 James P. Dugan
James P. Dugan Carrie B. Coleman
Carrie B. Coleman Bradley Haverkos
Bradley Haverkos- 1Division of Hematology, University of Colorado, Aurora, CO, United States
- 2Division of Immunology, University of Colorado, Aurora, CO, United States
Many lymphoproliferative disorders (LPDs) are considered “EBV associated” based on detection of the virus in tumor tissue. EBV drives proliferation of LPDs via expression of the viral latent genes and many pre-clinical and clinical studies have shown EBV-associated LPDs can be treated by exploiting the viral life cycle. After a brief review of EBV virology and the natural life cycle within a host we will discuss the importance of the viral gene programs expressed during specific viral phases, as well as within immunocompetent vs. immunocompromised hosts and corresponding EBV-associated LPDs. We will then review established and emerging treatment approaches for EBV-associated LPDs based on EBV gene expression programs. Patients with EBV-associated LPDs can have a poor performance status, multiple comorbidities, and/or are immunocompromised from organ transplantation, autoimmune disease, or other congenital or acquired immunodeficiency making them poor candidates to receive intensive cytotoxic chemotherapy. With the emergence of EBV-directed therapy there is hope that we can devise more effective therapies that confer milder toxicity.
Introduction
Epstein-Barr Virus (EBV), also called human herpesvirus 4 (HHV-4), is a lymphotropic gamma-herpes virus that infects >90% of adults worldwide (1). EBV is defined by a discrete viral life cycle with primary infection, latency, and lytic reactivation phases (2). There are two peaks of EBV infection as measured by seroconversion, age 2–4 years and 15 years (3). In children the primary infection may go undetected or present as an upper respiratory infection. In adults the symptoms of primary infection can be more severe, leading to a syndrome known as infectious mononucleosis. After primary infection the virus remains dormant in latency with memory B cells serving as the primary reservoir for persistence (4). For the vast majority of individuals latent EBV infection does not seem to have any serious health consequences. However, dysregulation of latency or inability to control lytic infection can lead to the development of lymphoproliferative diseases (LPDs) and lymphoma.
EBV was originally discovered in the context of African endemic Burkitt lymphoma (5, 6) and is classified as a Class I carcinogen by the International Agency for Cancer Research (7). EBV-associated malignancies are well characterized in individuals with suppressed immune systems such as after solid organ transplantation or in the setting of HIV/AIDS but are also recognized in patients without overt immunodeficiencies (8, 9). EBV is associated with nearly all nasopharyngeal carcinoma (NPC), approximately 10% of gastric carcinomas, 30–40% of classical Hodgkin lymphoma, a subset of diffuse large B-cell lymphoma (DLBCL), and other T/NK cell LPDs (8, 10–13). After primary infection EBV latency is defined by distinct gene expression programs (14–16). Viral latency is mediated through promoter silencing, characterized by limited protein expression, and categorized by four latency types (latency 0-III) (17–19). Expanded knowledge around the manipulation of DNA methylation and histone acetylation has led to a better understanding of the virus' ability to facilitate viral persistence in healthy individuals (20, 21), alter transcription factor accessibility (22, 23), silence tumor suppressor genes (24–26), and ultimately potentiate tumor development and growth (27–29). In the first part of our review we will discuss the regulation of the latent and lytic phases of EBV. In the second part we will show how researchers have capitalized on these mechanisms to target EBV and treat associated malignancy. EBV may prove to be the Achilles' heel of EBV-associated tumorigenesis and targeting the viral life cycle may help patients avoid toxic chemotherapeutics and receive more tailored and effective therapy.
Regulation of EBV Gene Expression
EBV latency is defined by a restricted, but variable protein expression that is specific to the host cell type (e.g., lymphoid or epithelial) or tumor origin. In vitro, B-cell immortalization is mediated by the viral latency III program in which all six EBV nuclear antigens (EBNAs 1, 2, 3A, 3B, 3C, and –LP), three latent membrane proteins (LMPs 1, 2A, and 2B), and viral non-coding RNAs (EBERs, miRNAs, and BARTs) are expressed and lead to the establishment of lymphoblastoid cell lines (1). The latency III program is the least restrictive latency type and is seen in LPDs associated with immunosuppression such as AIDS-associated DLBCL and post-transplant lymphoproliferative diseases (PTLDs) (30). Latency II is defined by expression of EBNA1, LMP1, and LMP2A/B and is most closely associated with Hodgkin lymphoma, NPC, and T/NK cell lymphomas (31–34). Only EBNA1 is expressed during latency I, which is associated with Burkitt lymphoma, as well as gastric carcinoma (35, 36). Latency 0 refers to the persistence of the viral genome in the absence of viral gene expression, which is associated with non-dividing memory B-cells (37).
The EBNA family of genes is among the viral genes differentially regulated by epigenetic modification during EBV latency. The six EBNA gene products expressed during the latency III program are constructed from one extensively spliced latency transcript. The EBV latency C promoter (Cp) is the origin for transcription of the EBNA latency proteins (38). CpG methylation of Cp plays an important role in regulating viral latency and limiting viral gene expression in normal lymphocytes and in certain malignancies including Burkitt, Hodgkin, AIDS associated DLBCL, and NK cell lymphomas, as well as NPC (39–42). During latent phase within the host cell the EBV genome is maintained as a circular episome that undergoes replication once per cycle, initiating from a region called oriP. EBNA1 is the only viral protein required to replicate from oriP and segregate the EBV episomal genomes in latency (43). EBNA1 has been shown to be responsible for promoting and maintaining latency (44). Compared to other EBV gene products EBNA1 is poorly recognized by CD8+ T-lymphocytes (45). The down regulation of the more immunogenic EBNA antigens (EBNA2, 3A-C), LMP1, early lytic antigens, and lytic viral kinases contributes to the virus' ability to evade the immune system during latency (46).
EBNA2 is critical for the transformation of B-cells (47) and is directly responsible for the initiation of transcription of EBV proteins associated with latency III like LMP1 and LMP2A/B (1, 48). EBNA2 is implicated in the Notch pathway, which contributes to viral latency by downregulating LMP1 and preventing the expression of BZLF1 (49). EBNA2 has been seen to target the c-Myc oncogene, which is important for EBV-induced B-cell immortalization in vitro (50). The EBNA3 family of proteins are also involved in B-cell transformation and essential for EBV persistence (51). EBNA2 and EBNA3 work together to regulate the expression of cellular and viral gene expression (52). EBNA3 may have a direct impact on progression through the cell cycle disrupting G2/M checkpoint (53) and has been shown to interact directly with human histone deacetylases influencing epigenetic regulation (54, 55). The EBNA family of proteins have been shown to work together in concert with host cellular machinery to affect histone acetylation and DNA methylation, directly impacting transcription of EBV related proteins to maintain latency (56–59).
LMP 1 and LMP 2A/2B are found in latency II and latency III EBV infected cells. LMP1 is essential for B lymphocyte growth transformation and for the survival of EBV transformed B-cells (60). LMP1 mimics CD40 signaling, which is a key B-cell costimulatory receptor (61). LMP1 behaves as a prototypical oncogene in vitro and is associated with upregulation of antiapoptotic proteins (62, 63) and stimulation of cytokine production (64). Specifically, constitutive activation of NF-kB and mitogen-activated protein kinase (MAPK) are supported by LMP1 and critical to lymphoblastoid cell line survival (65, 66). Knockdown of LMP1 downregulates NF-kB signaling and induces apoptosis (67). Expression of LMP1 in transgenic mice induces the development of B-cell lymphomas (68). LMP2A/B support LMP1 functions, as well as suppress B-cell receptor signaling (69). The inhibition of B-cell receptor signaling regulates EBV latency by preventing B-cell differentiation to plasma cells and effectively blocking the switch from latent to lytic replication (70). LMP1 and LMP2A signaling can induce expression of DNA methyltransferases (DNMT1, 3A, and 3B), which impacts major cellular pathway signaling. PARP1 mediates EBV replication during latency and LMP1 has been shown to alter expression of tumor-promoting genes by blocking histone methylation via PARP1 activation (71). LMP1 and LMP2A have been associated with hypermethylation and silencing of the PTEN gene in gastric carcinoma (72, 73). LMP1 induces the expression of the histone demethylase KDM6B, which has been associated with the pathogenesis of Hodgkin lymphoma (74). LMP2A is also implicated in the development of Hodgkin lymphoma via specific alterations in gene transcription (75). These examples highlight how EBV machinery can subvert the cell's normal epigenetic mechanisms thereby promoting viral latency and subsequent tumorigenesis.
EBV encodes many small non-coding RNAs (EBER1, EBER2, and viral miRNAs) that are widely expressed in infected cells (76, 77). Non-coding RNAs are expressed during all forms of EBV latency and also during the lytic cycle (78). Epigenetic manipulation by non-coding RNAs is thought to occur via recruitment of host transcription factors and chromatin regulators that modulate viral and host gene expression (79). Recruitment and thus alterations to host gene expression is mediated by viral RNA targeting of complementary sequences on cellular mRNA (80, 81). For example, EBER2 has been shown to target the B-cell transcription factor PAX5 via an RNA:RNA interaction (82). EBER1 has been shown to increase the expression of insulin growth factor-1 (IGF-1) and potentiate cellular proliferation in EBV associated gastric cancer (83). In fact, the EBERs, and in particular EBER1, have been shown to contribute to lymphoid hyperplasia and lymphoma on their own (84). There is evidence to suggest EBERs can increase IL-6 expression leading to the downstream activation of STAT3. This interaction may have a direct impact on host cell chemoresistance and migration (85).
The viral miRNAs are differentially expressed depending on the infected cell or tumor type. EBV miRNAs are involved with early B-cell proliferation and suppression of apoptosis (86, 87). The miRNAs are subdivided into two groups, Bam HI fragment H rightward open reading frame I microRNAs (BHRF1 miRNAs) and Bam HI-A rightward transcripts microRNAs (BART miRNAs), based on their locations (76, 88). The BARTs are a group of stable viral RNAs represented in every EBV infected cell type. Their expression is regulated by promoter methylation and treatment with a DNA methyltransferase increased the expression of BART miRNA transcripts (89). The BART promoter region is hypomethylated in NPC, which may explain why BART miRNAs are highly expressed in this tumor type (90, 91). Whether the BART miRNAs are translated to protein products remains controversial but is an important area of research for targeting EBV in malignancy (90, 92, 93). Expression of EBV miRNAs has been observed in gastric carcinoma (94), peripheral and cutaneous T-cell lymphoma (95–97), B-cell lymphoma cell lines, and NPC EBV-infected cells (76, 88) implicating EBV miRNAs in tumorigenesis.
When an EBV infected B-cell terminally differentiates to the plasma cell lineage the virus activates the lytic cycle genes and generates viral progeny (98). The switch from latency to the lytic cycles is mediated by two viral transactivator proteins, BZLF1 and BRLF1 (99). These two genes are influenced by DNA methylation of the viral genome. BZLF1 binds to methylated DNA and interacts with histone acetyltransferases to instigate expression of lytic promoters (100, 101). Expression of BZLF1 leads to a cascade of over 80 EBV gene products that results in viral replication and ultimately host-cell lysis. BRLF1 activates some early lytic genes through a direct binding mechanism (102, 103), while other gene activation is mediated through interactions with cellular transcription factors (104). BRLF1 activates phosphatidylinositol 3 kinase (PI3K), which is required for BRLF-mediated induction of lytic gene expression (105). During reactivation in an immunocompetent host, EBV-primed CD4+ and CD8+ memory T-cells can respond within hours and destroy virally infected cells before viral replication is finished (46). EBV expresses several gene products during lytic reactivation to directly counteract the T-cell response. Expression of BNLF2a, for example, prevents peptide presentation by MHC class I molecules through direct inhibition of antigen processing (106) and BGLF5, another lytic protein, augments the expression of MHC class I and II molecules (107). In addition, these lytic viral proteins can suppress pro-inflammatory cytokine release and temper the innate immune response including natural killer cell killing of EBV-infected B-cells (108).
This detailed knowledge of the viral life cycle provides opportunities to target features of the virus that promote lymphoproliferation. In the following section we will show how treatment strategies have capitalized on this to target EBV driven lymphoproliferation, thus providing novel treatment options. Ultimately though, we expect and look forward to future treatment approaches that will be more specific based on the improved understanding of the viral life cycle, including epigenetic modifications, outlined above.
EBV Targeted Therapy
The various patterns of viral gene expression in LPDs has treatment ramifications (Table 1). For lymphomas like Burkitt and classical Hodgkin only a small subset of the latent gene profile is expressed offering limited and poorly immunogenic antigens to target. Alternatively, EBV-associated LPDs that arise as a result of immunosuppression generally express more gene products and are thus susceptible to antiviral directed therapy and reduction in immunosuppression, as is the case for EBV-associated PTLD after solid organ transplantation (109). In tumor types like EBV-associated PTLD or DLBCL with expanded viral protein motifs, constitutive activation of lytic proteins, such as viral thymidine kinases (vTKs) BXLF1 and BGLF4, has been demonstrated (110–113). Activation in vTKs result in phosphorylation of the nucleoside analogs ganciclovir (GCV), acyclovir, and zidovudine (AZT) (114–116). As a result, these antivirals can be effective in tumor types that demonstrate constitutive activation of lytic phase proteins; however, they are inactive against latent infection since there is no expression of the lytic kinases. There has been an evolving interest in developing techniques for inducing the lytic phase of the virus, sometimes referred to as the “kick and kill” strategy. In this scenario the virus is pushed into replicating so that phosphorylation of the nucleoside analogs can occur.
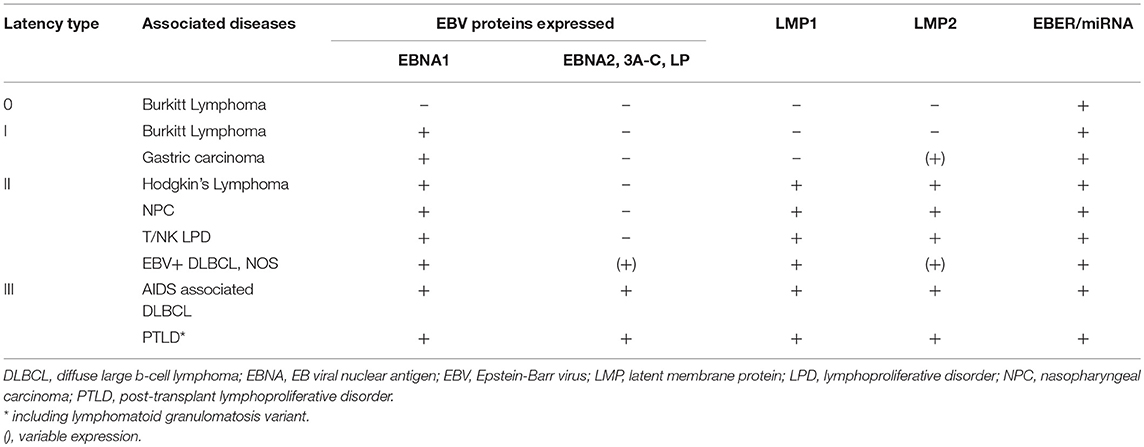
Table 1. Latent viral protein expression patterns in EBV-associated malignancy.
The use of AZT and GCV in patients with immunosuppression-related EBV-associated B-cell lymphoma was originally investigated by the late Dr. William J. Harrington at the University of Miami in the 1990s, based on in vitro data showing that AZT and GCV additively induced apoptosis in EBV+ cell lines (117) and on anecdotal reports of disease regression in patients with HIV-associated lymphomas after exposure to AZT (118, 119). Harrington and colleagues reported rapid clinical responses in 4 of 5 patients using a regimen of intravenous AZT, GCV and interleukin (IL)-2 for 2–3 weeks, without any antineoplastic chemotherapy or radiation (117). This regimen was adopted by the AIDs Malignancy Consortium in a prospective study (AMC-019, NCT00006264) (120), and in 1998 a Phase II clinical trial of the AZT/GCV combination, based on the Harrington schedule, was opened for patients with primary CNS PTLD, a B-cell neoplasm that shares a number of clinical and biologic features with HIV-associated PCNSL, including near universal association with EBV, inconsistent response to immune restoration, and poor prognosis (121). The original Harrington regimen was amended to eliminate use of IL-2 in recipients of solid organ transplant and to include an extended 2-year maintenance phase or oral AZT and GCV following the initial intravenous 14-day “induction” phase. The Phase II trial was eventually closed due to difficulties with accrual associated in part with the rarity of the indication and with the severity and acuity of the target population.
Other strategies have been employed to induce the lytic phase of the EBV lifecycle and make the associated malignancies susceptible to antiviral therapy regardless of latency subtype. Preclinical studies have shown that pharmacologic induction with dexamethasone and rituximab induces lytic protein expression and renders EBV infected B-lymphocytes sensitive to ganciclovir (122). In a phase I/II study investigating EBV lytic induction in nasopharyngeal carcinoma, patients were given a combination of gemcitabine and valproic acid to induce lytic gene expression and then treated with valganciclovir. The authors were able to demonstrate safety of this regimen, as well as increases in EBV-DNA loads in the blood (123). Other chemotherapies like 5-fluororuacil, platinum agents, or paclitaxel in conjunction with antiviral medication have also induced lytic activation and increased sensitivity to antiviral therapy in NPC (124). Aspirin can induce lytic gene expression via suppression of NF-kB, which allows downregulation of LMP1 and subsequent expression of BZLF1. Treatment of EBV+ cells in vitro with aspirin and ganciclovir shows significantly improved cytotoxic effect than with either drug alone (125). In many cases investigators have been able to show synergistic effects between traditional chemotherapies and antiviral therapies, as well as increased levels of viral DNA in blood. Clinically, these patients seem to have transient and/or moderate side effects and improved quality of life, which are important benchmarks for a patient population whom would not have tolerated traditional high dose chemotherapy and radiation.
Exposure of latently infected B-cells in vitro to histone deacetylase inhibitors (HDACi) alters the promoter sequences or disrupts the gene silencing of BZLF1 and BRLF1 genes thereby inducing lytic reactivation (126). Support for this approach was first demonstrated in a lung transplant recipient with an EBV-associated immunoblastic lymphoma 4 months following transplantation (127). Based on work demonstrating that butyrate congeners could induce EBV lytic genes, including vTKs (128, 129), a cell line derived from the patient's tumor was exposed to arginine butyrate resulting in induction of EBV TK transcription. The combination of arginine butyrate and ganciclovir resulted in inhibition of cell proliferation and cell death. Arginine butyrate was added to the patient's existing treatment with ganciclovir with no apparent increase in toxicity. Though the patient succumbed to a systemic aspergillus infection that had preceded the administration of arginine butyrate therapy, pathologic examination of the tumor demonstrated substantial necrosis compared to pre-therapy histology (127). Additional support for the use of an HDAC inhibitor to sensitize EBV infected tumor cells to nucleoside antivirals was demonstrated in a phase I/II trial of arginine butyrate combined with ganciclovir in 15 patients with EBV-associated LPDs previously treated with chemotherapy and/or radiation (130). Arginine butyrate was administered by daily continuous IV infusion on an escalating dose schedule for 21 days of a 28-day treatment course, combined with a fixed dose of daily continuous ganciclovir. Eleven patients received at least 28 days of arginine butyrate and ganciclovir, and all 15 patients were evaluable for response. Significant antitumor activity was seen in 10 patients, with 4 complete responses (CRs) and 6 partial responses (PRs). Several HDAC inhibitors in addition to arginine butyrate can induce expression of EBV lytic phase genes in vitro, leading to the sensitization of EBV infected lymphoma cells to nucleoside antivirals (127, 131). Ongoing studies are evaluating the ability of HDAC inhibitors to sensitize EBV infected lymphoma cells, irrespective of latency subtype, to ganciclovir in vivo (NCT0339770).
As discussed, CpG promoter methylation is used by EBV to silence lytic phase genes and promote latency. CpG methylation can be reversed via pharmacologic demethylation promoting the expression of EBV lytic genes in latently infected cells. Drugs targeting this epigenetic mechanism include the DNA hypomethylators 5-azacitadine and decitabine. 5-azacitadine inhibits DNA methyltransferase, reduces methylation of CpG promoter regions, and activates transcription of EBNA2 (132). EBNA2 is directly associated with regulation of the latency III program and transcription of proteins LMP1 and LMP2A/B (1, 48) In one study, EBV promoter methylation was evaluated in patients with NPC and AIDS associated lymphomas before and after receiving 5-azacitadine. The authors were able to demonstrate demethylation to varying degrees in the latent and early lytic EBV promoters evaluated (133). Reducing CpG methylation and stimulating the more immunogenic EBV proteins may facilitate immune-mediated destruction of the tumor cells and make them sensitive to antiviral therapy. Ultimately, methylation of viral DNA may become a clinically useful tool to characterize the associated latency subtype and treat the corresponding associated LPD (134).
Lastly, we would like to discuss the role of adoptive T-cell immunotherapy. In a seropositive immunocompetent person EBV is actively monitored by EBV-specific cytotoxic T-lymphocytes (CTLs). In patients that are immunocompromised either via iatrogenic means as in the case of organ transplantation or via infectious means like HIV, EBV is permitted to proliferate due to the depletion of these CTLs. Adoptive T-cell immunotherapy involves infusing EBV-specific CTLs generated in vitro with the aim of reconstituting the EBV immunity and influence targeted destruction of EBV infected tumor cells. Importantly, native T-cells are HLA-restricted meaning a patient's T-cell will only recognize antigen presented by HLA molecules of their own allelic type. This has implications for sourcing EBV-specific CTLs. For example, in bone marrow transplant recipients PTLD is almost exclusively of donor origin, so attempts have been made to treat PTLD in this scenario with donor derived EBV-specific CTLs. Preparing non-specific populations of CTLs (i.e., donor lymphocyte infusions) can have serious consequences, such as graft vs. host disease, related to the alloreactivity of T-lymphocytes (135). Severe and even fatal graft vs. host disease have been seen after administration of non-specific allogeneic CTLs, which is why most new therapies use polyclonal EBV-specific HLA matched CTL lines prepared in vitro (136). In 1995 Rooney et al. introduced gene-modified EBV-specific T-lymphocytes to allotransplant recipients with EBV-associated LPDs. The authors showed that EBV-CTLs could generate a CR without associated infusional complications (137). In a total of 101 patients given EBV-CTLs for prophylaxis, none of the patients developed an EBV-associated LPD and 11/13 patients with an EBV-associated LPD had a CR (138). The majority of research on the clinical application of EBV-CTLs has focused on PTLD given the immunogenicity of these tumors and the full expression of EBV latency III antigens. In 2007 Bollard and colleagues showed that by increasing the frequency of LMP2-specific CTLs responses in immunocompetent patients were dramatically improved. They concluded that it was possible to generate immune responses to weak tumor antigens like LMP2 by in vitro manipulation of CTL antigen recognition (139). In a 2014 study Bollard et al. looked at LMP1/2 specific EBV-CTLs in patients with a variety of latency II associated malignancy including Hodgkin lymphoma, DLBCL, T/NK cell lymphomas, and NPC. Twenty-nine of 50 patients treated with autologous CTLs in remission from high-risk or multiple-relapsed disease had an EFS of 82% and 11 of 21 patients treated with active disease experienced a CR (140). In a another trial, patients with extranodal NK cell lymphoma were given LMP1/2a specific CTLs and 9 of 10 patients showed a sustained remission (141). There are multiple active trials evaluating EBV-specific and EBV-antigen specific CTLs for use in patients with EBV-associated LPDs (see Table 2 for a list of trials).
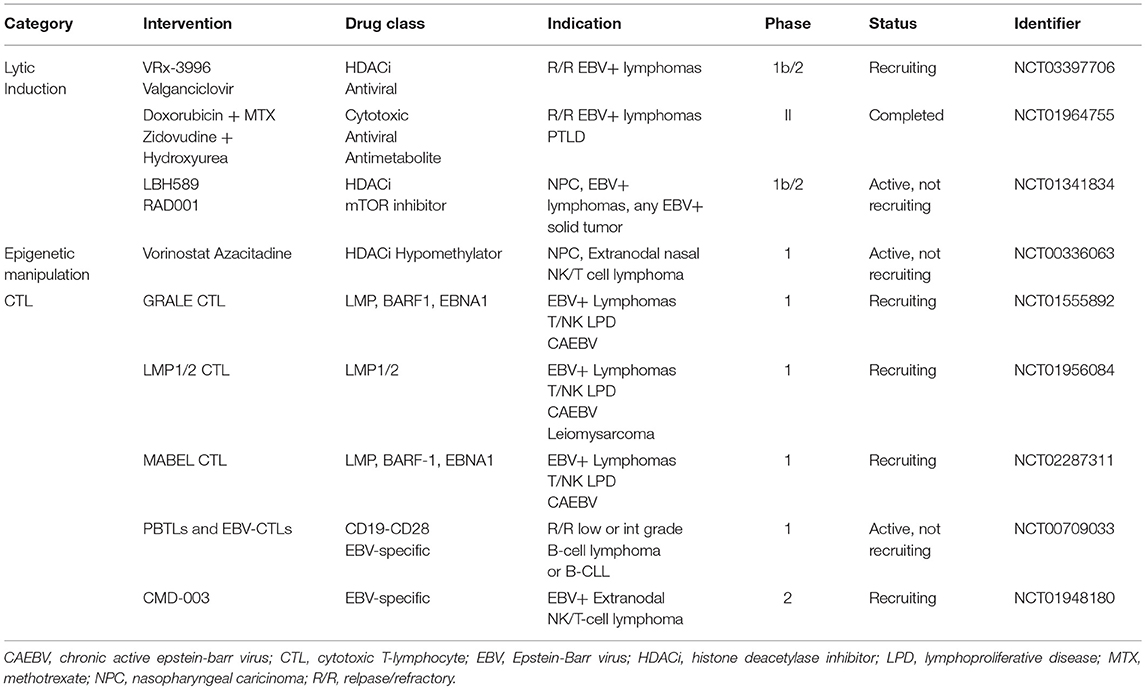
Table 2. Clinical trials targeting EBV in malignancy.
Another T-cell mediated strategy that has shown some promise in EBV-mediated malignancy involves checkpoint blockade. EBV-infected lymphoma cells express the inhibitory ligand PD-L1 (142). PD-L1 is also found in EBV-associated gastric cancer (143) and T/NK-cell lymphomas (144). Green et al. showed that EBV infection induced PD-L1 expression in classical Hodgkin lymphoma (145). Preclinical work by Ma et al. has shown that inhibition of PD-1 and CTLA-4 dramatically reduces lymphomas induced by EBV in a mouse model (142). The PD1 antibody pembrolizumab has been effective in relapsed/refractory NK cell lymphomas. In their case series, Kwong et al. showed 5 of 7 patients retained a CR after a median follow-up of 6 months after failing l-asparaginase containing therapy (144). Research into the role of checkpoint inhibitors in EBV-mediated malignancy is ongoing (NCT03586024). Combinations of checkpoint inhibitors with lytic induction may prove to be a promising strategy in the future.
Future Directions
Induction of EBV from latency to the lytic phase of viral replication has become an attractive method for treating EBV mediated malignancy. As discussed and outlined in Table 2, there are a variety of methods for achieving lytic induction. The methods for induction range from traditional chemotherapies to steroids to HDAC inhibitors. The optimal method for inducing lytic activation has not yet been defined. Many of the patients who receive EBV directed therapy have multiple comorbidities, are immunosuppressed, and have already received more traditional cytotoxic chemotherapy-based regimens. Identifying the ideal regimen to induce lytic reactivation and target EBV while reducing the degree of treatment related morbidity and mortality associated with traditional cytotoxic chemotherapy is an active area of research. Combining antiviral targeted approaches with immune based therapies that permit a functional adaptive immune system (or an “off the shelf” allogeneic CTL) may be a potentially rational synergist approach. Perhaps combination of the “kick and kill” approach with checkpoint blockade and/or CTLs (auto or allo) will be a simultaneous or sequential treatment strategy of the future.
Author Contributions
JD wrote the first draft of the manuscript and contributed to every draft thereafter. CC contributed to later drafts of the manuscript and provided expertise in virology. BH contributed to every iteration of the manuscript and provided expertise in EBV mediated malignancy.
Funding
BH was supported by the National Cancer Institute of the National Institutes of Health grant L30 CA189070.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kieff E, Rickinson AB. Epstein-Barr Virus and it Replication. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins (2007).
2. Cohen JI. Epstein–Barr Virus Infection. N Engl J Med. (2000) 343:481–92. doi: 10.1056/NEJM200008173430707
3. Henle G, Henle W. Observations on childhood infections with the Epstein-Barr virus. J Infect Dis. (1970) 121:303–10. doi: 10.1093/infdis/121.3.303
4. Moghaddam A, Rosenzweig M, Lee-Parritz D, Annis B, Johnson RP, Wang F. An animal model for acute and persistent Epstein-Barr virus infection. Science. (1997) 276:2030–3. doi: 10.1126/science.276.5321.2030
5. Wright DH. Burkitt's lymphoma: a review of the pathology, immunology, and possible etiologic factors. Pathol Ann. (1971)6:337–63.
6. de-The G, Geser A, Day NE, Tukei PM, Williams EH, Beri DP, et al. Epidemiological evidence for causal relationship between Epstein-Barr virus and Burkitt's lymphoma from Ugandan prospective study. Nature. (1978) 274:756–61. doi: 10.1038/274756a0
7. IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Epstein-Barr Virus and Kaposi's Sarcoma Herpesvirus/Human Herpesvirus 8. Lyon: International Agency for Research on Cancer (1997). (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 70.) Epstein-Barr virus.
8. Rezk SA, Weiss LM. Epstein-Barr virus-associated lymphoproliferative disorders. Hum Pathol. (2007) 38:1293–304. doi: 10.1016/j.humpath.2007.05.020
9. Greenspan JS, Greenspan D, Lennette ET, Abrams DI, Conant MA, Petersen V, et al. Replication of Epstein–Barr Virus within the epithelial cells of oral hairy leukoplakia, an AIDS-associated lesion. N Engl J Med. (1985) 313:1564–71. doi: 10.1056/NEJM198512193132502
10. Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. (2004) 4:757–68. doi: 10.1038/nrc1452
11. Ambinder RF. Epstein-barr virus and hodgkin lymphoma. Hematology. (2007) 2007:204–9. doi: 10.1182/asheducation-2007.1.204
12. Gru AA, Haverkos BH, Freud AG, Hastings J, Nowacki NB, Barrionuevo C, et al. The Epstein-Barr Virus (EBV) in T cell and NK cell lymphomas: time for a reassessment. Curr Hematol Malig Rep. (2015) 10:456–67. doi: 10.1007/s11899-015-0292-z
13. Haverkos BM, Pan Z, Gru AA, Freud AG, Rabinovitch R, Xu-Welliver M, et al. Extranodal NK/T cell lymphoma, nasal type (ENKTL-NT): an update on epidemiology, clinical presentation, and natural history in north american and european cases. Curr Hematol Malig Rep. (2016) 11:514–27. doi: 10.1007/s11899-016-0355-9
14. Lieberman PM. Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol. (2013) 11:863–75. doi: 10.1038/nrmicro3135
15. Takacs M, Banati F, Koroknai A, Segesdi J, Salamon D, Wolf H, et al. Epigenetic regulation of latent Epstein-Barr virus promoters. Biochimica et biophysica acta. (2010) 1799:228–35. doi: 10.1016/j.bbagrm.2009.10.005
16. Tempera I, Lieberman PM. Epigenetic regulation of EBV persistence and oncogenesis. Seminars Cancer Biol. (2014) 26:22–9. doi: 10.1016/j.semcancer.2014.01.003
17. Rowe M, Lear AL, Croom-Carter D, Davies AH, Rickinson AB. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J Virol. (1992) 66:122–31.
18. Tierney RJ, Steven N, Young LS, Rickinson AB. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J Virol. (1994) 68:7374–85.
19. Woisetschlaeger M, Yandava CN, Furmanski LA, Strominger JL, Speck SH. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc Natl Acad Sci USA. (1990) 87:1725–9. doi: 10.1073/pnas.87.5.1725
20. Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, et al. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe. (2012) 12:233–45. doi: 10.1016/j.chom.2012.06.008
21. Paulson EJ, Speck SH. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J Virol. (1999) 73:9959–68.
22. Tempera I, Lieberman PM. Chromatin organization of gammaherpesvirus latent genomes. Biochimica et biophysica acta. (2010) 1799:236–45. doi: 10.1016/j.bbagrm.2009.10.004
23. Wensing B, Stuhler A, Jenkins P, Hollyoake M, Karstegl CE, Farrell PJ. Variant chromatin structure of the oriP region of Epstein-Barr virus and regulation of EBER1 expression by upstream sequences and oriP. J Virol. (2001) 75:6235–41. doi: 10.1128/JVI.75.13.6235-6241.2001
24. Zhang T, Ma J, Nie K, Yan J, Liu Y, Bacchi CE, et al. Hypermethylation of the tumor suppressor gene PRDM1/Blimp-1 supports a pathogenetic role in EBV-positive Burkitt lymphoma. Blood Cancer J. (2014) 4:e261. doi: 10.1038/bcj.2014.75
25. Tian F, Yip SP, Kwong DL, Lin Z, Yang Z, Wu VW. Promoter hypermethylation of tumor suppressor genes in serum as potential biomarker for the diagnosis of nasopharyngeal carcinoma. Cancer Epidemiol. (2013) 37:708–13. doi: 10.1016/j.canep.2013.05.012
26. Saha A, Jha HC, Upadhyay SK, Robertson ES. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc Natl Acad Sci USA. (2015) 112:E5199–207. doi: 10.1073/pnas.1503806112
27. Macdiarmid J, Stevenson D, Campbell DH, Wilson JB. The latent membrane protein 1 of Epstein-Barr virus and loss of the INK4a locus: paradoxes resolve to cooperation in carcinogenesis in vivo. Carcinogenesis. (2003) 24:1209–18. doi: 10.1093/carcin/bgg070
28. Chong JM, Sakuma K, Sudo M, Ushiku T, Uozaki H, Shibahara J, et al. Global and non-random CpG-island methylation in gastric carcinoma associated with Epstein-Barr virus. Cancer Sci. (2003) 94:76–80. doi: 10.1111/j.1349-7006.2003.tb01355.x
29. Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, et al. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. (2002) 160:787–94. doi: 10.1016/S0002-9440(10)64901-2
30. Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. (2004) 350:1328–37. doi: 10.1056/NEJMra032015
31. Roughan JE, Thorley-Lawson DA. The intersection of Epstein-Barr virus with the germinal center. J Virol. (2009) 83:3968–76. doi: 10.1128/JVI.02609-08
32. Pallesen G, Hamilton-Dutoit SJ, Rowe M, Young LS. Expression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin's disease. Lancet. (1991) 337:320–2. doi: 10.1016/0140-6736(91)90943-J
33. Deacon EM, Pallesen G, Niedobitek G, Crocker J, Brooks L, Rickinson AB, et al. Epstein-Barr virus and Hodgkin's disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med. (1993) 177:339–49. doi: 10.1084/jem.177.2.339
34. Brooks L, Yao QY, Rickinson AB, Young LS. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol. (1992) 66:2689–97.
35. Rowe M, Rowe DT, Gregory CD, Young LS, Farrell PJ, Rupani H, et al. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. (1987) 6:2743–51. doi: 10.1002/j.1460-2075.1987.tb02568.x
36. Sugiura M, Imai S, Tokunaga M, Koizumi S, Uchizawa M, Okamoto K, et al. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency in the tumour cells. Br J Cancer. (1996) 74:625–31. doi: 10.1038/bjc.1996.412
37. Babcock GJ, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity. (2000) 13:497–506. doi: 10.1016/S1074-7613(00)00049-2
38. Woisetschlaeger M, Strominger JL, Speck SH. Mutually exclusive use of viral promoters in Epstein-Barr virus latently infected lymphocytes. Proc Natl Acad Sci USA. (1989) 86:6498–502. doi: 10.1073/pnas.86.17.6498
39. Robertson KD, Manns A, Swinnen LJ, Zong JC, Gulley ML, Ambinder RF. CpG methylation of the major Epstein-Barr virus latency promoter in Burkitt's lymphoma and Hodgkin's disease. Blood. (1996) 88:3129–36.
40. Tao Q, Robertson KD, Manns A, Hildesheim A, Ambinder RF. Epstein-Barr virus (EBV) in endemic Burkitt's lymphoma: molecular analysis of primary tumor tissue. Blood. (1998) 91:1373–81.
41. Ambinder RF, Robertson KD, Tao Q. DNA methylation and the Epstein-Barr virus. Semin Cancer Biol. (1999) 9:369–75. doi: 10.1006/scbi.1999.0137
42. Tao Q, Swinnen LJ, Yang J, Srivastava G, Robertson KD, Ambinder RF. Methylation status of the Epstein-Barr virus major latent promoter C in iatrogenic B cell lymphoproliferative disease. Application of PCR-based analysis. Am J Pathol. (1999) 155:619–25. doi: 10.1016/S0002-9440(10)65157-7
43. Frappier L. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr Opin Virol. (2012) 2:733–9. doi: 10.1016/j.coviro.2012.09.005
44. Mansouri S, Pan Q, Blencowe BJ, Claycomb JM, Frappier L. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J Virol. (2014) 88:11166–77. doi: 10.1128/JVI.01785-14
45. Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald-Mullen PM, Klein G, et al. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. (1995) 375:685–8. doi: 10.1038/375685a0
46. Ressing ME, Horst D, Griffin BD, Tellam J, Zuo J, Khanna R, et al. Epstein-Barr virus evasion of CD8(+) and CD4(+) T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol. (2008) 18:397–408. doi: 10.1016/j.semcancer.2008.10.008
47. Rabson M, Gradoville L, Heston L, Miller G. Non-immortalizing P3J-HR-1 Epstein-Barr virus: a deletion mutant of its transforming parent, Jijoye. J Virol. (1982) 44:834–44.
48. Zimber-Strobl U, Kremmer E, Grasser F, Marschall G, Laux G, Bornkamm GW. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. (1993) 12:167–75. doi: 10.1002/j.1460-2075.1993.tb05642.x
49. Rowe M, Raithatha S, Shannon-Lowe C. Counteracting effects of cellular Notch and Epstein-Barr virus EBNA2: implications for stromal effects on virus-host interactions. J Virol. (2014) 88:12065–76. doi: 10.1128/JVI.01431-14
50. Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol. (1999) 73:4481–4.
51. Allday MJ, Bazot Q, White RE. The EBNA3 Family: two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr Top Microbiol Immunol. (2015) 391:61–117. doi: 10.1007/978-3-319-22834-1_3
52. Allday MJ, Farrell PJ. Epstein-Barr virus nuclear antigen EBNA3C/6 expression maintains the level of latent membrane protein 1 in G1-arrested cells. J Virol. (1994) 68:3491–8.
53. Krauer KG, Burgess A, Buck M, Flanagan J, Sculley TB, Gabrielli B. The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene. (2004) 23:1342–53. doi: 10.1038/sj.onc.1207253
54. Radkov SA, Touitou R, Brehm A, Rowe M, West M, Kouzarides T, et al. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J Virol. (1999) 73:5688–97.
55. Portal D, Rosendorff A, Kieff E. Epstein-Barr nuclear antigen leader protein coactivates transcription through interaction with histone deacetylase 4. Proc Natl Acad Sci USA. (2006) 103:19278–83. doi: 10.1073/pnas.0609320103
56. Wang L, Grossman SR, Kieff E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc Natl Acad Sci USA. (2000) 97:430–5. doi: 10.1073/pnas.97.1.430
57. Wang S, Frappier L. Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol. (2009) 83:11704–14. doi: 10.1128/JVI.00931-09
58. Paschos K, Bazot Q, Ho G, Parker GA, Lees J, Barton G, et al. Core binding factor (CBF) is required for Epstein-Barr virus EBNA3 proteins to regulate target gene expression. Nucleic Acids Res. (2017) 45:2368–83. doi: 10.1093/nar/gkw1167
59. Zhou H, Schmidt SC, Jiang S, Willox B, Bernhardt K, Liang J, et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe. (2015) 17:205–16. doi: 10.1016/j.chom.2014.12.013
60. Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci USA. (1993) 90:9150–4. doi: 10.1073/pnas.90.19.9150
61. Wang LW, Jiang S, Gewurz BE. Epstein-Barr virus LMP1-mediated oncogenicity. J Virol. (2017) 91:e01718–16. doi: 10.1128/JVI.01718-16
62. Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, et al. Induction of bcl-2 expression by epstein-barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. (1991) 65:1107–15. doi: 10.1016/0092-8674(91)90007-L
63. Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem. (1992) 267:24157–60.
64. Eliopoulos AG, Gallagher NJ, Blake SM, Dawson CW, Young LS. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J Biol Chem. (1999) 274:16085–96. doi: 10.1074/jbc.274.23.16085
65. Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci USA. (1997) 94:12592–7. doi: 10.1073/pnas.94.23.12592
66. Huen DS, Henderson SA, Croom-Carter D, Rowe M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. (1995) 10:549–60.
67. Guasparri I, Bubman D, Cesarman E. EBV LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood. (2008) 111:3813–20. doi: 10.1182/blood-2007-03-080309
68. Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V, Raab-Traub N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc Natl Acad Sci USA. (1998) 95:11963–8. doi: 10.1073/pnas.95.20.11963
69. Longnecker R. Epstein-Barr virus latency: LMP2, a regulator or means for Epstein-Barr virus persistence? Adv Cancer Res. (2000) 79:175–200. doi: 10.1016/S0065-230X(00)79006-3
70. Miller CL, Lee JH, Kieff E, Longnecker R. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc Natl Acad Sci USA. (1994) 91:772–6. doi: 10.1073/pnas.91.2.772
71. Martin KA, Lupey LN, Tempera I. Epstein-Barr Virus Oncoprotein LMP1 Mediates Epigenetic Changes in Host Gene Expression through PARP1. J Virol. (2016) 90:8520–30. doi: 10.1128/JVI.01180-16
72. Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. (2009) 69:2766–74. doi: 10.1158/0008-5472.CAN-08-3070
73. Peng H, Chen Y, Gong P, Cai L, Lyu X, Jiang Q, et al. Higher methylation intensity induced by EBV LMP1 via NF-kappaB/DNMT3b signaling contributes to silencing of PTEN gene. Oncotarget. (2016) 7:40025–37. doi: 10.18632/oncotarget.9474
74. Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's Lymphoma. Oncogene. (2011) 30:2037–43. doi: 10.1038/onc.2010.579
75. Portis T, Dyck P, Longnecker R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood. (2003) 102:4166–78. doi: 10.1182/blood-2003-04-1018
76. Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, et al. Identification of virus-encoded microRNAs. Science. (2004) 304:734–6. doi: 10.1126/science.1096781
77. Skalsky RL, Cullen BR. Viruses, microRNAs, and host interactions. Ann Rev Microbiol. (2010) 64:123–41. doi: 10.1146/annurev.micro.112408.134243
78. Greifenegger N, Jager M, Kunz-Schughart LA, Wolf H, Schwarzmann F. Epstein-Barr virus small RNA (EBER) genes: differential regulation during lytic viral replication. J Virol. (1998) 72:9323–8.
79. Lee N, Steitz JA. Noncoding RNA-guided recruitment of transcription factors: A prevalent but undocumented mechanism? BioEssays. (2015) 37:936–41. doi: 10.1002/bies.201500060
80. Xia T, O'Hara A, Araujo I, Barreto J, Carvalho E, Sapucaia JB, et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1–3. Cancer Res. (2008) 68:1436–42. doi: 10.1158/0008-5472.CAN-07-5126
81. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
82. Lee N, Moss WN, Yario TA, Steitz JA. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell. (2015) 160:607–18. doi: 10.1016/j.cell.2015.01.015
83. Iwakiri D, Eizuru Y, Tokunaga M, Takada K. Autocrine growth of Epstein-Barr virus-positive gastric carcinoma cells mediated by an Epstein-Barr virus-encoded small RNA. Cancer Res. (2003) 63:7062–7.
84. Repellin CE, Tsimbouri PM, Philbey AW, Wilson JB. Lymphoid hyperplasia and lymphoma in transgenic mice expressing the small non-coding RNA, EBER1 of Epstein-Barr virus. PLoS ONE. (2010) 5:e9092. doi: 10.1371/journal.pone.0009092
85. Banerjee AS, Pal AD, Banerjee S. Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology. (2013) 443:294–305. doi: 10.1016/j.virol.2013.05.020
86. Feederle R, Haar J, Bernhardt K, Linnstaedt SD, Bannert H, Lips H, et al. The members of an Epstein-Barr virus microRNA cluster cooperate to transform B lymphocytes. J Virol. (2011) 85:9801–10. doi: 10.1128/JVI.05100-11
87. Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. (2010) 6:e1001063. doi: 10.1371/journal.ppat.1001063
88. Cai X, Schafer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, et al. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. (2006) 2:e23. doi: 10.1371/journal.ppat.0020023
89. Kim DN, Song YJ, Lee SK. The role of promoter methylation in Epstein-Barr virus (EBV) microRNA expression in EBV-infected B cell lines. Exp Mol Med. (2011) 43:401–10. doi: 10.3858/emm.2011.43.7.044
90. Marquitz AR, Raab-Traub N. The role of miRNAs and EBV BARTs in NPC. Semin Cancer Biol. (2012) 22:166–72. doi: 10.1016/j.semcancer.2011.12.001
91. Al-Mozaini M, Bodelon G, Karstegl CE, Jin B, Al-Ahdal M, Farrell PJ. Epstein-Barr virus BART gene expression. J Gen Virol. (2009) 90:307–16. doi: 10.1099/vir.0.006551-0
92. Smith PR, de Jesus O, Turner D, Hollyoake M, Karstegl CE, Griffin BE, et al. Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J Virol. (2000) 74:3082–92. doi: 10.1128/JVI.74.7.3082-3092.2000
93. Kienzle N, Sculley TB, Poulsen L, Buck M, Cross S, Raab-Traub N, et al. Identification of a cytotoxic T-lymphocyte response to the novel BARF0 protein of Epstein-Barr virus: a critical role for antigen expression. J Virol. (1998) 72:6614–20.
94. Kim DN, Chae HS, Oh ST, Kang JH, Park CH, Park WS, et al. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol. (2007) 81:1033–6. doi: 10.1128/JVI.02271-06
95. Jun SM, Hong YS, Seo JS, Ko YH, Yang CW, Lee SK. Viral microRNA profile in Epstein-Barr virus-associated peripheral T cell lymphoma. Br J Haematol. (2008) 142:320–3. doi: 10.1111/j.1365-2141.2008.07186.x
96. Haverkos BM, Huang Y, Gru A, Pancholi P, Freud AG, Mishra A, et al. Frequency and clinical correlates of elevated plasma Epstein-Barr virus DNA at diagnosis in peripheral T-cell lymphomas. Int J Cancer. (2017) 140:1899–906. doi: 10.1002/ijc.30566
97. Haverkos BM, Gru AA, Geyer SM, Bingman AK, Hemminger JA, Mishra A, et al. Increased levels of plasma Epstein Barr Virus DNA identify a poor-risk subset of patients with advanced stage cutaneous T-cell lymphoma. Clin Lymph Myel Leukemia. (2016) 16(Suppl.):S181–S90.e4. doi: 10.1016/j.clml.2016.02.014
98. Sun CC, Thorley-Lawson DA. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J Virol. (2007) 81:13566–77. doi: 10.1128/JVI.01055-07
99. Sinclair AJ. bZIP proteins of human gammaherpesviruses. J Gen Virol. (2003) 84:1941–9. doi: 10.1099/vir.0.19112-0
100. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet. (2004) 36:1099–104. doi: 10.1038/ng1424
101. Dickerson SJ, Xing Y, Robinson AR, Seaman WT, Gruffat H, Kenney SC. Methylation-dependent binding of the epstein-barr virus BZLF1 protein to viral promoters. PLoS Pathog. (2009) 5:e1000356. doi: 10.1371/journal.ppat.1000356
102. Gruffat H, Manet E, Rigolet A, Sergeant A. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. (1990) 18:6835–43. doi: 10.1093/nar/18.23.6835
103. Quinlivan EB, Holley-Guthrie EA, Norris M, Gutsch D, Bachenheimer SL, Kenney SC. Direct BRLF1 binding is required for cooperative BZLF1/BRLF1 activation of the Epstein-Barr virus early promoter, BMRF1. Nucleic Acids Res. (1993) 21:1999–2007. doi: 10.1093/nar/21.8.1999
104. Adamson AL, Darr D, Holley-Guthrie E, Johnson RA, Mauser A, Swenson J, et al. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J Virol. (2000) 74:1224–33. doi: 10.1128/JVI.74.3.1224-1233.2000
105. Darr CD, Mauser A, Kenney S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J Virol. (2001) 75:6135–42. doi: 10.1128/JVI.75.13.6135-6142.2001
106. Croft NP, Shannon-Lowe C, Bell AI, Horst D, Kremmer E, Ressing ME, et al. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. (2009) 5:e1000490. doi: 10.1371/journal.ppat.1000490
107. Rowe M, Glaunsinger B, van Leeuwen D, Zuo J, Sweetman D, Ganem D, et al. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc Natl Acad Sci USA. (2007) 104:3366–71. doi: 10.1073/pnas.0611128104
108. Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. (2012) 8:e1002704. doi: 10.1371/journal.ppat.1002704
109. Paya CV, Fung JJ, Nalesnik MA, Kieff E, Green M, Gores G, et al. Epstein-Barr virus-induced posttransplant lymphoproliferative disorders. ASTS/ASTP EBV-PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting. Transplantation. (1999) 68:1517–25. doi: 10.1097/00007890-199911270-00015
110. Dugan JP, Haverkos BM, Villagomez L, Martin LK, Lustberg M, Patton J, et al. Complete and durable responses in primary central nervous system posttransplant lymphoproliferative disorder with zidovudine, ganciclovir, rituximab, and dexamethasone. Clin Cancer Res. (2018) 24:3273–81. doi: 10.1158/1078-0432.CCR-17-2685
111. Fink SE, Gandhi MK, Nourse JP, Keane C, Jones K, Crooks P, et al. A comprehensive analysis of the cellular and EBV-specific microRNAome in primary CNS PTLD identifies different patterns among EBV-associated tumors. Am J Trans. (2014) 14:2577–87. doi: 10.1111/ajt.12858
112. Porcu P, Eisenbeis CF, Pelletier RP, Davies EA, Baiocchi RA, Roychowdhury S, et al. Successful treatment of posttransplantation lymphoproliferative disorder (PTLD) following renal allografting is associated with sustained CD8+ T-cell restoration. Blood. (2002) 100:2341–8. doi: 10.1182/blood-2002-01-0210
113. Roychowdhury S, Peng R, Baiocchi RA, Bhatt D, Vourganti S, Grecula J, et al. Experimental treatment of Epstein-Barr virus-associated primary central nervous system lymphoma. Cancer Res. (2003) 63:965–71.
114. Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. The Epstein-Barr Virus (EBV)-Encoded Protein Kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol. (2010) 84:4534–42. doi: 10.1128/JVI.02487-09
115. Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, et al. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J Virol. (2012) 86:4752–61. doi: 10.1128/JVI.06768-11
116. Woellmer A, Arteaga-Salas JM, Hammerschmidt W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. (2012) 8:e1002902. doi: 10.1371/journal.ppat.1002902
117. Raez L, Cabral L, Cai JP, Landy H, Sfakianakis G, Byrne GE Jr, et al. Treatment of AIDS-related primary central nervous system lymphoma with zidovudine, ganciclovir, and interleukin 2. AIDS Res Hum Retrovir. (1999) 15:713–9. doi: 10.1089/088922299310809
118. Harrington WJ Jr, Cabral L, Cai JP, Chan ASS, Wood C. Azothymidine and interferon-alpha are active in AIDS-associated small non-cleaved cell lymphoma but not large-cell lymphoma. Lancet. (1996) 348:833. doi: 10.1016/S0140-6736(05)65260-9
119. Baselga J, Krown SE, Telzak EE, Filippa DA, Straus DJ. Acquired immune deficiency syndrome-related pulmonary non-Hodgkin lymphoma regressing after zidovudine therapy. Cancer. (1993) 71:2332–4. doi: 10.1002/1097-0142(19930401)71:7<2332::AID-CNCR2820710726>3.0.CO;2-P
120. Aboulafia DM, Ratner L, Miles SA, Harrington WJ Jr. Consortium AAMCT. Antiviral and immunomodulatory treatment for AIDS-related primary central nervous system lymphoma: AIDS Malignancies Consortium pilot study 019. Clin Lymph Myel. (2006) 6:399–402. doi: 10.3816/CLM.2006.n.017
121. Evens AM, Choquet S, Kroll, Desrosiers AR, Jagadeesh D, Smith SM, et al. Primary CNS posttransplant lymphoproliferative disease (PTLD): an international report of 84 cases in the modern era. Am J Trans. (2013) 13:1512–22. doi: 10.1111/ajt.12211
122. Daibata M, Bandobashi K, Kuroda M, Imai S, Miyoshi I, Taguchi H. Induction of lytic Epstein-Barr virus (EBV) infection by synergistic action of rituximab and dexamethasone renders EBV-positive lymphoma cells more susceptible to ganciclovir cytotoxicity in vitro and in vivo. J Virol. (2005) 79:5875–9. doi: 10.1128/JVI.79.9.5875-5879.2005
123. Stoker SD, Novalic Z, Wildeman MA, Huitema AD, Verkuijlen SA, Juwana H, et al. Epstein-Barr virus-targeted therapy in nasopharyngeal carcinoma. J Cancer Res Clin Oncol. (2015) 141:1845–57. doi: 10.1007/s00432-015-1969-3
124. Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. (2002) 62:1920–6.
125. Liu SF, Wang H, Li ZJ, Deng XY, Xiang H, Tao YG, et al. Aspirin induces lytic cytotoxicity in Epstein-Barr virus-positive cells. Eur J Pharmacol. (2008) 589:8–13. doi: 10.1016/j.ejphar.2008.04.025
126. Li H, Hu J, Luo X, Bode AM, Dong Z, Cao Y. Therapies based on targeting Epstein-Barr virus lytic replication for EBV-associated malignancies. Cancer Sci. (2018) 109:2101–8. doi: 10.1111/cas.13634
127. Mentzer SJ, Fingeroth J, Reilly JJ, Perrine SP, Faller DV. Arginine butyrate-induced susceptibility to ganciclovir in an Epstein-Barr-virus-associated lymphoma. Blood Cells Mol Dis. (1998) 24:114–23. doi: 10.1006/bcmd.1998.0178
128. Anisimova E, Prachova K, Roubal J, Vonka V. Effects of n-butyrate and phorbol ester (TPA) on induction of Epstein-Barr virus antigens and cell differentiation. Arch Virol. (1984) 81:223–37. doi: 10.1007/BF01309995
129. Saemundsen AK, Kallin B, Klein G. Effect of n-butyrate on cellular and viral DNA synthesis in cells latently infected with Epstein-Barr virus. Virology. (1980) 107:557–61. doi: 10.1016/0042-6822(80)90326-8
130. Perrine SP, Hermine O, Small T, Suarez F, O'Reilly R, Boulad F, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood. (2007) 109:2571–8. doi: 10.1182/blood-2006-01-024703
131. Ghosh SK, Perrine SP, Williams RM, Faller DV. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood. (2012) 119:1008–17. doi: 10.1182/blood-2011-06-362434
132. Robertson KD, Hayward SD, Ling PD, Samid D, Ambinder RF. Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol Cell Biol. (1995) 15:6150–9. doi: 10.1128/MCB.15.11.6150
133. Chan AT, Tao Q, Robertson KD, Flinn IW, Mann RB, Klencke B, et al. Azacitidine induces demethylation of the Epstein-Barr virus genome in tumors. J Clin Oncol. (2004) 22:1373–81. doi: 10.1200/JCO.2004.04.185
134. Shamay M, Hand N, Lemas MV, Koon HB, Krown SE, Wrangle J, et al. CpG methylation as a tool to characterize cell-free Kaposi sarcoma herpesvirus DNA. J Infect Dis. (2012) 205:1095–9. doi: 10.1093/infdis/jis032
135. Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. (1994) 330:1185–91. doi: 10.1056/NEJM199404283301703
136. Riddell, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. (1992) 257:238. doi: 10.1126/science.1352912
137. Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. (1995) 345:9–13. doi: 10.1016/S0140-6736(95)91150-2
138. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. (2010) 115:925. doi: 10.1182/blood-2009-08-239186
139. Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood. (2007) 110:2838–45. doi: 10.1182/blood-2007-05-091280
140. Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. (2014) 32:798–808. doi: 10.1200/JCO.2013.51.5304
141. Cho S-G, Kim N, Sohn H-J, Lee SK, Oh ST, Lee H-J, et al. Long-term outcome of extranodal NK/T cell lymphoma patients treated with postremission therapy using EBV LMP1 and LMP2a-specific CTLs. Mol Ther. (2015) 23:1401–9. doi: 10.1038/mt.2015.91
142. Ma S-D, Xu X, Jones R, Delecluse H-J, Zumwalde NA, Sharma A, et al. PD-1/CTLA-4 Blockade inhibits Epstein-Barr Virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. (2016) 12:e1005642-e.
143. Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget. (2016) 7:32925–32. doi: 10.18632/oncotarget.9076
144. Kwong YL, Chan TSY, Tan D, Kim SJ, Poon LM, Mow B, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood. (2017) 129:2437–42. doi: 10.1182/blood-2016-12-756841
145. Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O'Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. (2012) 18:1611–8. doi: 10.1158/1078-0432.CCR-11-1942
Keywords: EBV (Epstein-Barr virus), EBV-associated cancers, Lymphoproliferative disease (LPD), EBV latency, viral life cycle inhibitors
Citation: Dugan JP, Coleman CB and Haverkos B (2019) Opportunities to Target the Life Cycle of Epstein-Barr Virus (EBV) in EBV-Associated Lymphoproliferative Disorders. Front. Oncol. 9:127. doi: 10.3389/fonc.2019.00127
Received: 03 December 2018; Accepted: 13 February 2019;
Published: 15 March 2019.
Edited by:
Massimo Libra, Università degli Studi di Catania, ItalyReviewed by:
Valli De Re, Centro di Riferimento Oncologico di Aviano (IRCCS), ItalyYago Nieto, University of Texas MD Anderson Cancer Center, United States
Copyright © 2019 Dugan, Coleman and Haverkos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bradley Haverkos, YnJhZGxleS5oYXZlcmtvc0B1Y2RlbnZlci5lZHU=