 Kshama Gupta*
Kshama Gupta* Terry C. Burns*
Terry C. Burns*- Department of Neurologic Surgery, Mayo Clinic, Rochester, MN, United States
Glioblastoma (GBM) is uniformly fatal with a median survival of just over 1 year, despite best available treatment including radiotherapy (RT). Impacts of prior brain RT on recurrent tumors are poorly understood, though increasing evidence suggests RT-induced changes in the brain microenvironment contribute to recurrent GBM aggressiveness. The tumor microenvironment impacts malignant cells directly and indirectly through stromal cells that support tumor growth. Changes in extracellular matrix (ECM), abnormal vasculature, hypoxia, and inflammation have been reported to promote tumor aggressiveness that could be exacerbated by prior RT. Prior radiation may have long-term impacts on microglia and brain-infiltrating monocytes, leading to lasting alterations in cytokine signaling and ECM. Tumor-promoting CNS injury responses are recapitulated in the tumor microenvironment and augmented following prior radiation, impacting cell phenotype, proliferation, and infiltration in the CNS. Since RT is vital to GBM management, but substantially alters the tumor microenvironment, we here review challenges, knowledge gaps, and therapeutic opportunities relevant to targeting pro-tumorigenic features of the GBM microenvironment. We suggest that insights from RT-induced changes in the tumor microenvironment may provide opportunities to target mechanisms, such as cellular senescence, that may promote GBM aggressiveness amplified in previously radiated microenvironment.
Introduction
Glioblastoma multiforme (GBM) is the most common and lethal adult primary brain malignancy (1, 2). Interactions within the brain extracellular matrix (ECM) facilitate diffuse infiltration, making GBM surgically incurable (3–5). After standard treatment involving maximal safe resection, RT, and chemotherapy, most tumors recur within 18 months. Eighty percent of recurrences occur at the resection margin, wherein highest radiation doses are delivered (6–8). Following recurrence, patients are managed only with palliative care and clinical trials, which rarely achieve prolonged remission of recurrent lesions (9, 10).
Since GBM recurrences typically occur within previously radiated brain parenchyma, understanding altered biology of the radiated microenvironment is critical. We here review evidence suggesting RT has lasting effects on structure and milieu of the GBM microenvironment, facilitating tumor aggressiveness upon recurrence. Such alterations include hypoxia, innate immune activation, and ECM changes. The extent these changes may impact drug penetration, pharmacokinetics, pharmacodynamics, and facilitate cellular resistance requires further investigation (11–13). Radiation, aging, and DNA damage promote senescence—a cellular state defined by a senescence-associated secretory profile (SASP), characterized by pro-inflammatory cytokine and ECM-degrading enzyme production. Given recent advances in therapies targeting senescence, we hypothesize senolytics may attenuate pro-tumorigenic features of GBM microenvironments.
Glioblastoma Microenvironment
GBM cells coexist with genetically normal stromal cells in a dynamic tumor microenvironment (TME) (14). Within the TME, the extracellular matrix (ECM) provides scaffolding for inter-cellular communication and cell migration. The GBM ECM is notable for altered ECM synthesis/degradation and aberrant cell surface profiles (15). GBM heterogeneity adds to GBM microenvironment complexity, spanning hypoxic, proliferative, and infiltrative regions, superimposed upon genetically and transcriptionally heterogeneous cells (16). With hypoxic and other hyper-perfused tumor regions, redox state gradients, oxygen tensions, and pro-inflammatory cytokines, a dynamic GBM microenvironment is formed, which promotes cell proliferation, and tumor infiltration (3–5).
Tumor heterogeneity is a formidable impediment to identifying effective GBM therapies. Variable distribution of soluble factors throughout tumors creates chemotactic gradients (5). Furthermore, infiltrating cells in surrounding brain tissue differ in type and number of genetic alterations compared to cells harvested from the ischemic core or proliferative zone of the tumor (16). Tumor hypoxia promotes matrix production and remodeling, facilitating cell resilience and crosstalk between ECM and the pro-tumorigenic microenvironment (17).
The GBM ECM Promotes Tumor Growth and Cell Invasion
Distinct from normal brain and other solid tumors, the GBM ECM is comprised of a diverse array of glycoproteins, proteoglycans, and polysaccharides that form specialized structures that signal through cell surface receptors (18). The GBM ECM is mechanically rigid compared to normal brain. Fibrillar proteins, like fibronectin, laminin, and collagen, contribute to rigidity (19) and promote proliferation and migration (20). Upregulation of specific glycoproteins (collagen-IV, fibronectin and vitronectin) and proteoglycans (lecticans), promote surface receptor interactions with other molecules as hyaluronan, CD44, tenascins promoting cell invasion pathways (21–31) and can promote neo-angiogenesis, causing vessel leakiness, which facilitates macrophage entry and microglial activation (32). GBM ECM degradative changes are caused by matrix metalloproteinases (MMPs) as MMP-2, MMP-9, which are essential for cell invasion (33). Increased MMP expression alters cell attachment, allowing GBM cells to spread on myelin pathways, facilitating parenchymal disruption and tumor infiltration (34, 35). Furthermore, immune-active proteoglycans as heparan sulfate proteoglycans (HSPGs) are upregulated in GBM (21, 36) and can act as co-receptors for chemokines, cytokines and growth factors (such as CCL2, IL-1β; tumor necrosis factor-α, TNF-α; transforming growth factor-β, TGF- β), harnessing pathways of progenitor proliferation, cell migration, and axonal pathfinding to facilitate tumor cell proliferation and infiltration (21, 37). Elevated extracellular adenosine in GBM also promotes proliferation, metastasis, microglial phagocytic activity, and adaptive tumor immune responses (38, 39). As critical mediators of tumor growth and cell invasion, various ECM components and associated receptors are hypothesized as therapeutic targets for GBM, including glycoproteins (e.g., tenascin-c; collagen, and its receptor DDR-1, discoidin domain receptor-1) (28, 29, 31) proteoglycans (brevican) (30), and extracellular nucleotides (adenosine triphosphate, ATP) (36, 40–44). Potential therapeutic targets of the radiated brain and GBM microenvironment are enlisted in Table 1.
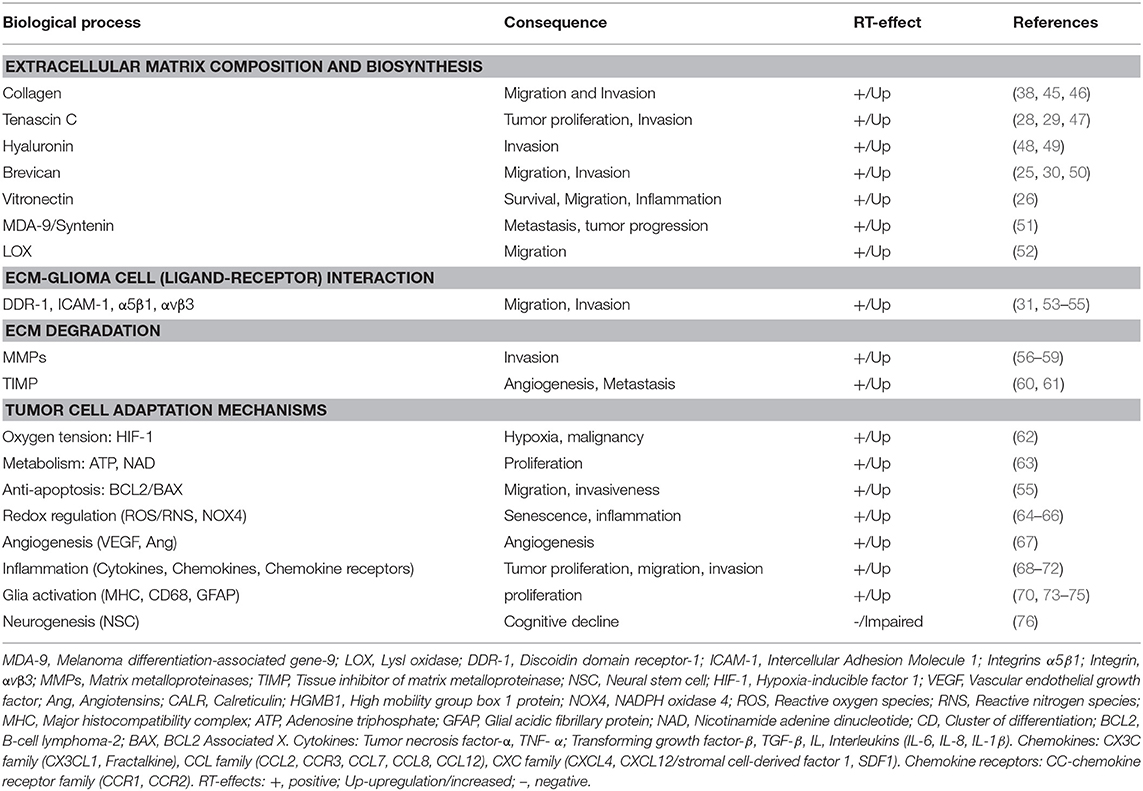
Table 1. Radiation-induced alterations to the GBM micro-environment.
Stromal Cell Populations and Functions in GBM
Various non-neoplastic stromal cells are important in tumor maintenance and recurrence. Resident microglia and infiltrating macrophages are attracted to GBM and comprise approximately 30% of cells in the tumor (77, 78) and with other CNS stromal cells spanning neurons, endothelial cells, astrocytes, and oligodendroglia, create favorable milieu for glioma proliferation and infiltration. Research on impacts of prior radiation on these cells has only just begun.
Resident microglia orchestrate behavior of other immune cells that enter the brain by secreting cytokines and chemokines upregulated in GBM, leading to chronic inflammation. These immune-modulatory factors include cytokines as TNF- α, TGF-β, chemokines as CX3CL1/Fractalkine, CCL2, CCL5 and growth factors as fibroblast growth factor, FGF-2, and granulocyte-monocyte colony stimulating factor, GM-CSF (68, 69, 79–82). Since microglia are difficult to distinguish from bone marrow-derived macrophages, the term “TAM,” for tumor associated macrophages, is often a blanket-term, describing all monocytic cells within the tumor, regardless of specific origin (83, 84). TAMs release various growth factors and cytokines in response to GBM-secreted factors or microenvironment-associated factors, facilitating tumor proliferation, survival, and invasion (83, 84). TAMs can express markers for pro-inflammatory/tumor-suppressing M1 or anti-inflammatory/tumor-promoting M2 phenotypes (83, 85). M1 macrophages are mostly found in oxygenated glioma regions and M2-polarized macrophages are increased in hypoxic areas (86, 87). Hypoxia causes recruitment of macrophages and M2 differentiation through Sema3/Nrp1 signaling (88). Colony stimulating factor 1 receptor (CSF1R) is a key regulator of monocyte/macrophage survival and proliferation and is upregulated in GBM and encourages M2 polarization (89, 90). CSF-1 inhibitors have shown to deter glioma recurrence after radiation in vivo (91), prevent radiation-induced cognitive impairment in preclinical models (92, 93), and are well-tolerated in clinical trials (94).
Neurons can communicate with astrocytes, oligodendrocytes, and microglia via signal molecule release from pre-synaptic terminals. Similarly, tumor growth is stimulated by factors such as electrical activity (95), release of neuroligin-3 (96, 97), neurotransmitters, and neurotrophins (96, 98). Within the TME, glioma cells release microvesicles, transporting miRNAs, mRNAs, angiogenic, and oncogenic factors which promote TAMs, inducing proliferation, infiltration, and immune detection evasion (99).
Astrocytes within the GBM TME exhibit a reactive phenotype, characterized by increased expression of glial acidic fibrillary protein (GFAP) (100). Reactive astrocytes release cytokines, matrix metalloproteinases, stromal cell-derived factor 1 (SDF-1) (101), and upregulate survival genes via gap junction communication with glioma cells, promoting tumor invasiveness and growth (102, 103).
The multiplicity of mechanisms by which the TME promotes glioma growth creates both challenges and opportunities. Although the various pro- tumorigenic activities of TAMs have prompted interest in eliminating them from the TME via CSF1 inhibition (104), the importance of monocytes for innate defense is suggested through increased gliomas in preclinical CSF1 mutants (105) and pharmacologic activation of macrophages to promote glioma phagocytosis (106, 107). Pro-tumorigenic features of the TME such as hypoxia, ECM changes, and neuroinflammation may be augmented following RT. Despite standard clinical use, certain impacts of prior radiation on the TME likely nurture growth of recurrent glioma.
Effects of Radiation Therapy on the GBM Microenvironment
Although RT remains a first-line GBM therapy, dose-dependent risk of devastating neurologic effects precludes doses sufficient for disease eradication, making recurrence inevitable (12, 108, 109). Over 90% of GBM patients experience recurrence at original lesions and 5% develop multiple lesions after RT (110–112). In contrast to intracranial metastatic tumors, which can be eliminated via localized or whole-brain radiation, glioma stem cells survive RT and eventual recurrence is fostered by radiation-induced changes in the TME (113, 114).
Many radiation-induced changes in the TME have been documented (115, 116). Ionizing radiation (IR) generates reactive oxygen and nitrogen species (ROS/RNS) that directly damage DNA, proteins, and phospholipid membranes (64). RT is partially dependent upon double-stranded DNA breaks that overwhelms cellular repair mechanisms, triggering apoptosis in proliferating cells (117). GSCs that escape apoptosis persist in a relatively non-proliferative state until recurrence. Changes in irradiated TME include increased oxidative stress, hypoxia, neuroinflammation, altered cell adhesion molecule expression, changes in ECM, stem/progenitor cell death, cellular senescence induction, and impaired neurogenesis (70, 118–121), followed by neo-angiogenesis, vasculogenesis (114), and tumor recurrence. The extent that these radiation-induced impacts may augment aggressiveness of recurrent tumors is a growing research area. However, the extent that radiation-induced senescence may exacerbate pro-tumorigenic microenvironment following RT is an underexplored avenue. We here discuss the key pathophysiologic changes of the irradiated brain and GBM microenvironment, and the central role ECM/cell-matrix interactions play in manifesting these processes. Their collective involvement in the establishment of a reactive TME that is supportive of aggressive tumor growth, is illustrated in Figure 1.
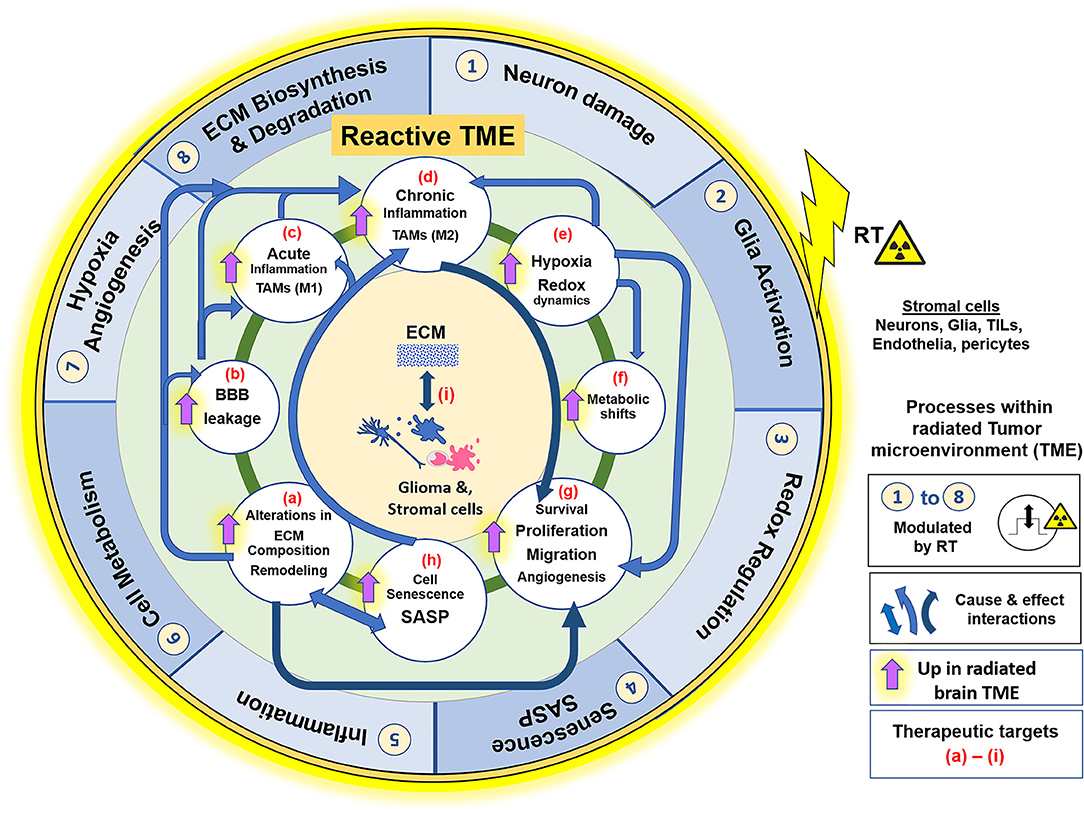
Figure 1. Effects of RT on the glioblastoma (GBM) tumor microenvironment (TME). ECM and its interaction with cellular components such as Glia (microglia and astrocytes), glioma cells, endothelia, pericytes and peripherally derived tumor infiltrating leukocytes (TILs), play a central role in the GBM TME, which contributes to tumor cell survival, proliferation, migration, and invasion. The illustration represents the key pathophysiological processes and their interactions within the radiated TME. Outer blue circle represents biological phenomenon (1–8) that are directly impacted by RT. The inner green circle represents the reactive GBM TME, with its various processes, alterations or adaptive mechanisms that are upregulated in effect of RT, described from (a–i). Blue arrows indicate the “cause and effect” interactions between these processes facilitating GBM pathology and, dark blue arrows signify the primary role of the respective alterations in facilitating cell motility and invasiveness and thus aggressive tumor recurrence. Radiation injury leads to neuronal damage, overactivation of M1 microglia, and elicits acute inflammatory response, with high ROS production in neurons and glia cells. There is alteration in ECM composition, and ECM-cell interactions. MMP/TIMP disbalance degrades Col-IV in basement membranes, which leads to blood brain barrier leakage. Pronounced inflammation causes infiltration of leukocytes (monocyte derived cell populations, macrophages), which along with activated microglia form the TAMs. TME of progressive tumors favor M2 phenotype and establishment of chronic inflammation. Redox dysregulation in effect of RT causes exacerbation of SASP phenotype and tumor cell adaptive processes, like Hypoxia, metabolic shifts, and redox regulation (ROS/NO production), leading to neo-angiogenesis and ECM remodeling. These alterations collectively make the TME permissive to glioma cell migration and invasion, thereby contributing to resistance and an aggressive tumor recurrence. All these biological processes in the reactive TME are potential therapeutic targets for improved glioblastoma care, with having ECM-cell interactions central to the manifestation of each of these phenomenon. RT, Radiation therapy; ECM, extracellular matrix; TAM, Tumor associated macrophages; TILs, Tumor infiltrating leukocytes; MMP, matrix metalloproteases; TIMP, Tissue inhibitor of metalloproteases; Col-IV, collagen-type IV; BBB, blood brain barrier; ROS, reactive oxygen species; NO, Nitric oxide; SASP, Senescence associated secretory phenotype; M1/M2, proinflammatory or immune suppressive phenotypes of TAMs, respectively.
Radiation-Induced Cellular Senescence
Senescence is a defensive mechanism in response to stress that arrests cells at risk for malignant transformation. Radiation-induced DNA damage and oxidative stress, cytotoxic exposure, and/or aging, prompts apoptosis, unless upregulation of cyclin-dependent kinase inhibitors such as p16 or p21 allows senescence induction (122). GBM cells harbor a heterogeneous array of mutations, leading to constitutive activation of repair mechanisms that prevent apoptosis in response to RT, despite damage that would otherwise render cells unviable (123). Such mutations may facilitate upregulation of oncogene-induced senescence (124, 125), prompting a baseline level of senescence-associated signaling in GBM that is amplified following radiation in a dose- and time-dependent manner (126, 127).
A hallmark feature of senescent cells is the “senescence-associated secretory phenotype” (SASP), characterized by release of proinflammatory signaling molecules, proteolytic enzymes, and ECM components (128). Together with MMPs, proinflammatory SASP components are thought to create a microenvironment that promotes survival, proliferation, and dissemination of neoplastic cells across brain parenchyma (129, 130). Such adaptations may contribute to increased aggression of recurrent GBM and have been shown in multiple cancers to promote tumor progression and metastatic spread (124). SASP also has paracrine effects, spreading the phenotype to neighboring cells (131). Discovery of senolytic drugs and their therapeutic combinations, such as dasatinib and quercetin (D+Q) has raised hopes that senescent cell-induced diseases may soon be curable (132, 133). Since radiation is among the most reliable experimental strategies to induce senescence, it is reasonable that the previously radiated tumor will be exposed to SASP. Prior work has demonstrated that co- implantation of radiated with non-radiated cells increases tumor aggressiveness. We (134), and others (133, 135–137), have observed increased tumor aggressiveness after implantation of glioma cells into previously radiated hosts. Given the potential for senescent cells to induce tissue dysfunction and inflammation, several studies have addressed the potential of metabolically active senescent cells and SASP factors to exacerbate recurrences of various cancers (124, 138, 139). Though much work remains to establish mechanisms, cellular senescence after radiation likely has important implications for recurrent GBM.
Elevated levels of ROS cause matrix dysfunction through remodeling and fragmentation of collagen and proteoglycans, pronounced protease activity, and altering cytoskeletal contractility (by modulating actin, and tubulin), fueling senescent phenotypes marked by irregular collagen meshworks and ECM degradation (140–142). These phenomena may reduce tension and elasticity of affected tissue, supporting invasion and metastasis. Oxidative stress and associated mitochondrial function can further propagate RT-induced senescence (143–145).
Radiation-induced bystander effects contribute to cellular senescence, as well as tumor promotion and recurrence (146). Bystander effects are defined by a cell's reaction to its neighboring irradiated cell, with consequences of damage to nearby healthy brain regions. Irradiated GBM cells have shown to induce bystander effects including increased cell growth, micronucleus formation, and apoptosis in non-irradiated tumor cells (147–149). These bystander processes can ignite ROS production and mitochondrial dysfunction, leading to persistent or irreparable DNA damage, activation of DNA damage responses, irreversible cell cycle arrest, and culmination of cellular senescence (128).
Radiation-Induced Adaptations Facilitate Aggressiveness of Recurrent GBMs
It has been said that what doesn't kill you makes you stronger, and in certain respects GBM exemplifies such adaptive behavior. Tumors adapt to radiation-induced oxidative stress through several mechanisms, including metabolic shifts (63), elevated antioxidant peptide production, and intra-tumoral hypoxia generation (150, 151).
Hypoxic conditions facilitate tumor radio-resistance and recurrence, as ROS are insufficient for apoptosis induction (152). Radiation-induced stabilization and activation of hypoxia-inducible factor 1 (HIF-1) elicits protective processes through regulating downstream target genes, such as nitric oxide (NO), which can stimulate immunosuppressive and anti-apoptotic responses (62, 67).
Vascular remodeling is a hallmark of IR injury. Radiation affects vascular integrity, causing vasculopathy, vascular depletion, hypoxia, and neo-angiogenesis (153–155). Levels of vascular endothelial growth factor (VEGF) and angiotensin are increased in GBM post-radiation, contributing to angiogenesis and tumor growth (67, 156), while SDF promotes vasculogenesis (114, 156). Radiation induces changes in cell density, tight junctions, and increased BBB permeability to inflammatory cells, and perhaps pharmacologic agents (11, 70, 154, 157).
Radiation-induced alterations in ECM composition have incompletely understood impacts on cognitive function and tumor infiltration and proliferation. Specific proteins involved in ECM biosynthesis (brevican, vitronectin, tenascin C, hyaluronin, lysyl oxidase) (25–27, 47, 52), degradation (matrix metalloproteinases) (56–59), signal transduction (melanoma differentiation-associated gene 9/Syntenin) (51), and ECM-glioma cell interactions (ICAM-1, DDR-1, integrins) (53–55, 158) are upregulated following radiation. These alterations may facilitate tumor cell infiltration and further exacerbate impacts of radiation on tumor cells themselves. These alterations include induction of a pro-migratory, p53-mediated mesenchymal phenotype (159). Additional p53-independent changes have been reported, including transcriptional regulation (45), integrin expression, MMP expression and activity (56, 57, 59, 160), altered membrane type 1 MMP and tissue inhibitor of MMP-2 expression, and increased BCL-2/BAX expression (55), resulting in apoptosis resistance and enhanced migration.
Effects of Radiation on Inflammation in the GBM Microenvironment
Radiation-induced vascular permeability leads to infiltration of immune cells into brain parenchyma. RT-induced chronic inflammation promotes high intracellular NO in glioma cells (161), causing stabilization of HIF-1 and inhibition of tissue inhibitor of matrix metalloproteases-1 (TIMP-1) (162, 163). HIF-1-induced expression of stromal-derived factor 1 (SDF-1) promotes recruitment of macrophages following RT (71, 164). Elevated NO also inhibits tissue TIMP-1, contributing to ECM remodeling and tumor cell invasion (60, 165).
Radiation-induced cellular damage induces astrocyte gliosis, characterized by increased GFAP expression (73, 74), mediated by oxidative stress and microglial release of prostaglandins (74). Radiation-induced redox signaling profoundly alters microglial function. Differential expression of iNOS and arginase-1 in M1 vs. M2 profiles provides the basis of redox control in TAM phenotypes (166). Nuclear factor (erythroid-derived 2)-like 2 (NRF2) regulates redox dynamics and favors an M2 phenotype with cytoprotective effects (167, 168). Additionally, glioma cells and stromal cells maintain a pool of antioxidant peptides, such as glutathione and thioredoxin, protecting them against radiation- induced redox (169, 170). Differential ability of glioma cells to modulate redox reactions may be a mechanism by which certain cancer cells are more radioresistant than others. Combinatorial effects of RT on the TME lead to increased aggressiveness of recurrent GBM.
The extent that M1 vs. M2 polarization states relate to radiation-induced changes in microglia remains unclear. The M1 polarization state is most characteristically defined as that exhibited by systemic monocytes upon in vivo exposure to lipopolysaccharide, compared to M2 phenotype following IL4 exposure. Though this terminology has been widely extended to microglia, the actual microglia activation states are almost certainly more complex. To date, the transcriptional profiles of radiated mouse microglia have been described 24 h and 1 month after whole brain radiation, yielding phenotypes unique from, but partially overlapping with published M1 and, to a lesser extent, M2 phenotypes. Notably, the degree of ECM changes induced in radiated microglia exceeded both M1 and M2, while closely approximating changes observed in aged microglia (171). How this pertains to radiation-induced changes in the radiated TME is unclear, however, our unpublished observations demonstrate strongest enrichment of radiated microglial genes in the mesenchymal GBM subtype, as well as patients with the worst prognosis. Poorer prognosis of aged patients with GBM is well-documented. Whether the more radiation-like polarization state of aged microglia contributes to such poorer outcomes remains unknown. However, given the recurrent theme of chronic inflammation, in GBM, radiation, aging, and neurodegeneration, efforts to modulate the inflammatory microenvironment of both primary and recurrent GBM are of broad interest to attenuate tumorigenesis and enhance cognitive outcomes following radiation (172, 173). Given the differences in the neuroinflammatory phenotype of rodents and humans, mechanistic studies specifically interrogating human disease will be paramount (174).
Therapeutic Implications and Future Directions
Most mortalities are due to GBM recurrence. Most recurrent tumors arise from the previously radiated location. As such, understanding and combating mechanisms via which RT may augment pro-tumorigenic mechanisms in the GBM microenvironment is necessary to facilitate long-term survival. GBM invasiveness is induced by radiation and facilitates distant failures in the event of prolonged local control. Systemic radiosensitizers in combination with RT are being explored to enhance the effects of radiation with aims of lowering radiation and chemotherapy doses with improved efficacy (175). Whether senolytics may complement other classes of radiation sensitizers remains unknown.
RT-induced ECM alterations for GBM infiltration are potentially important therapeutic targets. Prior studies on inhibition of ECM biosynthetic and degradative processes, and receptor blockade to prevent ECM-cell interactions for cancer prevention, further support this idea (176, 177). Targeting cytoskeletal dynamics is also a proposed therapeutic strategy (178). Targeting microglial-ECM interactions that promote pro-tumorigenic phenotypes in GBM may also offer opportunities for therapeutic intervention. Developing technologies to anatomically direct targeted therapies to a radiated region may provide capabilities akin to limit off-target effects through use of radiation sensitizers, senolytics, or other agents selectively active in radiated tissue (179).
This work has discussed a variety of challenges for recurrent GBM management, highlighting important roles of the TME and associated matrix-cell interactions in instigating pathophysiological processes. Radiation-induced alterations in the microenvironment that can serve as targets for therapeutic intervention are summarized in Table 1, whereas the immunostimulatory role of hypofractionated radiation as an immunotherapy component is described by others (180) and us (Rajani et al, article under preparation). Importantly, it should be emphasized that no single therapy will likely be sufficient for tumors as heterogeneous as GBM and combinatorial treatment may be required. Balancing pros and cons of RT in concert with targeted therapies will provide an ongoing focus of therapeutic efforts for glioblastoma. Translational strategies are needed that can yield mechanistic biomarkers of efficacy for optimization of multi-drug approaches.
Conclusions
Understanding and targeting the GBM microenvironment is no less important than targeting the biology of GBM cells. GBM infiltration depends on unique features of the CNS microenvironment. Several lines of evidence suggest long-term sequelae of radiation can exacerbate recurrent glioma. Understanding lasting impacts of radiation and other therapies on the TME will be necessary to overcome recurrent disease. Two other take-home points are worth emphasizing:
1) Unlike heterogeneous GBM cells that can out-mutate targeted therapies, the genetic stability and thus inherently greater predictability of tumor stroma should offer a tangible focal point for targeted therapies
2) The many mechanisms by which GBM cells harness the TME to their advantage should encourage multidisciplinary efforts to develop and translate synergistic multi-target therapies optimized to the unique biology of the CNS.
Author Contributions
KG proposed the presented concept and idea, wrote and edited the manuscript, and designed the figures. TB proposed the presented concept and idea, reviewed the manuscript and supervised and supported KG. All authors have contributed to the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors.
Acknowledgments
Funding support (TB) was provided by NIH K12 NRDCP, Brains Together for a Cure, the Mayo Clinic Grand Forks Career Development Program, and Regenerative Medicine Minnesota. The authors acknowledge the drafting, research, and editing assistance of Superior Medical Experts, and the editorial assistance of Kirsten Burns, Psy. D.
References
1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. (2008) 359:492–507. doi: 10.1056/NEJMra0708126
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
3. Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia (2006) 53:799–808. doi: 10.1002/glia.20334
4. Zagzag D, Esencay M, Mendez O, Yee H, Smirnova I, Huang Y, et al. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor- 1alpha/CXCR4 expression in glioblastomas: one plausible explanation of Scherer's structures. Am J Pathol. (2008) 173:545–60. doi: 10.2353/ajpath.2008.071197
5. Manini I, Caponnetto F, Bartolini A, Ius T, Mariuzzi L, Di Loreto C, et al. Role of microenvironment in glioma invasion: what we learned from in vitro models. Int J Mol Sci. (2018) 19:E147. doi: 10.3390/ijms19010147
6. Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. (2007) 25:4127–36. doi: 10.1200/JCO.2007.11.8554
7. Linz U. Commentary on Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459–466). Cancer (2010) 116:1844–6. doi: 10.1002/cncr.24950
8. Minniti G, Amelio D, Amichetti M, Salvati M, Muni R, Bozzao A, et al. Patterns of failure and comparison of different target volume delineations in patients with glioblastoma treated with conformal radiotherapy plus concomitant and adjuvant temozolomide. Radiother Oncol. (2010) 97:377–81. doi: 10.1016/j.radonc.2010.08.020
9. Seystahl K, Wick W, Weller M. Therapeutic options in recurrent glioblastoma—An update. Crit Rev Oncol Hematol. (2016) 99:389–408. doi: 10.1016/j.critrevonc.2016.01.018
10. Amelio D, Amichetti M. Radiation therapy for the treatment of recurrent glioblastoma: an overview. Cancers (2012) 4:257–80. doi: 10.3390/cancers4010257
11. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
12. Bhowmik A, Khan R, Ghosh MK. Blood brain barrier: a challenge for effectual therapy of brain tumors. Biomed Res Int. (2015) 2015:320941. doi: 10.1155/2015/320941
13. Lee WH, Warrington JP, Sonntag WE, Lee YW. Irradiation alters MMP-2/TIMP-2 system and collagen type IV degradation in brain. Int J Radiat Oncol Biol Phys. (2012) 82:1559–66. doi: 10.1016/j.ijrobp.2010.12.032
14. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer (2009) 9:239–52. doi: 10.1038/nrc2618
15. Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive oxygen species (ROS)—A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur Cell Mater. (2012) 24:249–65. doi: 10.22203/eCM.v024a18
16. De Vleeschouwer S, Bergers G. Glioblastoma: to target the tumor cell or the microenvironment? In: De Vleeschouwer S, editor. Glioblastoma. Brisbane, QLD: Codon Publications (2017), pp. 315–40.
17. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer (2014) 14:430–9. doi: 10.1038/nrc3726
18. Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The evolution of extracellular matrix. Mol Biol Cell (2010) 21:4300–5. doi: 10.1091/mbc.E10-03-0251
19. Mahesparan R, Tysnes BB, Read TA, Enger PO, Bjerkvig R, Lund-Johansen M. Extracellular matrix-induced cell migration from glioblastoma biopsy specimens in vitro. Acta Neuropathol. (1999) 97:231–9. doi: 10.1007/s004010050979
20. Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. (2009) 69:4167–74. doi: 10.1158/0008-5472.CAN-08-4859
21. Wade A, Robinson AE, Engler JR, Petritsch C, James CD, Phillips JJ. Proteoglycans and their roles in brain cancer. FEBS J. (2013) 280:2399–417. doi: 10.1111/febs.12109
22. Sim H, Hu B, Viapiano MS. Reduced expression of the hyaluronan and proteoglycan link proteins in malignant gliomas. J Biol Chem. (2009) 284:26547–56. doi: 10.1074/jbc.M109.013185
23. Yadavilli S, Hwang EI, Packer RJ, Nazarian J. The role of NG2 proteoglycan in glioma. Transl Oncol. (2016) 9:57–63. doi: 10.1016/j.tranon.2015.12.005
24. Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. (2011) 15:1013–31. doi: 10.1111/j.1582-4934.2010.01236.x
25. Gladson CL, Wilcox JN, Sanders L, Gillespie GY, Cheresh DA. Cerebral microenvironment influences expression of the vitronectin gene in astrocytic tumors. J Cell Sci. (1995) 108 (Pt 3):947–56.
26. Nutt CL, Matthews RT, Hockfield S. Glial tumor invasion: a role for the upregulation and cleavage of BEHAB/brevican. Neuroscientist (2001) 7:113–22. doi: 10.1177/107385840100700206
27. Milner R, Campbell IL. The extracellular matrix and cytokines regulate microglial integrin expression and activation. J Immunol. (2003) 170:3850–8. doi: 10.4049/jimmunol.170.7.3850
28. Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. (2006) 66:11771–80. doi: 10.1158/0008-5472.CAN-05-0470
29. Sarkar S, Zemp FJ, Senger D, Robbins SM, Yong VW. ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor- initiating cells. Neuro Oncol. (2015) 17:1095–105. doi: 10.1093/neuonc/nou362
30. Hu B, Kong LL, Matthews RT, Viapiano MS. The proteoglycan brevican binds to fibronectin after proteolytic cleavage and promotes glioma cell motility. J Biol Chem. (2008) 283:24848–59. doi: 10.1074/jbc.M801433200
31. Ram R, Lorente G, Nikolich K, Urfer R, Foehr E, Nagavarapu U. Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J Neurooncol. (2006) 76:239–48. doi: 10.1007/s11060-005-6874-1
32. Xu Y, Stamenkovic I, Yu Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. (2010) 70:2455–64. doi: 10.1158/0008-5472.CAN-09-2505
33. Wang M, Wang T, Liu S, Yoshida D, Teramoto A. The expression of matrix metalloproteinase-2 and−9 in human gliomas of different pathological grades. Brain Tumor Pathol. (2003) 20:65–72. doi: 10.1007/BF02483449
34. Chintala SK, Rao JK. Invasion of human glioma: role of extracellular matrix proteins. Front Biosci. (1996) 1:d324–39.
35. Amberger VR, Hensel T, Ogata N, Schwab ME. Spreading and migration of human glioma and rat C6 cells on central nervous system myelin in vitro is correlated with tumor malignancy and involves a metalloproteolytic activity. Cancer Res. (1998) 58:149–58.
36. Xiong A, Kundu S, Forsberg-Nilsson K. Heparan sulfate in the regulation of neural differentiation and glioma development. FEBS J. (2014) 281:4993–5008. doi: 10.1111/febs.13097
37. O'Callaghan P, Zhang X, Li JP. Heparan Sulfate Proteoglycans as Relays of Neuroinflammation. J Histochem Cytochem. (2018) 66:305–19. doi: 10.1369/0022155417742147
38. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13:842–57. doi: 10.1038/nrc3613
39. Rocha R, Torres A, Ojeda K, Uribe D, Rocha D, Erices J, et al. The adenosine A(3) receptor regulates differentiation of glioblastoma stem-like cells to endothelial cells under hypoxia. Int J Mol Sci. (2018) 19:E1228. doi: 10.3390/ijms19041228
40. Vihinen P, Kahari VM. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer (2002) 99:157–66. doi: 10.1002/ijc.10329
41. Wiranowska M, Ladd S, Moscinski LC, Hill B, Haller E, Mikecz K, et al. Modulation of hyaluronan production by CD44 positive glioma cells. Int J Cancer (2010) 127:532–42. doi: 10.1002/ijc.25085
42. Payne LS, Huang PH. The pathobiology of collagens in glioma. Mol Cancer Res. (2013) 11:1129–40. doi: 10.1158/1541-7786.MCR-13-0236
43. Seifert S, Sontheimer H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J Physiol. (2014) 592:5109–27. doi: 10.1113/jphysiol.2014.274498
44. Xia S, Lal B, Tung B, Wang S, Goodwin CR, Laterra J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. (2016) 18:507–17. doi: 10.1093/neuonc/nov171
45. Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T, et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. (2014) 16:671–85. doi: 10.1093/neuonc/not239
46. Young AR, Saveland H, Pickard JD, Perry S, Brandt L, Ljunggren B. Early effects of tetraethylammonium chloride on the contractile properties of isolated rabbit basilar arteries. J Cereb Blood Flow Metab. (1987) 7:237–47. doi: 10.1038/jcbfm.1987.49
47. Riekki R, Jukkola A, Oikarinen A, Kallioinen M. Radiation therapy induces tenascin expression and angiogenesis in human skin. Acta Derm Venereol. (2001) 81:329–33. doi: 10.1080/000155501317140025
48. Yoo KC, Suh Y, An Y, Lee HJ, Jeong YJ, Uddin N, et al. Proinvasive extracellular matrix remodeling in tumor microenvironment in response to radiation. Oncogene (2018) 37:3317–28. doi: 10.1038/s41388-018-0199-y
49. Jaworski DM, Kelly GM, Piepmeier JM, Hockfield S. BEHAB (brain enriched hyaluronan binding) is expressed in surgical samples of glioma and in intracranial grafts of invasive glioma cell lines. Cancer Res. (1996) 56:2293–8.
50. Donson AM, Erwin NS, Kleinschmidt-DeMasters BK, Madden JR, Addo-Yobo SO, Foreman NK. Unique molecular characteristics of radiation-induced glioblastoma. J Neuropathol Exp Neurol. (2007) 66:740–9. doi: 10.1097/nen.0b013e3181257190
51. Kegelman TP, Wu B, Das SK, Talukdar S, Beckta JM, Hu B, et al. Inhibition of radiation-induced glioblastoma invasion by genetic and pharmacological targeting of MDA-9/Syntenin. Proc Natl Acad Sci USA. (2017) 114:370–5. doi: 10.1073/pnas.1616100114
52. Shen CJ, Sharma A, Vuong DV, Erler JT, Pruschy M, Broggini-Tenzer A. Ionizing radiation induces tumor cell lysyl oxidase secretion. BMC Cancer (2014) 14:532. doi: 10.1186/1471-2407-14-532
53. Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Essential role of cooperative NF-kappaB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene (2013) 32:5144–55. doi: 10.1038/onc.2012.546
54. Ongusaha PP, Kim JI, Fang L, Wong TW, Yancopoulos GD, Aaronson SA, et al. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. (2003) 22:1289–301. doi: 10.1093/emboj/cdg129
55. Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. (2001) 61:2744–50.
56. Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. (2006) 66:8511–9. doi: 10.1158/0008-5472.CAN-05-4340
57. Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene (2006) 25:7009–18. doi: 10.1038/sj.onc.1209706
58. Wang JL, Sun Y, Wu S. Gamma-irradiation induces matrix metalloproteinase II expression in a p53-dependent manner. Mol Carcinog. (2000) 27:252–8. doi: 10.1002/(SICI)1098-2744(200004)27:4<252::AID-MC2>3.0.CO;2-3
59. Pei J, Park IH, Ryu HH, Li SY, Li CH, Lim SH, et al. Sublethal dose of irradiation enhances invasion of malignant glioma cells through p53-MMP 2 pathway in U87MG mouse brain tumor model. Radiat Oncol. (2015) 10:164. doi: 10.1186/s13014-015-0475-8
60. Gardner J, Ghorpade A. Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J Neurosci Res. (2003) 74:801–6. doi: 10.1002/jnr.10835
61. Nirmala C, Jasti SL, Sawaya R, Kyritsis AP, Konduri SD, Ali-Osman F, et al. Effects of radiation on the levels of MMP-2, MMP-9 and TIMP-1 during morphogenic glial-endothelial cell interactions. Int J Cancer (2000) 88:766–71. doi: 10.1002/1097-0215(20001201)88:53.0.CO;2-Y
62. Kim YH, Yoo KC, Cui YH, Uddin N, Lim EJ, Kim MJ, et al. Radiation promotes malignant progression of glioma cells through HIF-1alpha stabilization. Cancer Lett. (2014) 354:132–41. doi: 10.1016/j.canlet.2014.07.048
63. Spehalski EI, Peters C, Camphausen KA, Tofilon P. Distinctions between the metabolic changes in glioblastoma cells and glioma stem-like cells following irradiation. Int J Radiat Oncol Biol Phys. (2017) 99:E617. doi: 10.1016/j.ijrobp.2017.06.2088
64. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. (2012) 327:48–60. doi: 10.1016/j.canlet.2011.12.012
65. Ahamed J, Laurence J. Role of platelet-derived transforming growth factor-beta1 and reactive oxygen species in radiation-induced organ fibrosis. Antioxid Redox Signal (2017) 27:977–88. doi: 10.1089/ars.2017.7064
66. Collins-Underwood JR, Zhao W, Sharpe JG, Robbins ME. NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic Biol Med. (2008) 45:929–38. doi: 10.1016/j.freeradbiomed.2008.06.024
67. Kil WJ, Tofilon PJ, Camphausen K. Post-radiation increase in VEGF enhances glioma cell motility in vitro. Radiat Oncol. (2012) 7:25. doi: 10.1186/1748-717X-7-25
68. Matias D, Balça-Silva J, da Graça GC, Wanjiru CM, Macharia LW, Nascimento CP, et al. Microglia/astrocytes-glioblastoma crosstalk: crucial molecular mechanisms and microenvironmental factors. Front Cell Neurosci. (2018) 12:235. doi: 10.3389/fncel.2018.00235
69. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol. (2012) 189:444–53. doi: 10.4049/jimmunol.1103248
70. Lumniczky K, Szatmari T, Safrany G. Ionizing radiation-induced immune and inflammatory reactions in the brain. Front Immunol. (2017) 8:517. doi: 10.3389/fimmu.2017.00517
71. Tabatabai G, Frank B, Mohle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-beta-dependent HIF-1alpha-mediated induction of CXCL12. Brain (2006) 129:2426–35. doi: 10.1093/brain/awl173
72. Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol. (2012) 2:89. doi: 10.3389/fonc.2012.00089
73. Chiang CS, McBride WH, Withers HR. Radiation-induced astrocytic and microglial responses in mouse brain. Radiother Oncol. (1993) 29:60–8. doi: 10.1016/0167-8140(93)90174-7
74. Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, et al. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis. (2006) 21:457–67. doi: 10.1016/j.nbd.2005.08.006
75. Park B, Yee C, Lee KM. The effect of radiation on the immune response to cancers. Int J Mol Sci. (2014) 15:927–43. doi: 10.3390/ijms15010927
76. Acharya MM, Lan ML, Kan VH, Patel NH, Giedzinski E, Tseng BP, et al. Consequences of ionizing radiation-induced damage in human neural stem cells. Free Radic Biol Med. (2010) 49:1846–55. doi: 10.1016/j.freeradbiomed.2010.08.021
77. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia (2011) 59:1169–80. doi: 10.1002/glia.21136
78. Watters JJ, Schartner JM, Badie B. Microglia function in brain tumors. J Neurosci Res. (2005) 81:447–55. doi: 10.1002/jnr.20485
79. Albulescu R, Codrici E, Popescu ID, Mihai S, Necula LG, Petrescu D, et al. Cytokine patterns in brain tumour progression. Mediators Inflamm. (2013) 2013:979748. doi: 10.1155/2013/979748
80. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
81. Zhao L, Wang Y, Xue Y, Lv W, Zhang Y, He S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim Biophys Sin (2015) 47:890–8. doi: 10.1093/abbs/gmv095
82. Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia (2000) 29:305–15. doi: 10.1002/(SICI)1098-1136(20000215)29:4<305::AID-GLIA2>3.0.CO;2-V
83. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. (2016) 19:20–7. doi: 10.1038/nn.4185
84. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
85. Kennedy BC, Showers CR, Anderson DE, Anderson L, Canoll P, Bruce JN, et al. Tumor-associated macrophages in glioma: friend or foe? J Oncol. (2013) 2013:486912. doi: 10.1155/2013/486912
86. Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget (2016) 7:80521–42. doi: 10.18632/oncotarget.11825
87. Leblond MM, Gerault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, et al. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology (2016) 5:e1056442. doi: 10.1080/2162402X.2015.1056442
88. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24:695–709. doi: 10.1016/j.ccr.2013.11.007
89. De I, Steffen MD, Clark PA, Patros CJ, Sokn E, Bishop SM, et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. (2016) 76:2552–60. doi: 10.1158/0008-5472.CAN-15-2386
90. Naess F, Nesbakken A, Pillgram-Larsen J, Solheim K, Stadaas JO. [Diaphragmatic injuries]. Tidsskr Nor Laegeforen (1991) 111:1845–6.
91. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. (2016) 18:797–806. doi: 10.1093/neuonc/nov272
92. Feng X, Jopson TD, Paladini MS, Liu S, West BL, Gupta N, et al. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J Neuroinflamm. (2016) 13:215. doi: 10.1186/s12974-016-0671-y
93. Acharya MM, Green KN, Allen BD, Najafi AR, Syage A, Minasyan H, et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci Rep. (2016) 6:31545. doi: 10.1038/srep31545
94. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. (2016) 18:557–64. doi: 10.1093/neuonc/nov245
95. Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature (1993) 361:258–60. doi: 10.1038/361258a0
96. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell (2015) 161:803–16. doi: 10.1016/j.cell.2015.04.012
97. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high- grade glioma. Nature (2017) 549:533–7. doi: 10.1038/nature24014
98. Gillespie S, Monje M. An active role for neurons in glioma progression: making sense of Scherer's structures. Neuro Oncol. (2018) 20:1292–9. doi: 10.1093/neuonc/noy083
99. Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. (2018) 6:18. doi: 10.3389/fcell.2018.00018
100. Gagliano N, Costa F, Cossetti C, Pettinari L, Bassi R, Chiriva-Internati M, et al. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol Rep. (2009) 22:1349–56. doi: 10.3892/or_00000574
101. Barbero S, Bajetto A, Bonavia R, Porcile C, Piccioli P, Pirani P, et al. Expression of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1 in human brain tumors and their involvement in glial proliferation in vitro. Ann N Y Acad Sci. (2002) 973:60–9. doi: 10.1111/j.1749-6632.2002.tb04607.x
102. Guan X, Hasan MN, Maniar S, Jia W, Sun D. Reactive astrocytes in glioblastoma multiforme. Mol Neurobiol. (2018) 55:6927–38. doi: 10.1007/s12035-018-0880-8
103. Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, et al. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci. (2003) 23:4034–43. doi: 10.1523/JNEUROSCI.23-10-04034.2003
104. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
105. Bender AM, Collier LS, Rodriguez FJ, Tieu C, Larson JD, Halder C, et al. Sleeping beauty-mediated somatic mutagenesis implicates CSF1 in the formation of high-grade astrocytomas. Cancer Res. (2010) 70:3557–65. doi: 10.1158/0008-5472.CAN-09-4674
106. Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. (2017) 9:eaaf2968. doi: 10.1126/scitranslmed.aaf2968
107. Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS ONE (2016) 11:e0153550. doi: 10.1371/journal.pone.0153550
108. Chao ST, Ahluwalia MS, Barnett GH, Stevens GH, Murphy ES, Stockham AL, et al. Challenges with the diagnosis and treatment of cerebral radiation necrosis. Int J Radiat Oncol Biol Phys. (2013) 87:449–57. doi: 10.1016/j.ijrobp.2013.05.015
109. Yang H, Chopp M, Schallert T. Functional issues in brain tumor treatment. J Neurol Neurophysiol. (2011) 1:S5–002. doi: 10.4172/2155-9562.S5-002
110. Roy S, Lahiri D, Maji T, Biswas J. Recurrent glioblastoma: where we stand. South Asian J Cancer (2015) 4:163–73. doi: 10.4103/2278-330X.175953
111. Gaspar LE, Fisher BJ, Macdonald DR, LeBer DV, Halperin EC, Schold SC Jr, et al. Supratentorial malignant glioma: patterns of recurrence and implications for external beam local treatment. Int J Radiat Oncol Biol Phys. (1992) 24:55–7. doi: 10.1016/0360-3016(92)91021-E
112. Loeffler JS, Alexander E III, Hochberg FH, Wen PY, Morris JH, Schoene WC, et al. Clinical patterns of failure following stereotactic interstitial irradiation for malignant gliomas. Int J Radiat Oncol Biol Phys. (1990) 19:1455–62. doi: 10.1016/0360-3016(90)90358-Q
113. Rizzo AE, Yu JS. Radiation therapy for glioma stem cells. Adv Exp Med Biol. (2015) 853:85–110. doi: 10.1007/978-3-319-16537-0_6
114. Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. (2010) 120:694–705. doi: 10.1172/JCI40283
115. Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment—tumorigenesis and therapy. Nat Rev Cancer (2005) 5:867–75. doi: 10.1038/nrc1735
116. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer (2015) 15:409–25. doi: 10.1038/nrc3958
117. Yoshimura M, Itasaka S, Harada H, Hiraoka M. Microenvironment and radiation therapy. Biomed Res Int. (2013) 2013:685308. doi: 10.1155/2013/685308
118. Di Maggio FM, Minafra L, Forte GI, Cammarata FP, Lio D, Messa C, et al. Portrait of inflammatory response to ionizing radiation treatment. J Inflamm. (2015) 12:14. doi: 10.1186/s12950-015-0058-3
119. Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, et al. Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res. (2002) 104:159–69. doi: 10.1016/S0169-328X(02)00353-4
120. Lee WH, Sonntag WE, Mitschelen M, Yan H, Lee YW. Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int J Radiat Biol. (2010) 86:132–44. doi: 10.3109/09553000903419346
121. Tseng D, Vasquez-Medrano DA, Brown JM. Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas. Br J Cancer (2011) 104:1805–9. doi: 10.1038/bjc.2011.169
122. Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. (1999) 19:2109–17. doi: 10.1128/MCB.19.3.2109
123. Lindsay KJ, Coates PJ, Lorimore SA, Wright EG. The genetic basis of tissue responses to ionizing radiation. Br J Radiol. (2007) 80 (Spec No 1):S2–6. doi: 10.1259/bjr/60507340
124. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
125. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
126. Panganiban RA, Snow AL, Day RM. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int J Mol Sci. (2013) 14:15931–58. doi: 10.3390/ijms140815931
127. He X, Yang A, McDonald DG, Riemer EC, Vanek KN, Schulte BA, et al. MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget (2017) 8:69797–807. doi: 10.18632/oncotarget.19267
128. Jeon HY, Kim JK, Ham SW, Oh SY, Kim J, Park JB, et al. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol. (2016) 37:5857–67. doi: 10.1007/s13277-015-4439-2
129. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. (2016) 7:11762. doi: 10.1038/ncomms11762
130. Rao SG, Jackson JG. SASP: tumor suppressor or promoter? Yes! Trends Cancer (2016) 2:676–87. doi: 10.1016/j.trecan.2016.10.001
131. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
132. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell (2015) 14:644–58. doi: 10.1111/acel.12344
133. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc. (2017) 65:2297–301. doi: 10.1111/jgs.14969
134. Gupta K, Burns T. RBIO-01. Extracellular milieu in recurrent human gliomas: a repertoire for novel drug development. Neuro Oncol. (2017) 19:vi217. doi: 10.1093/neuonc/nox168.879
135. Desmarais G, Fortin D, Bujold R, Wagner R, Mathieu D, Paquette B. Infiltration of glioma cells in brain parenchyma stimulated by radiation in the F98/Fischer rat model. Int J Radiat Biol. (2012) 88:565–74. doi: 10.3109/09553002.2012.692495
136. Thomas JG, Parker Kerrigan BC, Hossain A, Gumin J, Shinojima N, Nwajei F, et al. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J Neurosurg. (2018) 128:287–95. doi: 10.3171/2016.9.JNS16278
137. Duan C, Yang R, Yuan L, Engelbach JA, Tsien CI, Rich KM, et al. Late effects of radiation prime the brain microenvironment for accelerated tumor growth. Int J Radiat Oncol Biol Phys. (2018). doi: 10.1016/j.ijrobp.2018.08.033. [Epub ahead of print].
138. Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. (2005) 118:485–96. doi: 10.1242/jcs.01635
139. Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. (2017) 7:165–76. doi: 10.1158/2159-8290.CD-16-0241
140. Sprenger CC, Plymate SR, Reed MJ. Aging-related alterations in the extracellular matrix modulate the microenvironment and influence tumor progression. Int J Cancer (2010) 127:2739–48. doi: 10.1002/ijc.25615
141. Dasgupta J, Kar S, Liu R, Joseph J, Kalyanaraman B, Remington SJ, et al. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. J Cell Physiol. (2010) 225:52–62. doi: 10.1002/jcp.22193
142. Wilson C, González-Billault C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front Cell Neurosci. (2015) 9:381. doi: 10.3389/fncel.2015.00381
143. Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis (2006) 21:361–7. doi: 10.1093/mutage/gel048
144. Guntuku L, Naidu VG, Yerra VG. Mitochondrial dysfunction in gliomas: pharmacotherapeutic potential of natural compounds. Curr Neuropharmacol. (2016) 14:567–83. doi: 10.2174/1570159X14666160121115641
145. Kim KS, Kim JE, Choi KJ, Bae S, Kim DH. Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int J Radiat Biol. (2014) 90:71–80. doi: 10.3109/09553002.2014.859763
146. Prise KM, O'Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer (2009) 9:351–60. doi: 10.1038/nrc2603
147. Shao C, Aoki M, Furusawa Y. Bystander effect on cell growth stimulation in neoplastic HSGc cells induced by heavy-ion irradiation. Radiat Environ Biophys. (2003) 42:183–7. doi: 10.1007/s00411-003-0202-y
148. Shao C, Stewart V, Folkard M, Michael BD, Prise KM. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. (2003) 63:8437–42.
149. Shao C, Aoki M, Furusawa Y. Bystander effect in lymphoma cells vicinal to irradiated neoplastic epithelial cells: nitric oxide is involved. J Radiat Res. (2004) 45:97–103. doi: 10.1269/jrr.45.97
150. Sabatino ME, Grondona E, Sosa LDV, Mongi Bragato B, Carreno L, Juarez V, et al. Oxidative stress and mitochondrial adaptive shift during pituitary tumoral growth. Free Radic Biol Med. (2018) 120:41–55. doi: 10.1016/j.freeradbiomed.2018.03.019
151. Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. (2012) 2012:762825. doi: 10.1155/2012/762825
152. Rey S, Schito L, Koritzinsky M, Wouters BG. Molecular targeting of hypoxia in radiotherapy. Adv Drug Deliv Rev. (2017) 109:45–62. doi: 10.1016/j.addr.2016.10.002
153. van Vulpen M, Kal HB, Taphoorn MJ, El-Sharouni SY. Changes in blood-brain barrier permeability induced by radiotherapy: implications for timing of chemotherapy? (Review). Oncol Rep. (2002) 9:683–8. doi: 10.3892/or.9.4.683
154. Trnovec T, Kallay Z, Bezek S. Effects of ionizing radiation on the blood brain barrier permeability to pharmacologically active substances. Int J Radiat Oncol Biol Phys. (1990) 19:1581–7. doi: 10.1016/0360-3016(90)90376-U
155. Short SC, Buffa FM, Bourne S, Koritzinsky M, Wouters BG, Bentzen SM. Dose- and time-dependent changes in gene expression in human glioma cells after low radiation doses. Radiat Res. (2007) 168:199–208. doi: 10.1667/RR0940.1
156. Januel E, Ursu R, Alkhafaji A, Marantidou A, Doridam J, Belin C, Levy-Piedbois C, Carpentier AF. Impact of renin-angiotensin system blockade on clinical outcome in glioblastoma. Eur J Neurol. (2015) 22:1304–9. doi: 10.1111/ene.12746
157. Baumann BC, Kao GD, Mahmud A, Harada T, Swift J, Chapman C, et al. Enhancing the efficacy of drug-loaded nanocarriers against brain tumors by targeted radiation therapy. Oncotarget (2013) 4:64–79. doi: 10.18632/oncotarget.777
158. Cordes N, Meineke V. Cell adhesion-mediated radioresistance (CAM-RR). Extracellular matrix-dependent improvement of cell survival in human tumor and normal cells in vitro. Strahlenther Onkol. (2003) 179:337–44. doi: 10.1007/s00066-003-1074-4
159. Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, et al. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci USA. (2014) 111:5248–53. doi: 10.1073/pnas.1321014111
160. Dong Z, Zhou L, Han N, Zhang M, Lyu X. Wnt/beta-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. (2015) 191:672–80. doi: 10.1007/s00066-015-0858-7
161. Kim RK, Suh Y, Cui YH, Hwang E, Lim EJ, Yoo KC, et al. Fractionated radiation-induced nitric oxide promotes expansion of glioma stem-like cells. Cancer Sci. (2013) 104:1172–7. doi: 10.1111/cas.12207
162. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. (2005) 7:134–53. doi: 10.1215/S1152851704001115
163. Fahey JM, Emmer JV, Korytowski W, Hogg N, Girotti AW. Antagonistic effects of endogenous nitric oxide in a glioblastoma photodynamic therapy model. Photochem Photobiol. (2016) 92:842–53. doi: 10.1111/php.12636
164. Wang SC, Yu CF, Hong JH, Tsai CS, Chiang CS. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS ONE (2013) 8:e69182. doi: 10.1371/journal.pone.0069182
165. Thomas DD, Heinecke JL, Ridnour LA, Cheng RY, Kesarwala AH, Switzer CH, et al. Signaling and stress: the redox landscape in NOS2 biology. Free Radic Biol Med. (2015) 87:204–25. doi: 10.1016/j.freeradbiomed.2015.06.002
166. Zhang W, Chen L, Ma K, Zhao Y, Liu X, Wang Y, et al. Polarization of macrophages in the tumor microenvironment is influenced by EGFR signaling within colon cancer cells. Oncotarget (2016) 7:75366–78. doi: 10.18632/oncotarget.12207
167. Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. (2010) 3:23–34. doi: 10.4161/oxim.3.1.10095
168. Sebens S, Bauer I, Geismann C, Grage-Griebenow E, Ehlers S, Kruse ML, et al. Inflammatory macrophages induce Nrf2 transcription factor-dependent proteasome activity in colonic NCM460 cells and thereby confer anti-apoptotic protection. J Biol Chem. (2011) 286:40911–21. doi: 10.1074/jbc.M111.274902
169. Erdi F, Kaya B, Esen H, Karatas Y, Findik S, Keskin F, et al. New clues in the malignant progression of glioblastoma: can the thioredoxin system play a role? Turk Neurosurg. (2018) 28:7–12. doi: 10.5137/1019-5149.JTN.18991-16.2
170. Salazar-Ramiro A, Ramirez-Ortega D, Perez de la Cruz V, Hernandez-Pedro NY, Gonzalez-Esquivel DF, Sotelo J, et al. Role of redox status in development of glioblastoma. Front Immunol. (2016) 7:156. doi: 10.3389/fimmu.2016.00156
171. Li MD, Burns TC, Kumar S, Morgan AA, Sloan SA, Palmer TD. Aging-like changes in the transcriptome of irradiated microglia. Glia (2015) 63:754–67. doi: 10.1002/glia.22782
172. Li MD, Burns TC, Morgan AA, Khatri P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Commun. (2014) 2:93. doi: 10.1186/s40478-014-0093-y
173. Burns TC, Awad AJ, Li MD, Grant GA. Radiation-induced brain injury: low-hanging fruit for neuroregeneration. Neurosurg Focus (2016) 40:E3. doi: 10.3171/2016.2.FOCUS161
174. Burns TC, Li MD, Mehta S, Awad AJ, Morgan AA. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: a systematic bioinformatics-based critique of preclinical models. Eur J Pharmacol. (2015) 759:101–17. doi: 10.1016/j.ejphar.2015.03.021
175. Chang JE, Khuntia D, Robins HI, Mehta MP. Radiotherapy and radiosensitizers in the treatment of glioblastoma multiforme. Clin Adv Hematol Oncol. (2007) 5:894–902, 907–15.
176. Hijova E. Matrix metalloproteinases: their biological functions and clinical implications. Bratisl Lek Listy (2005) 106:127–32.
177. Harisi R, Jeney A. Extracellular matrix as target for antitumor therapy. Onco Targets Ther. (2015) 8:1387–98. doi: 10.2147/OTT.S48883
178. Gandalovicova A, Rosel D, Fernandes M, Vesely P, Heneberg P, Cermak V, et al. Migrastatics-anti-metastatic and anti-invasion drugs: promises and challenges. Trends Cancer (2017) 3:391–406. doi: 10.1016/j.trecan.2017.04.008
179. Appelboom G, Detappe A, LoPresti M, Kunjachan S, Mitrasinovic S, Goldman S, et al. Stereotactic modulation of blood-brain barrier permeability to enhance drug delivery. Neuro Oncol. (2016) 18:1601–9. doi: 10.1093/neuonc/now137
Keywords: glioblastoma, radiotherapy, tumor microenvironment, extracellular matrix, recurrence
Citation: Gupta K and Burns TC (2018) Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 8:503. doi: 10.3389/fonc.2018.00503
Received: 02 August 2018; Accepted: 15 October 2018;
Published: 08 November 2018.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
Thomas Daubon, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceVijay Pandey, National University of Singapore, Singapore
Copyright © 2018 Gupta and Burns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kshama Gupta, Z3VwdGEua3NoYW1hQG1heW8uZWR1
Terry C. Burns, YnVybnMudGVycnlAbWF5by5lZHU=