 Anne Slomp
Anne Slomp Victor Peperzak
Victor Peperzak- Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
Apoptosis plays a key role in protection against genomic instability and maintaining tissue homeostasis, and also shapes humoral immune responses. During generation of an antibody response, multiple rounds of B-cell expansion and selection take place in germinal centers (GC) before high antigen affinity memory B-cells and long-lived plasma cells (PC) are produced. These processes are tightly regulated by the intrinsic apoptosis pathway, and malignant transformation throughout and following the GC reaction is often characterized by apoptosis resistance. Expression of pro-survival BCL-2 family protein MCL-1 is essential for survival of malignant PC in multiple myeloma (MM). In addition, BCL-2 and BCL-XL contribute to apoptosis resistance. MCL-1, BCL-2, and BCL-XL expression is induced and maintained by signals from the bone marrow microenvironment, but overexpression can also result from genetic lesions. Since MM PC depend on these proteins for survival, inhibiting pro-survival BCL-2 proteins using novel and highly specific BH3-mimetic inhibitors is a promising strategy for treatment. This review addresses the role and regulation of pro-survival BCL-2 family proteins during healthy PC differentiation and in MM, as well as their potential as therapeutic targets.
Introduction
Multiple myeloma (MM) is a malignancy of clonal long-lived plasma cells (PC) residing in the bone marrow (BM) (1). The malignancy arises as a result of genetic changes that occur during differentiation of B-cells into PC (2, 3). MM is characterized by resistance against the intrinsic apoptosis pathway, which is regulated by proteins of the BCL-2 family (4).
The BCL-2 protein family consists of pro-survival BCL-2-like proteins (BCL-2, BCL-B, BCL-W, BCL-XL, BFL-1/A1, and MCL-1), pro-apoptotic BH3-only proteins (initiators), and pro-apoptotic effectors BAX, BAK (5), and possibly BOK (6–8). Cytotoxic stimuli such as DNA damage, chemotherapeutic agents, or cytokine deprivation promote upregulation of BH3-only proteins, which inhibit pro-survival BCL-2 family members (5). In addition, post-translational modification of BH3-only proteins can affect their stability, activity, and subcellular localization (9). BH3-only proteins vary in their affinities for different pro-survival proteins. For instance, BAD only binds with high affinity to BCL-2, BCL-XL, and BCL-W, while NOXA selectively inhibits MCL-1 and BFL-1/A1. BIM, PUMA, and BID have high affinity for all pro-survival proteins (10, 11). If all available pro-survival proteins are sequestered by BH3-only proteins, BAX and BAK can disrupt the mitochondrial outer membrane, leading to cytochrome C release, caspase activation, and execution of apoptosis (12). In addition, some BH3-only proteins, including BIM, PUMA, and BID, can directly bind to BAX or BAK and induce conformational changes that contribute to BAX/BAK activation (13–15). Regulation of apoptosis is essential for generation and selection of high-affinity PC, and malignant transformation of cells in this process often coincides with defects in apoptosis.
Healthy PC Differentiation
Long-lived PC originate from germinal centers (GC), which are dynamic structures that develop in secondary lymphoid organs upon antigen stimulation and helper T-cell activation. Clonal expansion, somatic hypermutation, class switch recombination, and affinity-based selection of B-cells take place in GCs, resulting in the production of high-affinity antibodies (16). GCs contain a dark zone (DZ), consisting of dividing B-cells, and a light zone (LZ), in which B-cells are selected based on antigen affinity through B-cell receptor (BCR) signaling and CD40-CD40L interactions (17–19). B-cells with low antigen affinity undergo apoptosis, and B-cells with high antigen affinity either return to the DZ for another round of mutation and expansion, or differentiate and move out of the GC as memory B-cells or PC. Somatic hypermutation and class switch recombination take place during proliferation in the DZ and are mediated by activation-induced cytidine deaminase (AID) (20). Most GC-derived PC are recruited into the BM, where stromal cells provide signals for long-term survival (21, 22).
The BCL-2 Family in PC Differentiation
Apoptosis regulation plays a central role in the cycle of expansion, selection, and differentiation that eventually produces mature PC. Expression of BCL-2 family proteins during PC differentiation and after malignant transformation of (post-) GC B-cells is highly variable and shown in Figure 1. MCL-1 is essential for GC formation and maintenance, memory B-cell development (23), and survival of existing PC (24). In fact, B-cells are dependent on MCL-1 throughout development (25). BCL-2, which is important for naïve and memory B-cells, is downregulated in the GC (26, 27). In contrast, MCL-1, BCL-XL, and BFL-1 are upregulated. BH3-only proteins BIM and BIK are also upregulated in the GC, but this upregulation was shown to be countered by MCL-1 and BCL-XL, respectively (27). Apoptosis of low-affinity B-cells in the GC is dependent on the interplay between pro-survival and pro-apoptotic BCL-2 proteins. In mice, overexpression of Bcl-2 was shown to disrupt GC selection of memory B-cells, but not of high-affinity plasmablasts (28). Knockout of Bim (29) or Noxa (30) resulted in increased amounts of low-affinity B-cells, suggesting that these BH3-only proteins play a critical role in elimination of low-affinity B-cell and PC clones. Puma was shown to be essential for regulation of memory formation in mice, since its loss resulted in accumulation of memory B-cells (31). Fully differentiated GC-derived PC are characterized by high expression of transcriptional regulator BLIMP-1, which promotes MCL-1 expression and represses BIM (32).
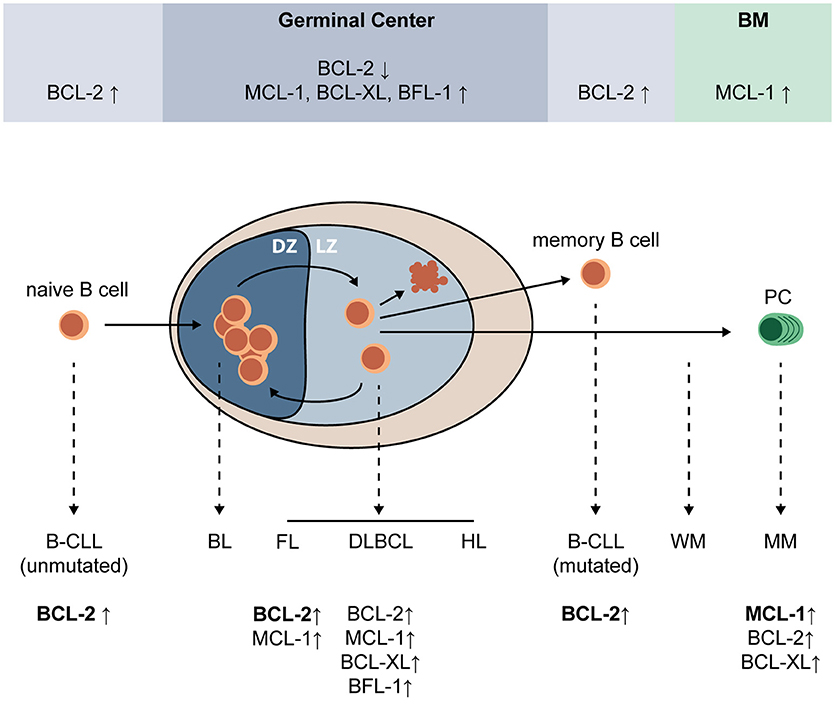
Figure 1. Expression of pro-survival BCL-2 family proteins during PC differentiation and after malignant transformation of (post-) GC B-cells. Upon encounter of a naïve B-cell with its cognate antigen, and in the presence of adequate T cell help, a germinal center (GC) is formed where the B-cell undergoes multiple cycles of expansion and hypermutation in the dark zone (DZ), and affinity-based selection in the light zone (LZ). Low-affinity B-cells undergo apoptosis, while high-affinity B-cells can undergo further selection, or exit the GC as a memory B-cell or plasma cell (PC). In the GC, BCL-2 expression is strongly repressed and expression of MCL-1, BCL-XL, and BFL-1 is increased. MCL-1, but not BCL-XL, was shown to be essential for survival of GC B-cells. Naïve and memory B-cells have high expression of BCL-2 and are sensitive to its inhibition, and PC depend on MCL-1 expression for survival. Erroneous targeting of activation-induced cytidine deaminase (AID) during somatic hypermutation and class switch recombination can lead to mutations that promote malignant transformation, resulting in a variety of GC-derived malignancies (dashed lines). Multiple GC-derived malignancies, such as follicular lymphoma (FL), diffuse-large B-cell lymphoma (DLBCL), some B-cell chronic lymphocytic leukemias (B-CLL), and multiple myeloma (MM) depend on overexpression of BCL-2 family proteins for survival. BL, Burkitt's lymphoma; BM, bone marrow; CLL, chronic lymphocytic leukemia; DLBCL, diffuse-large cell B-cell lymphoma; DZ, dark zone; FL, follicular lymphoma; HL, Hodgkin lymphoma; LZ, light zone; MCL, mantle cell lymphoma; MM, multiple myeloma; PC, plasma cell; WM, Waldenström macroglobulinemia.
Malignant Transformation of GC B-Cells
In the GC, somatic hypermutation and class switch recombination are mediated by AID, which functions by deaminating cytidine residues to uracil (20). AID is targeted to the variable immunoglobulin (Ig) regions, as well as the Ig switch regions. As a result of AID activity, the mutation rate in the variable Ig regions is estimated to increase to between 10−2 and 10−3 mutations per bp (33). In addition to its function in the Ig gene, AID can also be erroneously targeted to other genomic loci, introducing mutations and Ig translocations that can contribute to malignant transformation (34, 35). Many different malignancies, some of which dependent on BCL-2 family proteins for survival, arise from (post-) GC B-cells (Figure 1). These include B-cell chronic lymphocytic leukemia (CLL) (36), follicular lymphoma (37), diffuse-large B-cell lymphoma (DLBCL) (38), Waldenström macroglobulinemia (WM) (39), and multiple myeloma (MM) (40).
Pro-survival BCL-2 Proteins in GC B-Cell Malignancies
Pro-survival BCL-2 proteins contribute to apoptosis resistance of malignant B-cells, and their overexpression can be regulated in different ways. In 60–65% of CLL cases, the BCR is hypermutated, indicating that the malignancy originates from post-GC B-cells. Conversely, in the remaining 35–40% of cases, the BCR lacks signs of hypermutation and the disease presumably originates from B-cells that have differentiated independently of the GC (36). In both types, apoptosis resistance is mediated by overexpression of BCL-2 (41). This overexpression is due to BCL2 gene hypomethylation and genetic loss of microRNA loci that normally inhibit BCL-2 expression (42, 43). Inhibition of BCL-2 using specific BH3-mimetic inhibitor Venetoclax efficiently induces apoptosis in CLL cells in circulation, and is also promising for other BCL-2 dependent malignancies such as follicular lymphoma and a subset of DLBCL (44–46).
Follicular lymphoma originates from GC B-cells and is characterized by the hallmark chromosomal translocation t(14;18), which is present in 85% of patients and results in overexpression of BCL-2 due to juxtaposition of the Ig heavy chain (IGH) and BCL2 loci (37). In addition, MCL-1 is highly expressed in some follicular lymphomas, and its expression correlates with disease grade (47).
DLBCL has distinct subtypes, including germinal center B-cell-like (GCB-) DLBCL, which is derived from normal GC B-cells; and activated B-cell-like (ABC-) DLBCL, originating from B-cells that have completed the GC reaction (48). T(14;18) is present in 45% of GCB-DLBCL, but does not occur in ABC-DLBCL (49). Still, BCL2 expression is high in many cases of ABC-DLBCL, as a result of gain or amplification of the 18q chromosome arm on which BCL2 is located (50). MCL-1 expression is also frequently high in ABC-DLBCL and sometimes in GCB-DLBCL, possibly as a result of chromosomal amplification or transcriptional regulation (51). In addition, ABC-DLBCL is characterized by constitutively high NF-κB activity. Among the targets of NF-κB are BCL-XL, BFL-1/A1, and possibly BCL-2, whose high expression as a result of NF-κB signaling may contribute to apoptosis resistance in ABC-DLBCL (52–54).
MM and WM are malignancies that contain a clonal PC population residing in the bone marrow. Both are preceded by monoclonal gammopathy of undetermined significance (MGUS), which is characterized by presence of <10% clonal PC in the BM, presence of monoclonal Ig in the blood, and lack of clinical symptoms (55, 56). WM originates from post-GC B-cells that have undergone somatic hypermutation but did not undergo class switching, whereas MM originates from post-GC B-cells after class switching (39). As a result, the serum Ig in WM is of the IgM type, and IgH translocations do not occur (57). The cellular phenotype is mixed, ranging from B-cells to PC (58). Possibly, malignancy is acquired during the B-cell or plasmablast stage, with some malignant cells continuously differentiating into PC. MM, on the other hand, consists of fully differentiated PC and is characterized by frequent IgH translocations and genomic instability (59). MM cells most frequently produce IgG or IgA, although IgM or IgD have been observed in rare cases (60). In WM, pro-apoptotic and pro-survival BCL-2 family proteins are expressed at low levels similar to non-malignant B-cells and PC. It is therefore expected that WM will only be sensitive to BH3-mimetic drugs if these are combined with other treatments that increase pro-apoptotic protein levels and mitochondrial priming (61). In contrast, MM cells are highly dependent on BCL-2 family proteins for survival, with MCL-1 as the essential player (62, 63).
The BCL-2 Family in Multiple Myeloma
MCL-1 protein expression is increased in newly diagnosed MM compared to healthy PC, and protein levels are even higher at relapse (64). In addition, overexpression of MCL-1 is associated with shorter patient survival (64). Using RNA interference lethality screening in cell lines, MCL-1 was also identified as one of the most important and selective survival genes for MM (65). In subsets of MM cell lines and patient samples, BCL-2 and BCL-XL expression is also high (66), suggesting that these three proteins may act redundantly in preventing apoptosis. Since expression of both pro-survival and pro-apoptotic BCL-2 family members is heterogeneous, and the interplay between them is complex and dynamic, dependence on MCL-1, BCL-2, and BCL-XL is likely to differ between patients (66–68). Signals and cellular processes that may lead to overexpression of MCL-1, BCL-2, and BCL-XL in MM are indicated in Figure 2.
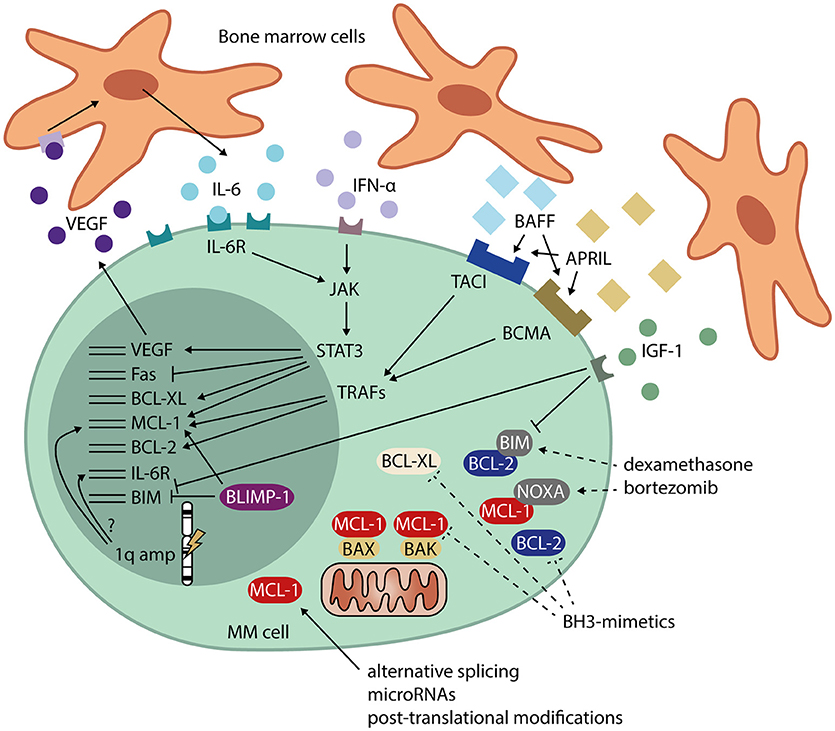
Figure 2. Signals and cellular processes that mediate apoptosis resistance in MM. MM cells receive signals from the bone marrow microenvironment that stimulate their survival. These signals include IL-6 and IFN-α, leading to JAK/STAT signaling and expression of MCL-1, BCL-XL, and VEGF. VEGF, in turn, promotes IL-6 production by neighboring cells. Other signals from the bone marrow microenvironment include BAFF and APRIL, which signal via TRAFs and induce expression of MCL-1 and BCL-2. IGF-1 signaling downregulates BIM, transcriptionally as well as post-translationally. MM cells also have high expression of the PC transcriptional regulator BLIMP-1, which promotes MCL-1 and represses BIM expression. Amplification of the 1q chromosome arm often occurs in MM. The genes for both MCL-1 and the IL-6 receptor (IL-6R) are present on this locus, possibly leading to overexpression in 1q-amplified MM. In addition to transcriptional regulation, MCL-1 is heavily regulated post-transcriptionally, which may contribute to the high MCL-1 protein levels found in MM. Dashed lines represent methods for interference in apoptosis resistance by MM drugs dexamethasone and bortezomib, and by BH3-mimetics. APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; BCMA, B-cell maturation antigen; BH-3, BCL-2 homolog 3; BLIMP-1, B lymphocyte-induced maturation protein 1; IFN-α, interferon alpha; IGF-1, insulin-like growth factor 1; IL-6, interleukin 6; IL-6R, interleukin 6 receptor; JAK, janus kinase; STAT3, signal transducer and activator of transcription 3; TACI, transmembrane activator and calcium-modulating ligand interactor; TRAF, TNF receptor-associated factor; VEGF, vascular endothelial growth factor.
Survival Signals From the BM Microenvironment
MM cells reside in the BM, where they interact with extracellular matrix proteins and cells from the BM microenvironment, which include stromal cells, osteoblasts, osteoclasts, endothelial cells, fibroblasts, adipocytes, and cells of hematopoietic origin (40). MM cells promote neighboring cells to produce IL-6 (69), which induces JAK/STAT3 signaling in MM, leading to transcription of MCL-1 and BCL-XL (70–73). MCL-1 expression in MM can also be IL-6-independent (74), or occur via other signals from the BM microenvironment (75). For instance, signaling through BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand), whose levels are increased in MM patients compared to healthy controls, induces expression of both MCL-1 and BCL-2 and promotes PC survival (24, 76). Other survival signals from the bone marrow environment include interferon α (IFN-α), which induces MCL-1 in a STAT3-dependent manner (77), and insulin-like growth factor 1 (IGF-1), which downregulates expression of BIM (78).
Genetic Lesions
MM is characterized by recurrent chromosomal aberrations, some of which may be linked to apoptosis pathways. Translocations or chromosomal amplifications and gains involving 18q are rare in MM (79), suggesting that BCL2 overexpression is not a key event in malignant transformation. No other genetic lesions in MM have directly been correlated to overexpression of a BCL-2 family member. Nevertheless, gain or amplification of 1q21, the chromosome region containing the MCL1 gene, occurs in approximately 40% of MM cases and correlates with poor disease prognosis (80). Notably, IL6R, the gene encoding the IL-6 receptor, is also located on 1q21, as are several other candidate drivers of high-risk disease (81).
T(4;14), which is present in 10–15% of MM patients (80), may lead to disruption and subsequent overexpression of fibroblast growth receptor 3 (FGFR3), which is considered an oncogene (79). In a murine IL-6-dependent hybridoma cell line, FGFR3 was shown to signal through STAT3 and substitute IL-6 signaling, leading to increased BCL-XL expression and decreased apoptosis (82). Correspondingly, specific tyrosine kinase inhibitors with known anti-FGFR3 activity induced apoptosis in t(4;14)-positive cell lines (83).
MCL-1 Stabilization
Unlike for BCL-2 and BCL-XL (66), transcriptional activity of MCL1 does not directly correlate to protein levels. MCL-1 is unique within the BCL-2 family because it has a large N-terminal domain that allows for post-translational modification (84, 85). Proteasomal degradation of MCL-1 occurs upon phosphorylation and subsequent poly-ubiquitination of this N-terminal region. Kinases associated with phosphorylation of MCL-1 include JNK, GSK-3, and ERK-1 (86). Ubiquitin ligases Mule, SCFβ−TrCP, SCFFbw7, and APC/CCdc20 were shown to target MCL-1 for proteasomal degradation after recognizing specific phosphorylated residues (87). This process can be reversed by deubiquitinases, such as USP9X (88). The contribution of these kinases and ubiquitin modifiers to MCL-1 regulation in MM is currently unknown. If the key players in MCL-1 regulation can be identified for MM, these MCL-1-modifying proteins may be interesting targets for therapeutic intervention.
Overcoming Apoptosis Resistance: BCL-2 Proteins as Therapeutic Targets in MM
As apoptosis resistance in B-cell malignancies often results from overexpression of pro-survival BCL-2 family proteins, inhibiting these proteins is a promising strategy for development of targeted therapeutics. Several BCL-2 family inhibitors, also named BH3-mimetics because of their structural and functional resemblance to the BH3 domain of BH3-only proteins, are currently in clinical development. BCL-2 inhibitor Venetoclax is the first BH3-mimetic approved by the Food and Drug Administration. It was approved in 2016 for treatment of CLL with a 17p deletion (46). Additionally, Venetoclax was tested in phase I clinical trials with relapsed and refractory MM patients, where monotherapy was particularly effective when the t(11;14) translocation was present (89). T(11;14) is associated with an increased BCL2/MCL1 mRNA ratio, but the mechanism behind this is unknown (90). When MM patients were treated with Venetoclax in combination with conventional MM drugs bortezomib (a proteasome inhibitor) and dexamethasone, it was well tolerated and the response rate was highest in patients with high BCL2 expression (91). Experiments in cell lines even indicate more-than-additive effects when Venetoclax is combined with proteasome inhibitor carfilzomib or dexamethasone, due to upregulation of NOXA and BIM, respectively (92). If conventional treatment increases availability of BH3-only proteins and their distribution toward pro-survival target proteins, this may increase sensitivity to BH3-mimetic drugs.
While the results of MM treatment with Venetoclax underline the potential of using BH3-mimetics in MM, they also suggest that Venetoclax may only be effective in a subset of patients, namely those who have relatively high BCL-2 and relatively low MCL-1. Based on in vitro and xenograft experiments, MCL-1 is often shown to be essential for MM survival and its generally high expression may confer resistance to Venetoclax (66, 93, 94). Therefore, MCL-1 itself is a very promising therapeutic target in MM, and multiple MCL-1 inhibitors are currently under development (95). MCL-1 inhibitor S63845 efficiently kills MM and other MCL-1-dependent cancer cell lines (96). Its derivate S64315/MIK665 is currently being tested in phase I clinical trials by Servier for acute myeloid leukemia and myelodysplastic syndrome (NCT02979366), and by Novartis for MM and DLBCL (NCT02992483). In addition, clinical testing in MM patients has started with MCL-1 inhibitors developed by Amgen, named AMG 176 and AMG 397 (NCT02675452 and NCT03465540, respectively) (97), and by AstraZeneca, named AZD5991 (NCT03218683) (98).
Simultaneous targeting of multiple BCL-2 family proteins may be a solution to resistance in case of redundancy between MCL-1, BCL-2, and BCL-XL in MM. Before the development of Venetoclax, BH3-mimetics with broader protein specificity have been studied, such as Navitoclax (99). Navitoclax (ABT-263) mimics the selectivity of BAD, thereby inhibiting only BCL-2, BCL-XL, and BCL-W. When tested in CLL patients, results were promising, but dose-limiting thrombocytopenia was observed as a result of BCL-XL inhibition (100–102). This led to the development of BCL-2-selective BH3-mimetic Venetoclax (44). Other putative BCL-2 family inhibitors with broad target specificity, such as Obatoclax (GX15-070), were shown to function partly or completely in a BAX/BAK-independent manner, and are therefore no longer considered BH3-mimetics (103). The results with Navitoclax indicate that potential side-effects of BCL-2 family inhibitors may be dose-limiting, and that combined inhibition of BCL-2 family members may only be possible if the concentration of each specific inhibitor remains below the threshold of toxicity.
MCL-1 is not only essential for B-cells and PC, it is also essential in other cell types, including hematopoietic stem cells (104), cardiomyocytes (105), and neural precursor cells (106). In contrast to healthy cells, increased expression of pro-apoptotic molecules (“priming”) renders malignant cells more susceptible to apoptosis upon inactivation of pro-survival proteins (107). Since MCL-1 is the most dominant pro-survival protein in MM, its inhibition leads to release of a large proportion of pro-apoptotic proteins present in MM cells, thereby promoting apoptosis induction. In mice, MCL-1 inhibitor S63845 was tolerated well at concentrations that killed cancer cells (96), even when murine Mcl-1 was replaced by its human ortholog, thereby increasing inhibitor sensitivity of all cells (108). This may yield a therapeutic window for targeting MCL-1, especially if MCL-1 inhibitors are combined with existing treatments that increase pro-apoptotic protein expression.
Conclusion
High expression of pro-survival BCL-2 family proteins contributes to outgrowth and drug resistance of malignant B-cell clones. While beneficial for cell survival, addiction to high levels of specific pro-survival BCL-2 proteins also makes cells vulnerable to BCL-2 family inhibition using BH3-mimetic drugs. MM is characterized by high expression of MCL-1, and overexpression of BCL-2 and BCL-XL is observed in subsets of patients. Constitutive overexpression of these pro-survival proteins in MM results from a range of microenvironmental signals and different genetic lesions. This complex regulation of MCL-1, BCL-2, and BCL-XL offers multiple direct and indirect targets for therapeutic intervention. Recent development of BH3-mimetic drugs, that specifically target MCL-1, BCL-2, or BCL-XL, may contribute to overcoming apoptosis resistance and improving treatment for MM.
Author Contributions
AS and VP wrote the manuscript and designed the figures. Both authors read and approved the final manuscript.
Funding
This work was supported in part by a Bas Mulder Award from the Dutch Cancer Foundation (KWF)/Alpe d'HuZes foundation (No. UU 2015-7663) and grants from the Dutch Cancer Foundation (KWF)/Alpe d'HuZes foundation (No. 11108 and No. 11270). The funding agencies played no role in the design, reviewing or writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABC-DLBCL, activated B-cell-like diffuse-large B-cell lymphoma; AID, activation-induced cytidine deaminase; APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; BCL-2, B-cell lymphoma 2; BCR, B-cell receptor; BH3, BCL-2 homology 3; BM, bone marrow; CLL, (B-cell) chronic lymphocytic leukemia; DLBCL, diffuse-large B-cell lymphoma; DZ, dark zone; FGFR3, fibroblast growth factor receptor 3; GC, germinal center; GCB-DLBCL, germinal center B-cell-like diffuse-large B-cell lymphoma; IFN-α, interferon α; Ig, immunoglobulin; IGF-1, insulin-like growth factor 1; IgH, immunoglobulin heavy chain; IL-6, interleukin 6; LZ, light zone; MCL-1, myeloid cell leukemia 1; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; PC, plasma cell; WM, Waldenström macroglobulinemia.
References
2. Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene (2001) 20:5580–94. doi: 10.1038/sj.onc.1204640
3. Küppers R, Klein U, Hansmann M-L, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. (1999) 341:1520–9. doi: 10.1056/NEJM199911113412007
4. Peperzak V, Vikstrom IB, Tarlinton DM. Through a glass less darkly: apoptosis and the germinal center response to antigen. Immunol Rev. (2012) 247:93–106. doi: 10.1111/j.1600-065X.2012.01123.x
5. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. (2014) 15:49–63. doi: 10.1038/nrm3722
6. Haschka MD, Villunger A. There is something about BOK we just don't get yet. FEBS J. (2017) 284:708–10. doi: 10.1111/febs.14031
7. Zheng JH, Grace CR, Guibao CD, McNamara DE, Llambi F, Wang YM, et al. Intrinsic instability of BOK enables membrane permeabilization in apoptosis. Cell Rep. (2018) 23:2083–94. doi: 10.1016/j.celrep.2018.04.060
8. Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors, B. A X, BAK, and BOK. Cell (2018) 173:1217–30. doi: 10.1016/j.cell.2018.04.036
9. Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene (2008) 27:S2–19. doi: 10.1038/onc.2009.39
10. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell (2005) 17:393–403. doi: 10.1016/j.molcel.2004.12.030
11. Happo L, Strasser A, Cory S. BH3-only proteins in apoptosis at a glance. J Cell Sci. (2012) 125:1081–7. doi: 10.1242/jcs.090514
12. Wong RSY. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. (2011) 30:87. doi: 10.1186/1756-9966-30-87
13. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell (2002) 2:183–92. doi: 10.1016/S1535-6108(02)00127-7
14. Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell (2011) 44:517–31. doi: 10.1016/j.molcel.2011.10.001
15. Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell (2013) 152:519–31. doi: 10.1016/j.cell.2012.12.031
16. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
17. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
18. Adem J, Hämäläinen A, Ropponen A, Eeva J, Eray M, Nuutinen U, et al. ERK1/2 has an essential role in B cell receptor- and CD40-induced signaling in an in vitro model of germinal center B cell selection. Mol Immunol. (2015) 67:240–7. doi: 10.1016/j.molimm.2015.05.017
19. Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, et al. T Follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity (2018) 48:702–15. doi: 10.1016/j.immuni.2018.03.027
20. Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded, D. N A J Exp Med. (2003) 197:1291–6. doi: 10.1084/jem.20030481
21. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KGC, Dörner T, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. (2006) 6:741–50. doi: 10.1038/nri1886
22. Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunol Rev. (2013) 251:177–88. doi: 10.1111/imr.12011
23. Vikström I, Carotta S, Lüthje K, Peperzak V, Jost PJ, Glaser S, et al. Mcl-1 Is essential for germinal center formation and B cell memory. Science (2010) 330:1095–100. doi: 10.1126/science.1191793
24. Peperzak V, Vikström I, Walker J, Glaser SP, Lepage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. (2013) 14:290–7. doi: 10.1038/ni.2527
25. Vikström I, Slomp A, Carrington EM, Moesbergen LM, Chang C, Kelly GL, et al. MCL-1 is required throughout B-cell development and its loss sensitizes specific B-cell subsets to inhibition of BCL-2 or BCL-XL. Cell Death Dis. (2016) 7:e2345. doi: 10.1038/cddis.2016.237
26. Shen Y, Iqbal J, Huang JZ, Zhou G, Chan WC. BCL2 protein expression parallels its mRNA level in normal and malignant B cells. Blood (2004) 104:2936–9. doi: 10.1182/blood-2004-01-0243
27. Peperzak V, Slinger E, Burg J, Ter, Eldering E. Functional disparities among BCL-2 members in tonsillar and leukemic B-cell subsets assessed by BH3-mimetic profiling. Cell Death Differ. (2017) 24:111–9. doi: 10.1038/cdd.2016.105
28. Smith KGC, Light A, O'Reilly LA, Ang S-M, Strasser A, Tarlinton D. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory b cells and bone marrow antibody-forming cells. J Exp Med. (2000) 191:475–84. doi: 10.1084/jem.191.3.475
29. Fischer SF, Bouillet P, O'Donnell K, Light A, Tarlinton DM, Strasser A. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood (2007) 110:3978–84. doi: 10.1182/blood-2007-05-091306
30. Wensveen FM, Slinger E, Attekum MHA, Van, Brink R, Eldering E. Antigen-affinity controls pre- germinal center B cell selection by promoting Mcl-1 induction through BAFF receptor signaling. Sci. Rep. (2016) 6:35673. doi: 10.1038/srep35673
31. Clybouw C, Fischer S, Auffredou MT, Hugues P, Alexia C, Bouillet P, et al. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood (2011) 118:4120–8. doi: 10.1182/blood-2011-04-347096
32. Lin FR, Kuo HK, Ying HY, Yang FH, Lin KI. Induction of apoptosis in plasma cells by B lymphocyte-induced maturation protein-1 knockdown. Cancer Res. (2007) 67:11914–23. doi: 10.1158/0008-5472.CAN-07-1868
33. Maul RW, Gearhart PJ. AID and somatic hypermutation. Adv Immunol. (2010) 105:159–91. doi: 10.1016/S0065-2776(10)05006-6
34. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature (2008) 451:841–6. doi: 10.1038/nature06547
35. Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell (2010) 141:27–38. doi: 10.1016/j.cell.2010.03.016
36. Gaidano G, Foà R, Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J. Clin. Invest. (2012) 122:3432–8. doi: 10.1172/JCI64101
37. Kridel R, Sehn LH, Gascoyne RD. Pathogenesis of follicular lymphoma. J Clin Invest. (2012) 122:3424–31. doi: 10.1172/JCI63186
38. Pasqualucci L, Dalla-Favera R. Genetics of diffuse large B-cell lymphoma. Blood (2018) 131:2307–19. doi: 10.1182/blood-2017-11-764332
39. Ghobrial IM, Gertz MA, Fonseca R. Waldenström macroglobulinaemia. Lancet Oncol. (2003) 4:679–85. doi: 10.1016/S1470-2045(03)01246-4
40. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood (2015) 125:3049–58. doi: 10.1182/blood-2014-11-568881
41. Buggins AGS, Pepper CJ. The role of Bcl-2 family proteins in chronic lymphocytic leukaemia. Leuk Res. (2010) 34:837–42. doi: 10.1016/j.leukres.2010.03.011
42. Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood (1993) 82:1820–8.
43. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Wojcik SE, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA (2005) 102:13944–9. doi: 10.1073/pnas.0506654102
44. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
45. Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. (2016) 17:768–78. doi: 10.1016/S1470-2045(16)30019-5
46. Zhu H, Almasan A. Development of venetoclax for therapy of lymphoid malignancies. Drug Des Devel Ther. (2017) 11:685–94. doi: 10.2147/DDDT.S109325
47. Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, et al. MCL-1 expression in B-cell non-Hodgkin's lymphomas. Hum Pathol. (2004) 35:1095–100. doi: 10.1016/j.humpath.2004.04.018
48. Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. (2005) 87:163–208. doi: 10.1016/S0065-2776(05)87005-1
49. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large B-cell lymphoma. N Engl J Med. (2002) 346:1937–47. doi: 10.1056/NEJMoa012914
50. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
51. Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia (2013) 27:1381–90. doi: 10.1038/leu.2012.367
52. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. (2001) 194:1861–74. doi: 10.1084/jem.194.12.1861
53. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, et al. NFκB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood (2005) 106:1392–9. doi: 10.1182/blood-2004-12-4901
54. Vogler M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. (2012) 19:67–74. doi: 10.1038/cdd.2011.158
55. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. (2006) 354:1362–9. doi: 10.1056/NEJMoa054494
56. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. (2014) 15:e538–e548. doi: 10.1016/S1470-2045(14)70442-5
57. Schop RFJ, Kuehl WM, Wier SA, Van Ahmann GJ, Price-Troska T, Bailey RJ, et al. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood (2002) 100:2996–3001. doi: 10.1182/blood.V100.8.2996
58. Gutiérrez NC, Ocio EM, Las Rivas J, De Maiso P, Delgado M, Fermi-an E, et al. Gene expression profiling of B lymphocytes and plasma cells from Waldenström's macroglobulinemia: comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia (2007) 21:541–9. doi: 10.1038/sj.leu.2404520
59. Nieuwenhuijzen N, Van Spaan I, Raymakers R, Peperzak V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. (2018) 78:2449–57. doi: 10.1158/0008-5472.CAN-17-3115
60. Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. (2003) 78:21–33. doi: 10.4065/78.1.21
61. Gaudette BT, Dwivedi B, Chitta KS, Poulain S, Powell D, Vertino P, et al. Low expression of pro-apoptotic Bcl-2 family proteins sets the apoptotic threshold in Waldenström Macroglobulinemia. Oncogene (2016) 35:479–90. doi: 10.1038/onc.2015.103
62. Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood (2002) 99:1885–93. doi: 10.1182/blood.V99.6.1885
63. Derenne S, Monia B, Dean NM, Taylor JK, Rapp M, Harousseau J, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x L is an essential survival protein of human myeloma cells. Blood (2002) 100:194–9. doi: 10.1182/blood.V100.1.194
64. Wuillème-Toumi S, Robillard N, Gomez P, Moreau P, Gouill S, Le Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia (2005) 19:1248–52. doi: 10.1038/sj.leu.2403784
65. Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, et al. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. (2012) 72:757–68. doi: 10.1158/0008-5472.CAN-11-2781
66. Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. (2016) 15:1132–44. doi: 10.1158/1535-7163.MCT-15-0730
67. Bodet L, Ménoret E, Descamps G, Pellat-Deceunynck C, Bataille R, Gouill S, Le, et al. BH3-only protein Bik is involved in both apoptosis induction and sensitivity to oxidative stress in multiple myeloma. Br. J. Cancer (2010) 103:1808–14. doi: 10.1038/sj.bjc.6605981
68. Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL / Bcl-2 in Mcl-1 – expressing myeloma cells. Blood (2011) 118:1329–39. doi: 10.1182/blood-2011-01-327197
69. Dankbar B, Padró T, Leo R, Feldmann B, Kropff M, Mesters RM, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood (2000) 95:2630–6.
70. Puthier D, Derenne S, Barill,é S, Moreau P, Harousseau, J.-L., Bataille R, et al. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br. J. Haematol. (1999) 107:392–395. doi: 10.1046/j.1365-2141.1999.01705.x
71. Puthier D, Bataille R, Amiot M. IL-6 up-regulates Mcl-1 in human myeloma cells through JAK/STAT rather than Ras/MAP kinase pathway. Eur J Immunol. (1999) 29:3945–50. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O
72. Jourdan M, Veyrune JL, Vos J, De Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene (2003) 22:2950–9. doi: 10.1038/sj.onc.1206423
73. Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment–derived signals induce Mcl-1 dependence in multiple myeloma. Blood (2017) 129:1969–79. doi: 10.1182/blood-2016-10-745059
74. Zhang B, Potyagaylo V, Fenton RG. IL-6-independent expression of Mcl-1 in human multiple myeloma. Oncogene (2003) 22:1848–59. doi: 10.1038/sj.onc.1206358
75. Gouill S, Le Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle (2004) 3:1259–62. doi: 10.4161/cc.3.10.1196
76. Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by IL-6 deprivation and dexamethasone. Blood (2004) 103:3148–57. doi: 10.1182/blood-2003-06-1984
77. Jourdan M, Vos J, De Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. (2000) 7:1244–52. doi: 10.1038/sj.cdd.4400758
78. Bruyne E, De Bos TJ, Schuit F, Valckenborgh E, Van, Menu E, Thorrez L, et al. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood (2010) 115:2430–40. doi: 10.1182/blood-2009-07-232801
79. Pratt G. Molecular aspects of multiple myeloma. J Clin Pathol Mol Pathol. (2002) 55:273–83. doi: 10.1136/mp.55.5.273
80. Shah V, Sherborne AL, Walker BA, Johnson DC, Boyle EM, Ellis S, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia (2017) 32:102–10. doi: 10.1038/leu.2017.179
81. Pawlyn C, Morgan GJ. Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer (2017) 17:543–56. doi: 10.1038/nrc.2017.63
82. Plowright EE, Li Z, Bergsagel PL, Chesi M, Barber DL, Branch DR, et al. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood (2000) 95:992–8.
83. Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NCP. Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia (2004) 18:962–6. doi: 10.1038/sj.leu.2403347
84. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA (1993) 90:3516–20. doi: 10.1073/pnas.90.8.3516
85. Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol. (1995) 128:1173–84. doi: 10.1083/jcb.128.6.1173
86. Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. (2010) 584:2981–9. doi: 10.1016/j.febslet.2010.05.061
87. Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells (2014) 3:418–37. doi: 10.3390/cells3020418
88. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature (2010) 463:103–7. doi: 10.1038/nature08646
89. Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood (2017) 130:2401–9. doi: 10.1182/blood-2017-06-788786
90. Touzeau C, Dousset C, Gouill S, Le Sampath D, Leverson JD, Souers AJ, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia (2014) 28:210–12. doi: 10.1038/leu.2013.216
91. Moreau P, Chanan-Khan A, Roberts AW, Agarwal AB, Facon T, Kumar S, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood (2017) 130:2392–400. doi: 10.1182/blood-2017-06-788323
92. Matulis SM, Gupta VA, Nooka AK, Hollen HV, Kaufman JL, Lonial S, et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia (2016) 30:1086–93. doi: 10.1038/leu.2015.350
93. Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood (2016) 128:1834–44. doi: 10.1182/blood-2016-03-704908
94. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma (2017) 58:2026–39. doi: 10.1080/10428194.2017.1283032
95. Chen L, Fletcher S. Mcl-1 inhibitors: a patent review. Expert Opin Ther Pat. (2017) 27:163–78. doi: 10.1080/13543776.2017.1249848
96. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Toumelin-Braizat G, Le, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature (2016) 538:477–82. doi: 10.1038/nature19830
97. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematological cancer models alone and in combination with established therapies. Cancer Discov. (2018) doi: 10.1158/2159-8290.CD-18-0387. [Epub ahead of print].
98. Hird AW, Secrist JP, Adam A, Belmonte MA, Gangl E, Gibbons F, et al. Abstract DDT01-02: AZD5991: A potent and selective macrocyclic inhibitor of Mcl-1 for treatment of hematologic cancers. Cancer Res. (2017) 77:DDT01–02. doi: 10.1158/1538-7445.AM2017-DDT01-02
99. Opferman JT. Attacking cancer's Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J. (2016) 283:2661–75. doi: 10.1111/febs.13472
100. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. (2008) 68:3421–9. doi: 10.1158/0008-5472.CAN-07-5836
101. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell (2007) 128:1173–86. doi: 10.1016/j.cell.2007.01.037
102. Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. (2010) 11:1149–59. doi: 10.1016/S1470-2045(10)70261-8
103. Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJS, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. (2009) 16:1030–9. doi: 10.1038/cdd.2009.48
104. Opferman JT, Iwasaki H, Ong CC. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science (2005) 307:1101–5. doi: 10.1126/science.1106114
105. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. (2013) 27:1365–77. doi: 10.1101/gad.215871.113
106. Malone CD, Hasan SMM, Roome RB, Xiong J, Furlong M, Opferman JT, et al. Mcl-1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci. (2012) 49:439–47. doi: 10.1016/j.mcn.2012.02.003
107. Certo M, Gaizo Moore V, Del Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9:351–65. doi: 10.1016/j.ccr.2006.03.027
Keywords: apoptosis, B-cell malignancy, BCL-2 family, BH3-mimetic, germinal center, MCL-1, multiple myeloma, plasma cell differentiation
Citation: Slomp A and Peperzak V (2018) Role and Regulation of Pro-survival BCL-2 Proteins in Multiple Myeloma. Front. Oncol. 8:533. doi: 10.3389/fonc.2018.00533
Received: 07 September 2018; Accepted: 30 October 2018;
Published: 20 November 2018.
Edited by:
Eric Eldering, Academic Medical Center (AMC), NetherlandsReviewed by:
Stephen Tait, University of Glasgow, United KingdomThomas Kaufmann, Universität Bern, Switzerland
Copyright © 2018 Slomp and Peperzak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor Peperzak, di5wZXBlcnpha0B1bWN1dHJlY2h0Lm5s