. DmWwox mutants (DmWwox1) were therefore generated and characterized (49). The DmWwox1 mutants were surprisingly viable and exhibited no obvious phenotype. Neverthless, a higher sensitivity and a decrease in the survival rate of DmWwox1 mutant third instar larvae was observed following dose–dependent irradiation when compared with WT. Protective role of WWOX in Drosophila was confirmed when DmWwox1 mutants rescued the WT phenotype with both DmWwox and human WWOX constructs. Therefore, based on these results, authors concluded that CFSs and the genes within its vicinity would be involved in protective mechanisms against environmental perturbations (49). Later on, it was reported that background mutation as a result of insertion of piggyBag transposon into the second intron of DmWwox was responsible for this phenotype. Hence, the authors proposed that a thorough screening of alleles generated by targeted mutagenesis via homologous recombination is mandatory to control the genetic background in Drosophila (107). To overcome this drawback and to determine the biological functions of WWOX, a distinct combinatorial approach by using biochemical screening, such as proteomic and microarray screens, as well as genetic analysis to explain functionality of the interactions was performed in Drosophila. It was surprising that proteomic analysis did not show any physical interactions of WWOX through its WW domain with PPxY motifs containing proteins. However, functional partners of DmWWOX protein were identified by mass spectrometry and shed light on novel functional characteristics of WWOX contributing to cellular metabolism. The analysis identified significantly altered 13 candidate proteins in DmWwox mutants as well as 16 proteins in WWOX ectopically over–expressed adult flies most of which were known to be involved in metabolic pathways. It was striking that two candidate interactors, catalytic enzymes–isocitrate dehydrogenase (CG6439) and malate dehydrogenase (CG7998) involved in Tri–Carboxylic Acid (TCA) cycle were identified indicating an important contribution of WWOX toward aerobic metabolism. Also several candidates involved in glucose and lipid metabolism were identified. Additionally, enzymes invovled in oxidative stress pathways, such as superoxide dismutase (Sod) enzymes were identified as candidate WWOX interactors. This later finding could propose a direct role of the oxidoreductase domain of WWOX in biology of reactive oxygen species (ROS), a known byproduct of oxidative phosphorylation (41).
Using Drosophila as a model system, WWOX was also reported to be a modulator of apoptosis via modulation of Caspase-3 and regulation of ROS activity. During Drosophila development, ectopic over–expression of low level expression construct for TNFα/Egr caused disruption of repeated ommatidial unit patterning on the eye surface and reduced overall eye size. Introduction of DmWwox knock down construct rescued this TNFα/Egr mediated rough eye phenotype as ommatidial patterning across the surface of the eye was restored and the size of the eye was increased. Further, it was shown that WWOX activity is essential for the removal of tumorigenic cells from a developing epithelial tissue (42). Altogether, it is obvious that modeling WWOX loss in Drosophila identified conserved WWOX functions in cellular metabolism and apoptosis that has also been reported, at least in part, in mammalian cells (37, 40).
Zebrafish
The biology of WWOX was also studied in zebrafish as an animal model. Tsuruwaka and co–workers generated antisense morpholinos (MO) and siRNA oligos against wwox gene sequence in Danio rerio and studied their phenotypes. Embryos harboring genetic manipulated wwox were characterized by pericardial edema causing post–natal lethality within a week of age. Wwox-deficient zebrafish embryos display growth retardation, as revealed by small eye and head size as well as short body lengths combined with impaired bone development and intracellular Ca2+ levels (108). Altogether, wwox–mutant zebrafish embryos recapitulate many of the phenotypes of Wwox–null rodent models further implicating the conservation of WWOX function across species.
Summary and Future Perspective
Modeling of WWOX gene in mice, rats, Drosophila, and zebrafish have not only advanced our understanding of its tumor suppressor functions but also have identified its emerging physiological roles in different human pathologies (Figure 1). Targeted ablation of WWOX in these established animal models facilitated the characterization of cellular pathways controlled by WWOX and that its deregulation exhibited severe pathological conditions like cancer, CNS related pathologies, metabolic syndromes, and reproductive defects (Table 1).
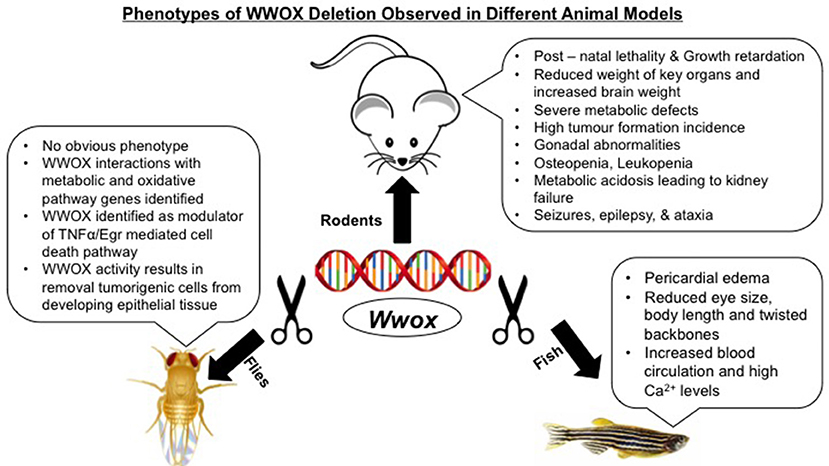
Figure 1. Phenotypes of WWOX deletion observed in different animal models.
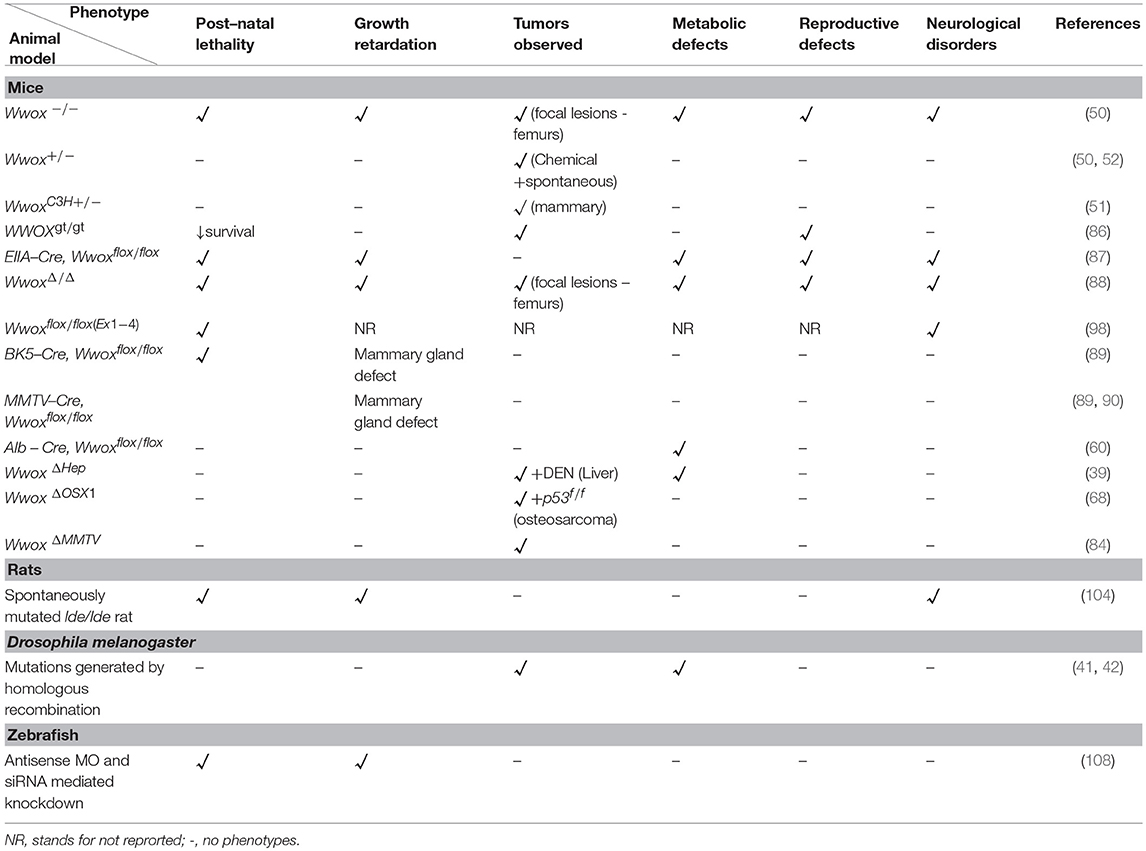
Table 1. Common and unique phenotypes for WWOX animal models.
The phenotypes of Wwox null mice include early post–natal lethality, significant growth retardation and dwarfism, severe metabolic defects, osteopenia, and gonadal abnormalities (62, 83, 109). Along with the above mentioned complex phenotypes, Wwox null mice also displayed spontaneous and audiogenic seizures as well as showed aggregation of plaque forming protein in the brain cortex indicating that WWOX has an importnat role in the CNS and its loss could lead to epilepsy, ataxia and AD (45, 83). Similar epileptogenic and ataxic phenotype was observed in spontaneously mutated lde rats along with male rat hypogonadism (104).
Despite of early post–natal lethality, juvenile Wwox null mice phenotype demonstrated the occurrence of lesions resembling osteosarcomas, the first direct evidence of tumor suppressor functions of WWOX (50, 63). Although, this later finding was not detected in all null models but a conserved anti-oncosuppressor function was documented in heterozygous or hypomorph mutant mice (50, 52, 86). Moreover, carcinogen experiments in Wwox-heterozygous mutant mice showed higher tumor incidence and multiplicity, compared with control mice, of different tumor types implying WWOX haploinsufficiency (50, 52), a hallamrk of many known tumor suppressors. Conditional Wwox KO mouse models recapitulated many of the Wwox null phenotypes (87, 88). These conditional mouse models facilitated and will be instrumental in studying the distinct physiological functions of WWOX in a tissue-specific manner. Conditional deletion of Wwox in murine mammary epithelium revealed mammary developmental defects suggesting that WWOX expression is indispensable for proper normal breast development (89, 90). More recently, somatic deletion of Wwox in mammary epithelium of C3H mice resulted in high penetrance of mammry tumorigenesis that is associated with p53 perturbations (84). Liver tissue-specific conditional Wwox KO mouse model revealed WWOX involvement in the complex network of cholesterol homeostasis, HDL and lipoprotein metabolism highlighting a role of WWOX in regulating lipid metabolic pathways and underscoring a therapeutic use of WWOX for atherosclerosis and cardiovascular diseases (60). Somatic Wwox deletion in hepatocytes combined with DEN–treatment and/or western diet have been also reported to result in increased HCC incidence further elucidating the tumor suppressor role of WWOX (39).
Drosophila models of WWOX orthologues identified candidate interactors of WWOX in metabolic as well as oxidative pathways (47). Also, WWOX function for the removal of tumorigenic cells from developing epithelial cells was also reported in the Drosophila model (42). Lastly, WWOX ablation within zebrafish embryos displayed a non–cell–autonomous phenotype characterized by the symptoms of pericardial edema, reduced eye size and body length as well as twisted backbones along with high blood circulation and Ca2+ levels at ventral–dorsal–posterior regions (108).
Taken together, modeling WWOX in different animal models established WWOX functions in cancer as well as revealed its contribution in metabolic syndromes and neuropathy. These studies identified numerous interacting protein partners of WWOX and indicated new and possible physiological functions (110). The emerging roles of WWOX in DNA damage response (34, 35, 111) and in cellular metabolism (36, 41, 46) could have far reaching effects on the disorders identified in animal models and human patients. Complete understanding of the physiological functions of WWOX is yet under explored and much still remains to be learned about the enzymatic functions of SDR domain. Further characterization of these phenotypic defects, observed both at molecular and cellular levels in WWOX animal models, would be essential for designing novel diagnostic, prognostic, and therapeutic tools.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The Aqeilan lab is funded by the European Research Council (ERC)-Consolidator Grant (grant agreement No. 682118), Israel science foundation grant (grant agreement No. 15/1574) and the Alex U. Soyka Pancreatic Cancer Research grant (CFHU).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank all the current and alumni members of the Aqeilan group who contributed to better understanding of WWOX functions.
References
1. Sutherland GR. Heritable fragile sites on human chromosomes I. Factors affecting expression in lymphocyte culture. Am J Hum Genet. (1979) 31:125–35.
2. Sutherland GR, Baker E, Seshadri RS. Heritable fragile sites on human chromosomes. V. A new class of fragile site requiring BrdU for expression. Am J Hum Genet. (1980) 32:542–8.
3. Sutherland GR, Jacky PB, Baker EG. Heritable fragile sites on human chromosomes. XI. Factors affecting expression of fragile sites at 10q25, 16q22, and 17p12. Am J Hum Genet. (1984) 36:110–22.
4. Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. (1984) 67:136–42. doi: 10.1007/BF00272988
5. Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. (2007) 41:169–92. doi: 10.1146/annurev.genet.41.042007.165900
6. Schrock MS, Batar B, Lee J, Druck T, Ferguson B, Cho JH, et al. Wwox-Brca1 interaction: role in DNA repair pathway choice. Oncogene (2017) 36:2215–27. doi: 10.1038/onc.2016.389
7. Dekaban A. Persisting clone of cells with an abnormal chromosome in a woman previously irradiated. J Nucl Med. (1965) 6:740–6.
8. Magenis RE, Hecht F, Lovrien EW. Heritable fragile site on chromosome 16: probable localization of haptoglobin locus in man. Science (1970) 170:85–7. doi: 10.1126/science.170.3953.85
9. Giraud F, Ayme S, Mattei JF, Mattei MG. Constitutional chromosomal breakage. Hum Genet. (1976) 34:125–36. doi: 10.1007/BF00278880
10. Harvey J, Judge C, Wiener S. Familial X-linked mental retardation with an X chromosome abnormality. J Med Genet. (1977) 14:46–50. doi: 10.1136/jmg.14.1.46
12. Turner G, Till R, Daniel A. Marker X chromosomes, mental retardation and macro-orchidism. N Engl J Med. (1978) 299:1472. doi: 10.1056/NEJM197812282992624
13. Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science (1977) 197:265–6. doi: 10.1126/science.877551
14. Schwartz M, Zlotorynski E, Kerem B. The molecular basis of common and rare fragile sites. Cancer Lett. (2006) 232:13–26. doi: 10.1016/j.canlet.2005.07.039
15. Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. a genome-wide study. Oncogene (2008) 27:3256–64. doi: 10.1038/sj.onc.1210989
16. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature (2005) 434:864–70. doi: 10.1038/nature03482
17. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature (2005) 434:907–13. doi: 10.1038/nature03485
18. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science (2008) 319:1352–5. doi: 10.1126/science.1140735
19. Sarni D, Kerem B. The complex nature of fragile site plasticity and its importance in cancer. Curr Opin Cell Biol. (2016) 40:131–6. doi: 10.1016/j.ceb.2016.03.017
20. Hecht F, Glover TW. Cancer chromosome breakpoints and common fragile sites induced by aphidicolin. Cancer Genet Cytogenet. (1984) 13:185–8. doi: 10.1016/0165-4608(84)90060-8
21. Hecht F, Sutherland GR. Fragile sites and cancer breakpoints. Cancer Genet Cytogenet. (1984) 12:179–81. doi: 10.1016/0165-4608(84)90132-8
22. LeBeau MM, Rowley JD. Heritable fragile sites in cancer. Nature (1984) 308:607–8. doi: 10.1038/308607a0
23. Yunis JJ, Soreng AL. Constitutive fragile sites and cancer. Science (1984) 226:1199–204. doi: 10.1126/science.6239375
24. Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. (2000) 60:2140–5.
25. Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, et al. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet. (2000):1651–63.
26. Del Mare S, Salah Z, Aqeilan RI. WWOX: its genomics, partners, and functions. J Cell Biochem. (2009) 108:737–45. doi: 10.1002/jcb.22298
27. Chang NS, Pratt N, Heath J, Schultz L, Sleve D, Carey GB, et al. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J Biol Chem. (2001) 276:3361–70. doi: 10.1074/jbc.M007140200
28. Salah Z, Alian A, Aqeilan RI. WW domain-containing proteins: retrospectives and the future. Front Biosci. (2012)17:331–48. doi: 10.2741/3930
29. Salah Z, Aqeilan R, Huebner K. WWOX gene and gene product: tumor suppression through specific protein interactions. Future Oncol. (2010) 6:249–59. doi: 10.2217/fon.09.152
30. Mangelsdorf M, Ried K, Woollatt E, Dayan S, Eyre H, Finnis M, et al. Chromosomal fragile site FRA16D and DNA instability in cancer. Cancer Res. (2000) 60:1683–9.
31. Paige AJ, Taylor KJ, Stewart A, Sgouros JG, Gabra H, Sellar GC, et al. A 700-kb physical map of a region of 16q23.2 homozygously deleted in multiple cancers and spanning the common fragile site FRA16D. Cancer Res. (2000) 60:1690–7.
32. Gardenswartz A, Aqeilan RI. WW domain-containing oxidoreductase's role in myriad cancers: clinical significance and future implications. Exp Biol Med. (2014) 239:253–63. doi: 10.1177/1535370213519213
33. Donati V, Fontanini G, Dell'Omodarme M, Prati MC, Nuti S, Lucchi M, et al. WWOX expression in different histological types and subtypes of non-small cell lung cancer. Clin Cancer Res. (2007) 13:884–91. doi: 10.1158/1078-0432.CCR-06-2016
34. Abu-Odeh M, Hereema NA, Aqeilan RI. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget (2016) 7:4344–55. doi: 10.18632/oncotarget.6571
35. Abu-Odeh M, Salah Z, Herbel C, Hofmann TG, Aqeilan RI. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci USA. (2014) 111:E4716–25. doi: 10.1073/pnas.1409252111
36. Abu-Remaileh M, Aqeilan RI. Tumor suppressor WWOX regulates glucose metabolism via HIF1alpha modulation. Cell Death Differ. (2014) 21:1805–14. doi: 10.1038/cdd.2014.95
37. Abu-Remaileh M, Aqeilan RI. The tumor suppressor WW domain-containing oxidoreductase modulates cell metabolism. Exp Biol Med (Maywood). (2015) 240:345–50. doi: 10.1177/1535370214561956
38. Abu-Remaileh M, Joy-Dodson E, Schueler-Furman O, Aqeilan RI. Pleiotropic functions of tumor suppressor WWOX in normal and cancer cells. J Biol Chem. (2015) 290:30728–35. doi: 10.1074/jbc.R115.676346
39. Abu-Remaileh M, Khalaileh A, Pikarsky E, Aqeilan RI. WWOX controls hepatic HIF1alpha to suppress hepatocyte proliferation and neoplasia. Cell Death Dis. (2018) 9:511. doi: 10.1038/s41419-018-0510-4
40. Abu-Remaileh M, Seewaldt VL, Aqeilan RI. WWOX loss activates aerobic glycolysis. Mol Cell Oncol. (2015) 2:e965640. doi: 10.4161/23723548.2014.965640
41. O'Keefe LV, Colella A, Dayan S, Chen Q, Choo A, Jacob R, et al. Drosophila orthologue of WWOX, the chromosomal fragile site FRA16D tumour suppressor gene, functions in aerobic metabolism and regulates reactive oxygen species. Hum Mol Genet. (2011) 20:497–509. doi: 10.1093/hmg/ddq495
42. O'Keefe LV, Lee CS, Choo A, Richards RI. Tumor suppressor WWOX contributes to the elimination of tumorigenic cells in drosophila melanogaster. PLoS ONE (2015) 10:e0136356. doi: 10.1371/journal.pone.0136356
43. Chang NS, Hsu LJ, Lin YS, Lai FJ, Sheu HM. WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol Med. (2007) 13:12–22. doi: 10.1016/j.molmed.2006.11.006
44. El-Hage P, Petitalot A, Monsoro-Burq AH, Maczkowiak F, Driouch K, Formstecher E, et al. The tumor-suppressor WWOX and HDAC3 inhibit the transcriptional activity of the beta-catenin coactivator BCL9-2 in breast cancer cells. Mol Cancer Res. (2015) 13:902–12. doi: 10.1158/1541-7786.MCR-14-0180
45. Chang NS. Introduction to a thematic issue for WWOX. Exp Biol Med (Maywood). (2015) 240:281–4. doi: 10.1177/1535370215574226
46. Aqeilan RI, Abu-Remaileh M, Abu-Odeh M. The common fragile site FRA16D gene product WWOX: roles in tumor suppression and genomic stability. Cell Mol Life Sci. (2014) 71:4589–99. doi: 10.1007/s00018-014-1724-y
47. Richards RI, Choo A, Lee CS, Dayan S, O'Keefe L. WWOX, the chromosomal fragile site FRA16D spanning gene: its role in metabolism and contribution to cancer. Exp Biol Med (Maywood). (2015) 240:338–44. doi: 10.1177/1535370214565990
48. Krummel KA, Denison SR, Calhoun E, Phillips LA, Smith DI. The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1. Genes Chromosomes Cancer (2002) 34:154–67. doi: 10.1002/gcc.10047
49. O'Keefe LV, Liu Y, Perkins A, Dayan S, Saint R, Richards RI. FRA16D common chromosomal fragile site oxido-reductase (FOR/WWOX) protects against the effects of ionizing radiation in Drosophila. Oncogene (2005) 24:6590–6. doi: 10.1038/sj.onc.1208806
50. Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci USA. (2007) 104:3949–54. doi: 10.1073/pnas.0609783104
51. Abdeen SK, Salah Z, Maly B, Smith Y, Tufail R, Abu-Odeh M, et al. Wwox inactivation enhances mammary tumorigenesis. Oncogene (2011) 30:3900–6. doi: 10.1038/onc.2011.115
52. Aqeilan RI, Hagan JP, Aqeilan HA, Pichiorri F, Fong LY, Croce CM. Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. (2007) 67:5606–10. doi: 10.1158/0008-5472.CAN-07-1081
53. Mallaret M, Synofzik M, Lee J, Sagum CA, Mahajnah M, Sharkia R, et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain (2014) 137:411–9. doi: 10.1093/brain/awt338
54. Abdel-Salam G, Thoenes M, Afifi HH, Korber F, Swan D, Bolz HJ. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J Rare Dis. (2014) 9:12. doi: 10.1186/1750-1172-9-12
55. Lee JC, Weissglas-Volkov D, Kyttala M, Dastani Z, Cantor RM, Sobel EM, et al. WW-domain-containing oxidoreductase is associated with low plasma HDL-C levels. Am J Hum Genet. (2008) 83:180–92. doi: 10.1016/j.ajhg.2008.07.002
56. Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. (2008) 40:161–9. doi: 10.1038/ng.76
57. Wang K, Li WD, Zhang CK, Wang Z, Glessner JT, Grant SF, et al. A genome-wide association study on obesity and obesity-related traits. PLoS ONE (2011) 6:e18939. doi: 10.1371/journal.pone.0018939
58. Chang YC, Chiu YF, Liu PH, Shih KC, Lin MW, Sheu WH, et al. Replication of genome-wide association signals of type 2 diabetes in Han Chinese in a prospective cohort. Clin Endocrinol. (2012) 76:365–72. doi: 10.1111/j.1365-2265.2011.04175.x
59. Aqeilan RI, Hassan MQ, de Bruin A, Hagan JP, Volinia S, Palumbo T, et al. The WWOX tumor suppressor is essential for postnatal survival and normal bone metabolism. J Biol Chem. (2008) 283:21629–39. doi: 10.1074/jbc.M800855200
60. Iatan I, Choi HY, Ruel I, Reddy MV, Kil H, Lee J, et al. The WWOX gene modulates high-density lipoprotein and lipid metabolism. Circ Cardiovasc Genet. (2014) 7:491–504. doi: 10.1161/CIRCGENETICS.113.000248
61. Aqeilan RI, Hagan JP, de Bruin A, Rawahneh M, Salah Z, Gaudio E, et al. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology (2009) 150:1530–5. doi: 10.1210/en.2008-1087
62. Del Mare S, Kurek KC, Stein GS, Lian JB, Aqeilan RI. Role of the WWOX tumor suppressor gene in bone homeostasis and the pathogenesis of osteosarcoma. Am J Cancer Res. (2011) 1:585–94.
63. Kurek KC, Del Mare S, Salah Z, Abdeen S, Sadiq H, Lee SH, et al. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. (2010) 70:5577–86. doi: 10.1158/0008-5472.CAN-09-4602
64. Yang J, Zhao L, Tian W, Liao Z, Zheng H, Wang G, et al. Correlation of WWOX, RUNX2 and VEGFA protein expression in human osteosarcoma. BMC Med Genomic (2013) 6:56. doi: 10.1186/1755-8794-6-56
65. Zheng QW, Zhou YL, You QJ, Shou F, Pang QF, Chen JL. WWOX inhibits the invasion of lung cancer cells by downregulating RUNX2. Cancer Gene Ther. (2016) 23:433–8. doi: 10.1038/cgt.2016.59
66. Yang J, Cogdell D, Yang D, Hu L, Li H, Zheng H, et al. Deletion of the WWOX gene and frequent loss of its protein expression in human osteosarcoma. Cancer Lett. (2010) 291:31–8. doi: 10.1016/j.canlet.2009.09.018
67. Liu P, Wang M, Li L, Jin T. Correlation between osteosarcoma and the expression of WWOX and p53. Oncol Lett. (2017) 14:4779–83. doi: 10.3892/ol.2017.6747
68. Del Mare S, Husanie H, Iancu O, Abu-Odeh M, Evangelou K, Lovat F, et al. WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res. (2016) 76:6107–17. doi: 10.1158/0008-5472.CAN-16-0621
69. Yendamuri S, Kuroki T, Trapasso F, Henry AC, Dumon KR, Huebner K, et al. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res. (2003) 63:878–81.
70. Kimura M, Takenobu H, Akita N, Nakazawa A, Ochiai H, Shimozato O, et al. Bmi1 regulates cell fate via tumor suppressor WWOX repression in small-cell lung cancer cells. Cancer Sci. (2011) 102:983–90. doi: 10.1111/j.1349-7006.2011.01891.x
71. Baykara O, Demirkaya A, Kaynak K, Tanju S, Toker A, Buyru N. WWOX gene may contribute to progression of non-small-cell lung cancer (NSCLC). Tumour Biol. (2010) 31:315–20. doi: 10.1007/s13277-010-0039-3
72. Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan RI, Rabson AB, et al. The tumor suppressor gene WWOX links the canonical and noncanonical NF-kappaB pathways in HTLV-I Tax-mediated tumorigenesis. Blood (2011) 117:1652–61. doi: 10.1182/blood-2010-08-303073
73. Roy D, Sin SH, Damania B, Dittmer DP. Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood (2011) 118:e32–9. doi: 10.1182/blood-2010-12-323659
74. Handa H, Sasaki Y, Hattori H, Alkebsi L, Kasamatsu T, Saitoh T, et al. Recurrent alterations of the WW domain containing oxidoreductase gene spanning the common fragile site FRA16D in multiple myeloma and monoclonal gammopathy of undetermined significance. Oncol Lett. (2017) 14:4372–8. doi: 10.3892/ol.2017.6672
75. Huang SS, Su WP, Lin HP, Kuo HL, Wei HL, Chang NS. Role of WW domain-containing oxidoreductase WWOX in driving T cell acute lymphoblastic leukemia maturation. J Biol Chem. (2016) 291:17319–31. doi: 10.1074/jbc.M116.716167
76. Shi Y, Du M, Fang Y, Tong N, Zhai X, Sheng X, et al. Identification of a novel susceptibility locus at 16q23.1 associated with childhood acute lymphoblastic leukemia in Han Chinese. Hum Mol Genet. (2016) 25:2873–80. doi: 10.1093/hmg/ddw112
77. Guler G, Himmetoglu C, Jimenez RE, Geyer SM, Wang WP, Costinean S, et al. Aberrant expression of DNA damage response proteins is associated with breast cancer subtype and clinical features. Breast Cancer Res Treat (2011) 129:421–32. doi: 10.1007/s10549-010-1248-6
78. Guler G, Iliopoulos D, Guler N, Himmetoglu C, Hayran M, Huebner K. Wwox and Ap2gamma expression levels predict tamoxifen response. Clin Cancer Res. (2007) 13:6115–21. doi: 10.1158/1078-0432.CCR-07-1282
79. Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, et al. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer (2004) 100:1605–14. doi: 10.1002/cncr.20137
80. Guler G, Uner A, Guler N, Han SY, Iliopoulos D, McCue P, et al. Concordant loss of fragile gene expression early in breast cancer development. Pathol Int. (2005) 55:471–8. doi: 10.1111/j.1440-1827.2005.01855.x
81. Iliopoulos D, Guler G, Han SY, Johnston D, Druck T, McCorkell KA, et al. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene (2005) 24:1625–33. doi: 10.1038/sj.onc.1208398
82. Aqeilan RI, Donati V, Gaudio E, Nicoloso MS, Sundvall M, Korhonen A, et al. Association of Wwox with ErbB4 in breast cancer. Cancer Res. (2007) 67:9330–6. doi: 10.1158/0008-5472.CAN-07-2147
83. Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta (2014) 1846:188–200. doi: 10.1016/j.bbcan.2014.06.001
84. Abdeen SK, Ben-David U, Shweiki A, Maly B, Aqeilan RI. Somatic loss of WWOX is associated with TP53 perturbation in basal-like breast cancer. Cell Death Dis. (2018) 9:832. doi: 10.1038/s41419-018-0896-z
85. Kuroki T, Trapasso F, Shiraishi T, Alder H, Mimori K, Mori M, et al. Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res. (2002) 62:2258–60.
86. Ludes-Meyers JH, Kil H, Nunez MI, Conti CJ, Parker-Thornburg J, Bedford MT, et al. WWOX hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosomes Cancer (2007) 46:1129–36. doi: 10.1002/gcc.20497
87. Ludes-Meyers JH, Kil H, Parker-Thornburg J, Kusewitt DF, Bedford MT, Aldaz CM. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS ONE (2009) 4:e7775. doi: 10.1371/journal.pone.0007775
88. Abdeen SK, Del Mare S, Hussain S, Abu-Remaileh M, Salah Z, Hagan J, et al. Conditional inactivation of the mouse Wwox tumor suppressor gene recapitulates the null phenotype. J Cell Physiol. (2013) 228:1377–82. doi: 10.1002/jcp.24308
89. Ferguson BW, Gao X, Kil H, Lee J, Benavides F, Abba MC, et al. Conditional Wwox deletion in mouse mammary gland by means of two cre recombinase approaches. PLoS ONE (2012) 7:e36618. doi: 10.1371/journal.pone.0036618
90. Abdeen SK, Salah Z, Khawaled S, Aqeilan RI. Characterization of WWOX inactivation in murine mammary gland development. J Cell Physiol. (2013) 228:1391–6. doi: 10.1002/jcp.24310
91. Del Mare S, Aqeilan RI. Tumor Suppressor WWOX inhibits osteosarcoma metastasis by modulating RUNX2 function. Sci Rep. (2015) 5:12959. doi: 10.1038/srep12959
92. Martin JW, Zielenska M, Stein GS, van Wijnen AJ, Squire JA. The Role of RUNX2 in Osteosarcoma Oncogenesis. Sarcoma (2011) 2011:282745. doi: 10.1155/2011/282745
93. van der Deen M, Akech J, Lapointe D, Gupta S, Young DW, Montecino MA, et al. Genomic promoter occupancy of runt-related transcription factor RUNX2 in Osteosarcoma cells identifies genes involved in cell adhesion and motility. J Biol Chem. (2012) 287:4503–17. doi: 10.1074/jbc.M111.287771
94. Hazan I, Abu Odeh M, Hofmann TG, Aqeilan RI. WWOX guards genome stability by activating ATM. Molecular and cellular oncology. (2015) 2:e1008288. doi: 10.1080/23723556.2015.1008288
95. Chen ST, Chuang JI, Wang JP, Tsai MS, Li H, Chang NS. Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience (2004) 124:831–9. doi: 10.1016/j.neuroscience.2003.12.036
96. Nunez MI, Ludes-Meyers J, Aldaz CM. WWOX protein expression in normal human tissues. J Mol Histol. (2006) 37:115–25. doi: 10.1007/s10735-006-9046-5
97. Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, et al. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer's disease. J Biol Chem. (2004) 279:30498–506. doi: 10.1074/jbc.M401399200
98. Chang JY, Lee MH, Lin SR, Yang LY, Sun HS, Sze CI, et al. Trafficking protein particle complex 6A delta (TRAPPC6ADelta) is an extracellular plaque-forming protein in the brain. Oncotarget (2015) 6:3578–89. doi: 10.18632/oncotarget.2876
99. Ben-Salem S, Al-Shamsi AM, John A, Ali BR, Al-Gazali L. A novel whole exon deletion in WWOX gene causes early epilepsy, intellectual disability and optic atrophy. J Mol Neurosci. (2015) 56:17–23. doi: 10.1007/s12031-014-0463-8
100. Elsaadany L, El-Said M, Ali R, Kamel H, Ben-Omran T. W44X mutation in the WWOX gene causes intractable seizures and developmental delay: a case report. BMC Med Genet. (2016) 17:53. doi: 10.1186/s12881-016-0317-z
101. Mignot C, Lambert L, Pasquier L, Bienvenu T, Delahaye-Duriez A, Keren B, et al. WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J Med Genet. (2015) 52:61–70. doi: 10.1136/jmedgenet-2014-102748
102. Tabarki B, AlHashem A, AlShahwan S, Alkuraya FS, Gedela S, Zuccoli G. Severe CNS involvement in WWOX mutations: description of five new cases. Am J Med Genet A. (2015) 167A:3209–13. doi: 10.1002/ajmg.a.37363
103. Valduga M, Philippe C, Lambert L, Bach-Segura P, Schmitt E, Masutti JP, et al. WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J Hum Genet. (2015) 60:267–71. doi: 10.1038/jhg.2015.17
104. Suzuki H, Katayama K, Takenaka M, Amakasu K, Saito K, Suzuki K. A spontaneous mutation of the Wwox gene and audiogenic seizures in rats with lethal dwarfism and epilepsy. Genes Brain Behav. (2009) 8:650–60. doi: 10.1111/j.1601-183X.2009.00502.x
105. Suzuki H, Takenaka M, Suzuki K. Phenotypic characterization of spontaneously mutated rats showing lethal dwarfism and epilepsy. Comp Med. (2007) 57:360–9.
106. Li MY, Lai FJ, Hsu LJ, Lo CP, Cheng CL, Lin SR, et al. Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE (2009) 4:e7820. doi: 10.1371/journal.pone.0007820
107. O'Keefe LV, Smibert P, Colella A, Chataway TK, Saint R, Richards RI. Know thy fly. Trends Genet. (2007) 23:238–42. doi: 10.1016/j.tig.2007.03.007
108. Tsuruwaka Y, Konishi M, Shimada E. Loss of wwox expression in zebrafish embryos causes edema and alters Ca(2+) dynamics. PeerJ. (2015) 3:e727. doi: 10.7717/peerj.727
109. Aqeilan RI, Croce CM. WWOX in biological control and tumorigenesis. J Cell Physiol. (2007) 212:307–10. doi: 10.1002/jcp.21099
110. Abu-Odeh M, Bar-Mag T, Huang H, Kim T, Salah Z, Abdeen SK, et al. Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J Biol Chem. (2014) 289:8865–80. doi: 10.1074/jbc.M113.506790
Keywords: model organisms, knockout, mouse, Drosophila, zebrafish, cancer, metabolism, epilepsy
Citation: Tanna M and Aqeilan RI (2018) Modeling WWOX Loss of Function in vivo: What Have We Learned? Front. Oncol. 8:420. doi: 10.3389/fonc.2018.00420
Received: 31 May 2018; Accepted: 10 September 2018;
Published: 10 October 2018.
Edited by:
Barbara Zavan, Università degli Studi di Padova, ItalyReviewed by:
Valentina Tosato, AREA Science Park, ItalyLuisa Lanfrancone, Istituto Europeo di Oncologia s.r.l., Italy
Copyright © 2018 Tanna and Aqeilan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rami I. Aqeilan, YXFlaWxhbkBjYy5odWppLmFjLmls