 Khalid O. Alfarouk1,2,3,4*
Khalid O. Alfarouk1,2,3,4* Adil H. H. Bashir2,5Ahmed N. Aljarbou6,7
Adil H. H. Bashir2,5Ahmed N. Aljarbou6,7 AbdelRahman M. Ramadan8Abdel Khalig Muddathir2,9Sari T. S. AlHoufie3,10Abdelhamid Hifny11
AbdelRahman M. Ramadan8Abdel Khalig Muddathir2,9Sari T. S. AlHoufie3,10Abdelhamid Hifny11 Gamal O. Elhassan12Muntaser E. Ibrahim5
Gamal O. Elhassan12Muntaser E. Ibrahim5 Saad S. Alqahtani13
Saad S. Alqahtani13 Shakir D. AlSharari14,15Claudiu T. Supuran16
Shakir D. AlSharari14,15Claudiu T. Supuran16 Cyril Rauch17Rosa Angela Cardone18
Cyril Rauch17Rosa Angela Cardone18 Stephan J. Reshkin18
Stephan J. Reshkin18 Stefano Fais19
Stefano Fais19 Salvador Harguindey20
Salvador Harguindey20- 1Alfarouk Biomedical Research LLC, Tampa, FL, United States
- 2Hala Alfarouk Cancer Center, Khartoum, Sudan
- 3Al-Ghad International College for Applied Medical Sciences, Medina, Saudi Arabia
- 4American Biosciences, Inc., New York City, NY, United States
- 5Institute of Endemic Diseases, University of Khartoum, Khartoum, Sudan
- 6College of Pharmacy, Qassim University, Buraydah, Saudi Arabia
- 7Al-Ghad International College for Applied Medical Sciences, Jeddah, Saudi Arabia
- 8College of Dentistry, Taibah University, Medina, Saudi Arabia
- 9Department of Pharmacognosy, Faculty of Pharmacy, University of Khartoum, Khartoum, Sudan
- 10Department of Clinical Laboratory Sciences, Faculty of Applied Medical Sciences, Taibah University, Medina, Saudi Arabia
- 11Al-Azhar University, Cairo, Egypt
- 12Unaizah College of Pharmacy, Qassim University, Unaizah, Saudi Arabia
- 13Clinical Pharmacy Department, College of pharmacy, Jazan University, Jazan, Saudi Arabia
- 14Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
- 15Department of Pharmacology and Toxicology, Medical College of Virginia, Virginia Commonwealth University, Richmond, VA, United States
- 16University of Florence, Florence, Italy
- 17School of Veterinary Medicine and Science, University of Nottingham, Nottingham, United Kingdom
- 18Department of Biosciences, Biotechnologies and Biopharmaceutics, University of Bari, Bari, Italy
- 19Department of Oncology and Molecular Medicine, National Institute of Health, Rome, Italy
- 20Institute for Clinical Biology and Metabolism, Vitoria, Spain
Helicobacter pylori (HP) is a facultative anaerobic bacterium. HP is a normal flora having immuno-modulating properties. This bacterium is an example of a microorganism inducing gastric cancer. Its carcinogenicity depends on bacteria-host related factors. The proper understanding of the biology of HP inducing gastric cancer offers the potential strategy in the managing of HP rather than eradicating it. In this article, we try to summarize the biology of HP-induced gastric cancer and discuss the current pharmacological approach to treat and prevent its carcinogenicity.
Historical Background
In 1866, Kussmaul suggested the use of bismuth salts for the treatment of peptic ulcers (1). As bismuth has an oligodynamic effect (toxic to bacteria in a minuscule amount), that was probably the first published evidence of the bacterial role in causing peptic ulcers. After more than 100 years scientists use Bismuth subsalicylate in treating gastritis after conventional antibiotics have failed in improving its symptoms (2). In 1875, Bottcher and Letulle hypothesized that ulcers are caused by bacteria (1). Over a century later Marshal and Warren isolated a spiral bacteria that causes gastritis in 1983 and 1984 (3, 4). After that, the prevailing medical dogma of gastritis was shifted from stress-induced gastritis to be defined as an infectious disease. In 1997, Tomb et al. published the whole genome sequence of that spiral bacterium that was termed by Campylobacter pylori did (5) and later became Helicobacter pylori (HP) (6).
Helicobacter pylori (HP) was identified as a common cause of chronic gastritis (3, 4). Chronic gastritis, perhaps through mucosal pH changes and activation of chemical carcinogens, may lead not only to gastric ulcers but also to stomach cancer and other malignancies of the gastrointestinal tract (7). In this context, we discuss the role of HP in gastric cancer and new possible measures of prevention and treatment.
Clinical Perspective
HP originated in Africa over 100,000 ago and has become a very useful tool to monitor human migration and detect human ancestry (8–11). More than 50% of the world's population is infected with HP in their stomachs, with a higher prevalence rate in developing countries as compared to developed countries (12). Although <20% of those infected may develop any symptoms, many develop wide-ranging symptomatology. Moreover, HP can be acquired during childhood (13), and its transmission can be associated with childhood episodes of gastroenteritis (13, 14).
HP infection often results in deficiencies in micronutrients such as vitamin A, C, E, Iron, Copper, and B12 (15). Also, HP alters nocturnal melatonin secretion, which might perturb its gastroprotective effects and lead to disturbances of the upper digestive tract (16). Also, HP alters the antioxidant properties of melatonin (17). HP viability may be impaired by ascorbic acid, as its growth in the stomach is increased in patients with low ascorbic acid, while the disappearance of the bacterium increases stomach ascorbic acid (18, 19). Therefore, HP alters the redox status of the organism by scavenging the anti-oxidants from the body.
HP is correlated to many aging-related diseases, and also it increases the susceptibility to other infectious diseases such as cholera (20). HP is also a causative agent of acne vulgaris and Polycystic Ovarian Syndrome (PCOS) due to its ability to induce hyperprolactinemia (21). HP also raises blood pressure (22) and increases the risk of ischemic heart disease (23). Its infection may also increase the incidence of diabetes (24), yet evidence suggests that HP eradication may result in weight gain (25). HP has an essential role in preventing diseases, such as asthma (26, 27) or as an immunomodulator against infectious agents such as Mycobacterium tuberculosis (28).
HP has been designated by the WHO as a carcinogen (20, 29) because it can develop: (i) gastric adenocarcinoma and (ii) MALT lymphoma (mucosa-associated lymphoid tissue) (30). However, not every infected individual will develop gastric cancer due to (1) the nature of HP and (2) host vulnerability (31, 32). Surprisingly, HP has also been shown to play a critical role in the prevention of esophageal carcinoma (33). Indeed, in western countries, lowering the prevalence rate of HP has been associated with an increase in the incidence of esophageal adenocarcinomas (14), possibly due to a hygienic culture that excludes naturally occurring defense mechanisms. The eradication of HP not only results in an increased incidence of developing esophageal adenocarcinoma but also appears to decrease its ability to delay or prevent gastric cancer (34–36). However, HP has no relationship with esophageal squamous cell carcinomas (37).
Helicobacter Pylori-Induced Gastric Cancer
HP expresses a variety of genes involved in its pathogenicity and remodeling of the microenvironment. Here, we review several of these factors that may be involved in HP-mediated carcinogenesis.
Urease
Urease enzyme plays a critical role in maintaining the HP niche as it hydrolyzes urea into ammonia. This leads to a neutralization of the acidity around the bacteria to create a suitable microhabitat (38). It also facilitates diffusion through mucus by reducing its viscoelasticity (39) and modulates the host's immune response against HP (40, 41). Therefore, urease enzyme is a critical factor that determines HP fitness (42) but not its pathogenesis (43).
Urease catalyzes the breakdown of urea into NH3 and CO2 (44, 45), which provide both acid-neutralizing and acid-buffering capacities. It appears conceivable that urease is a cytoplasmic enzyme, since the urease activity increases in media where the pH was progressively lowered, without detectable changes of the bacterial cytoplasmic pH, and without evidence of bacterial membrane damage.To support this finding, a transporter has been identified encoded by the ureI gene capable of delivering urea to the cytoplasm (46), where urease enables neutralization and buffering capacities (47). It is now apparent that the activities of the transporter and enzyme are coupled not only functionally but physically as well. Under acidic conditions a neosynthesis of bacterial proteins (e.g., arginase and carbonic anhydrase) has been shown, many of them directly or indirectly involved in pH regulation of both cytosol and cytoplasmic vacuoles. Arginase is involved in providing the substrate to urease to produce L-ornithine and urea (48).
Carbonic Anhydrase
Carbonic anhydrase (CA) is a form of family of zinc-containing metalloenzymes that catalyze the interconversion of carbon dioxide (CO2) and water (H2O) to form carbonic acid that dissociates to form bicarbonate () and Hydrogen ion (H+). It is expressed in both prokaryote as well as eukaryote (49), and even within the eukaryotic cells, it is found in many subcellular compartments, such as the cytosol, mitochondria or anchored to membranes (50, 51). CAs are ubiquitously expressed in many tissues of human body that reflects the importance of their physiological functions in maintaining the buffering capacity of the biological systems as well as in biosynthetic processes (52). It is associated with many other diseases including cancer.
Regard the HP, HP has two forms of CA, an alpha-type enzyme (HpαCA) and the Hp beta CA (HpβCA) (53, 54). While HpαCA supports urease activity, HpβCA supports bacterial growth at acidic pH (54). Therefore, HpCAs plays crucial role in adapting of HP to gastric environment and supports its fitness.
Lewis Antigen
HP expresses Lewis antigens on their surface as part of their lipopolysaccharide components (55, 56). The Lewis antigen system is a human blood group system based upon genes on chromosome 19 p13.3 (FUT3 or Lewis gene). HP NCTC11637 expresses a lipopolysaccharide (LPS) that comprises an O-antigen side chain with structural homology to the human blood group antigen Lewis X [Le(x)] (57). Therefore, expression of the Lewis antigen could be for mimicry (anti-predation strategy to avoid immune system). Also, Lewis antigens expressed on the bacterial surface facilitate adherence of HP to gastric epithelial cells (tissue tropism) (58). Therefore, humans carrying blood group A, B are relatively resistant to HP adherence to their epithelium (57, 59). HP strains differ in their expression of Lewis antigen into Lex, Ley, both, Lea, sialyl-Lex or negative for both (60). Lex and Ley are correlated with cagA+ and s1/m1 VacA. Among the western population, the dominant phenotypes are LeX and Ley while Lea and Leb are found in a smaller proportion (61). Possessing of Lex and Ley leads to higher HP internalization rates by gastric epithelium as compared to Lea and Leb or non-expressing Lewis antigen (62).
Lewis antigen and urease are inversely related because urease promotes HP survival and colonization (63) while inhibiting internalization (64). Moreover, we are not sure if the concomitant loss of urease activity occurs due to phenotypic changes from mucus to adherent epithelial phenotype nor if this loss of urease activity and acquiring of Lex and Ley is beneficial for immune evasion (65). Therefore, HP internalization might occur during the development of gastric atrophy as an unfavorable habitat. Adaptation in hostile habitats that affect organismal multiplication is called “cost of adaptation.” Therefore, the acquisition of Lewis antigen is associated with the cost of adaptation. In other words, acquiring of Lewis antigen will support HP' survival, but it affects negatively on the proliferation rate.
VacA
VacA (vacuolating cytotoxin) is a secreted protein encoded by the vacA gene. All HP strains have the vacA gene, but they differ in their expressivity (66). VacA secretion is more frequent in patients with gastric cancer as compared to patients with gastritis (alone) (67), indicating a connection between expressivity and pathogenicity. Characterization of 59 different HP isolates revealed the existence of three distinct families of vacA sequences (s1a, s1b, and s2) and two separate families of middle-region alleles (m1 and m2) (68). The sub-strain that has the m1/s1 allele is the most virulent isoform regarding its ability to induce inflammation (69). VacA is composed of the P55 and P33 proteins. P55 is responsible for producing pores within the gastric epithelium while P33 disrupts mitochondrial fission machinery upon its inoculation (70), inducing cellular death of the epithelium (71). VacA also alters the maturation and trafficking of lysosomal enzymes (72). Also, VacA inhibits T-cells population expansion (73), and so it may promote survival of various phenotypes.
CagA
CagA is a 40 kbp horizontally acquired gene (6, 74) that encodes for the Type IV Protein Secretion System (T4SS). T4SS delivers cagA-oncoprotein (75–77) to suppress apoptosis (78). Outer membrane proteins (OMPs) and cagA target mitochondria. In this regard, development of gastric cancer due to mitochondrial injury is compatible, or at least parallel, with what Otto Warburg had earlier hypothesized on the respiratory impairment of cancer cells (79).
HP is heterogeneous in inducing pathogenesis (12). If cagA+ is associated with developing gastric adenocarcinoma, either cagA+ or cagA− can induce β-Lymphoma (30). The growth of MALTomas (MALT-NHL) is a more immune-dependent growth. It is clear that HP interacts with the immune system with a “gold panning” strategy to increase IL-2 expression through T-cells (80). In summary, eradication of HP might represent a potential strategy for treating these tumors (81), either low-grade β-cell lymphoma and to some extent higher grades of this tumor (82–84).
CagA is associated with a higher production rate of cytokines (85, 86). HP seropositivity has been classified to be either cagA+ or cagA− sub-strains. The presence of cagA+ modulates epithelial activity that acts as a phosphatase enzyme (dephosphorylation) resulting in pro-inflammation by releasing IL-8 (87), MAPK (88), and NF-KB (89–91). Hence, cagA+ might be considered to be a hallmark of HP carcinogenicity. Some studies have shown that the cagA+ strain significantly increases epithelial proliferation rate either directly (92, 93) or through the induction of hypergastrinemia (increased gastrin level) (94, 95) while another study shows that cagA+ apoptosis indices are increased (96, 97). Such contradictions might reflect that epithelial proliferation and apoptosis are contagious processes that are controlled by the cagA+ strain in a way that expresses a multistage progression of epithelial transformation.
Outer Proteins (BabA2)
The outer membrane protein (OMP) of HP, babA2, is associated with an increased risk of gastric cancer. BabA2 is a member of a family of highly conserved OMP, is encoded by strain-specific gene babA2 and binds the Lewisb (Le b). BabA2 is commonly found in phenotypes that adhere to the epithelium (14, 98). The BabA2+ sub-strain in Leb expressing mice results in developing atrophy anti-parietal antibodies, i.e., the development of gastric atrophy is considered an autoimmune disease (98–100) and so results in chronic inflammation. The combination in a triple positive strain of the above proteins (BabA2, CagA+, and VacA) is a reliable indicator of the possibility of inducing carcinogenicity (101).
In conclusion, if urease is responsible for establishing a suitable platform for HP colonization, Lewis antigen is an essential protein that serves HP fitness under unfavorable habitat conditions followed by CagA, Bab, and VacA that makes HP occupy the gastric epithelium and induce inflammation. In summary, all these genes and their subsequent proteins are working orchestrally in a cascade manner to serve the evolutionary trajectory of HP.
Human Response
Some of the keys elements that interact with HP to induce gastric cancer include.
β–Catenin
Beta-catenin (β-catenin) is a protein encoded by CTNNB1 gene on band p12 placed on the short arm of chromosome 3, a region that is affected by a somatic alteration in the tumor (102). β-catenin is a protein that plays a paramount role in the coordination of both cell-cell adhesion and gene transcription.
β-catenin is a proto-oncogene that has been found to accumulate inside the nucleus in precancerous lesions of gastric cancer (103). β-catenin is associated with several types of tumors including primary hepatocellular carcinoma, ovarian carcinoma, breast cancer, lung cancer, colorectal cancer, basal cell carcinoma, prostate cancer, pilomatrixoma, medulloblastoma, Head and neck squamous cell carcinoma and glioblastoma (104–108).
HP activates β-catenin expression (76, 109, 110) such that β-catenin activates its expression via HP as a positive feedback mechanism to induce intestinal metaplasia. Initially, intestinal metaplasia is dependent on cagA (111) and remains latently independent even after HP eradication (112).
EGFR
EGFR is a member of the ErB family which is structurally related to tyrosine kinase receptors Her1 (EGFR or ErB-1), Her2 (ErB-2), Her3 (ErB-3), and Her4 (ErB-4) (113). Epidermal growth factor receptors (EGFR) are cell surface expressed proteins. EGFR is a target for Epidermal Growth Factor (EGF) and Transforming Growth Factor–alpha (TGF-alpha) ligands to stimulate cellular proliferation. EGFR is expressed in several carcinomas and induces cellular transformation (114). Those carcinomas are EGFR-dependent in their survival and growth (114).
HP keeps in balance an epithelial proliferation/apoptosis ratio (96, 115, 116). HP increases such ratio via activation of EGFR (117, 118) i.e., HP increases cellular proliferation through activation of EGFR.
Targeting EGFR, perhaps as a too potent strategy for prevention of HP-inducing gastric cancer; due to its ability to induce excessive apoptosis and/or “oncogenic shock” (113, 119).
Inflammation as a Result of Helicobacter Pylori—Human Interactions
The correlation between gastric inflammation and ulcers, gastritis, and gastric cancer had been first explored by Stahl in 1728 and Nevpeu as soon as 1821 (1) and later on by many other groups [for a review see: Harguindey, (7)]. HP is a chronic infection that leads to chronic inflammation (41, 120) and so promotes tumorigenesis (gastric cancer) (121, 122). The inflammatory response induced by HP leads to the release of mutagenic substances e.g., metabolites of inducible nitric oxide synthase (iNOS). Nitric oxide can result in a change in reactive nitrogen species that are found in DNA, proteins, etc. (14). Therefore, HP releases free radicals and removes antioxidant agents.
Phospholipase A2 (PLA2)
Phospholipase A2 is an enzyme that catalyzes the production of arachidonic acid from fatty acids. Arachidonic acid is further converted to prostaglandins and Leukotriene by cyclooxygenase and lipoxygenases enzymes, respectively. HP activates the Phospholipase A2 enzyme (PLA2) (123, 124) (see Figure 1).
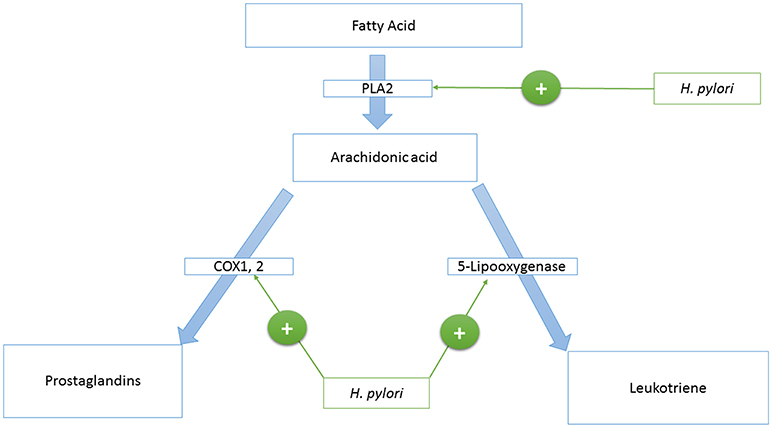
Figure 1. A model that represents the H. pylori—host’s eicosanoids interactions.
Cyclooxygenase Enzyme 1 and 2 (COX-1,−2)
Cyclooxygenase 1, and 2 (COX-1,−2) are enzymes that catalyze the conversion of prostanoid to prostaglandins. Prostanoid synthesis is thought to be cytoprotective to the stomach and increases production of the pro-aggregatory prostanoid, thromboxane, by platelets (125) (see Figure 1). Alternatively, COX-2 induction by TNF-alpha, INF-gamma, and IL-1 (126–131) is associated with colorectal cancer (132), and HP activates PLA2 and TNF-alpha expression (133–135). Also, COX-2 is present in the atrophic area and malignant gastric lesions (125, 136). Overexpression of COX-2 prevents apoptosis (137). In this way, COX-2 might support the tumorigenic potential of HP.
Leukotriene
Leukotriene is produced from arachidonic acid by the activity of the 5-Lipooxygenase enzyme, and this is accompanied by the release of histamine (see Figure 1). Leukotriene is associated with gastritis (138, 139), and its receptors have been found to be overexpressed in gastric cancer (140). Leukotriene receptors are persistent in the epithelium of the stomach even after eradication of HP (141).
In summary, HP is related to eicosanoids at multiple levels to create a sustainable chronic inflammatory environment that leads to initiation, development, and progression of gastric cancer.
Acid-Base Considerations in Etiopathogenesis and Therapy
Last but not least, IL-1β is a TH1 cytokine having a strong acid production inhibitory property (14, 142). Thus, it is not surprising that IL-1β has a pivotal role in initiating the development of gastric adenocarcinomas (7, 143) since inhibition of acid production is a significant step in developing gastric cancer (144). Together with cagA+, IL-1β overexpression, it also increases the prevalence of gastric adenocarcinomas (14). TNF-α also inhibits gastric acid secretion (145), and this is also correlated with gastric cancer (146). All these situations can be inserted within the concept of pH-direct and pH-indirect gastrointestinal oncogenesis [for a review, see Harguindey, (7); Table 1].

Table 1. Shows some factors that govern H. pylori—human (host) that mediates carcinogenesis.
Notes in the Management of HP Infection
Antibiotics represent the backbone of the currently used strategy of triple therapy. This “triple therapy” strategy consists of two antibiotics (Clarithromycin and Amoxicillin) for HP eradication plus an acid-suppressing agent, usually a proton pump inhibitor to alter the bacterial microenvironment and reduce gastric pain.
Antibacterial administration not only leads to post-elimination complications [e.g., the development of esophageal adenocarcinoma, of asthma, of metabolic disorders, etc. (147)]. Furthermore, the efficacy of antibiotics are questionable due to (i) uncertainty of antibiotic ability to create stomach sterility (38), (ii) prevention of HP's tumorigenicity (120), or even (iii) induction cost of resistance rather than diminishing microbial-host co-evolution (i.e., perturbation of ecological interactions) (25, 148).
- Clarithromycin is a semi-synthetic macrolide antibiotic derived from erythromycin which at the same time is originated from Streptomyces erythreus (112, 149). It binds to the part of the ribosome (50s ribosomal subunit), and so it affects peptide translation. In addition to its bactericidal activity, Clarithromycin inhibits the production of superoxide that is released from neutrophils and other white blood cells (150). Clarithromycin has a membrane stabilizer property and, so, inhibits the release of pro-inflammatory mediators (151). Clarithromycin inhibits IL-8 production through affecting AP-1 and NF-kappa B expression. So, it is useful in treating HP infection (152, 153). That is why Clarithromycin represents the core of the three-pronged therapy approach. However, if Clarithromycin failed, Levofloxacin could be used.
- Levofloxacin is a suggestive substitue for Clarithromycin resistant strains (154, 155), although Levofloxacin might induce localized inflammation in the form of tendinitis or tendon rupture (156–158).
- Amoxicillin is a semi-synthetic Beta-Lactam antibiotic derived from Penicillium notatum. It inhibits bacterial cell wall synthesis. Amoxicillin has a high therapeutic index (high safety profile), and it can be combined with Clavulanic acid if some strains develop Beta-lactamase enzymatic inhibition. Metronidazole is a substitute in the case of penicillin hypersensitivity.
- Metronidazole is a nitroimidazole compound that acts against anaerobic bacteria and protozoa. Metronidazole is a prodrug activated by an unusual enzymatic system, the Pyruvate: Ferredoxin oxidoreductase (PFO) found in hydrogenosome (an anaerobic version of mitochondrion) (159). PFO leads to activation of Metronidazole, and the product of this reaction leads to the destruction of the helical structure of the DNA of the microorganism.
- Acid-suppressive agents: In the past, clinicians used H2-receptor antagonists, e.g., Cimetidine, which was later substituted by Famotidine because of drug side effects of the former. Currently, using proton pump inhibitors (PPIs) have become most popular. Moreover, it has also been shown that omeprazole and its analogs may behave as Helicobacter pylori urease inhibitors (160). Two very recent reports add much to the use of proton pumps inhibitors in HP eradication. One shows that the genotypic polymorphisms of HP may be predictive of the of the optimum PPI dose to improve the therapeutic outcome (161). The other highly support the use of proton pump inhibitors in the HP-mediated gastric atrophy eradication approaches (162). However, it appears clear actually that the use of PPI should be mandatory in the prevention of gastric cancer relapses after endoscopic submucosal dissection for early gastric cancer (163), and this is of course of paramount importance. A role of pH and proton pumps for the growth of a variety of infectious agents has been provided, and it is still under a challenging debate (164). However, it is impressive how the involvement of proton pumps exert a central role a specificity in infectivity of so many microbes and parasites, in a way that proton pump inhibition has proven to induce an apparent anti-infective effect. This has been shown in many bacteria such as of course M. Tuberculosis, where the PPI lansoprazole seems to exert its action through a specific target to cytochrome bc1 (165); but also Salmonella enteric infection, where Omeprazole interferes with virulence and inflammation in infected cells (166); in experimental Clostridium difficile infection in mice (167). Some research on new compounds based on biologically active peptide against proton pumps seems to generically support these data (168). Moreover, PPI has been shown to be effective against a series of protozoa, yeasts, and amoebas, including Giardia lamblia (169), Trichomonas vaginalis (170), Plasmodium falciparum (171). The use of appears promising in yeasts (172), and Dictyostelium discoideum (173, 174) infections, where a role of proton pumps in intracellular replication has been shown. Therefore, PPIs have a direct bactericidal activity (175–179). This represented an additional antibiotic-like measure within the triple therapy strategy.
- This suppression is beneficial for gastric atrophy. Therefore, in this respect, PPIs might be equivalent to IL-1β and TNF-α. So, it will be wise if someone raises that PPIs perhaps could support gastric atrophy and hypochlorida (180). However, this is not a case, because PPIs show a promising potential effect in treating cancer (181–184).
Recently, the concept of quadruple therapy has also been introduced to the development of drug resistance (185), which reflects the unusual ability of HP to adapt.
Administering of NSAIDs as COX-inhibitors is considered as a potential strategy for preventing the development of gastric cancer because they decrease and attenuate the inflammatory environments and so prevent and/or delay tumor progression. However, cyclooxygenase inhibition will prevent mucous formation that leads to a decrease of the M/A ratio and so increases adherence phenotypes that result in malignant transformed consequences. That is why the administration of NSAIDs to prevent gastric cancer becomes questionable. Part of this idea has been considered previously (186). More comments on anti-inflammatory drugs include:
1) Prevention of PGE-synthesis might alter population dynamics through suppression of M phenotypes as well as alters the entire population by its bactericidal activity (97, 187–190).
2) HP infection potentiates Aspirin-induced gastric injury (191).
3) Administration of Aspirin results in increasing of Leukotriene production (192). In this regard, the role of Aspirin in preventing carcinogenesis becomes questionable unless certain tumors rely on PGs rather than Leukotriene.
4) Most probably, administration of steroids will be beneficial due to (i) it decreases incidence of inflammation and so becomes more similar to malaria-selection for populations (193), (ii) the fact that steroids (immunosuppressive therapy) in mice does not alter HP colonization (194, 195) and so it is superior to NSAIDs, (iii) steroid as immunosuppressant is beneficial in delaying and/or preventing gastric atrophy, and finally, (iv) HP elicits and activates Phospholipase-A2; where PL-A2 is a critical player in producing the inflammatory mediators, e.g., Prostaglandins, Leukotrienes, etc. (123, 124). Interestingly, down-regulation of inflammatory response might, represent a potential preventive strategy for developing gastric cancer. However, chronic administration of steroids might lead to bone marrow suppression and adrenal atrophy.
Rendering HP to acquire the cagA Island, and/or reversing the HP population to become cagA- might represent potential preventive strategies against HP induction of gastric adenocarcinoma as well as serving a protective role against developing esophageal adenocarcinoma and other diseases.
Does HP Behave Like a Cancer Cell?
Tissue acidification is a framework of activities from the internal compartments of the cells to the extracellular microenvironment. The cellular events include the multifunction cascade of internal vesicles, from endosome to phagosome, but this process is highly dependent on the cytosolic pH, that has to be considered a key factor is dictating the vesicle fission and/or fusion. We know for instance that the lipid composition of both the internal and extracellular vesicles may change depending on the pH, and this is particularly true under tumor condition (196). The nature and function of acidic vesicles markedly change dependently on the pH condition, thus conditioning the level of maturation of the vesicles themselves. From a mechanistic point of view the role of proton pumps is critical in orchestrating the pH control in both the intracellular and extracellular microenvironment, and this, in turn, has a crucial role in regulating both the intracellular pathway following membrane receptors triggering (e.g., apoptotic pathways) and the activation of a series of enzymatic cascades, such as caspases. Between the proton pumps, the V-ATPase seem to exert a central role as pH sensors (197). V-ATPase has a crucial involvement in maintaining the intracellular pH gradients in normal condition, through a continuous H+ transferring from the cytosol to the acidic vesicles. This activity, on the one hand, avoids acidification of the cytosol, on the other hand, maintain an acidic pH within the internal vacuoles (198). This background may explain a mechanism that pathogenic bacteria use to evade internal killing through alterations in pH homeostasis. The pathogenetic mechanism of HP, in fact, include a pH modulator such as urease. However, to date, it is not entirely clear whether the acidification derangement is aimed at targeting the pH regulation of the bacterium itself or the pH of the gastric environment where the bacterium lives. By analogy with the M. tuberculosis, we know that the normal phagosomal acidification fails to develop because the bacterium interferes with internal vesicles maturation. However, we don't focus enough on both the mycobacterial effectors that inhibit vacuoles maturation and the real mechanism of action.
Similarities Between HP and Cancer Cells
This, on the one hand, supports the use of PPI as a new class of anti-infectious agents without a clear antibiotic activity, on the other hand, raises the interesting hypothesis that microbial agents may behave like tumor cells in their ability to select cells able to survive in the acidic microenvironment. Some analogies between unicellular microorganisms and malignant tumors cells have been provided in the last decade. The most striking evidence of this amoeba-like behavior of cancer cells came from the way tumor cells feed on other cells when starved or placed in low nutrient supply conditions. This was called tumor cannibalism (199, 200). Moreover, a protein and a gene in common between cancer cells and amoebas have been described (TM9SF4) and recalled Tumor Cannibalism Associated Protein 1 (TUCAP-1) (201). That behavior was very similar to the one used by unicellular microorganisms against bacteria (202). Intriguingly, TUCAP-1 has been shown to be involved in tumor acidification by modulating the activity of proton pumps, such as V-ATPases (203). All in all these results led to hypothesize that cancer cells came back to a very primitive condition (atavistic state) where the cells aimed to survive against the other living beings (204).
Moreover, it has been shown as proton pump inhibitors exert a particular antibacterial activity against HP in vitro, and it was clearly pH dependent. In fact, the bactericidal activity specifically against both resting (in the buffer) and growing (in broth) HP was significantly higher at pH 5 as compared to that at pH 7. On the other hand, we know that not only cancer neoplasms are acidic (205) but that both cancer cells and tumors are extremely sensitive to proton pump inhibitors, at pre-clinical and clinical levels (181), and there is a list of proton exchangers inhibitors that have been proven highly effective against cancer (198).
All in all, there are many analogies in general between microbes and cancer cells, but most of all between HP and cancer cells. A question, however, remains open actually. Does HP behave like a cancer cell? Or does the cancer cell that indeed acts the HP?
Conclusion
HP could be seen as a naturally occurring, “acquired normal flora” and immunomodulating bacterium. It is evolutionarily designed for co-existence with rather than extinction of other bacterial flora. HP induced inflammation might, under certain circumstances, stimulate Prostaglandin E2 (PGE-2) which restores the mucous membrane of the stomach to normal homeostasis. This habitat restoration re-populates the mucous adherent phenotype (M) and so re-structures levels of M phenotypes into the already existing epithelial adherent phenotypes (A) In other words, the presence of mucus helps to maintain HP diversity as well as to stabilize the population size of HP in the stomach. Perhaps the presence of mucus can represent a proxy for the M-phenotype distribution as it relates to (i) the provision of L-lactic acid as a growth enhancer (206) and (ii) how HP obtains benefits from the mucosal flora which is necessary for its survival (207). Further, the normal epithelium is a suitable habitat for the A phenotype (208) instead of an epithelium that is undergoing intestinal metaplasia (fragmentized habitat).
Chronic inflammation results in the creation of unfavorable habitat, by altering pH, around normal cells which instigated on their malignant transformation (209). This environmental change leads to (i) selection and/or elicits phenotypic plasticity of human cells to become acidophilic phenotypes (i.e., the “cave fish's principle”) (210) and (ii) promotion of transformed phenotypes' fitness using spite strategy (211). However, HP suppresses acid production. Therefore, could cancer be considered a strategic defense against HP? In other words, could cancer be represented as an aggressive immune response against HP? We believe that it is time to change the monolithic model that misrepresents HP as an inducer of gastric carcinogenesis as a purpose.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Mercedes Castresana Foundation, Vitoria, Spain; The Association for Proton Cancer Research and Treatment (APCRT), Madrid, Spain (SH); and American BioSciences, Inc., NY, USA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge Robert A. Gatenby, Mark C. Lloyd, Charles E. Bailey, Carlo C. Maley for their comments and discussion. Also, the authors would like to thank the Deanship of Scientific Research, and Research Center, College of pharmacy King Saud University, Riyadh, Kingdom of Saudi Arabia.
References
1. Kidd M, Modlin IM. A century of Helicobacter pylori: paradigms lost-paradigms regained. Digestion (1998) 59:1–15. doi: 10.1159/000007461
2. Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. (1987) 82:192–9.
3. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet (1983) 1:1273–5.
4. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet (1984) 1:1311–5.
5. Waghorn DJ. Campylobacter pyloridis: a new organism to explain an old problem? Postgrad Med J. (1987) 63:533–7.
6. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature (1997) 388:539–47. doi: 10.1038/41483
7. Harguindey S, Pedraz JL, García Cañero R, Pérez de Diego J, Cragoe EJ. Hydrogen ion-dependent oncogenesis and parallel new avenues to cancer prevention and treatment using a H(+)-mediated unifying approach: pH-related and pH-unrelated mechanisms. Crit Rev Oncog. (1995) 6:1–33.
8. Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science (1999) 284:1328–33. doi: 10.1126/science.284.5418.1328
9. Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature (2007) 445:915–8. doi: 10.1038/nature05562
10. Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. (2009) 119:2475–87. doi: 10.1172/JCI38605
11. Latifi-Navid S, Ghorashi SA, Siavoshi F, Linz B, Massarrat S, Khegay T, et al. Ethnic and geographic differentiation of Helicobacter pylori within Iran. PLoS ONE (2010) 5:e9645. doi: 10.1371/journal.pone.0009645
12. Blaser MJ. Helicobacter pylori eradication and its implications for the future. Aliment Pharmacol Ther. (1997) 11(Suppl. 1):103–7.
13. Mitchell A, Silva TMJ, Barrett LJ, Lima AAM, Guerrant RL. Age-specific Helicobacter pylori seropositivity rates of children in an impoverished urban area of northeast Brazil. J Clin Microbiol. (2003) 41:1326–8. doi: 10.1128/JCM.41.3.1326-1328.2003
14. Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer (2002) 2:28–37. doi: 10.1038/nrc703
15. Yakoob J, Jafri W, Abid S. Helicobacter pylori infection and micronutrient deficiencies. World J Gastroenterol. (2003) 9:2137–9. doi: 10.3748/wjg.v9.i10.2137
16. Klupinska G, Wiśniewska-Jarosinska M, Harasiuk A, Chojnacki C, Stec-Michalska K, Błasiak J, et al. Nocturnal secretion of melatonin in patients with upper digestive tract disorders. J Physiol Pharmacol. (2006) 57(Suppl. 5):41–50.
17. Tan D-X, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. (2007) 42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x
18. Sobala GM, Schorah CJ, Shires S, Lynch DA, Gallacher B, Dixon MF, et al. Effect of eradication of Helicobacter pylori on gastric juice ascorbic acid concentrations. Gut (1993) 34:1038–41. doi: 10.1136/gut.34.8.1038
19. Correa P, Malcom G, Schmidt B, Fontham E, Ruiz B, Bravo JC, et al. Review article: antioxidant micronutrients and gastric cancer. Aliment Pharmacol Ther. (1998) 12(Suppl. 1):73–82. doi: 10.1111/j.1365-2036.1998.00006.x
20. Correa P, Fox J, Fontham E, Ruiz B, Lin YP, Zavala D, et al. Helicobacter pylori and gastric carcinoma. Serum antibody prevalence in populations with contrasting cancer risks. Cancer (1990) 66:2569–74.
21. Bashir AHH, Yousif SM, Mahmoud MOA. Clinicoepidemiological study in Sudanese patients : prevalence and effect of eradicative triple therapy on extra digestive Helicobacter pylori skin manifestations , EdHpSm. Clin Rev Opin. (2011) 3:14–19.
22. Migneco A, Ojetti V, Specchia L, Franceschi F, Candelli M, Mettimano M, et al. Eradication of Helicobacter pylori infection improves blood pressure values in patients affected by hypertension. Helicobacter (2003) 8:585–9. doi: 10.1111/j.1523-5378.2003.00180.x
23. Strachan DP. Non-gastrointestinal consequences of Helicobacter pylori infection. Br Med Bull. (1998) 54:87–93. doi: 10.1093/oxfordjournals.bmb.a011684
24. Jeon CY, Haan MN, Cheng C, Clayton ER, Mayeda ER, Miller JW, et al. Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care (2012) 35:520–5. doi: 10.2337/dc11-1043
25. Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. (2004) 113:321–33. doi: 10.1172/JCI20925
26. Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. (2008) 198:553–60. doi: 10.1086/590158
27. Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest. (2011) 121:3088–93. doi: 10.1172/JCI45041
28. Perry S, de Jong BC, Solnick JV, de la Luz Sanchez M, Yang S, Lin PL, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE (2010) 5:e8804. doi: 10.1371/journal.pone.0008804
29. Sipponen P, Hyvärinen H. Role of Helicobacter pylori in the pathogenesis of gastritis, peptic ulcer and gastric cancer. Scand J Gastroenterol Suppl. (1993) 196:3–6.
30. de Jong D, van der Hulst RW, Pals G, van Dijk WC, van der Ende A, Tytgat GN, et al. Gastric non-Hodgkin lymphomas of mucosa-associated lymphoid tissue are not associated with more aggressive Helicobacter pylori strains as identified by CagA. Am J Clin Pathol. (1996) 106:670–5. doi: 10.1093/ajcp/106.5.670
31. Blaser MJ. Intrastrain differences in Helicobacter pylori: a key question in mucosal damage? Ann Med (1995) 27:559–63.
32. Blaser MJ. Heterogeneity of Helicobacter pylori. Eur J Gastroenterol Hepatol. (1997) 9(Suppl. 1):S3–6; discussion: S6–7.
33. Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res. (2008) 1:329–38. doi: 10.1158/1940-6207.CAPR-08-0109
34. Correa P, Fontham ET, Bravo JC, Bravo LE, Ruiz B, Zarama G, et al. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-helicobacter pylori therapy. J Natl Cancer Inst. (2000) 92:1881–8. doi: 10.1093/jnci/92.23.1881
35. Wong BCY, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA (2004) 291:187–94. doi: 10.1001/jama.291.2.187
36. Wu CY, Kuo KN, Wu MS, Chen YJ, Wang CB, Lin JT. Early Helicobacter pylori eradication decreases risk of gastric cancer in patients with peptic ulcer disease. Gastroenterology (2009) 137:1641–8.e1–2. doi: 10.1053/j.gastro.2009.07.060
37. Blaser MJ. Disappearing microbiota: Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res. (2008) 1:308–11. doi: 10.1158/1940-6207.CAPR-08-0170
38. Kirschner DE, Blaser MJ. The dynamics of Helicobacter pylori infection of the human stomach. J Theor Biol. (1995) 176:281–90. doi: 10.1006/jtbi.1995.0198
39. Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, et al. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci USA. (2009) 106:14321–6. doi: 10.1073/pnas.0903438106
40. Fan X, Gunasena H, Cheng Z, Espejo R, Crowe SE, Ernst PB, et al. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J Immunol. (2000) 165:1918–24. doi: 10.4049/jimmunol.165.4.1918
41. Mortazavi M, Akbarzadeh A, Farhangi A, Mehrabi M, Hosseinian Z, Mokhtari MJ, et al. Immunosuppressive proteins isolated from spiral and coccoid cytoplasmic solutions of Helicobacter pylori. Pakistan J Biol Sci. (2011) 14:128–32. doi: 10.3923/pjbs.2011.128.132
42. Figura N, Valassina M. Helicobacter pylori determinants of pathogenicity. J Chemother. (1999) 11:591–600. doi: 10.1179/joc.1999.11.6.591
43. Ghiara P, Marchetti M, Blaser MJ, Tummuru MK, Cover TL, Segal ED, et al. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect Immun. (1995) 63:4154–60.
44. Mobley HL, Hu LT, Foxal PA. Helicobacter pylori urease: properties and role in pathogenesis. Scand J Gastroenterol Suppl. (1991) 187:39–46. doi: 10.3109/00365529109098223
45. Mobley HL, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev. (1995) 59:451–80.
46. Weeks DL, Eskandari S, Scott DR, Sachs G. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science (2000) 287:482–5. doi: 10.1126/science.287.5452.482
47. Scott DR, Marcus EA, Weeks DL, Lee A, Melchers K, Sachs G. Expression of the Helicobacter pylori ureI gene is required for acidic pH activation of cytoplasmic urease. Infect Immun. (2000) 68:470–7. doi: 10.1128/IAI.68.2.470-477.2000
48. McGee DJ, Radcliff FJ, Mendz GL, Ferrero RL, Mobley HL. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J Bacteriol. (1999) 181:7314–22.
49. Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites (2017) 7:E56. doi: 10.3390/metabo7040056
50. Ferreira-Martins D, McCormick SD, Campos A, Lopes-Marques M, Osório H, Coimbra J, et al. A cytosolic carbonic anhydrase molecular switch occurs in the gills of metamorphic sea lamprey. Sci Rep. (2016) 6:33954. doi: 10.1038/srep33954
51. Dodgson SJ, Forster RE, Storey BT, Mela L. Mitochondrial carbonic anhydrase. Proc Natl Acad Sci USA. (1980) 77:5562–66.
52. Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. (2008) 7:168–81. doi: 10.1038/nrd2467
53. Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des. (2008) 14:622–30. doi: 10.2174/138161208783877875
54. Buzás GM, Supuran CT. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In memoriam Ioan Puşcaş (1932-2015). J Enzyme Inhib Med Chem. (2016) 31:527–33. doi: 10.3109/14756366.2015.1051042
55. Monteiro MA, Chan KH, Rasko DA, Taylor DE, Zheng PY, Appelmelk BJ, et al. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. Molecular mimicry between H. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. J Biol Chem. (1998) 273:11533–43.
56. Aspinall GO, Monteiro MA. Lipopolysaccharides of Helicobacter pylori strains P466 and MO19: structures of the O antigen and core oligosaccharide regions. Biochemistry (1996) 35:2498–504. doi: 10.1021/bi951853k
57. Edwards NJ, Monteiro MA, Faller G, Walsh EJ, Moran AP, Roberts IS, et al. Lewis X structures in the O antigen side-chain promote adhesion of Helicobacter pylori to the gastric epithelium. Mol Microbiol. (2000) 35:1530–9. doi: 10.1046/j.1365-2958.2000.01823.x
58. Hildebrandt E, McGee DJ. Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol. (2009) 9:258. doi: 10.1186/1471-2180-9–258
59. Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science (1993) 262:1892–5.
60. Wirth HP, Yang M, Karita M, Blaser MJ. Expression of the human cell surface glycoconjugates Lewis x and Lewis y by Helicobacter pylori isolates is related to cagA status. Infect Immun. (1996) 64:4598–605.
61. Monteiro MA, Appelmelk BJ, Rasko DA, Moran AP, Hynes SO, MacLean LL, et al. Lipopolysaccharide structures of Helicobacter pylori genomic strains 26695 and J99, mouse model H. pylori Sydney strain, H. pylori P466 carrying sialyl Lewis X, and H. pylori UA915 expressing Lewis B classification of H. pylori lipopolysaccharides into glycotype families. Eur J Biochem. (2000) 267:305–20. doi: 10.1046/j.1432-1327.2000.01007.x
62. Lozniewski A, Haristoy X, Rasko DA, Hatier R, Plénat F, Taylor DE, et al. Influence of Lewis antigen expression by Helicobacter pylori on bacterial internalization by gastric epithelial cells. Infect Immun. (2003) 71:2902–6. doi: 10.1128/IAI.71.5.2902-2906.2003
63. Schoep TD, Fulurija A, Good F, Lu W, Himbeck RP, Schwan C, et al. Surface properties of Helicobacter pylori urease complex are essential for persistence. PLoS ONE (2010) 5:e15042. doi: 10.1371/journal.pone.0015042
64. Makristathis A, Rokita E, Pasching E, Apfalter P, Willinger B, Rotter ML, et al. Urease prevents adherence of Helicobacter pylori to Kato III gastric epithelial cells. J Infect Dis. (2001) 184:439–45. doi: 10.1086/322776
65. Appelmelk BJ, Simoons-Smit I, Negrini R, Moran AP, Aspinall GO, Forte JG, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun. (1996) 64:2031–40.
66. Perez-Perez GI, Peek RM, Atherton JC, Blaser MJ, Cover TL. Detection of anti-VacA antibody responses in serum and gastric juice samples using type s1/m1 and s2/m2 Helicobacter pylori VacA antigens. Clin Diagn Lab Immunol. (1999) 6:489–93.
67. Miehlke S, Kirsch C, Agha-Amiri K, Günther T, Lehn N, Malfertheiner P, et al. The Helicobacter pylori vacA s1, m1 genotype and cagA is associated with gastric carcinoma in Germany. Int J Cancer (2000) 87:322–7. doi: 10.1002/1097-0215(20000801)87:3<322::AID-IJC3>3.0.CO;2-M
68. Atherton JC, Cao P, Peek RM, Tummuru MKR, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. (1995) 270:17771–7.
69. Atherton JC, Peek RM, Tham KT, Cover TL, Blaser MJ. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology (1997) 112:92–9. doi: 10.1016/S0016-5085(97)70223-3
70. Jain P, Luo ZQ, Blanke SR. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci USA. (2011) 108:16032–7. doi: 10.1073/pnas.1105175108
71. Cover TL, Krishna US, Israel DA, Peek RM. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. (2003) 63:951–7.
72. Satin B, Norais N, Telford J, Rappuoli R, Murgia M, Montecucco C, et al. Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J Biol Chem. (1997) 272:25022–8. doi: 10.1074/jbc.272.40.25022
73. Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer (2010) 10:403–14. doi: 10.1038/nrc2857
74. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature (1999) 397:176–80. doi: 10.1038/16495
75. Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. (2008) 10:1573–81. doi: 10.1111/j.1462-5822.2008.01156.x
76. Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. (2005) 102:10646–51. doi: 10.1073/pnas.0504927102
77. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. (2008) 105:1003–8. doi: 10.1073/pnas.0711183105
78. Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe (2007) 2:250–63. doi: 10.1016/j.chom.2007.09.005
79. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
80. Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet (1993) 342:571–4. doi: 10.1016/0140-6736(93)91408-E
81. Lee SK, Lee YC, Chung JB, Chon CY, Moon YM, Kang JK, et al. Low grade gastric mucosa associated lymphoid tissue lymphoma: treatment strategies based on 10 year follow-up. World J Gastroenterol. (2004) 10:223–226. doi: 10.3748/wjg.v10.i2.223
82. Salam I, Durai D, Murphy JK, Sundaram B. Regression of primary high-grade gastric B-cell lymphoma following Helicobacter pylori eradication. Eur J Gastroenterol Hepatol. (2001) 13:1375–8. doi: 10.1097/00042737-200111000-00018
83. Montalban C, Santon A, Boixeda D, Bellas C. Regression of gastric high grade mucosa associated lymphoid tissue (MALT) lymphoma after Helicobacter pylori eradication. Gut (2001) 49:584–7. doi: 10.1136/gut.49.4.584
84. Gretschel S, Hünerbein M, Foss HD, Krause M, Schlag PM. Regression of high-grade gastric B-cell lymphoma after eradication of Helicobacter pylori. Endoscopy (2001) 33:805–807.
85. Blaser MJ, Crabtree JE. CagA and the outcome of Helicobacter pylori infection. Am J Clin Pathol. (1996) 106:565–7. doi: 10.1093/ajcp/106.5.565
86. Atherton JC, Tham KT, Peek RM, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis. (1996) 174:552–6. doi: 10.1093/infdis/174.3.552
87. Sharma SA, Tummuru MK, Miller GG, Blaser MJ. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. (1995) 63:1681–87.
88. Keates S, Keates AC, Warny M, Peek RM, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. (1999) 163:5552–9.
89. Glocker E, Lange C, Covacci A, Bereswill S, Kist M, Pahl HL. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect Immun. (1998) 66:2346–8.
90. Saha A, Hammond CE, Beeson C, Peek RM, Smolka AJ. Helicobacter pylori represses proton pump expression and inhibits acid secretion in human gastric mucosa. Gut (2010) 59:874–81. doi: 10.1136/gut.2009.194795
91. Smolka AJ, Backert S. How Helicobacter pylori infection controls gastric acid secretion. J Gastroenterol. (2012) 47:609–18. doi: 10.1007/s00535-012-0592-1
92. Peek RM, Moss SF, Tham KT, Pérez-Pérez GI, Wang S, Miller GG, et al. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. (1997) 89:863–8.
93. Rokkas T, Ladas S, Liatsos C, Petridou E, Papatheodorou G, Theocharis S, et al. Relationship of Helicobacter pylori CagA status to gastric cell proliferation and apoptosis. Dig Dis Sci. (1999) 44:487–93. doi: 10.1023/A:1026636803101
94. Levi S, Beardshall K, Haddad G, Playford R, Ghosh P, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet (1989) 1:1167–8. doi: 10.1016/S0140-6736(89)92752-9
95. Galdós Ayala J, Basterra Romo G, Martínez Blázquez C, Díaz de Otazu R, Alvarez Rubio M, Katin M, et al. Duodenal carcinoid tumor and esophageal carcinoma associated with a high chronic intake of sodium bicarbonate. Possible pathophysiological mechanisms. Oncology (2003) 26:123–7.
96. Moss SF, Sordillo EM, Abdalla AM, Makarov V, Hanzely Z, Perez-Perez GI, et al. Increased gastric epithelial cell apoptosis associated with colonization with cagA + Helicobacter pylori strains. Cancer Res. (2001) 61:1406–11.
97. Shirin H, Sordillo EM, Oh SH, Yamamoto H, Delohery T, Weinstein IB, et al. Helicobacter pylori inhibits the G1 to S transition in AGS gastric epithelial cells. Cancer Res. (1999) 59:2277–81.
98. Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, et al. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci USA. (1998) 95:3925–30. doi: 10.1073/pnas.95.7.3925
99. Ito M, Haruma K, Kaya S, Kamada T, Kim S, Sasaki A, et al. Role of anti-parietal cell antibody in Helicobacter pylori-associated atrophic gastritis: evaluation in a country of high prevalence of atrophic gastritis. Scand J Gastroenterol. (2002) 37:287–93. doi: 10.1080/003655202317284183
100. Mahdi BM, Hassan RM, Maki B, Ghanim N, Ghalog L. Atrophic gastritis by Helicobacter pylori and antiparietal antibodies. Rawal Med J. (2011) 36:74–78.
101. Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA. (1999) 96:12778–83.
102. Kraus C, Liehr T, Hülsken J, Behrens J, Birchmeier W, Grzeschik KH, et al. Localization of the human beta-catenin gene (CTNNB1) to 3p21: a region implicated in tumor development. Genomics (1994) 23:272–4.
103. Tsukashita S, Kushima R, Bamba M, Nakamura E, Mukaisho K, Sugihara H, et al. Beta-catenin expression in intramucosal neoplastic lesions of the stomach. Oncology (2003) 64:251–8. doi: 10.1159/000069310
104. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. (2011) 39:D945–50. doi: 10.1093/nar/gkq929
105. Hassanein AM, Glanz SM, Kessler HP, Eskin TA, Liu C. β-catenin is expressed aberrantly in tumors expressing shadow cells. Pilomatricoma, craniopharyngioma, and calcifying odontogenic cyst. Am J Clin Pathol. (2003) 120:732–6. doi: 10.1309/EALEG7LD6W7167PX
106. Saldanha G, Ghura V, Potter L, Fletcher A. Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation. Br J Dermatol. (2004) 151:157–64. doi: 10.1111/j.1365-2133.2004.06048.x
107. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol. (2005) 23:7951–7. doi: 10.1200/JCO.2005.01.5479
108. Kypta RM, Waxman J. Wnt/β-catenin signalling in prostate cancer. Nat Rev Urol. (2012) 9:418–28. doi: 10.1038/nrurol.2012.116
109. Kurashima Y, Murata-Kamiya N, Kikuchi K, Higashi H, Azuma T, Kondo S, et al. Deregulation of beta-catenin signal by Helicobacter pylori CagA requires the CagA-multimerization sequence. Int J Cancer (2008) 122:823–31. doi: 10.1002/ijc.23190
110. El-Etr SH, Mueller A, Tompkins LS, Falkow S, Merrell DS. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J Infect Dis. (2004) 190:1516–23. doi: 10.1086/424526
111. Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene (2007) 26:4617–26. doi: 10.1038/sj.onc.1210251
112. Hung PP, Marks CL, Tardrew PL. The biosynthesis and metabolism of erythromycins by streptomyces erythreus. J Biol Chem. (1965) 240:1322–6.
113. Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. (2007) 117:2051–8. doi: 10.1172/JCI32278
114. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene (2006) 366:2–16. doi: 10.1016/j.gene.2005.10.018
115. Crabtree JE, Court M, Aboshkiwa MA, Jeremy AHT, Dixon MF, Robinson PA. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J Pathol. (2004) 202:197–207. doi: 10.1002/path.1498
116. Ernst PB, Crowe SE, Reyes VE. How does Helicobacter pylori cause mucosal damage? The inflammatory response. Gastroenterology (1997) 113:S35–42; discussion: S50. doi: 10.1016/S0016-5085(97)80009-1
117. Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology (2009) 136:1297–307.e1-3. doi: 10.1053/j.gastro.2008.12.059
118. Tabassam FH, Graham DY, Yamaoka Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol. (2009) 11:70–82. doi: 10.1111/j.1462-5822.2008.01237.x
119. Sharma S V, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7:169–81. doi: 10.1038/nrc2088
120. Feldman RA. Review article: would eradication of Helicobacter pylori infection reduce the risk of gastric cancer? Aliment Pharmacol Ther. (2001) 15(Suppl. 1):2–5. doi: 10.1046/j.1365-2036.2001.00109.x
121. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process–First American Cancer Society Award Lecture on cancer epidemiology and prevention. Cancer Res. (1992) 52:6735–40.
122. Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. (2006) 72:1605–21. doi: 10.1016/j.bcp.2006.06.029
123. Pomorski T, Meyer TF, Naumann M. Helicobacter pylori-induced prostaglandin E(2) synthesis involves activation of cytosolic phospholipase A(2) in epithelial cells. J Biol Chem. (2001) 276:804–10. doi: 10.1074/jbc.M003819200
124. Nardone G, Holicky EL, Uhl JR, Sabatino L, Staibano S, Rocco A, et al. In vivo and in vitro studies of cytosolic phospholipase A2 expression in Helicobacter pylori infection. Infect Immun. (2001) 69:5857–63. doi: 10.1128/IAI.69.9.5857-5863.2001
125. Ristimäki A, Honkanen N, Jänkälä H, Sipponen P, Härkönen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. (1997) 57:1276–80.
126. Lin CC, Hsiao LD, Chien CS, Lee CW, Hsieh JT, Yang CM. Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression in human tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-kappaB. Cell Signal. (2004) 16:597–607. doi: 10.1016/j.cellsig.2003.10.002
127. Ke J, Long X, Liu Y, Zhang YF, Li J, Fang W, et al. Role of NF-kappaB in TNF-alpha-induced COX-2 expression in synovial fibroblasts from human TMJ. J Dent Res. (2007) 86:363–7. doi: 10.1177/154405910708600412
128. Barrios-Rodiles M, Chadee K. Novel regulation of cyclooxygenase-2 expression and prostaglandin E2 production by IFN-gamma in human macrophages. J Immunol. (1998) 161:2441–8.
129. Lee CS, Shin YJ, Won C, Lee YS, Park CG, Ye SK, et al. Simvastatin acts as an inhibitor of interferon gamma-induced cycloxygenase-2 expression in human THP-1 cells, but not in murine RAW264.7 cells. Biocell (2009) 33:107–14.
130. Mifflin RC, Saada JI, Di Mari JF, Adegboyega PA, Valentich JD, Powell DW. Regulation of COX-2 expression in human intestinal myofibroblasts: mechanisms of IL-1-mediated induction. Am J Physiol Cell Physiol. (2002) 282:C824–34. doi: 10.1152/ajpcell.00388.2001
131. Tsuzaki M, Guyton G, Garrett W, Archambault JM, Herzog W, Almekinders L, et al. IL-1 beta induces COX2, MMP-1,−3 and−13, ADAMTS-4, IL-1 beta and IL-6 in human tendon cells. J Orthop Res. (2003) 21:256–64. doi: 10.1016/S0736-0266(02)00141-9
132. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology (1994) 107:1183–8. doi: 10.1016/0016-5085(94)90246-1
133. Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, et al. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. (2001) 276:44856–64. doi: 10.1074/jbc.M105381200
134. Su B, Ceponis PJM, Lebel S, Huynh H, Sherman PM. Helicobacter pylori activates toll-like receptor 4 expression in gastrointestinal epithelial cells. Infect Immun. (2003) 71:3496–502. doi: 10.1128/IAI.71.6.3496-3502.2003
135. Tang CL, Hao B, Zhang GX, Shi RH, Cheng WF. Helicobacter pylori tumor necrosis factor-α inducing protein promotes cytokine expression via nuclear factor-κB. World J Gastroenterol. (2013) 19:399–403. doi: 10.3748/wjg.v19.i3.399
136. Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, et al. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol. (2000) 157:729–35. doi: 10.1016/S0002-9440(10)64586-5
137. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell (1995) 83:493–501. doi: 10.1016/0092-8674(95)90127-2
138. Fukuda T, Kimura S, Arakawa T, Kobayashi K. Possible role of leukotrienes in gastritis associated with Campylobacter pylori. J Clin Gastroenterol. (1990) 12(Suppl. 1):S131–4. doi: 10.1097/00004836-199001001-00022
139. Kimura S, Fukuda T, Arakawa T, Kobayashi K. [Leukotriene levels of Helicobacter pylori-infected gastric mucosa]. Nihon Rinsho (1993) 51:3159–62.
140. Venerito M, Kuester D, Harms C, Schubert D, Wex T, Malfertheiner P. Upregulation of leukotriene receptors in gastric cancer. Cancers (2011) 3:3156–68. doi: 10.3390/cancers3033156
141. Venerito M, Kuester D, Wex T, Roessner A, Malfertheiner P, Treiber G. The long-term effect of Helicobacter pylori eradication on COX-1/2, 5-LOX and leukotriene receptors in patients with a risk gastritis phenotype–a link to gastric carcinogenesis. Cancer Lett. (2008) 270:218–28. doi: 10.1016/j.canlet.2008.05.015
142. Takashima M, Furuta T, Hanai H, Sugimura H, Kaneko E. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut (2001) 48:765–73. doi: 10.1136/gut.48.6.765
143. Beales IL, Calam J. Interleukin 1 beta and tumour necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut (1998) 42:227–34. doi: 10.1136/gut.42.2.227
144. El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature (2000) 404:398–402. doi: 10.1038/35006081
145. Crabtree JE, Shallcross TM, Heatley RV, Wyatt JI. Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut (1991) 32:1473–7. doi: 10.1136/gut.32.12.1473
146. El-Omar EM. The importance of interleukin 1beta in Helicobacter pylori associated disease. Gut (2001) 48:743–7. doi: 10.1136/gut.48.6.743
147. Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. (2006) 7:956–60. doi: 10.1038/sj.embor.7400812
148. Björkholm B, Sjölund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci USA. (2001) 98:14607–12. doi: 10.1073/pnas.241517298
149. Anadón A, Reeve-johnson L. Macrolide antibiotics, drug interactions and microsomal enzymes: implications for veterinary medicine. Res Vet Sci. (1999) 66:197–203.
150. Theron AJ, Feldman C, Anderson R. Investigation of the anti-inflammatory and membrane-stabilizing potential of spiramycin in vitro. J Antimicrob Chemother. (2000) 46:269–71. doi: 10.1093/jac/46.2.269
151. Anderson R, Theron AJ, Feldman C. Membrane-stabilizing, anti-inflammatory interactions of macrolides with human neutrophils. Inflammation (1996) 20:693–705. doi: 10.1007/BF01488805
152. Kohyama T, Takizawa H, Kawasaki S, Akiyama N, Sato M, Ito K. Fourteen-member macrolides inhibit interleukin-8 release by human eosinophils from atopic donors. Antimicrob Agents Chemother. (1999) 43:907–11. doi: 10.1128/AAC.43.4.907
153. Kikuchi T, Hagiwara K, Honda Y, Gomi K, Kobayashi T, Takahashi H, et al. Clarithromycin suppresses lipopolysaccharide-induced interleukin-8 production by human monocytes through AP-1 and NF-kappa B transcription factors. J Antimicrob Chemother. (2002) 49:745–55. doi: 10.1093/jac/dkf008
154. Perna F, Zullo A, Ricci C, Hassan C, Morini S, Vaira D. Levofloxacin-based triple therapy for Helicobacter pylori re-treatment: role of bacterial resistance. Dig Liver Dis. (2007) 39:1001–5. doi: 10.1016/j.dld.2007.06.016
155. Hsu PI, Wu DC, Chen A, Peng NJ, Tseng HH, Tsay FW, et al. Quadruple rescue therapy for Helicobacter pylori infection after two treatment failures. Eur J Clin Invest. (2008) 38:404–9. doi: 10.1111/j.1365-2362.2008.01951.x
156. Burkhardt O, Köhnlein T, Pap T, Welte T. Recurrent tendinitis after treatment with two different fluoroquinolones. Infect Dis. (2004) 36:315–6. doi: 10.1080/00365540410019390
157. Durey A, Baek YS, Park JS, Lee K, Ryu JS, Lee JS, et al. Levofloxacin-induced Achilles tendinitis in a young adult in the absence of predisposing conditions. Yonsei Med J. (2010) 51:454–6. doi: 10.3349/ymj.2010.51.3.454
158. Gold L, Igra H. Levofloxacin-induced tendon rupture: a case report and review of the literature. J Am Board Fam Pract. (2003) 16:458–60. doi: 10.3122/jabfm.16.5.458
159. Alfarouk KO, Shayoub MEA, Muddathir AK, Elhassan amal O, Bashir AHH. Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers (2011) 3:3002–17. doi: 10.3390/cancers3033002
160. Kühler TC, Fryklund J, Bergman NA, Weilitz J, Lee A, Larsson H. Structure-activity relationship of omeprazole and analogues as Helicobacter pylori urease inhibitors. J Med Chem. (1995) 38:4906–16.
161. Ormeci A, Emrence Z, Baran B, Soyer OM, Gokturk S, Evirgen S, et al. Can Helicobacter pylori be eradicated with high-dose proton pump inhibitor in extensive metabolizers with the CYP2C19 genotypic polymorphism? Eur Rev Med Pharmacol Sci. (2016) 20:1795–7.
162. Shinozaki S, Nomoto H, Kondo Y, Sakamoto H, Hayashi Y, Yamamoto H, et al. Comparison of vonoprazan and proton pump inhibitors for eradication of Helicobacter pylori. Kaohsiung J Med Sci. (2016) 32:255–60. doi: 10.1016/j.kjms.2016.04.009
163. Higuchi K, Takeuchi T, Uedo N, Takeuchi Y, Naito Y, Yagi N, et al. Efficacy and safety of 1-week Helicobacter pylori eradication therapy and 7-week rebamipide treatment after endoscopic submucosal dissection of early gastric cancer in comparison with 8-week PPI standard treatment: a randomized, controlled, prospective, multicenter study. Gastric Cancer (2015) 18:612–7. doi: 10.1007/s10120-014-0404-9
164. Huynh KK, Grinstein S. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev. (2007) 71:452–62. doi: 10.1128/MMBR.00003-07
165. Rybniker J, Vocat A, Sala C, Busso P, Pojer F, Benjak A, et al. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat Commun. (2015) 6:7659. doi: 10.1038/ncomms8659
166. Puiac S, Negrea A, Richter-Dahlfors A, Plant L, Rhen M. Omeprazole antagonizes virulence and inflammation in Salmonella enterica-infected RAW264.7 cells. Antimicrob Agents Chemother. (2009) 53:2402–9. doi: 10.1128/AAC.01483-08
167. Kaur S, Vaishnavi C, Prasad KK, Ray P, Kochhar R. Comparative role of antibiotic and proton pump inhibitor in experimental Clostridium difficile infection in mice. Microbiol Immunol. (2007) 51:1209–14. doi: 10.1111/j.1348-0421.2007.tb04016.x
168. Lu J, Chen Z, Wu Y, Zhang M, Ding JP, Cederlund E, et al. An ATPase inhibitory peptide with antibacterial and ion current effects. Biochem Biophys Res Commun. (2014) 446:519–22. doi: 10.1016/j.bbrc.2014.02.138
169. Reyes-Vivas H, de la Mora-de la Mora I, Castillo-Villanueva A, Yepez-Mulia L, Hernandez-Alcantara G, Figueroa-Salazar R, et al. Giardial triosephosphate isomerase as possible target of the cytotoxic effect of omeprazole in Giardia lamblia. Antimicrob Agents Chemother. (2014) 58:7072–82. doi: 10.1128/AAC.02900-14
170. Aksoy Gökmen A, Girginkardeşler N, Kilimcioglu AA, Sirin MC, Özbilgin A. [In vitro susceptibility of Trichomonas vaginalis to metronidazole, ornidazole and proton pump inhibitors pantoprazole and esomeprazole]. Mikrobiyol Bul. (2016) 50:133–9. doi: 10.5578/mb.10241
171. Kochar DK, Gupta V, Kochar A, Acharya J, Middha S, Sirohi P, et al. Comparison of quinine and rabeprazole with quinine monotherapy in the treatment of uncomplicated falciparum malaria. J Vector Borne Dis. (2010) 47:140–4.
172. Martinez-Munoz GA, Kane P. Vacuolar and plasma membrane proton pumps collaborate to achieve cytosolic pH homeostasis in yeast. J Biol Chem. (2008) 283:20309–19. doi: 10.1074/jbc.M710470200
173. Clarke M, Köhler J, Arana Q, Liu T, Heuser J, Gerisch G. Dynamics of the vacuolar H(+)-ATPase in the contractile vacuole complex and the endosomal pathway of Dictyostelium cells. J Cell Sci. (2002) 115:2893–905.
174. Padh H, Lavasa M, Steck TL. Endosomes are acidified by association with discrete proton-pumping vacuoles in Dictyostelium. J Biol Chem. (1991) 266:5514–20.
175. Iwahi T, Satoh H, Nakao M, Iwasaki T, Yamazaki T, Kubo K, et al. Lansoprazole, a novel benzimidazole proton pump inhibitor, and its related compounds have selective activity against Helicobacter pylori. Antimicrob Agents Chemother. (1991) 35:490–6. doi: 10.1128/AAC.35.3.490
176. Hirai M, Azuma T, Ito S, Kato T, Kohli Y. A proton pump inhibitor, E3810, has antibacterial activity through binding to Helicobacter pylori. J Gastroenterol. (1995) 30:461–4. doi: 10.1007/BF02347561
177. Sjøstrøm JE, Kühler T, Larsson H. Basis for the selective antibacterial activity in vitro of proton pump inhibitors against Helicobacter spp. Antimicrob Agents Chemother. (1997) 41:1797–801.
178. Gatta L, Perna F, Figura N, Ricci C, Holton J, D'Anna L, et al. Antimicrobial activity of esomeprazole versus omeprazole against Helicobacter pylori. J Antimicrob Chemother. (2003) 51:439–42. doi: 10.1093/jac/dkg085
179. Sharara AI. Rabeprazole: the role of proton pump inhibitors in Helicobacter pylori eradication. Expert Rev Anti Infect Ther. (2005) 3:863–70. doi: 10.1586/14787210.3.6.863
180. Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology (2010) 139:239–48. doi: 10.1053/j.gastro.2010.03.036
181. Fais S. Evidence-based support for the use of proton pump inhibitors in cancer therapy. J Transl Med. (2015) 13:368. doi: 10.1186/s12967-015-0735-2
182. Fais S. A nonmainstream approach against cancer. J Enzyme Inhib Med Chem. (2016) 31:882–9. doi: 10.3109/14756366.2016
183. Wang BY, Zhang J, Wang JL, Sun S, Wang ZH, Wang LP, et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J Exp Clin Cancer Res. (2015) 34:85. doi: 10.1186/s13046-015-0194-x
184. Walsh M, Fais S, Spugnini EP, Harguindey S, Abu Izneid T, Scacco L, et al. Proton pump inhibitors for the treatment of cancer in companion animals. J Exp Clin Cancer Res. (2015) 34:93. doi: 10.1186/s13046-015-0204-z
185. Lee BH, Kim N. Quadruple or triple therapy to eradicate H pylori. Lancet (2011) 377:877–8. doi: 10.1016/S0140-6736(11)60168-2
186. Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 in Helicobacter pylori- induced gastritis in Mongolian gerbils. Am J Physiol Gastrointest Liver Physiol. (2000) 279:G791–8. doi: 10.1152/ajpgi.2000.279.4.G791
187. Akhter T, Baqai R, Aziz M. Antibacterial effect of NSAIDS on clinical isolates of urinary tract infection and diabetic foot infection. Pak J Pharm Sci. (2010) 23:108–13.
188. Al-Bakri AG, Othman G, Bustanji Y. The assessment of the antibacterial and antifungal activities of aspirin, EDTA and aspirin-EDTA combination and their effectiveness as antibiofilm agents. J Appl Microbiol. (2009) 107:280–6. doi: 10.1111/j.1365-2672.2009.04205.x
189. Bartzatt R, Cirillo SLG, Cirillo JD, Donigan L. Bifunctional constructs of aspirin and ibuprofen (non-steroidal anti-inflammatory drugs; NSAIDs) that express antibacterial and alkylation activities. Biotechnol Appl Biochem. (2003) 37:273–82. doi: 10.1042/BA20020108
190. Umaru T, Nwamba CO, Kolo I NU. Antimicrobial activity of non-steroidal anti-inflammatory drugs with respect to immunological response: Diclofenac sodium as a case study. Afr J Biotechnol. (2009) 8:7332–9.
191. Yoshida N, Sugimoto N, Hirayama F, Nakamura Y, Ichikawa H, Naito Y, et al. Helicobacter pylori infection potentiates aspirin induced gastric mucosal injury in Mongolian gerbils. Gut (2002) 50:594–8. doi: 10.1136/gut.50.5.594
192. Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood (2012) 119:3790–8. doi: 10.1182/blood-2011-10-384826
193. Blaser MJ. Malaria and the natural history of Helicobacter pylori infection. Lancet (1993) 342:551. doi: 10.1016/0140-6736(93)91674-B
194. Conlan JW, KuoLee R, Webb A, Perry MB. Imunosuppression by a corticosteroid fails to exacerbate Helicobacter pylori infection in a mouse model of gastric colonization. Can J Microbiol. (1999) 45:975–80. doi: 10.1139/w99-101
195. Biewer W, Stolte M. [Helicobacter pylori colonization of the gastric mucosa in rheumatic patients]. Zeitschrift für Gastroenterol. (1991) 29:585–9.
196. Fais S, O'Driscoll L, Borras FE, Buzas E, Camussi G, Cappello F, et al. Evidence-based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano (2016) 10:3886–99. doi: 10.1021/acsnano.5b08015
197. Marshansky V. The V-ATPase a2-subunit as a putative endosomal pH-sensor. Biochem Soc Trans. (2007) 35:1092–9. doi: 10.1042/BST0351092
198. Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, et al. Proton channels and exchangers in cancer. Biochim Biophys Acta – Biomembr. (2014) 1848:2715–26. doi: 10.1016/j.bbamem.2014.10.015
199. Lugini L, Lozupone F, Matarrese P, Funaro C, Luciani F, Malorni W, et al. Potent phagocytic activity discriminates metastatic and primary human malignant melanomas: a key role of ezrin. Lab Invest. (2003) 83:1555–67. doi: 10.1097/01.LAB.0000098425.03006.42
200. Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. (2006) 66:3629–38. doi: 10.1158/0008-5472.CAN-05-3204
201. Lozupone F, Perdicchio M, Brambilla D, Borghi M, Meschini S, Barca S, et al. The human homologue of Dictyostelium discoideum phg1A is expressed by human metastatic melanoma cells. EMBO Rep. (2009) 10:1348–54. doi: 10.1038/embor.2009.236
202. Fais S, Fauvarque MO. TM9 and cannibalism: how to learn more about cancer by studying amoebae and invertebrates. Trends Mol Med. (2012) 18:4–5. doi: 10.1016/j.molmed.2011.09.001
203. Lozupone F, Borghi M, Marzoli F, Azzarito T, Matarrese P, Iessi E, et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene (2015) 34:5163–74. doi: 10.1038/onc.2014.437
204. Lozupone F, Fais S. Cancer cell cannibalism: a primeval option to survive. Curr Mol Med. (2015) 15:836–41. doi: 10.2174/1566524015666151026100916
205. Fais S, Venturi G, Gatenby B. Microenvironmental acidosis in carcinogenesis and metastases: new strategies in prevention and therapy. Cancer Metastasis Rev. (2014) 33:1095–108. doi: 10.1007/s10555-014-9531-3
206. Takahashi T, Matsumoto T, Nakamura M, Matsui H, Tsuchimoto K, Yamada H. L-lactic acid secreted from gastric mucosal cells enhances growth of Helicobacter pylori. Helicobacter (2007) 12:532–40. doi: 10.1111/j.1523-5378.2007.00524.x
207. Olson JW, Maier RJ. Molecular hydrogen as an energy source for Helicobacter pylori. Science (2002) 298:1788–90. doi: 10.1126/science.1077123
208. van Amsterdam K, van der Ende A. Nutrients released by gastric epithelial cells enhance Helicobacter pylori growth. Helicobacter (2004) 9:614–21. doi: 10.1111/j.1083-4389.2004.00272.x
209. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
210. Gatenby RA, Gillies RJ, Brown JS. Of cancer and cave fish. Nat Rev Cancer (2011) 11:237–8. doi: 10.1038/nrc3036
Keywords: Helicobacter pylori, gastric cancer, pH, inflammation, pharmacology
Citation: Alfarouk KO, Bashir AHH, Aljarbou AN, Ramadan AM, Muddathir AK, AlHoufie STS, Hifny A, Elhassan GO, Ibrahim ME, Alqahtani SS, AlSharari SD, Supuran CT, Rauch C, Cardone RA, Reshkin SJ, Fais S and Harguindey S (2019) The Possible Role of Helicobacter pylori in Gastric Cancer and Its Management. Front. Oncol. 9:75. doi: 10.3389/fonc.2019.00075
Received: 03 September 2018; Accepted: 28 January 2019;
Published: 22 February 2019.
Edited by:
Tiziana Venesio, Institute for Cancer Research and Treatment (IRCC), ItalyReviewed by:
Amit Gupta, All India Institute of Medical Sciences, Rishikesh, IndiaCristiana Laudi, Istituto di Candiolo (IRCCS), Italy
Copyright © 2019 Alfarouk, Bashir, Aljarbou, Ramadan, Muddathir, AlHoufie, Hifny, Elhassan, Ibrahim, Alqahtani, AlSharari, Supuran, Rauch, Cardone, Reshkin, Fais and Harguindey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalid O. Alfarouk, QWxmYXJvdWtASGFsYS1BbGZhcm91ay5vcmc=; S2hhbGlvbWVyQGdtYWlsLmNvbQ==