 Katarina K. Jovanović1
Katarina K. Jovanović1 Guillaume Escure2Jordane Demonchy2Alexandre Willaume2Zoe Van de Wyngaert2
Guillaume Escure2Jordane Demonchy2Alexandre Willaume2Zoe Van de Wyngaert2 Meryem Farhat2Paul Chauvet2Thierry Facon2
Meryem Farhat2Paul Chauvet2Thierry Facon2 Bruno Quesnel1,2
Bruno Quesnel1,2 Salomon Manier1,2*
Salomon Manier1,2*- 1IRCL, INSERM UMR-S1172, University of Lille, Lille, France
- 2Department of Hematology, CHU Lille, University of Lille, Lille, France
Multiple Myeloma (MM) is an incurable disease characterized by a clonal evolution across the course of the diseases and multiple lines of treatment. Among genomic drivers of the disease, alterations of the tumor suppressor TP53 are associated with poor outcomes. In physiological situation, once activated by oncogenic stress or DNA damage, p53 induces either cell-cycle arrest or apoptosis depending on the cellular context. Its inactivation participates to drug resistance in MM. The frequency of TP53 alterations increases along with the progression of the disease, from 5 at diagnosis to 75% at late relapses. Multiple mechanisms of regulation lead to decreased expression of p53, such as deletion 17p, TP53 mutations, specific microRNAs overexpression, TP53 promoter methylations, and MDM2 overexpression. Several therapeutic approaches aim to target the p53 pathway, either by blocking its interaction with MDM2 or by restoring the function of the altered protein. In this review, we describe the mechanism of deregulation of TP53 in MM, its role in MM progression, and the therapeutic options to interact with the TP53 pathway.
Introduction
Multiple Myeloma (MM) is an incurable hematological malignancy developing as a result of clonal proliferation of plasma cells originating from post–germinal-center B cells (1). Age-adjusted rates of new cases and deaths, covering the period from 2011–2015, show that new myeloma cases incidence was 6.6 per 100,000 people per year, while the number of deaths was 3.3 per 100,000 people per year (2). MM accounts for ~10% of all hematologic malignancies (3), and mostly affects elderly people, with 69 years being the median age at diagnosis (2).
One of the distinguished characteristics of MM is the clonal progression of the disease, which is reflected through precursor stages, named monoclonal gammopathy of undetermined significance (MGUS), and smoldering multiple myeloma (SMM) (4–6). Genomic events occurring across the course of the disease can be divided into primary and secondary genomic events, which altogether form a unique genomic landscape of the disease. Primary events are further divided into hyperdiploid (HRD) and non-HRD subtypes, which are mutually exclusive. Primary HRD events are usually trisomies of odd-numbered chromosomes 3, 5, 7, 9, 11, 15, 19, and/or 21 (7). Primary non-HRD events include translocations of the immunoglobulin (Ig) heavy chains (IGH), with five most frequently occurring translocations being t(11;14) (15%), t(4;14) (12%), t(14;16) (3%), t(14;20) (2%), and t(6;14) (1%), as well as del13q, which is the most frequent deletion in MM (59%) (8). All these primary events are usually detected at the MGUS stage of the disease development. Most frequent copy number gains and losses found in MM are del13q (45%), 1q+ (40%), del14q (39%), del16q (35%), del6q (33%), del1p (30%), and del8p (25%), all representing secondary mutation events, that can be also seen as driver events (9, 10).
TP53 gene is located at the chromosome 17p13.1, coding for p53 tumor suppressor protein, which is also known as guardian of the genome. The whole p53 signaling network is turned off under the normal physiological conditions, when p53 itself is expressed at low levels. In response to cellular stress, such as hypoxia, DNA damage, heat shock, p53 pathway becomes activated by means of protein stabilization through posttranslational mechanisms, which include phosphorylation and acetylation. The resulting accumulation of p53 protein in the nucleus triggers activation of various downstream pathways, which work in cooperation to keep the genomic integrity and homeostasis of the cell. This is achieved through several tumor suppressive mechanisms, including cell cycle arrest, apoptosis, and angiogenesis inhibition (11, 12).
In general, mutations in TP53 have been found in 50% of all human cancers (13). In MM, the frequency of TP53 alterations—by means of mutations and deletions—are more frequent in late stages of the disease and are associated with treatment resistance (7, 14). In this review we focus on the role of deregulated p53 in the progression of MM and the latest the therapeutic approaches designed to target specifically this tumor suppressor.
The TP53 Tumor Suppressor Gene
Ten years after the initial discovery of p53 in 1979 as a host cell protein bound to T antigen in SV40-transformed mouse cells (15), it was finally established that this 53 kDa protein performs its role as a tumor suppressor in cell culture, contrary to the popular belief that it functions as an oncogene (16). Immediately after this discovery inactivating TP53 mutations were confirmed to be a common event in the colorectal cancer tumorigenesis (17), and mutations occurring within this tumor suppressor were detected as a distinctive feature of Li-Fraumeni syndrome (18).
The p53 protein is structurally organized into several domains that are crucial for maintaining several of its functions. Two N-terminal transactivation domains, TAD1, and TAD2 respectively, are followed by a conserved proline-rich domain, which plays a role in DNA repair in response to γ-radiation (19). Positioned centrally, DNA binding domain is responsible for the site-specific DNA-binding function of p53 and represents a hot spot for the most of tumor-derived missense mutations (20). Within the C-terminal domain are located sequences necessary for nuclear localization and non-specific DNA binding, as well as an oligomerization domain mediating the formation of homo- and hetero-tetramers (21) (Figure 1).
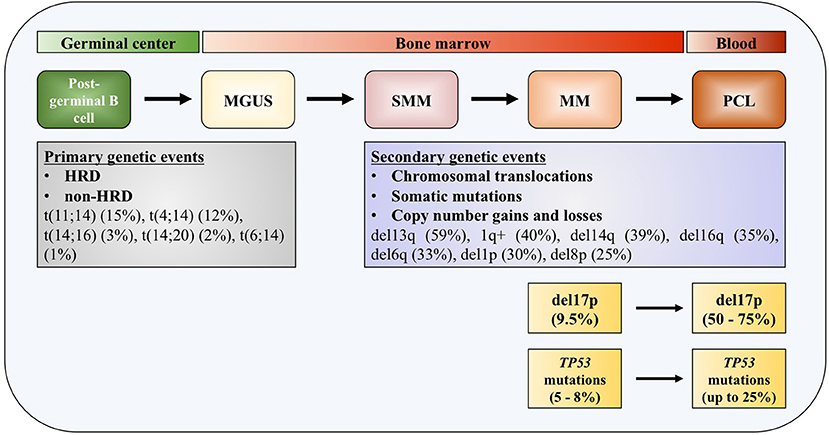
Figure 1. TP53 deregulation in multiple myeloma. Increased incidence of deletion 17p and TP53 mutations across the course of the disease. MGUS, monoclonal gamopathy of undetermined significance; SMM, smoldering multiple myeloma; MM, multiple myeloma; PCL, plasma cell leukemia; HDR, hyperdiploidy.
Since p53 is a transcription factor that can sense cellular stress and gets activated in response to the DNA damage and oncogene activation, during homeostasis its levels are kept low through interaction with E3 ubiquitin-protein ligase MDM2. MDM2 works on p53 inhibition in two ways, by interacting with its transactivation domain and by targeting p53 for proteasomal degradation, conferring a very short half-life of p53 in the range from 5 to 30 min (22, 23). Moreover, MDM2 expression is regulated by p53, meaning that low levels of tumor suppressor are maintained via negative feedback loop in normal physiological conditions. Depending on the nature of the cellular stress, mechanisms of activation of p53 can be different—DNA damage results in inhibition of MDM2-mediated degradation of p53 (24), whereas oncogenic signaling activates the ARF tumor suppressor, which also prevents MDM2 from degrading p53 (25).
During the response to oncogenic stress, ATM/ATR, CHK1, and CHK2 kinases phosphorylate p53, disrupting its binding to MDM2. At the same time, CBP and KAT5 acetyl-transferases induces acetylation of the tumor suppressor, specifically at lysine 120 (K120) within the DNA-binding domain (24, 26, 27).
Once activated, p53 induces either cell-cycle arrest or apoptosis depending on the cellular context, by transactivation of its downstream target genes, such as CDKN1A (coding for p21 protein), resulting in cell cycle arrest and senescence, and BAX, PUMA, and NOXA, triggering apoptosis (23). Recently it was reported that p53 is also involved in metabolism regulation by decreasing expression of the cystine/glutamate antiporter XCT2 (transcribed from SLC7A11 gene) and upregulating expression of glutaminase 2 (GLS2 gene) (28, 29).
Deregulation of TP53 in Multiple Myeloma
Deletion of 17p
Deletions of chromosome 17p13 region containing TP53 gene are usually monoallelic and associated with less favorable outcome in patients with MM (30–32). The adverse outcome is observed in patients at diagnosis with 17p deletion affecting more than 60% of their plasma cells, which translate in shorter median event-free survival (EFS) (14.6 months) and median overall survival (OS) (22.4 months), compared to patients without this genomic aberration or in <60% of plasma cells (31). In another study, 17p deletion was also associated with worse progression-free survival (PFS) and OS irrespective of the percentage of the tumor fraction of the alteration (33). In patients newly diagnosed with symptomatic myeloma (NDMM), loss of 17p was detected with a frequency of 9.5% (34). This number increases in the most aggressive forms of the disease, up to 50% in primary plasma cell leukemia (pPCL), and even up to 75% in secondary plasma cell leukemia (sPCL) (35). Together with serum β2-microglobulin and serum albumin levels (the International Staging System, ISS) and serum lactate dehydrogenase (LDH) level, 17p deletion—together with t(4;14) and t(14;16)—is included in the revised International Staging System (R-ISS) (32).
TP53 Mutations
Most of the TP53 mutations occurring in MM—as in human cancers—are missense mutations (Figure 1). Interestingly, in some cases it was found that protein products of missense mutant TP53 can obtain gain of function (GOF) activities, making such gene an oncogene which is promoting tumor progression, rather than being a tumor suppressor (36–38). Around 10% of TP53 mutations are found to be nonsense mutations resulting in truncated p53 proteins (39). Mutations in TP53 gene are reported in generally low frequencies in NDMM by different research groups, usually around 8% (40–42). In a recent report on 1,273 patients with NDMM, it was slightly lower, around 5% (8). On the contrary, in the later stages of the disease percentage increases significantly, to 25% in PCL, indicating role of these mutations in the disease progression and drug resistance (35, 43). The presence of mutations in TP53 gene is connected to adverse outcome in terms of PFS and OS, same as for 17p deletion (33). By analyzing tumor fractions of TP53 mutations in MM, it was found that this aberration appeared with higher frequency in subclonal, which implies preferential acquisition of these events later during clonal evolution of MM, as opposed to founder events. Analysis of co-occurrence of genomic events reveals that TP53 mutations almost always occur after or simultaneously with allelic 17p13 deletion (44). Inversely, TP53 mutations were found in about one third of MM patients carrying 17p deletion, with the tendency of increase to more than 50% in refractory disease (41, 45).
TP53 biallelic inactivation is a driver of progression in MM. At diagnosis, biallelic events in terms of loss or mutation of TP53 (called double hit myeloma) are detected in 3.7% of patients and represent a crucial marker of adverse prognosis for both PFS and OS, compared to wild-type or mono-allelic inactivation (46). In contrast, in a cohort of patients with relapsed MM, a TP53 abnormalities was identified 45% of the patients, and a double-hit events del(17p)/TP53mut or del(17p)/TP53del were observed in 15% of the cases. Those patients had a worse outcome (47).
Epigenomic Regulation
DNA methylation of the cytosines within CpG islands in promoter region of genes is an epigenetic modification known to decrease gene expression. Hypermethylation of the promoter of TP53 has been reported in MM cell lines and the expression of p53 protein was increased after treating cells by demethylating agents (48). In the context of TP53 haploinsufficiency in MM, it was shown that in cell lines without p53 protein expression the remaining allele was silenced by promoter hypermethylation (49). However, data on primary MM samples are lacking.
MiRNAs Regulation of TP53
Regardless of low mutation and deletion rates of TP53 in NDMM, the lack of functional protein is much more frequent than it would be expected based solely on detected genetic aberrations. This implies the existence of post-transcriptional mechanisms of regulation of p53 signaling in MM (50). The interaction between p53 and miRNAs is performed in both directions—p53 can act upon regulating transcription and maturation of several miRNAs, whilst miRNAs can perform either direct or indirect repression of p53 (51). The tumor suppressor miR-34 family is involved in repression of p53 pathway in MM through interaction with constituents of cell cycle and proliferation signaling pathways (52). MiR-34a expression is decreased in MM cells harboring 17p deletion/TP53 mutation (53) and its promoter region is often hypermethylated in MM (54). MiR-125b and miR-504 are direct negative regulators of p53 as they bind to the 3′UTR of TP53 mRNA, and their overexpression lead to downregulation of the endogenous level of p53 protein, inhibiting apoptosis in human neuroblastoma, and lung fibroblast cells (55, 56). Deregulation of miR-125b in MM pathogenesis has also been confirmed (50).
Post-translational Regulation
MDM2 is overexpressed in several MM cell lines and PCL patients, inducing a down-regulation of p53 (57). MDM2 gene localizes on 12q15. Gain if 12q is one of the most frequent copy number abnormalities in pan-cancer analysis (58, 59), however it is not often reported in MM.
Targeting p53 in Multiple Myeloma
Although the clinical outcome of patients with MM was significantly improved with the introduction of novel therapeutic strategies such as new-generation proteasome inhibitors, immunomodulatory drugs, anti-CD38 antibodies, and more recently CAR-T cells, MM remains incurable.
Patients with a 17p13 deletion demonstrated significantly lower response rate to lenalidomide treatment compared to patients not bearing this abnormality (60). Moreover, combinational treatment with lenalidomide, Adriamycin, and dexamethasone showed similar results in relapsed patients with 17p13 deletion (61). These results emphasize the need for novel therapeutic approaches that would overcome the 17p13 deletion related resistance to conventional treatment.
Two main approaches in reestablishing normal function of p53 in MM and in cancer in general are based inhibiting the interaction of the protein with its negative regulators (such as MDM2 and MDM4), and on restoring the function of protein product of the mutated TP53 gene (Figure 2).
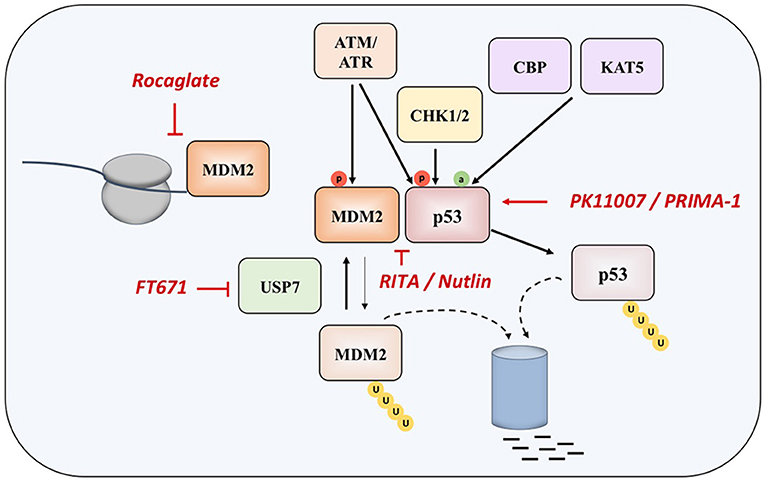
Figure 2. Interacting with TP53 pathway. Different drugs interacting with MDM2 and p53 (in red).
Nutlin was detected as the first potential inhibitor of p53-MDM2 interaction, as it was found that prevents binding of p53 to MDM2, which results in stabilization, accumulation, and activation of p53 signaling in cancer cells. However, nutlin works only in cells with preserved p53 signaling pathway and wt p53 (62). Nutlin-3 was showed to have significant activity in MM in experiments performed in vitro and ex vivo, as well as in synergy with drugs such as melphalan, etoposide, and bortezomib (63, 64), however the drug induces resistance and clonal selection.
Another compound called RITA (Reactivation of p53 and Induction of Tumor Cell Apoptosis) binds directly to the N-terminal domain of p53, which reduces its affinity for MDM2 as a consequence of conformational changes in the structure of p53 protein (65), although later study questioned this mechanism of action (66). It appears that RITA sensitivity correlates with induction of DNA damage as resistant cells show increased DNA cross-link repair. Inhibition of FancD2 restores RITA sensitivity (67). In MM cells, which acquired resistance to other therapeutic approaches, RITA induced cell cycle arrest and apoptosis (68). However, when the efficacies of nutlin-3a and RITA were compared in a panel of HMCLs with different TP53 statuses, it was found that, unlike nutlin-3a which exhibited toxicity only in HMCLs with wt-TP53, RITA killed 25% of HMCLs independently of the TP53 status, and showed efficacy independently of the presence or absence of 17p deletion in primary patient samples, implying that RITA could be a therapeutic approach of choice in patients with TP53 abnormalities, who showed resistance to current therapies (69).
When overexpressed in MM cells, MDM2 maintains low levels of p53, thus inhibiting its tumor suppressive functions (70, 71). Recently, a compound CMLD010509, which is a synthetic analog of the rocaglate, was found to inhibit the oncogenic translation program in MM cells and in mouse models of MM, targeting MDM2 oncoprotein among other targets (72). This can be explained by the fact that rocaglates are inhibitors of translation initiation factors and that MDM2 has a short half-life, suggesting that these proteins are preferentially affected in case of low translational flux.
Another approach to interact with MDM2 is to inhibit the ubiquitin-specific protease 7 (USP7), which is as a deubiquitinase removing ubiquitin specifically from MDM2. The inhibition of UPS7 results in the degradation of MDM2 and leads to re-activation of p53 (73). The small molecule FT671 is a potent USP7 inhibitor with a potent effect in vitro and in vivo in various cancers.
In was demonstrated that TP53-deficient cells are highly sensitive to the inhibition of Ataxia-Telangiectasia Rad3-related (ATR), one of the kinases responsible for DNA damage response (DDR) (74). Likewise, MM cells lines bearing TP53 mutation showed better response to ATR inhibition compared to TP53 wild-type MM cell lines, implying that, in the subset of MM clones with increased replicative stress, and DNA damage, inhibition of ATR could be potentially exploited as a synthetic lethal therapeutic approach (75).
PRIMA-1 (p53 reactivation and induction of massive apoptosis) has been introduced as therapeutic agent with aim of restoring the original function of mutant p53, through obtaining the wt conformation of the protein (76). The more effective derivative, PRIMA-1MET, except restoring mutant p53, is also acting in a TP53-independent manner by inducing reactive oxygen species (ROS) in cancer cells, with a good efficacy in MM (77–81). In fact, the accumulation of mutant-p53 protein suppresses the expression of SLC7A11, a component of the cystine/glutamate antiporter, through binding to the master antioxidant transcription factor NRF2. This diminishes glutathione synthesis, rendering mutant-p53 tumors susceptible to oxidative damage (82).
Other therapeutic approaches have been recently investigated (83, 84). A novel compound PK11007, belonging to the class of selective thiol alkylators 2-sulfonylpyrimidines, was found to alkylate surface-exposed cysteines 182 and 277 of p53, which results in stabilization of its DNA binding domain in vitro. This compound is particularly effective in cancer cell lines with null or mutant p53 background (83). An innovative approach consists of functional screening of phage display libraries for peptides, which carry the potential for reactivation of the mutant p53 (84).
Conclusion
TP53 alterations are of adverse prognosis in MM as in cancer in general. Recent publications of next generation sequencing on large cohort of patients with MM have enabled to better define the incidence of TP53 alteration across the course of the disease. In MM, TP53 is deregulated through different molecular mechanism, such as deletion17p, TP53 mutations, MDM2 overexpression, methylation of TP53 promoter, and deregulation of specific miRNAs. The knowledge of these molecular mechanisms of TP53 pathway alteration has helped the development of therapeutic strategies to target this pathway. Different approaches are currently tested: blocking the interaction between MDM2 and p53 with small molecules such as Nutlin or RITA, targeting MDM2 by inhibiting translation initiation with rocaglate or by blocking its deubiquitination by USP7 inhibitors, and restoring the function of altered p53 proteins. All these therapeutic strategies are currently being tested and need to be further validated for clinical use. Considering that TP53 alterations increase during progression of MM and induce drug resistance, it will be necessary to interact with TP53 pathway to be able to cure MM, or at least develop strategies that do not select TP53 subclones.
Author Contributions
KJ, GE, JD, AW, ZV, MF, PC, TF, BQ, and SM wrote the manuscript, generated the figures, and revised the review.
Funding
Funding through the JHCL (Journées d'Hématologie Clinique Lilloise) foundation of the Department of Hematology of Lille, France.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. (2011) 364:1046–60. doi: 10.1056/NEJMra1011442
2. Seer Cancer Statistics Review (2018). Seer Cancer Statistics Review. Bethesda, MD: National Cancer Institute. Available online at: https://seer.cancer.gov/csr/1975_2015/
3. Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. (2016) 91:719–34. doi: 10.1002/ajh.24402
4. Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood (2009) 113:5412–7. doi: 10.1182/blood-2008-12-194241
5. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood (2009) 113:5418–22. doi: 10.1182/blood-2008-12-195008
6. Jovanovic KK, Roche-Lestienne C, Ghobrial IM, Facon T, Quesnel B, Manier S. Targeting MYC in multiple myeloma. Leukemia (2018) 32:1295–306. doi: 10.1038/s41375-018-0036-x
7. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. (2017) 14:100–13. doi: 10.1038/nrclinonc.2016.122
8. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood (2018) 132:587–97. doi: 10.1182/blood-2018-03-840132
9. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood (2010) 116:e56–65. doi: 10.1182/blood-2010-04-279596
10. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer (2012) 12:335–48. doi: 10.1038/nrc3257
11. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature (2000) 408:307–10. doi: 10.1038/35042675
12. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer (2002) 2:594–604. doi: 10.1038/nrc864
13. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. (2013) 15:2–8. doi: 10.1038/ncb2641
14. Drach J, Ackermann J, Fritz E, Kromer E, Schuster R, Gisslinger H, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood (1998) 92:802–9.
15. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature (1979) 278:261–3. doi: 10.1038/278261a0
16. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell (1989) 57:1083–93. doi: 10.1016/0092-8674(89)90045-7
17. Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. (1990) 50:7717–22.
18. Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (1990) 250:1233–8. doi: 10.1126/science.1978757
19. Campbell HG, Mehta R, Neumann AA, Rubio C, Baird M, Slatter TL, et al. Activation of p53 following ionizing radiation, but not other stressors, is dependent on the proline-rich domain (PRD). Oncogene (2013) 32:827–36. doi: 10.1038/onc.2012.102
20. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. (2010) 2:a001008. doi: 10.1101/cshperspect.a001008
21. Meek DW. Regulation of the p53 response and its relationship to cancer. Biochem J. (2015) 469:325–46. doi: 10.1042/BJ20150517
22. Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. (2003) 1:1001–8. Available online at: http://mcr.aacrjournals.org/content/1/14/1001.article-info
23. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
24. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell (1997) 91:325–34. doi: 10.1016/S0092-8674(00)80416-X
25. Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell (1998) 92:713–23. doi: 10.1016/S0092-8674(00)81400-2
26. Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. (1999) 13:152–7. doi: 10.1101/gad.13.2.152
27. Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. (2000) 14:289–300. doi: 10.1101/gad.14.3.289
28. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
29. Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. (2016) 30:918–30. doi: 10.1101/gad.275891.115
30. Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood (2003) 101:4569–75. doi: 10.1182/blood-2002-10-3017
31. Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the intergroupe francophone du myelome. Blood (2007) 109:3489–95. doi: 10.1182/blood-2006-08-040410
32. Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol. (2015) 33:2863–9. doi: 10.1200/JCO.2015.61.2267
33. Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, De Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia (2018) 32:2604–16. doi: 10.1038/s41375-018-0037-9
34. Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. (2015) 6:6997. doi: 10.1038/ncomms7997
35. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell (2008) 13:167–80. doi: 10.1016/j.ccr.2008.01.007
36. Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. (2010) 107:246–51. doi: 10.1073/pnas.0908428107
37. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi: 10.1016/j.ccr.2014.01.021
38. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer (2018) 18:89–102. doi: 10.1038/nrc.2017.109
39. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. (2016) 37:865–76. doi: 10.1002/humu.23035
40. Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol. (2007) 20:571–96. doi: 10.1016/j.beha.2007.08.004
41. Lode L, Eveillard M, Trichet V, Soussi T, Wuilleme S, Richebourg S, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica (2010) 95:1973–6. doi: 10.3324/haematol.2010.023697
42. Boyd KD, Ross FM, Tapper WJ, Chiecchio L, Dagrada G, Konn ZJ, et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosomes Cancer (2011) 50:765–74. doi: 10.1002/gcc.20899
43. Lionetti M, Barbieri M, Manzoni M, Fabris S, Bandini C, Todoerti K, et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: an Italian cohort study and overview of the literature. Oncotarget (2016) 7:21353–61. doi: 10.18632/oncotarget.7241
44. Chin M, Sive JI, Allen C, Roddie C, Chavda SJ, Smith D, et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. (2017) 7:e610. doi: 10.1038/bcj.2017.76
45. Kortum KM, Mai EK, Hanafiah NH, Shi CX, Zhu YX, Bruins L, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood (2016) 128:1226–33. doi: 10.1182/blood-2016-02-698092
46. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia (2018). doi: 10.1038/s41375-018-0196-8. [Epub ahead of print].
47. Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood (2016) 128:1735–44. doi: 10.1182/blood-2016-06-723007
48. Hurt EM, Thomas SB, Peng B, Farrar WL. Reversal of p53 epigenetic silencing in multiple myeloma permits apoptosis by a p53 activator. Cancer Biol Ther. (2006) 5:1154–60. doi: 10.4161/cbt.5.9.3001
49. Teoh PJ, Chung TH, Sebastian S, Choo SN, Yan J, Ng SB, et al. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia (2014) 28:2066–74. doi: 10.1038/leu.2014.102
50. Murray MY, Rushworth SA, Zaitseva L, Bowles KM, Macewan DJ. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle (2013) 12:2144–53. doi: 10.4161/cc.25251
51. Feng Z, Zhang C, Wu R, Hu W. Tumor suppressor p53 meets microRNAs. J Mol Cell Biol. (2011) 3:44–50. doi: 10.1093/jmcb/mjq040
52. Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res. (2012) 18:6260–70. doi: 10.1158/1078-0432.CCR-12-1708
53. Stebner E, Neri P, Johnson J, Gratton KJ, Ren L, Duggan P, et al. Mir-34a sensitizes multiple myeloma (MM) cells to the proteasome inhibitor bortezomib. Blood (2011) 118:138. Available online at: http://www.bloodjournal.org/content/118/21/138?sso-checked=true
54. Okada N, Lin CP, Ribeiro MC, Biton A, Lai G, He X, et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. (2014) 28:438–50. doi: 10.1101/gad.233585.113
55. Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. (2009) 23:862–76. doi: 10.1101/gad.1767609
56. Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song JS, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell (2010) 38:689–99. doi: 10.1016/j.molcel.2010.05.027
57. Teoh G, Urashima M, Ogata A, Chauhan D, Decaprio JA, Treon SP, et al. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood (1997) 90:1982–92.
58. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi: 10.1038/nature08822
59. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. (2013) 45:1134–40. doi: 10.1038/ng.2760
60. Reece D, Song KW, Fu T, Roland B, Chang H, Horsman DE, et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: adverse effect of deletion 17p13. Blood (2009) 114:522–5. doi: 10.1182/blood-2008-12-193458
61. Knop S, Gerecke C, Liebisch P, Topp MS, Platzbecker U, Sezer O, et al. Lenalidomide, adriamycin, and dexamethasone (RAD) in patients with relapsed and refractory multiple myeloma: a report from the german myeloma study group DSMM (Deutsche Studiengruppe Multiples Myelom). Blood (2009) 113:4137–43. doi: 10.1182/blood-2008-10-184135
62. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. in vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (2004) 303:844–8. doi: 10.1126/science.1092472
63. Saha MN, Jiang H, Chang H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: evidence for p53-transcription-dependent and -independent pathways. Cancer Biol Ther. (2010) 10:567–78. doi: 10.4161/cbt.10.6.12535
64. Saha MN, Jiang H, Jayakar J, Reece D, Branch DR, Chang H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol Ther. (2010) 9:936–44. doi: 10.4161/cbt.9.11.11882
65. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. (2004) 10:1321–8. doi: 10.1038/nm1146
66. Krajewski M, Ozdowy P, D'silva L, Rothweiler U, Holak TA. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat Med. (2005) 11:1135–6; author reply 1136–7. doi: 10.1038/nm1105-1135
67. Wanzel M, Vischedyk JB, Gittler MP, Gremke N, Seiz JR, Hefter M, et al. CRISPR-Cas9-based target validation for p53-reactivating model compounds. Nat Chem Biol. (2016) 12:22–8. doi: 10.1038/nchembio.1965
68. Jones RJ, Bjorklund CC, Baladandayuthapani V, Kuhn DJ, Orlowski RZ. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol Cancer Ther. (2012) 11:2243–53. doi: 10.1158/1535-7163.MCT-12-0135
69. Surget S, Descamps G, Brosseau C, Normant V, Maiga S, Gomez-Bougie P, et al. RITA (Reactivating p53 and Inducing Tumor Apoptosis) is efficient against TP53abnormal myeloma cells independently of the p53 pathway. BMC Cancer (2014) 14:437. doi: 10.1186/1471-2407-14-437
70. Wade M, Li YC, Wahl GM. MDM2, MDMX, and p53 in oncogenesis and cancer therapy. Nat Rev Cancer (2013) 13:83–96. doi: 10.1038/nrc3430
71. Teoh PJ, Chng WJ. p53 abnormalities and potential therapeutic targeting in multiple myeloma. Biomed Res Int. (2014) 2014:717919. doi: 10.1155/2014/717919
72. Manier S, Huynh D, Shen YJ, Zhou J, Yusufzai T, Salem KZ, et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci Transl Med. (2017) 9:eaal2668. doi: 10.1126/scitranslmed.aal2668
73. Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature (2017) 550:481–6. doi: 10.1038/nature24451
74. Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. (2011) 18:721–7. doi: 10.1038/nsmb.2076
75. Cottini F, Hideshima T, Suzuki R, Tai YT, Bianchini G, Richardson PG, et al. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discov. (2015) 5:972–87. doi: 10.1158/2159-8290.CD-14-0943
76. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell (2009) 15:376–88. doi: 10.1016/j.ccr.2009.03.003
77. Rokaeus N, Shen J, Eckhardt I, Bykov VJ, Wiman KG, Wilhelm MT. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene (2010) 29:6442–51. doi: 10.1038/onc.2010.382
78. Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, et al. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. (2013) 4:e881. doi: 10.1038/cddis.2013.417
79. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther. (2013) 12:2331–41. doi: 10.1158/1535-7163.MCT-12-1166
80. Tessoulin B, Descamps G, Moreau P, Maiga S, Lode L, Godon C, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood (2014) 124:1626–36. doi: 10.1182/blood-2014-01-548800
81. Teoh PJ, Bi C, Sintosebastian C, Tay LS, Fonseca R, Chng WJ. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget (2016) 7:61806–19. doi: 10.18632/oncotarget.11241
82. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. (2017) 8:14844. doi: 10.1038/ncomms14844
83. Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc Natl Acad Sci USA. (2016) 113:E5271–80. doi: 10.1073/pnas.1610421113
Keywords: TP53, multiple myeloma, targeted therapy, clonal evolution, precision medicine
Citation: Jovanović KK, Escure G, Demonchy J, Willaume A, Van de Wyngaert Z, Farhat M, Chauvet P, Facon T, Quesnel B and Manier S (2019) Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 8:665. doi: 10.3389/fonc.2018.00665
Received: 14 October 2018; Accepted: 17 December 2018;
Published: 09 January 2019.
Edited by:
Martine Amiot, Center for the National Scientific Research (CNRS), FranceReviewed by:
Jerome Moreaux, UMR9002 Institut de Génétique Humaine (IGH), FranceMarta Coscia, Università degli Studi di Torino, Italy
Copyright © 2019 Jovanović, Escure, Demonchy, Willaume, Van de Wyngaert, Farhat, Chauvet, Facon, Quesnel and Manier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salomon Manier, c2Fsb21vbi5tYW5pZXJAaW5zZXJtLmZy