 An Thys
An Thys Tiphaine Douanne
Tiphaine Douanne Nicolas Bidère
Nicolas Bidère- Team SOAP, CRCINA, Institut National de la Santé et de la Recherche Médicale, CNRS, Université de Nantes, Université d'Angers, Nantes, France
Piracy of the NF-κB transcription factors signaling pathway, to sustain its activity, is a mechanism often deployed in B-cell lymphoma to promote unlimited growth and survival. The aggressive activated B-cell like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) exploits a multi-protein complex of CARMA1, BCL10, and MALT1 (CBM complex), which normally conveys NF-κB signaling upon antigen receptors engagement. Once assembled, the CBM also unleashes MALT1 protease activity to finely tune the immune response. As a result, ABC DLBCL tumors develop a profound addiction to NF-κB and to MALT1 enzyme, leaving open a breach for therapeutics. However, the pleiotropic nature of NF-κB jeopardizes the success of its targeting and urges us to develop new strategies. In this review, we discuss how post-translational modifications, such as phosphorylation and ubiquitination of the CBM components, as well as, MALT1 proteolytic activity, shape the CBM activity in lymphocytes and ABC DLBCL, and may provide new avenues to restore vulnerability in lymphoma.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most prevalent aggressive B-cell non-Hodgkin lymphoma (NHL) and accounts for 30–40% of diagnosed NHL (1, 2). The current standard treatment, called R-CHOP, consists of a combination of chemotherapies including cyclophosphamide, doxorubicin hydrochloride, vincristine and prednisone, supplemented with the CD20 antibody rituximab. Patient 3-years progression free survival upon R-CHOP treatment ranges from 30 to 70%, indicating that although all DLBCL are characterized by dense neoplastic B-blasts, the DLBCL group represents many different entities with diverse clinical outcomes (1, 2). Gene expression profiling studies from the early 2000's identified two major subtypes of DLBCL, i.e., germinal center B-cell like (GCB)-DLBCL and activated B-cell like (ABC)-DLBCL (3–6). GCB-DLBCL generally express genes that can be detected in germinal center B-cells and are thought to be derived from centroblasts, whereas ABC DLBCL's expression profile is closer to mature plasma cells and are assumed to originate from plasmablastic cells prior to germinal center exit (3, 7, 8). The importance of the ABC/GCB DLBCL classification has been acknowledged by the World Health Organization and the European Society for Medical Oncology, as clinical response to R-CHOP are divergent between subtypes, with ABC DLBCL having a significantly inferior response to treatment (1, 4, 9). However, gene expression profiling is an expensive method not customarily available in routine pathology laboratories and has universal standardized criteria for its use. Furthermore, determining cell of origin by the classical immunohistochemistry method has led to contradictory results (9).
The gene expression profile of ABC DLBCL is highly similar to that of normal B cells upon stimulation of their antigen receptors (3). Notably, nearly all cases of ABC DLBCL are characterized by a constitutive activation of the NF-κB transcription factors as a driver of lymphomagenesis (10, 11). The NF-κB heterodimers of Rel-family proteins are normally tethered to their cognate inhibitors, IκBs in the cytosol of cells. The ligation of the B-cell receptor (BCR) engages the signaling cascade of Burton's tyrosine kinase (BTK), phospholipase Cγ, which culminates in the activation of the protein kinase Cβ (PKCβ). This results in the assembly of a multi-protein complex that contains the scaffold Caspase recruitment domain family member 11 (CARD11, also called CARMA1), the adaptor B cell CLL/lymphoma-10 (BCL10), and the MALT1 paracaspase (MALT1) (CARMA1-BCL10-MALT1, CBM complex). The CBM organizes as a filamentous high-order structure that serves as a docking surface for the recruitment of signaling adaptors and of the inhibitor of NF-κB kinase (IKK), composed of the catalytic subunits IKKα and IKKβ, plus the regulatory subunit IKKγ (also known as NEMO) (12). IKK subsequently phosphorylates and thereby marks IκBs for proteasomal degradation (13–16) (Figure 1). IκB-free NF-κB translocates to the nucleus and initiates the transcription of specific target genes involved in inflammation, immune regulation, cell proliferation and apoptotic prevention (17). As a result, these lymphoma cells develop a profound addiction to NF-κB, opening up new opportunities for therapeutic treatments (18).
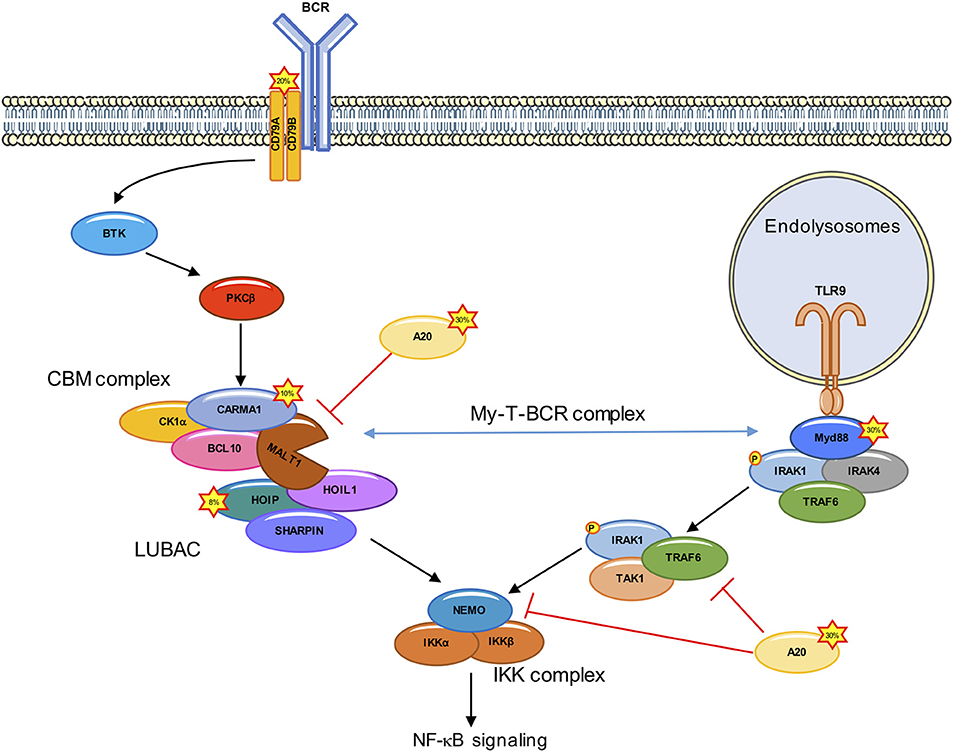
Figure 1. Deregulated NF-κB signaling in ABC DLBCL. In ABC DLBCL, a combination of genetic lesions leads to an effective hijacking of the BCR signaling cascade, which culminates with the assembly of the CARMA1-BCL10-MALT1 (CBM) complex, and the subsequent IKK/NF-κB activation. Mutations in ~20% of ABC DLBCL cases occur in the upstream CD79A/B. CARMA1 is mutated in ~10% of ABC DLBCL, leading to a constitutive recruitment of BCL10 and MALT1. Rare polymorphisms in the gene encoding for the LUBAC subunit HOIP (~8% of cases) increase the recruitment and activation of the IKK complex. Approximately 30% of ABC DLBCL have A20 loss of function. The Toll-like receptor adaptor MYD88 is also mutated in ~30% of ABC DLBCL. ABC DLBCL carrying both CD79A/B and MYD88 mutations form a signalosome at the endolysomome, consisting of the MYD88, TLR9, and BCR (My-T-BCR complex) which recruits the CBM.
In 2006, the Staudt laboratory engineered genomic-scale interference screens to search for genes required for the survival of ABC DLBCL-derived cell lines, and found that silencing the CBM core subunits was toxic (19). Further supporting a crucial role for this complex, gain-of-function mutations in the CARD11 gene encoding for CARMA1 were found in ~10% of ABC DLBCL tumors (11, 16). Those mutations were mapped to the CARMA1 coiled-coil domain, a region which normally locks the protein in an inactive conformation unless phosphorylated by PKCβ (20). CARMA1 mutants are constitutively activated, and form high-order signaling complexes (16). Nevertheless, the prevalence of CARD11 mutations was not sufficient to explain the addiction of all ABC DLBCL cell lines to the CBM complex (19). This was resolved with the discovery of mutations in the immune receptor tyrosine-based activation motive of CD79A or CD79B in 20% of ABC DLBCL, that lead to an effective hijacking of the downstream BCR signaling (13).
In addition to CD79A/CD79B and CARD11 mutations, ~40% of ABC DLBCL cases harbor a gain-of-function mutations (L265P, V217F, S219C, M232T, S243N, T294P amino acid substitution) in Myeloid differentiation primary response 88 (MYD88) (21). L265P is the most common MYD88 mutation found in ABC DLBCL (29%), while it is rare in the GCB subtype (21). This mutation enables the spontaneous assembly of IL-receptor-associated kinase-1 (IRAK1) and IRAK4, allowing IRAK4 to phosphorylate IRAK, which promotes the oligomerization and activation of the E3 ligase TNF receptor associated factor 6 (TRAF6). TRAF6 recruits TGFβ activated kinase1 (TAK1) and MAP3K7 binding protein 2 (TAB2) (22–24). TRAF6 then ubiquitinates TAK1 and NEMO. Ubiquitination of NEMO leads to a stabilization of the IKK complex, while the ubiquitination of TAK1 activates its kinase activity and leads to the phosphorylation of IKKα and downstream NF-κB activation (25, 26) (Figure 1). A clinical trial targeting ABC DLBCL with the BTK inhibitor Ibrutinib led to a response rate of 80% in ABC DLBCL carrying both CD79A/CD79B and MYD88 mutations (27). Recently, Phelan et al. explain this increased sensitivity by linking BCR and MyD88 signaling through the description of a new multiprotein supercomplex formed by MYD88, TLR9, and the BCR (My-T-BCR). This complex, located at the endolysosomes, recruits the CBM complex and drives NF-κB signaling in Ibrutinib-responsive cell lines and biopsies (28) (Figure 1). Nevertheless, the overall responsive rate to Ibrutinib in ABC DLBCL is only 37% (21), and this can partly be explained by mutations downstream of BCR or MYD88 (16, 29–33).
The CBM complex orchestrates a choreography of post-translational modifications. This signalosome forms a scaffolding platform for the recruitment of a variety of kinases and phosphatases, E3 ligases and deubiquitinating enzymes (DUBs), and unleashes MALT1 protease. A case study is provided with the linear ubiquitin assembly complex (LUBAC). This ternary complex consists of the E3 ligases HOIL1-interacting protein (HOIP) and RanBP-type and C3HC4-type zinc finger-containing protein 1 (HOIL1), together with the adaptor and SHANK-associated RH domain interactor (SHARPIN). The LUBAC binds the CBM to authorize the recruitment and phosphorylation of IKK in response to the engagement of antigen receptors, as well as in ABC DLBCL by conjugating linear chains to substrates (34–38). In addition, the LUBAC subunit HOIL1 is specifically cleaved by MALT1 to promote full NF-κB activation. Importantly, the silencing of the LUBAC components is toxic in ABC DLBCL (34, 38), a finding which was further validated in recent genome-wide loss-of-function CRISPR-Cas9 screens (28). Furthermore, two rare polymorphisms in the RNF31 gene encoding for HOIP were found enriched in ABC DLBCL with an overall frequency of 7.8% (38). Such variants increase the interaction between HOIP and HOIL1 and result in an enhanced activity of the LUBAC. Moreover, the introduction in ABC DLBCL cell lines of stapled α-helical peptides to dismantle the LUBAC reduced the aberrant NF-κB activation and was toxic (38).
Although interfering with NF-κB addiction in ABC DLBCL is appealing, its pleiotropic nature may jeopardize the success of its targeting and urges us to establish treatments that selectively kill tumor cells while sparing healthy cells. A clearer understanding of signaling mechanisms and post-translational modifications occurring in ABC DLBCL could provide new potential prognostic markers and putative targets for therapy. In this review, we therefore focus on the post-translational modifications, which can occur within the CBM complex, as this signalosome is instrumental to the survival of ABC DLBCL.
Phosphorylation of CARMA1 and BCL10 Shapes the CBM Complex Activity
Phosphorylation is considered to be the most prevalent reversible post-translational modification in eukaryotic cells and is crucial for cellular processes including cell cycle, growth, apoptosis and signaling transduction pathways (39). This modification principally occurs on Serine (S), Threonine (T), or Tyrosine (Y) residues to regulate protein/enzyme function by either activating or deactivating them (39). It is therefore not surprising that both CARMA1 and BCL10 are extensively phosphorylated upon antigen receptor engagement (20, 32, 40–55).
CARMA1 Phosphorylation
CARMA1 consists of a caspase recruitment domain (CARD), coiled-coil (CC), PSD95, DLG and ZO1 homology (PDZ), Src homology 3 (SH3) and guanylate kinase (GUK) domain (20). Phosphorylation of key Serine residues in the linker domain between CC and PDZ is a crucial step for the formation of the CBM complex upon antigen receptor engagement (20, 32, 40–42, 44, 56) (Figure 2). PKCβ/θ emerged as the most crucial kinase to phosphorylate CARMA1 in its linker domain on S552, and S645, causing CARMA1 to switch from an inactive “locked” conformation to an active “open” conformation by relieving the autoinhibition caused by the intramolecular association of the linker region to the CARD motif (32, 40). This conformational change in CARMA1 is crucial to enable the CARD-CARD-dependent recruitment of BCL10 and the subsequent activation of the downstream NF-κB pathway (32, 40–42, 57). The PKCθ-dependent phosphorylation of S645 is removed by the Serine/Threonine Protein phosphatases family member phosphatase 2A (PP2A), which constitutively binds CARMA1. By dephosphorylating CARMA1, PP2A finely tunes CARMA1 association with BCL10-MALT1 and therefore prevents excessive NF-κB activation (57).
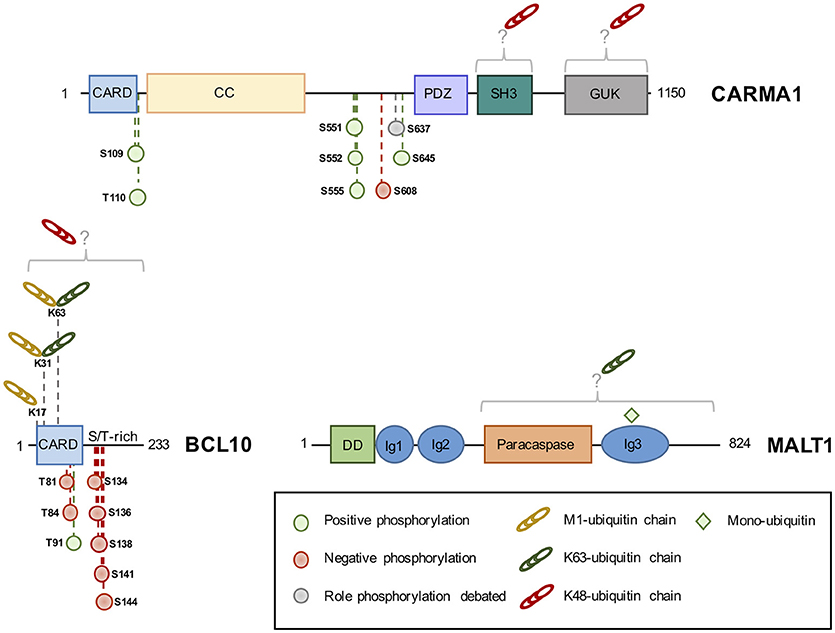
Figure 2. The domain structure of CBM components and their identified phosphorylation and ubiquitination sites. Phosphorylation of CBM components can positively (green circle) or negatively (red circle) influence NF-κB, although the role of some sites is debated (gray circle). K48-linked ubiquitin linked (red chains) chains lead to protein degradation, while K63 or M1-linked chains (green and yellow chains, respectively) modulate activity. Monoubiquitination (green square) activates MALT1 protease activity. CARD, Caspase recruitment domain; CC, Coiled-Coil; PDZ, PSD95-DLG-ZO1 homology domain; SH3, Src homology 3; GUK, guanylate kinase domain; S/T-rich, serine/threonine-rich domain; DD, death domain; Ig, immunoglobulin-like domain; K, lysine; M, methionine.
A large number of kinases, besides PKCβ/θ, have been shown to phosphorylate CARMA1 on a variety of sites, influencing the strength of BCL10-MALT1 recruitment and downstream NF-κB activation (32, 42, 55, 58). This includes protein kinase B (PKB or AKT), which also phosphorylates CARMA1 on S645, in addition to PKCβ, upon TCR stimulation (55). IKKβ and hematopoietic progenitor kinases 1 (HPK1) were shown to phosphorylate CARMA1 on S555 and S551, respectively, leading to an optimal NF-κB activation (32, 42, 58). In contrast, phosphorylation of CARMA1 on S608 by casein kinase 1α (CK1α) leads to a diminished NF-κB activation (44, 59). However, this negative role is overwhelmed by the crucial adaptor role of CK1α in bridging the CBM and IKK complexes in antigen receptor-stimulated lymphocytes (44). In ABC DLBCL cell lines, CK1α knockdown halts aberrant NF-κB signaling and is toxic (44). CK1α therefore emerges as a non-oncogenic addiction gene, a gene not involved in the initiation of the transformed phenotype but required for the tumorigenesis (60). In addition to S608, it was shown that CARMA1 S637 phosphorylation also reduced NF-κB activation in a DT40 chicken B cells model (59). AKT was later identified as the kinase in charge of phosphorylating this site. However, interfering with this modification only modestly influenced NF-κB activation in human T-cells. Rather, the phosphorylation of CARMA1 on S637 in combination with S645 may positively regulate NF-κB activation (55). Outside the linker domain, BCL10 recruitment and NF-κB activation were shown to be enhanced by phosphorylation of S109 by Ca2+-calmodulin-dependent kinase II (CaMKII) in T cells, and phosphorylation of T110 (T119 chicken ortholog) by an unknown kinase in chicken DT40-cells (42, 43).
Of note, gain of function mutations were found in the CC domain of CARMA1, in ~10% ABC DLBCL cases (16, 61). Although oncogenic CARMA1 mutants form cytosolic aggregates and spontaneously induces NF-κB signaling, whether they bypass the need for CARMA1 phosphorylation remains unclear (16, 61). In addition, the status of CARMA1 in ABC DLBCL is yet to be defined.
BCL10 Phosphorylation
Unlike CARMA1, whose phosphorylation mostly results in a positive regulation of the NF-κB pathway (see above), the phosphorylation of BCL10 essentially curbs its activity (45–48, 51–53). BCL10 is constitutively bound to MALT1 and consists of a CARD domain and a Serine/Threonine-rich domain (20) (Figure 2). First hints that BCL10 is a phosphoprotein came with the discovery that BCL10 equine herpesvirus 2 viral homolog v-E10 drives BCL10 hyperphosphorylation and redistribution from the cytosol to the plasma membrane (62). In lymphocytes, Wegener et al. identified five different sites (S134, S136, S138, S141, and S144) on BCL10, which are phosphorylated by IKKβ upon antigen receptor engagement (47). The recruitment of BCL10 to CARMA1 was shown to be seemingly necessary for its phosphorylation (47). IKKβ phosphorylation of BCL10 does not interfere with CARMA1-BCL10 association in activated T cells, but disrupts BCL10-MALT1 interaction, thereby attenuating NF-κB activation (47). Yet, the ability of IKKβ to phosphorylate BCL10 was rather weak, hinting at the involvement of other kinases (47). Indeed, BCL10 can also be phosphorylated on S138 by CaMKII in T cells. However, unlike CaMKII phosphorylation of CARMA1, which leads to and increased recruitment of BCL10 to CARMA1 and higher NF-κB activation, CaMKII phosphorylation of BCL10 on S138 recapitulates the results found by Wegener et al. and Ishiguro et al. (47, 48).
The majority of studies agree that BCL10 phosphorylation leads to BCL10 degradation, however, the mechanism and molecular players vary between studies and are a matter of debate (45–48, 51). Zeng et al. reported S138 BCL10 phosphorylation to signal for BCL10 degradation in a proteasome-independent manner, while Lobry et al. described IKKβ phosphorylation of BLC10 on T81 and T85 to lead to a proteasome-dependent degradation of BCL10 (45, 51). Conversely, experiments identifying Ca2+-dependent phosphatase Calcineurin as a specific S138 BCL10 phosphatase in T cells, did not show BCL10 degradation, nor the dissociation of BCL10 from MALT1 (52, 53). However, they confirm S138 BCL10 phosphorylation to be vital in curbing NF-κB activation (52, 53). Inhibition of Calcineurin leads to an increased BCL10 phosphorylation and decreases NF-κB activation, and in T-helper cells, the dephosphorylation of S138 by Calcineurin was shown to be essential for NF-κB activation (52, 53). Besides this negative role, BCL10 phosphorylation on S138 controls T-cell receptor and Fcγ-induced actin polymerization, in a MALT1- and CARMA1- independent manner (54). Finally, BCL10 phosphorylation can also positively regulate NF-κB signaling. For instance, both BCL10 phosphorylation by CaMKII, on T91, and GSK3β, on unmapped phosphorylation sites, promote NF-κB signaling in T cells (49, 50).
In essence, the CBM components CARMA1 and BCL10 are regulated by phosphorylation, and depending on the phosphorylation site and kinase involved; phosphorylation can lead to both positive and negative regulation of the NF-κB pathway.
Control of the CBM Activity by Ubiquitination
Ubiquitination is a reversible post-translational modification of Lysine (K) residues in proteins with ubiquitin, which governs nearly all cellular processes in eukaryotes (63). A plethora of polyubiquitin chains exist, as ubiquitin itself is a substrate of ubiquitination at eight different position (M1, K6, K11, K27, K29, K33, K48, and K63). This variety of ubiquitin linkages was proposed to form a code that leads to different cellular outcomes (63). Ubiquitination is a reversible process counteracted by a family of proteases called deubiquitinating enzymes (DUBs). A large body of literature now supports the idea that ubiquitination shapes the activity of the CBM in lymphocytes and lymphoma (64, 65).
CARMA1 Ubiquitination
The engagement of antigen receptors was shown to promote the K48-linked ubiquitination of CARMA1 and its subsequent degradation by the proteasome (66). Although not formally identified, the Lysine acceptors were mapped within the SH3 and GUK domains of CARMA1 (Figure 2). The substitution of the 29 Lysine residues with Arginines enhances both basal and TCR-mediated activation of NF-κB (66). Of note, the SH3 and GUK domains form intra- and intermolecular bounds necessary for CARMA1 recruitment at the immunological synapse, and for the activation of NF-κB both in lymphocytes and in ABC lymphoma (67). Yet, the abundance of CARMA1 was not found overtly decreased in ABC DLBCL (28, 34), suggesting that mechanisms that prevent ubiquitination and degradation may take place. Recently, a comprehensive analysis by quantitative mass spectrometry-based proteomics of the landscape of BCR-mediated ubiquitination revealed that CARMA1 is also subjected to linear ubiquitination (35). Future works are therefore required to elucidate the function of this modification.
BCL10 Ubiquitination
In sharp contrast to CARMA1, the ubiquitination status of BCL10 has received a large amount of attention. Although the exact nature of chains bound to BCL10 continues to be deciphered, it is now clear that a mixture of at least K48, K63 and M1 linkages can decorate the adaptor, leading to different cellular outcomes (Figure 2). BCL10 ubiquitination was initially linked to degradation as a mean of terminating NF-κB activation following antigen receptors ligation, and several hypotheses have been proposed. First, the E3 ligases β-TRCP and c-IAP2 were found to catalyze K48-linked ubiquitination of BCL10 following its phosphorylation by IKKβ, thus promoting its proteasomal degradation (45, 68). However, ubiquitinated BCL10 can also be eliminated by the lysosome, either in a NEDD4/Itch-dependent manner (46), or once recognized by the autophagy receptor p62 (69). K48-linked ubiquitin chains bound to BCL10 were proposed to be removed by the pleiotropic DUB USP9X in lymphocytes (70). Rather than preventing BCL10 degradation, the trimming of K48 chains would allow the CBM complex to assemble properly (70). However, the subsequent generation of mice knockout for USP9X in T or B cell compartments revealed a role for USP9X in the CBM restrained to B cells, possibly by selectively regulating PKCβ upstream of BCL10 (71, 72). BCL10 ubiquitination and stability is also under the control of the COP9 signalosome (CSN), a multiprotein complex of the ubiquitin-proteasome system, which participates to the CBM upon TCR engagement (73). Finally, the E3 ligase RNF181 adjusts the basal abundance of BCL10 and therefore, negatively regulates NF-κB signaling in lymphocytes and ABC lymphoma (74). Nevertheless, whether K48-linked ubiquitination operates in ABC DLBCL remains to be explored.
Pioneer work by Wu and Ashwell revealed that the binding of K63 chains to BCL10 precedes K48-linked ubiquitination in lymphocytes (15). In ABC DLBCL, experiments with tandem ubiquitin-binding entities (TUBE) to selectively enrich for polyubiquitinated proteins also showed that BCL10 is merely attached to K63-linked chains (75). This non-degradative ubiquitination requires an intact CBM complex and was proposed to recruit the LUBAC complex and allow IKK activation (15, 34, 36, 75, 76). Mutagenesis experiments identified the Lysine residues K31 and K63 as the main acceptors of ubiquitin in TCR-stimulated Jurkat cells as well as in ABC DLBCL (15, 36, 75, 76). Their substitution to Arginine hampered TCR-mediated NF-κB activation (15, 75). Yang et al. elegantly demonstrated that the E3 ligases cIAP1/2 were integral to the CBM complex and promoted BCL10 K63-linked ubiquitination in ABC DLBCL (75). The treatment of ABC DLBCL with the bivalent SMAC mimetic Birinapant to eliminate cIAP1/2 was toxic, as it disrupted the constitutive CBM-LUBAC signalosome and hampered aberrant NF-κB activation (75). Moreover, Birinapant inhibited tumor growth in vivo in BCR-dependent ABC DLBCL xenografts models (75). Of note, the requirement for c-IAP1/2 may be more restricted to chronic or tonic stimulation of the BCR, as no overt defect in TCR-mediated NF-κB activation was observed in Birinapant-treated Jurkat T cells (our unpublished observations).
The discovery that the LUBAC binds to the CBM in lymphocytes and lymphoma suggested the existence of substrate(s) within this signalosome (34, 38). However, whether catalytic activity of the LUBAC is crucial was initially disputed, as NF-κB signaling can occur in antigen receptors-stimulated dead-HOIP cells and was not significantly altered by the silencing of OTULIN (34, 77). Nevertheless, a combination of TUBE, SILAC, and standard biochemistry revealed that BCR and oncogenic BCR stimulation led to linear ubiquitination of BCL10 (35, 75). In addition, the overexpression of oncogenic CARD11 mutations also drove the LUBAC to decorate BCL10 with M1-linked chains (36). Supporting a positive role for this linkage, a fusion protein of BCL10 conjugated to four linearly linked DUB-resistant ubiquitin moieties enhanced the activation of NF-κB when ectopically expressed (35). In Jurkat T cells, the ligation of TCR also leads to the conjugation of M1 chains on BCL10, which required the Lysine residues 17, 31, and 63 (36). Because K31 and K63 are also targeted for K63-linked ubiquitination (15), it is tempting to speculate that K63/M1 branched poly-ubiquitin chains bind BCL10 (78). In keeping with this idea, BCL10 K17R mutation was sufficient to impair linear and K63-linked ubiquitination of BCL10 and to dampen TCR-mediated NF-κB activation (36).
MALT1 Ubiquitination
MALT1 K63-linked poly-ubiquitination has been observed in lymphocytes shortly following antigen receptor stimulation, as well as, in ABC DLBCL (14, 34, 44, 73, 79, 80). In their seminal work, Oeckinghaus et al. identified 11 putative Lysine acceptors in the COOH-terminal segment of the protein, which abolished ubiquitination and recruitment of IKK when mutated to Arginines (14). MALT1−/− CD4+ T lymphocytes reconstituted with a ubiquitin-resistant MALT1 mutant display reduced NF-κB activation and diminished production of IL-2 (14). Several lines of evidence have linked the E3 ligase TRAF6 to MALT1 ubiquitination. First, TRAF6 can directly binds the COOH-terminal part of MALT1 via two consensus binding sites (81, 82). In addition, oligomers composed of BCL10 and MALT1 promote the binding and activation of TRAF6 enzyme activity (81). Moreover, siRNA-mediated silencing of TRAF6 in Jurkat cells abolished MALT1 ubiquitination and dampened TCR-mediated NF-κB activation (14, 83). However, T-cell-specific deletion of TRAF6 had no overt effect on TCR-mediated activation of NF-κB, IL-2 production or proliferation (84). Of note, the abundance of TRAF6 is rather low in naïve mouse CD4+ T cells and is rapidly upregulated following the engagement of antigen receptors, suggesting that TRAF6 may be more relevant in activated lymphocytes (84). It is also possible that TRAF6 action is redundant with additional E3 ligases, such as TRAF2 (81). Shortly following TCR stimulation in Jurkat cells, the DUB A20 is recruited to the CBM (our unpublished results), to selectively remove K63-linked ubiquitin attached to MALT1 and therefore ensuring optimal recruitment and activation of the IKK complex (79). In return, A20 is rapidly cleaved and inactivated by MALT1 (see below). Whether MALT1 poly-ubiquitination participates in its paracaspase activity is unknown. Interestingly, the deubiquination of MALT1 still occurred without A20, although delayed, suggesting that other DUBs may participate to this process (79). MALT1 is also monoubiquitinated on the Lysine residue 644 shortly after TCR stimulation in Jurkat, or in a constitutive manner in ABC DLBCL cell lines (Figure 2) (85). This monoubiquitination is driven by the dimerization of MALT1 and requires a functional CBM complex (85, 86). A monoubiquitination-resistant mutant was not able to completely unleash the protease activity of MALT1 (85–87). MALT1 K644R-expressing cells displayed reduced cleavage of MALT1 substrates and diminished NF-κB activation (85, 86). In ABC DLBCL, interfering with MALT1 monoubiquitination was toxic (85).
MALT1 Protease Activity Tunes the Immune Response and Is Required for Lymphomagenesis
Although MALT1 encompasses a COOH-terminal paracaspase domain that shares homology with caspases, it was initially thought to lack proteolytic activity and to only serve as a scaffold adaptor (88). In 2008 however, Margot Thome's and Rudi Beyaert's laboratories established MALT1 as a functional protease that cleaves substrates after an arginine residue embedded in a consensus S/P-R↓G/A site (89, 90). As previously mentioned, the activation of MALT1 results from a monoubiquitination on the Lysine residue 644 upon antigen receptor engagement (85). Recently, the generation of mice expressing a catalytically dead MALT1 uncovered the essential role of the protease in lymphocyte development and activation (91–94). Notably, MALT1 protease activity was demonstrated to control the development of regulatory T cells (TReg) and T helper 17 (TH17) cells, as well as, innate-like B cells. Furthermore, protease dead mature T lymphocytes displayed proliferation impairment and reduced TNFα and IL-2 secretion (91–94). Interestingly, MALT1 inactivation protected the mice against experimental autoimmune encephalomyelitis (EAE) and colitis, suggesting a critical role for MALT1 in the control of autoimmune diseases (91–94). Nevertheless, the deletion of MALT1 proteolytic activity is also deleterious, as mice expressing a catalytically dead MALT1 develop a lethal multi-organ inflammatory syndrome, arising from aberrant secretion of interferon gamma (IFNγ) by effector lymphocytes (91). Interestingly, several specific types of tissue damage have been reported upon protease inactivation ranging from neurodegeneration (91), gastric inflammation (91–94), lung immune infiltration (94) to eye inflammation (93). Furthermore, proteolytic dead MALT1 mice developed signs of autoimmune gastritis associated with high serum levels of IgE and IgG1 resulting from accumulation of activated T cells and a loss of TReg cells (92). In keeping with this, reconstitution of MALT1 protease dead mice with TReg overcame this defect and delayed the multi-organ inflammatory pathology and lethality (93). Strikingly, MALT1−/− mice lacking the paracaspase are immunodeficient but do not display any overt pathology (95, 96). In addition, several human patients with MALT1 deficiency have been reported to suffer from combined immunodeficiency disorders (CID) (97, 98). The differences observed in MALT1 deficiency and protease inactivation support a balanced function of MALT1 as a scaffold and as a protease cleaving a subset of substrates.
MALT1 Cleaves Regulators of NF-κB Activation
Over the last decade, our understanding of the landscape of MALT1 functions has improved with the discovery of ten substrates (99) (Table 1). Among them, a subset of substrates (A20, RelB, HOIL1, MALT1, and NIK) participates in the regulation of NF-κB signaling. As previously discussed, the DUB A20 negatively regulates NF-κB activation in response to various stimuli by specifically removing K63 ubiquitin chains from key signaling adaptors, such as TRAF6, NEMO, and MALT1 (89, 115). Upon TCR engagement, MALT1-mediated cleavage of A20 at the R439 residue promotes proteasomal degradation of the resulting fragments, and limits its negative action on NF-κB signaling (79, 89). However, no overt change in IKK-dependent phosphorylation of IκBα was observed in the absence of MALT1 activity (92). Further work is therefore needed to precisely pinpoint the effect of A20 processing, and to determine if the generated fragments could have a role on NF-κB activation independently of IKK. Interestingly, A20 has been shown to function as a tumor suppressor in multiple cancer like mantle cell lymphoma (MCL) (116), mucosa-associated lymphoid tissue (MALT) lymphoma (117) and in DLBCL (118). Because this DUB presents inactivating mutations in several hematological malignancies, studying the status and role of A20 processing could reveal some insight into the molecular mechanisms at hand and offer relevant therapeutic strategies.
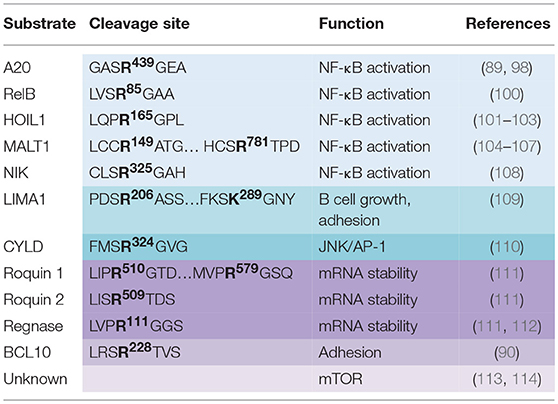
Table 1. MALT1 substrates, cleavage sites and their influence on cellular function.
Another MALT1 substrate involved in NF-κB signaling is the NF-κB family member RelB (100). The NF-κB transcription family is comprised of five members that all share a Rel homology domain (RHD) required for oligomerization and DNA binding. NF-κB activation can rise from the canonical pathway that relies on the formation of RelA-p50 or c-Rel-p50 complexes or the non-canonical pathway that leads to the formation of RelB-p50 heterodimers. RelB was proposed to negatively regulate canonical NF-κB activation by competing for DNA binding sites, as well as, by forming transcriptionally inactive complexes with RelA and c-Rel (100). RelB processing upon antigen receptor engagement in Raji B cells, lymphoblastoid Jurkat, and in human primary CD4+ T cells is followed by rapid proteasomal degradation of the produced fragment. The ectopic expression of a MALT1-resistant RelB mutant led to a decrease in NF-κB activation assessed by gene reporter assay in Jurkat, while RelB silencing increased IL-2 production in naïve primary T cells (100).
Three independent groups including ours, recently identified the LUBAC subunit HOIL1 as a substrate of MALT1 (101–103). Although the LUBAC has been shown to be an integral part of the CBM required for IKK activation in lymphocytes and in ABC DLBCL (34, 38), the role of HOIL1 has remained rather elusive. Our in-silico analysis of the known CBM partners led to the identification of HOIL1, as a putative MALT1 substrate (103). Experiments with Jurkat cells, human primary T lymphocytes and mouse primary T lymphocytes further validated that HOIL1 was processed by MALT1 at R165 upon antigen receptor engagement (102, 103). Of note, API2-MALT1 fusion oncoprotein resulting from the recurrent t(11;18)(q21;q21) in MALT lymphoma also hydrolyses HOIL1 at R165 (102). Concurrently, Klein et al. also identified HOIL1 as a substrate of the paracaspase by studying the B cells profile in a MALT1 deficient patient using 10-plex Tandem Tag TAILS N-Terminal peptide proteomics (101). The authors further propose that HOIL1 processing destabilizes the LUBAC, impairs linear ubiquitination, therefore reducing NF-κB signaling (101). However, we favor a model, in which MALT1 cleaves and alleviates HOIL1 negative regulation on NF-κB (103). Indeed, the expression of a MALT1-resistant HOIL1 mutant in Jurkat cells hampered NF-κB activation and subsequent IL-2 secretion in response to the ligation of TCR (103). This suggests that HOIL1, together with A20 and RelB, belongs to a subset of MALT1 substrates that negatively regulate NF-κB when left uncleaved. Nevertheless, further work needs to be done to precise the function of the fragments generated by HOIL1 processing.
Interestingly, recent work report that MALT1 also contributes to full NF-κB activation via its auto-proteolysis at R149 (104). Expression of a non-cleavable MALT1 mutant led to a decrease in IL-2 production in Jurkat T cells without affecting phosphorylation of IκBα or cleavage of other MALT1 substrates. Furthermore, a qRT-PCR approach revealed that MALT1 auto-processing is essential for expression of NF-κB target genes in activated Jurkat cells (104). Another study confirmed the function of MALT1 self-cleavage in driving IL-2 production in CD4+ T cells, and showed a subsequent function in controlling TReg homeostasis. Strikingly, overexpression of a MALT1 unable to self-process led to a defect in TReg number and an increase in anti-tumoral immunity reactivity (105). More recently, MALT1 was reported to also auto-cleave at R781 upon TCR engagement. Overexpression of a processing resistant mutant impaired NF-κB activation and consequent IL-2 production (106). Recently, TRAF6 was shown to induce MALT1 auto-proteolysis at R149 generating a fragment with high signaling properties whereas processing at R781 was shown to abrogate TRAF6 binding and subsequent NF-κB activation (107). Of note, MALT1 seems to control the non-canonical NF-κB signaling pathway as well, by processing the NF-κB induced kinase (NIK) in the context of the oncogenic fusion protein API2-MALT1 (108). NIK phosphorylates NF-κB p100 subunit to trigger its processing into the active p52 subunit (119). This pathway is tightly regulated by the short lifespan of NIK that is rapidly degraded by the proteasome. Strikingly, API2-MALT1 cleaves NIK at the R325 to generate a fragment containing the kinase activity and resistant to degradation. The subsequent NF-κB activation was shown to drive B cell adhesion and resistance to apoptosis (108). Importantly, MALT1 only cleaves NIK independently of the CBM complex in the context of API2-MALT1. This is also the case of the tumor suppressor LIM domain and actin-binding protein 1 (LIMA1), which promotes B-cell growth, adhesion and tumorigenicity when cleaved (109).
MALT1 Processes the Deubiquitinating Enzyme CYLD
In addition to its role in finely tuning NF-κB, MALT1 was also linked to JNK/AP-1 signaling. Purified T cells from MALT1-deficient mice displayed impairment in JNK activation upon antigen receptor engagement (95). Staal et al. showed that T cell receptor ligation as well as overexpression of the oncogenic API2-MALT1 fusion proteins led to the processing and inactivation of CYLD at R324 (110). CYLD is a deubiquitinating enzyme that acts as a negative regulator of the JNK and AP-1 pathways by removing ubiquitin from TAK1 (120). The overexpression of a MALT1-resistant CYLD mutant led to a decreased expression of JNK and AP-1 target genes like IL-2, IL-8, and c-Jun (110). Yet, siRNA-mediated silencing of the DUB only partially increased JNK signaling, suggesting other DUBs could be involved. Moreover, mice expressing a catalytically dead MALT1 do not present overt defects in JNK and AP-1 signaling, suggesting additional functions for CYLD processing (93).
MALT1 Promotes mRNA Editing
Another unexpected function of MALT1 was further characterized with the discovery that Regnase-1 and Roquin1/2, proteins involved in RNA stability, are substrates of the paracaspase (111, 112). Regnase-1 is an RNase that mediates the mRNA stability of several genes including c-Rel, OX40, and IL-2 and is essential for the prevention of aberrant CD4+ effectors T cells generation (112). In keeping with this, mice deficient for Regnase-1 develop severe systemic inflammation resulting from B and T cell activation (121). Uehala and colleagues reported that antigen receptor engagement induced the cleavage of Regnase-1 at R111 by MALT1 and subsequent proteasomal degradation of the generated fragment. Interfering with MALT1 protease activity led to the destabilization of a subset of mRNA and therefore to a deregulation in T cell activation (112). In addition to its role in CD4+ T effector differentiation, Regnase-1 has been shown to act together with the RNA binding proteins Roquin1 and Roquin2 to control TH17 effector differentiation and IL17 production (111). Roquin 1 and 2 were also demonstrated to be processed by MALT1 following antigen receptor signaling. Roquin deficient T cells have upregulated IκBζ and IκBNS transcripts, driving aberrant TH17 differentiation. This phenotype was reversed by expression of a MALT1-insensitive Roquin mutant (111).
MALT1, Adhesion, and Metabolic Signaling
The spectrum of MALT1's function was widened with the discovery of its involvement in adhesion, and metabolic signaling via mTORC1. Upon TCR engagement, MALT1 cleaves its binding partner BCL10 at R228 to ensure optimal adhesion of cells to fibronectin, through mechanisms that remain poorly understood (90). In 2014, Nayaka and colleagues reported that the CBM complex and MALT1 paracaspase activity are required for glutamine uptake and mTORC1 activation upon TCR engagement, independent of IKK (113). The use of z-VRPR.fmk led to a decrease in glutamine uptake in adition to phosphorylation of mTORC1 targets S6 and S6K1. The role of MALT1's protease activity in driving metabolic flux was concurrently reported in activated CD4+ T cells (114). In keeping with this, a recent work identified 4 rare hypomorphic dominant negative mutations in CARMA1 that interfere with the activation of MALT1 and mTORC1 (122). Conversely, mTORC1 signaling is increased in a B-cell intrinsinc expression of an activating mutation in CARMA1 model (123). How MALT1 enzyme links antigen receptors to mTORC1 activation and metabolic changes remains unclear, as the substrates involved are yet to be identified.
MALT1 Protease and ABC DLBCL
While dormant in resting lymphocytes unless stimulated through their antigen receptors, MALT1 protease is constitutively active in ABC DLBCL and contributes to pathogenesis (124, 125). Pioneer works by Margot Thome and Jurgen Ruland laboratories reported that inhibition of the catalytic activity of the paracaspase with the tetrapeptide inhibitor z-VRPR.fmk and overexpression of a catalytically dead MALT1 specifically decreased growth and survival of ABC DLBCL lines (124, 125). These results opened the path to several additional studies both in vitro and in vivo, providing a rationale for targeting the paracaspase in these types of lymphomas. Notably, additional molecules, such as the small compound MI2 or phenothiazines were shown to directly bind and suppress MALT1 protease activity, and were reported to be selectively toxic to ABC DLBCL cell lines in vitro as well as xenotransplanted ABC DLBCL in vivo without displaying toxicity in mice (111, 126). Nevertheless, the role of MALT1 substrates in ABC DLBCL survival remains poorly characterized. For instance, BCL10, A20, RelB, CYLD, HOIL1 and MALT1 itself were found constitutively processed in a panel of ABC DLBCL cell lines, and only the impact of RelB and MALT1 (R781) were extensively assessed. Indeed, Hailfinger et al. conclusively demonstrated that the overexpression of RelB was toxic in ABC DLBCL cell lines (85). Wu et al. showed that overexpression of a non-self-cleavable MALT1 (R781) mutant impaired cell viability in HBL-1 (106). Surprisingly, MALT1 substrates, such as Roquin1/2 and Regnase have not been studied in the context of ABC DLBCL and whether MALT1 aberrant activation modulates the stability of mRNAs in this subtype of lymphoma is unknown. Moreover, the exact roles of the tumor suppressors A20 and CYLD in ABC DLBCL remain elusive. Identifying if some substrates are differentially cleaved in ABC DLBCL and precising the function of MALT1 processing seems essential to better understand the pathology which could uncover relevant therapeutic targets. Hence, additional work is necessary to provide a comprehensive understanding of the precise role of each substrate in the pathology.
Concluding Remarks
Although in recent years we have witnessed a tremendous amount of progress in our understanding of the post-translational modifications of the CBM complex upon antigen receptor stimulation, key questions remain about their status and functions in ABC DLBCL: Are CARMA1 and BCL10 phosphorylated in ABC lymphoma? What, if anything, is the impact of their inhibition? In that view, the clinically available PKC inhibitor Sotrastaurin led to a diminished NF-κB activation and offered a significant anti-tumor effect in a subcutaneous CD79-mutated ABC DLBCL xenograft model (30). In keeping with this idea, interfering with the ubiquitination of the CBM subunits may be another attractive path to pursue. Indeed, the cIAP inhibitor Birinapant, which prevents K63-linked ubiquitination of BCL10 impedes tumor growth in ABC DLBCL xenograft models (75). One other approach may lie in the generation of small-molecule inhibitors highly specific of deubiquitinating enzymes, as recently demonstrated for USP7 (127, 128). Finally, strategies targeting the MALT1 enzyme in ABC DLBCL have shown promising results. However, genetic paracaspase dead mouse models suggests that systematically blocking MALT1 activity may be harmful. A better understanding of the landscape of MALT1 substrates and their specific roles is therefore required. Also, are all substrates cleaved in ABC DLBCL? The development of technologies, such as genome editing with CRISPR/Cas9, will be helpful to generate MALT1-resistant mutants, and therefore to pinpoint the exact function of each individual substrate (129–131).
Dysregulation in the activity of the CBM subunits is not restricted to ABC DLBCL, and has been linked to T-cell and B-cell lymphoma, cancer, lymphoproliferation, allergy, inflammatory diseases, and primary immunodeficiencies [for review, see (132)]. Greater understanding of the functions of post-translational modifications of the CBM in ABC DLBCL will be essential to develop new diagnostic, prognostic and therapeutic strategies.
Author Contributions
AT, TD, and NB designed and wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by Fondation pour la Recherche Medicale (Equipe labellisée DEQ20180339184), Fondation ARC pour la Recherche sur le Cancer, Ligue nationale contre le cancer comités de Loire-Atlantique, Maine et Loire, Vendée, Région Pays de la Loire et Nantes Métropole under Connect Talent Grant. AT holds a postdoctoral fellowship from Fondation ARC, and TD is a PhD fellow funded by Nantes Métropole. We thank Kathryn A. Jacobs for kindly proofreading this review.
References
1. Beham-Schmid C. Aggressive lymphoma 2016: revision of the WHO classification. Memo Mag Eur Med Oncol. (2017) 10:248–54. doi: 10.1007/s12254-017-0367-8
2. Lenz GL, Staudt LM. Aggressive lymphomas. N Engl J Med. (2010) 362:1417–29. doi: 10.1056/NEJMra0807082
3. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
4. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
5. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The Use of Molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. (2002) 346:1937–47. doi: 10.1056/NEJMoa012914
6. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. (2003) 100:9991–6. doi: 10.1073/pnas.1732008100
7. Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood (2015) 125:22–32. doi: 10.1182/blood-2014-05-577189
8. Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 87:163–208. doi: 10.1016/S0065-2776(05)87005-1
9. Tilly H, Gomes da Silva M, Vitolo U, Jack A, Meignan M, Lopez-Guillermo A, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2015) 26(Suppl. 5):v116–25. doi: 10.1093/annonc/mdv304
10. Davis RE, Brown KD, Siebenlist U, Staudt L. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. (2001) 194:1861–74. doi: 10.1084/jem.194.12.1861
11. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature (2009) 459:717–21. doi: 10.1038/nature07968
12. Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell (2013) 51:766–79. doi: 10.1016/j.molcel.2013.08.032
13. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature (2010) 463:88–92. doi: 10.1038/nature08638
14. Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SÇ, Ruland J, et al. Malt1 ubiquitination triggers NF-κB signaling upon T-cell activation. EMBO J. (2007) 26:4634–45. doi: 10.1038/sj.emboj.7601897
15. Wu C-J, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF- B activation. Proc Natl Acad Sci USA. (2008) 105:3023–8. doi: 10.1073/pnas.0712313105
16. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 Mutations in human diffuse large B cell lymphoma. Science (2008) 319:1676–9. doi: 10.1126/science.1153629
17. Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. (2004) 25:280–8. doi: 10.1016/j.it.2004.03.008
18. Shaffer AL III, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. (2012) 30:565–610. doi: 10.1146/annurev-immunol-020711-075027
19. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature (2006) 441:106–10. doi: 10.1038/nature04687
20. Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol. (2006) 6:799–812. doi: 10.1038/nri1944
21. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470:115–9. doi: 10.1038/nature09671
22. Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell (2000) 5:649–58. doi: 10.1016/S1097-2765(00)80244-0
23. Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the Interleukin-1-induced activation of NFκB. J Biol Chem. (2001) 276:41661–7. doi: 10.1074/jbc.M102262200
24. Ansell SM, Hodge LS, Secreto FJ, Manske M, Braggio E, Price-Troska T, et al. Activation of TAK1 by MYD88 L265P drives malignant B-cell Growth in non-Hodgkin lymphoma. Blood Cancer J (2014) 4:e183. doi: 10.1038/bcj.2014.4
25. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412:346–51. doi: 10.1038/35085597
26. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB Kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell (200) 103:351–61. doi: 10.1016/S0092-8674(00)00126-4
27. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. (2015) 21:922–6. doi: 10.1038/nm.3884
28. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
29. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. (2011) 43:830–7. doi: 10.1038/ng.892
30. Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, et al. Protein Kinase C inhibitor Sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. (2011) 71:2643–53. doi: 10.1158/0008-5472.CAN-10-2525
31. Yang Y, Shaffer AL, Emre NCT, Ceribelli M, Zhang M, Wright G, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell (2012) 21:723–7. doi: 10.1016/j.ccr.2012.05.024
32. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, et al. Phosphorylation of CARMA1 Plays a critical role in T cell receptor-mediated NF-κB activation. Immunity (2005) 23:575–85. doi: 10.1016/j.immuni.2005.10.007
33. McCully RR, Pomerantz JL. The protein kinase C-responsive inhibitory domain of CARD11 functions in NF- B activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol Cell Biol. (2008) 28:5668–86. doi: 10.1128/MCB.00418-08
34. Dubois SM, Alexia C, Wu Y, Leclair HM, Leveau C, Schol E, et al. A catalytic-independent role for the LUBAC in NF-kappaB activation upon antigen receptor engagement and in lymphoma cells. Blood (2014) 123:2199–203. doi: 10.1182/blood-2013-05-504019
35. Satpathy S, Wagner SA, Beli P, Gupta R, Kristiansen TA, Malinova D, et al. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol Syst Biol. (2015) 11:810. doi: 10.15252/msb.20145880
36. Yang Y-K, Yang C, Chan W, Wang Z, Deibel KE, Pomerantz JL. Molecular determinants of scaffold-induced linear Ubiquitinylation of B cell lymphoma/leukemia 10 (Bcl10) during T cell receptor and oncogenic caspase recruitment domain-containing protein 11 (CARD11) Signaling. J Biol Chem. (2016) 291:25921–36. doi: 10.1074/jbc.M116.754028
37. Teh CE, Lalaoui N, Jain R, Policheni AN, Heinlein M, Alvarez-Diaz S, et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat Commun. (2016) 7:13353. doi: 10.1038/ncomms13353
38. Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. (2014) 4:480–93. doi: 10.1158/2159-8290.CD-13-0915
39. Cohen P. The regulation of protein function by multisite phosphorylation – a 25 year update. Trends Biochem Sci. (2000) 25:596–601. doi: 10.1016/S0968-0004(00)01712-6
40. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-García ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 linker controls NF-κB activation. Immunity (2005) 23:561–74. doi: 10.1016/j.immuni.2005.09.014
41. Shinohara H, Yasuda T, Aiba Y, Sanjo H, Hamadate M, Watarai H, et al. PKCβ regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J Exp Med. (2005) 202:1423–31. doi: 10.1084/jem.20051591
42. Shinohara H, Maeda S, Watarai H, Kurosaki T. IκB kinase β-induced phosphorylation of CARMA1 contributes to CARMA1–Bcl10–MALT1 complex formation in B cells. J Exp Med. (2007) 204:3285–93. doi: 10.1084/jem.20070379
43. Ishiguro K, Green T, Rapley J, Wachtel H, Giallourakis C, Landry A, et al. Ca2+/Calmodulin-Dependent Protein Kinase II Is a Modulator of CARMA1-Mediated NF-κB Activation. Mol Cell Biol. (2006) 26:5497–508. doi: 10.1128/MCB.02469-05
44. Bidere N, Ngo VN, Lee J, Collins C, Zheng L, Wan F, et al. Casein kinase 1alpha governs antigen-receptor-induced NF-kappaB activation and human lymphoma cell survival. Nature (2009) 458:92–6. doi: 10.1038/nature07613
45. Lobry C, Lopez T, Israel A, Weil R. Negative feedback loop in T cell activation through IkappaB kinase-induced phosphorylation and degradation of Bcl10. Proc Natl Acad Sci USA. (2007) 104:908–13. doi: 10.1073/pnas.0606982104
46. Scharschmidt E, Wegener E, Heissmeyer V, Rao A, Krappmann D. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappaB signaling. Mol Cell Biol. (2004) 24:3860–73. doi: 10.1128/MCB.24.9.3860-3873.2004
47. Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U, et al. Essential Role for IκB Kinase β in Remodeling Carma1-Bcl10-Malt1 Complexes upon T Cell Activation. Mol Cell (2006) 23:13–23. doi: 10.1016/j.molcel.2006.05.027
48. Ishiguro K, Ando T, Goto H, Xavier R. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. Mol Immunol. (2007) 44:2095–100. doi: 10.1016/j.molimm.2006.09.012
49. Oruganti SR, Edin S, Grundström C, Grundström T. CaMKII targets Bcl10 in T-cell receptor induced activation of NF-κB. Mol Immunol. (2011)48:1448–60. doi: 10.1016/j.molimm.2011.03.020
50. Abd-Ellah A, Voogdt C, Krappmann D, Möller P, Marienfeld RB. GSK3β modulates NF-κB activation and RelB degradation through site-specific phosphorylation of BCL10. Sci Rep. (2018) 8:1352. doi: 10.1038/s41598-018-19822-z
51. Zeng H, Di L, Fu G, Chen Y, Gao X, Xu L, et al. Phosphorylation of Bcl10 Negatively Regulates T-Cell Receptor-Mediated NF- B Activation. Mol Cell Biol. (2007) 27:5235–45. doi: 10.1128/MCB.01645-06
52. Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, Marienfeld RB. The Ca2+-dependent Phosphatase Calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-κB activation. J Biol Chem. (2011) 286:7522–34. doi: 10.1074/jbc.M110.155895
53. Frischbutter S, Gabriel C, Bendfeldt H, Radbruch A, Baumgrass R. Dephosphorylation of Bcl-10 by calcineurin is essential for canonical NF-κB activation in Th cells. Eur J Immunol. (2011) 41:2349–57. doi: 10.1002/eji.201041052
54. Rueda D, Gaide O, Ho L, Lewkowicz E, Niedergang F, Hailfinger S, et al. Bcl10 controls TCR- and Fc R-induced Actin Polymerization. J Immunol. (2007) 178:4373–84. doi: 10.4049/jimmunol.178.7.4373
55. Cheng J, Hamilton KS, Kane LP. Phosphorylation of Carma1, but not Bcl10, by Akt regulates TCR/CD28-mediated NF-κB induction and cytokine production. Mol Immunol. (2014) 59:110–6. doi: 10.1016/j.molimm.2014.01.011
56. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet J-L, Srinivasula SM, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-κB. J Biol Chem. (2001) 276:11877–82. doi: 10.1074/jbc.M010512200
57. Eitelhuber AC, Warth S, Schimmack G, Düwel M, Hadian K, Demski K, et al. Dephosphorylation of Carma1 by PP2A negatively regulates T-cell activation: dephosphorylation of Carma1 by PP2A. EMBO J. (2011) 30:594–605. doi: 10.1038/emboj.2010.331
58. Brenner D, Brechmann M, Rohling S, Tapernoux M, Mock T, Winter D, et al. Phosphorylation of CARMA1 by HPK1 is critical for NF-κB activation in T cells. Proc Natl Acad Sci USA. (2009) 106:14508–13. doi: 10.1073/pnas.0900457106
59. Moreno-Garcia ME, Sommer K, Haftmann C, Sontheimer C, Andrews SF, Rawlings DJ. Serine 649 Phosphorylation within the Protein Kinase C-regulated domain down-regulates CARMA1 activity in lymphocytes. J Immunol. (2009) 183:7362–70. doi: 10.4049/jimmunol.0902438
60. Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell (2007) 130:986–8. doi: 10.1016/j.cell.2007.09.007
61. Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry (2010) 49:8240–50. doi: 10.1021/bi101052d
62. Thome M, Gaide O, Micheau O, Martinon F, Bonnet D, Gonzalez M, et al. Equine Herpesvirus protein E10 induces membrane recruitment and phosphorylation of its cellular homologue, Bcl-10. J Cell Biol. (2001) 152:1115–22. doi: 10.1083/jcb.152.5.1115
63. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
64. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. (2012) 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x
65. Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunol Rev. (2012) 246:107–24. doi: 10.1111/j.1600-065X.2012.01100.x
66. Moreno-Garcia ME, Sommer K, Shinohara H, Bandaranayake AD, Kurosaki T, Rawlings DJ. MAGUK-controlled ubiquitination of CARMA1 modulates lymphocyte NF-kappaB activity. Mol Cell Biol. (2010) 30:922–34. doi: 10.1128/MCB.01129-09
67. Hara H, Yokosuka T, Hirakawa H, Ishihara C, Yasukawa S, Yamazaki M, et al. Clustering of CARMA1 through SH3-GUK domain interactions is required for its activation of NF-kappaB signalling. Nat Commun. (2015) 6:5555. doi: 10.1038/ncomms6555
68. Hu S, Du M-Q, Park S-M, Alcivar A, Qu L, Gupta S, et al. cIAP2 is a ubiquitin protein ligase for BCL10 and is dysregulated in mucosa-associated lymphoid tissue lymphomas. J Clin Invest. (2006) 116:174–81. doi: 10.1172/JCI25641
69. Paul S, Kashyap AK, Jia W, He Y-W, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity (2012) 36:947–58. doi: 10.1016/j.immuni.2012.04.008
70. Park Y, Jin H, Liu Y-C. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc Natl Acad Sci USA. (2013) 110:9433–8. doi: 10.1073/pnas.1221925110
71. Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med. (2014) 211:1947–55. doi: 10.1084/jem.20140860
72. Naik E, Dixit VM. Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCbeta kinase activity. J Immunol. (2016) 196:3438–51. doi: 10.4049/jimmunol.1403165
73. Welteke V, Eitelhuber A, Duwel M, Schweitzer K, Naumann M, Krappmann D. COP9 signalosome controls the Carma1-Bcl10-Malt1 complex upon T-cell stimulation. EMBO Rep. (2009) 10:642–8. doi: 10.1038/embor.2009.64
74. Pedersen SM, Chan W, Jattani RP, Mackie deMauri S, Pomerantz JL. Negative regulation of CARD11 signaling and lymphoma cell survival by the E3 ubiquitin ligase RNF181. Mol Cell Biol. (2015) 36:794–808. doi: 10.1128/MCB.00876-15
75. Yang Y, Kelly P, Shaffer AL III, Schmitz R, Yoo HM, Liu X, et al. Targeting Non-proteolytic protein ubiquitination for the treatment of diffuse large B cell lymphoma. Cancer Cell (2016) 29:494–507. doi: 10.1016/j.ccell.2016.03.006
76. Alexia C, Poalas K, Carvalho G, Zemirli N, Dwyer J, Dubois SM, et al. The endoplasmic reticulum acts as a platform for ubiquitylated components of nuclear factor kappaB signaling. Sci Signal. (2013) 6:ra79. doi: 10.1126/scisignal.2004496
77. Sasaki Y, Sano S, Nakahara M, Murata S, Kometani K, Aiba Y, et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. EMBO J. (2013) 32:2463–76. doi: 10.1038/emboj.2013.184
78. Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA. (2013) 110:15247–52. doi: 10.1073/pnas.1314715110
79. Duwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. (2009) 182:7718–28. doi: 10.4049/jimmunol.0803313
80. Carvalho G, Le Guelte A, Demian C, Vazquez A, Gavard J, Bidere N. Interplay between BCL10, MALT1 and IkappaBalpha during T-cell-receptor-mediated NFkappaB activation. J Cell Sci. (2010) 123:2375–80. doi: 10.1242/jcs.069476
81. Sun L, Deng L, Ea C-K, Xia Z-P, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell (2004) 14:289–301. doi: 10.1016/S1097-2765(04)00236-9
82. Meininger I, Griesbach RA, Hu D, Gehring T, Seeholzer T, Bertossi A, et al. Alternative splicing of MALT1 controls signalling and activation of CD4(+) T cells. Nat Commun. (2016) 7:11292. doi: 10.1038/ncomms11292
83. Bidere N, Snow AL, Sakai K, Zheng L, Lenardo MJ. Caspase-8 regulation by direct interaction with TRAF6 in T cell receptor-induced. Curr Biol. (2006) 16:1666–71. doi: 10.1016/j.cub.2006.06.062
84. King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. (2006) 12:1088–92. doi: 10.1038/nm1449
85. Pelzer C, Cabalzar K, Wolf A, Gonzalez M, Lenz G, Thome M. The protease activity of the paracaspase MALT1 is controlled by monoubiquitination. Nat Immunol. (2013) 14:337–45. doi: 10.1038/ni.2540
86. Cabalzar K, Pelzer C, Wolf A, Lenz G, Iwaszkiewicz J, Zoete V, et al. Monoubiquitination and activity of the Paracaspase MALT1 requires Glutamate 549 in the dimerization interface. PLoS ONE (2013) 8:e72051. doi: 10.1371/journal.pone.0072051
87. Hachmann J, Edgington-Mitchell LE, Poreba M, Sanman LE, Drag M, Bogyo M, et al. Probes to monitor activity of the paracaspase MALT1. Chem Biol. (2015) 22:139–47. doi: 10.1016/j.chembiol.2014.11.011
88. Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, et al. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell (2000) 6:961–7. doi: 10.1016/S1097-2765(05)00086-9
89. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. (2008) 9:263–71. doi: 10.1038/ni1561
90. Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol. (2008) 9:272–81. doi: 10.1038/ni1568
91. Gewies A, Gorka O, Bergmann H, Pechloff K, Petermann F, Jeltsch KM, et al. Uncoupling Malt1 threshold function from Paracaspase activity results in destructive autoimmune inflammation. Cell Rep. (2014) 9:1292–305. doi: 10.1016/j.celrep.2014.10.044
92. Jaworski M, Marsland BJ, Gehrig J, Held W, Favre S, Luther SA, et al. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. (2014) 33:2765–81. doi: 10.15252/embj.201488987
93. Bornancin F, Renner F, Touil R, Sic H, Kolb Y, Touil-Allaoui I, et al. Deficiency of MALT1 Paracaspase activity results in unbalanced regulatory and effector T and B cell responses leading to multiorgan inflammation. J Immunol. (2015) 194:3723–34. doi: 10.4049/jimmunol.1402254
94. Yu JW, Hoffman S, Beal AM, Dykon A, Ringenberg MA, Hughes AC, et al. MALT1 Protease activity is required for innate and adaptive immune responses. PLoS ONE (2015) 10:e0127083. doi: 10.1371/journal.pone.0127083
95. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity (2003) 19:749–58. doi: 10.1016/S1074-7613(03)00293-0
96. Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science (2003) 302:1581–4. doi: 10.1126/science.1090769
97. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. (2013) 132:151–8. doi: 10.1016/j.jaci.2013.04.047
98. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined Immunodeficiency Due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol. (2015) 35:135–46. doi: 10.1007/s10875-014-0125-1
99. Jaworski M, Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell Mol Life Sci CMLS (2016) 73:459–73. doi: 10.1007/s00018-015-2059-z
100. Hailfinger S, Nogai H, Pelzer C, Jaworski M, Cabalzar K, Charton J-E, et al. Malt1-dependent RelB cleavage promotes canonical NF- B activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci USA. (2011) 108:14596–601. doi: 10.1073/pnas.1105020108
101. Klein T, Fung S-Y, Renner F, Blank MA, Dufour A, Kang S, et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat Commun. (2015) 6:8777. doi: 10.1038/ncomms9777
102. Elton L, Carpentier I, Staal J, Driege Y, Haegman M, Beyaert R. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-κB signaling. FEBS J. (2016) 283:403–12. doi: 10.1111/febs.13597
103. Douanne T, Gavard J, Bidère N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J Cell Sci. (2016) 129:1775–80. doi: 10.1242/jcs.185025
104. Baens M, Bonsignore L, Somers R, Vanderheydt C, Weeks SD, Gunnarsson J, et al. MALT1 auto-proteolysis is essential for NF-κB-dependent gene transcription in activated lymphocytes. PLoS ONE (2014) 9:e103774. doi: 10.1371/journal.pone.0103774
105. Baens M, Stirparo R, Lampi Y, Verbeke D, Vandepoel R, Cools J, et al. Malt1 self-cleavage is critical for regulatory T cell homeostasis and anti-tumor immunity in mice. Eur J Immunol. (2018) 48:1728–38. doi: 10.1002/eji.201847597
106. Wu C-H, Yang Y-H, Chen M-R, Tsai C-H, Cheng A-L, Doong S-L. Autocleavage of the paracaspase MALT1 at Arg-781 attenuates NF-kappaB signaling and regulates the growth of activated B-cell like diffuse large B-cell lymphoma cells. PLoS ONE (2018) 13:e0199779. doi: 10.1371/journal.pone.0199779
107. Ginster S, Bardet M, Unterreiner A, Malinverni C, Renner F, Lam S, et al. Two Antagonistic MALT1 Auto-cleavage mechanisms reveal a role for TRAF6 to unleash MALT1 activation. PLoS ONE (2017) 12:e0169026. doi: 10.1371/journal.pone.0169026
108. Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical. Science (2011) 331:468–72. doi: 10.1126/science.1198946
109. Nie Z, Du M-Q, McAllister-Lucas LM, Lucas PC, Bailey NG, Hogaboam CM, et al. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun. (2015) 6:5908. doi: 10.1038/ncomms6908
110. Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1: CYLD is cleaved by MALT1. EMBO J. (2011) 30:1742–52. doi: 10.1038/emboj.2011.85
111. Jeltsch KM, Hu D, Brenner S, Zöller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat Immunol. (2014) 15:1079–89. doi: 10.1038/ni.3008
112. Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-induced cleavage of Regnase-1 in CD4+ helper T cells regulates immune activation. Cell (2013) 153:1036–49. doi: 10.1016/j.cell.2013.04.034
113. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
114. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal. (2014) 7:ra55. doi: 10.1126/scisignal.2005169
115. Malinverni C, Unterreiner A, Staal J, Demeyer A, Galaup M, Luyten M, et al. Cleavage by MALT1 induces cytosolic release of A20. Biochem Biophys Res Commun. (2010) 400:543–7. doi: 10.1016/j.bbrc.2010.08.091
116. Honma K, Tsuzuki S, Nakagawa M, Karnan S, Aizawa Y, Kim WS, et al. TNFAIP3 is the target gene of chromosome band 6q23.3-q24.1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer (2008) 47:1–7. doi: 10.1002/gcc.20499
117. Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature (2009) 459:712–6. doi: 10.1038/nature07969
118. Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood (2009) 114:2467–75. doi: 10.1182/blood-2008-12-194852
119. Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell (2001) 7:401–9. doi: 10.1016/S1097-2765(01)00187-3
120. Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. (2007) 204:1475–85. doi: 10.1084/jem.20062694
121. Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, et al. The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR–IL-1R by controlling degradation of regnase-1. Nat Immunol. (2011) 12:1167–75. doi: 10.1038/ni.2137
122. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. (2017) 49:1192–201. doi: 10.1038/ng.3898
123. Wray-Dutra MN, Chawla R, Thomas KR, Seymour BJ, Arkatkar T, Sommer KM, et al. Activated CARD11 accelerates germinal center kinetics, promoting mTORC1 and terminal differentiation. J Exp Med. (2018). 215:2445–61. doi: 10.1084/jem.20180230
124. Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. (2009) 206:2313–20. doi: 10.1084/jem.20091167
125. Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large. Proc Natl Acad Sci USA. (2009) 106:19946–51. doi: 10.1073/pnas.0907511106
126. Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell (2012) 22:812–24. doi: 10.1016/j.ccr.2012.11.003
127. Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature (2017) 550:481–6. doi: 10.1038/nature24451
128. Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature (2017) 550:534–8. doi: 10.1038/nature24006
129. Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature (2016) 533:125–9. doi: 10.1038/nature17664
130. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. (2013) 8:2281–308. doi: 10.1038/nprot.2013.143
131. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi: 10.1126/science.1225829
Keywords: CARMA1, CARD11, BCL10, MALT1, CBM, post-translational modifications, diffuse large B-cell lymphoma, ABC DLBCL
Citation: Thys A, Douanne T and Bidère N (2018) Post-translational Modifications of the CARMA1-BCL10-MALT1 Complex in Lymphocytes and Activated B-Cell Like Subtype of Diffuse Large B-Cell Lymphoma. Front. Oncol. 8:498. doi: 10.3389/fonc.2018.00498
Received: 07 September 2018; Accepted: 15 October 2018;
Published: 09 November 2018.
Edited by:
Catherine Pellat-Deceunynck, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Andrew L. Snow, Uniformed Services University of the Health Sciences, United StatesFengyi Wan, Johns Hopkins University, United States
Copyright © 2018 Thys, Douanne and Bidère. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Bidère, bmljb2xhcy5iaWRlcmVAaW5zZXJtLmZy