 Catherine Vander Linden
Catherine Vander Linden Cyril Corbet
Cyril Corbet- Pole of Pharmacology and Therapeutics (FATH), Institut de Recherche Expérimentale et Clinique, UCLouvain, Brussels, Belgium
Cancer stem cells (CSC) or tumor-initiating cells represent a small subpopulation of cells within the tumor bulk that share features with somatic stem cells, such as self-renewal and pluripotency. From a clinical point of view, CSC are thought to be the main drivers of tumor relapse in patients by supporting treatment resistance and dissemination to distant organs. Both genome instability and microenvironment-driven selection support tumor heterogeneity and enable the emergence of resistant cells with stem-like properties, when therapy is applied. Besides hypoxia and nutrient deprivation, acidosis is another selection barrier in the tumor microenvironment (TME) which provides a permissive niche to shape more aggressive and fitter cancer cell phenotypes. This review describes our current knowledge about the influence of the “acidic niche” on the stem-like phenotypic features of cancer cells. In addition, we briefly survey new therapeutic options that may help eradicate CSC by integrating and/or exploiting the acidic niche, and thereby contribute to prevent the occurrence of therapy resistance as well as metastatic dissemination.
Introduction
Despite a broader arsenal of (targeted) therapies, prognosis is still very poor for several types of cancer. At present, most patients with advanced cancers die because tumor cells have a remarkable capacity to develop drug resistance, through both genetic and non-genetic mechanisms (1). Current therapeutic failures are thought to originate, at least partly, from the Darwinian nature of cancer according to which, both genetic alterations and highly selective local microenvironments (the so-called niches) help to develop tumor cell adaptive phenotypes to sustain malignant progression (2, 3). Indeed, while current clinical protocols aim to eradicate the tumor as quickly as possible [i.e., maximum-tolerated dose (MTD) strategies], they often lead to therapeutic failure due to the occurrence of tumor relapse and dissemination of cancer cells to distant organs, after an initial tumor response or the lack of effectiveness at the outset. This alarming observation is thought to arise from two neglected evolutionary concepts. First, phenotypic heterogeneity within a tumor makes it very likely that resistant cells are present before therapy regardless of the cancer genetic landscape (i.e., de novo drug resistance). Second, MTD-based therapy promotes the growth of resistant populations via the clonal selection of cancer cells with adapted phenotypes and elimination of all potentially competing populations (the so-called “competitive release”) (4).
Cancer stem cells (CSC), also referred to as tumor-initiating cells, have been thought to actively contribute to the so-called “minimal residual disease” which is a small population of cancer cells that survive drug treatment and re-initiate the malignant disease, with poor outcome, even some years later (Figure 1) (5, 6). Within the tumor mass, CSC are typically dormant (i.e., non- or slow-proliferating) but they have also the capacity to proliferate either for their maintenance (self-renewal) or for the generation of progenitor tumor cells (clonal tumor initiation and long-term repopulation) (Figure 1) (7). CSC are located in specific niches, determined by tumor microenvironment (TME) peculiarities, that enable them to be phenotypically better adapted and more prone to regain fitness (i.e., ability to survive and proliferate in a given environment) than other cancer cell populations within the tumor bulk (8, 9). Moreover, these niches are thought to help protect CSC from the immune system, resist conventional treatments by reducing their proliferation state and/or evading apoptosis, and facilitate their metastatic potential (9–11). Since most of the normal stem cell populations (e.g., hematopoietic, mesenchymal, and neural stem cells) are located in hypoxic niches, how hypoxia contributes to the maintenance and/or emergence of the CSC phenotype has been extensively studied and reviewed over the years (12–14). Moreover, the role of stromal cells (e.g., cancer-associated fibroblasts, adipocytes, endothelial cells, or immune cells), as cellular components of specific CSC-supportive niches, has been also reported elsewhere (15–18). In this review, we describe how acidosis, another hallmark of TME, may act as a permissive niche for adaptive stem-like cancer cell phenotypes. We also discuss the contribution of the acidic niche to tumor initiation and progression, as well as to therapy resistance and metastatic dissemination. This review finally explores potential therapeutic strategies that may help eradicate CSC by integrating and/or exploiting the acidosis-induced phenotypic alterations.
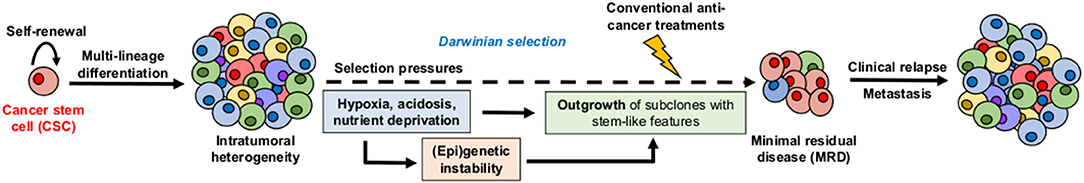
Figure 1. Hypothetical model for the role of cancer stem cells (CSC) and microenvironmental selection pressure in clinical relapse. CSC display both self-renewal capacity and multi-lineage differentiation potential, leading to intratumoral heterogeneity. Local TME peculiarities such as hypoxia, acidosis, and nutrient deprivation act as high selection pressures for adaptive stem-like phenotypes that participate to therapy resistance, minimal residual disease, and long-term clinical relapse.
Acidosis and CSC-Related Phenotypic Features
Glycolysis, Mitochondrial Respiration, and Tumor Acidosis
Acidosis is now considered as a hallmark of the microenvironment in solid tumors with mean values of extracellular pH (pHe) ranging from 6.2 to 6.8 (19, 20). Although initially described as a strict consequence of the exacerbated glycolysis in tumor cells and the disorganized tumor vasculature, accumulation of H+ ions in the TME also results from the mitochondrial respiration-derived CO2 hydration (Figure 2) (21, 22). Direct measurements of both intratumoral pO2 and pH have indeed revealed a spatial heterogeneity as well as an imperfect overlapping of hypoxia and acidosis gradients, with the existence of acidic areas that are also well-oxygenated (23, 24). Other studies have also shown that glycolysis-impaired or LDH-deficient tumor cell lines still have the ability to acidify the extracellular environment in vivo (25–27). More recently, Hulikova et al. (28) reported a role for stromal cells in the venting of hypoxia-induced acidosis, with gap junction-mediated connections that enable the cell-to-cell shuttling of cancer cell-derived H+ ions and their venting at far distance from the hypoxic regions.
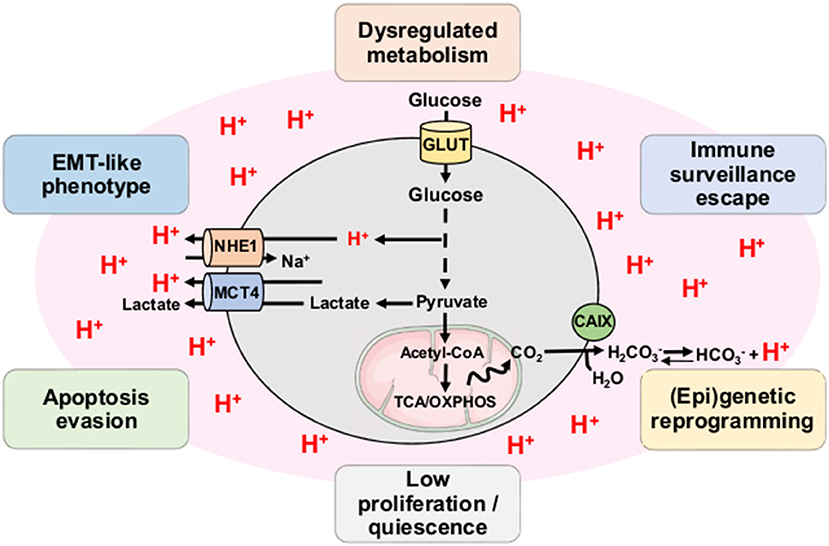
Figure 2. Tumor acidosis influences and maintains CSC-related phenotypic features. Both exacerbated glycolysis and mitochondrial respiration-derived CO2 hydration in tumor account for production of H+ ions and subsequent environment acidification. Tumor acidosis contributes to the emergence and/or maintenance of stem-like phenotypic features such as dysregulated metabolism, immune surveillance escape, (epi)genetic reprogramming, low proliferation, apoptosis evasion, and EMT-like phenotype. CAIX, carbonic anhydrase IX; MCT4, monocarboxylate transporter 4; NHE1, sodium-hydrogen antiporter 1; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid cycle.
Tumor Acidosis and CSC-Related Gene Reprogramming
Although the effects of acid exposure on stem cell phenotype have been under controversy (29), there are now several lines of evidence for the role of tumor acidosis in the emergence and/or maintenance of CSC phenotypic features (e.g., slow-proliferating state, invasive capacities, and therapy resistance) that may participate to the minimal residual disease and the long-term clinical dormancy/relapse (30, 31) (Figure 2). Nevertheless, the straightforward contribution of a transcriptional acidosis-responsive element that could mediate gene reprogramming has not been reported so far. Several studies have however identified the hypoxia-inducible factor 2α (HIF2α) as a master regulator of gene expression in cancer cells, under acidic conditions (32–35) (Figure 3). Besides an increase of HIF2α abundance, acidosis also enhances its transcriptional activity through the activation of NAD+-dependent histone deacetylases sirtuins 1 and 6 (SIRT1/6) (32, 36), thereby leading to the deacetylation of lysine residues in the HIF2α regulatory amino-terminal transactivation domain (N-TAD) region (32, 37). Another study has shown that highly acidic conditions (pH 5.8–6.2) can trigger nucleolar sequestration of the von Hippel-Lindau (VHL) tumor suppressor protein and subsequent HIF2α stabilization (35) (Figure 3). However, a recent report has observed that VHL-deficient renal carcinoma cells are still responsive to acidosis with an increase in HIF2α levels, and that the acidic pH-induced stabilization of HIF2α is mediated by the HSP90 chaperone protein (33). Acidic pH, under normoxia, was also found to induce L-2-hydroxyglutarate (L-2HG) production through several mechanisms including the activation of lactate dehydrogenase A and malate dehydrogenase 2 enzymes, the inhibition of the mitochondrial L-2HG removal enzyme L-2HG dehydrogenase and the stimulation of the reverse reaction of isocitrate dehydrogenase (carboxylation of α-ketoglutarate to isocitrate), thereby leading to stabilization of HIF-1α (38, 39). Although 2HG-mediated epigenetic changes have been thought to support a stem-like cell state (40–42), the direct implication of an acidosis/2HG/HIF-2α signaling axis in cancer stem cell biology remains to be determined (Figure 3). These data are however reminiscent of the occurrence of lactic acidosis in some 2-HG aciduric patients (43) and it could be hypothesized that acidotic episodes may induce 2HG accumulation.
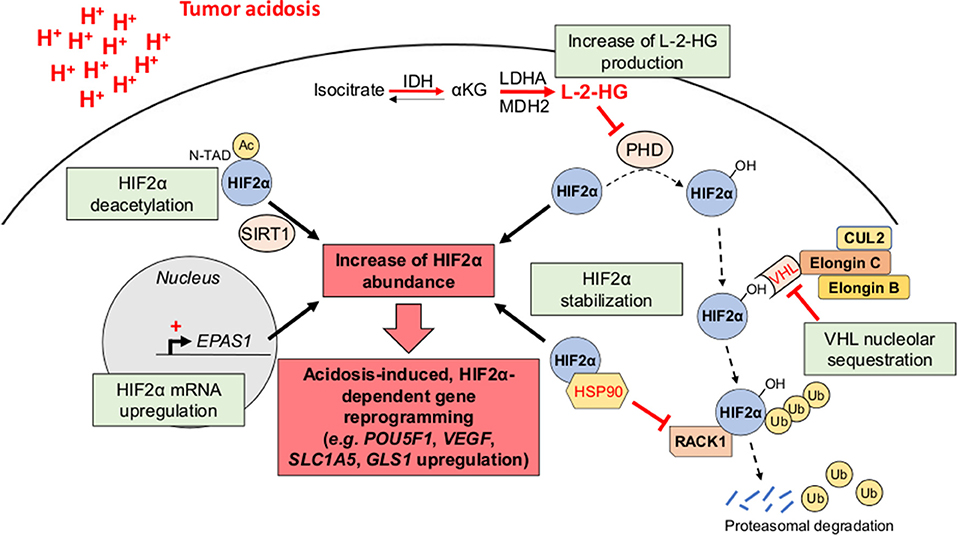
Figure 3. HIF2α as a transcriptional regulator of acidosis-induced gene reprogramming. Tumor acidosis leads to an increase of HIF2α abundance either by directly inducing EPAS1 gene transcription or by promoting HIF2α protein stabilization. For the latter, several mechanisms have been proposed, including the nucleolar sequestration of the von Hippel-Lindau (VHL) protein, the competitive binding of the chaperone heat shock protein 90 (HSP90) instead of the receptor of activated protein kinase C (RACK1), and an increased L-2-hydroxyglutarate (L-2-HG) production, thereby inhibiting HIF2α hydroxylation and subsequent proteasomal degradation. Besides its stabilization, acidosis also enhances HIF2α transcriptional activity through sirtuin-mediated deacetylation of lysine residues in its regulatory amino-terminal transactivation domain (N-TAD). αKG, α-ketoglutarate; Ac, acetyl group; CUL2, cullin-2; IDH, isocitrate dehydrogenase; LDHA, lactate dehydrogenase A; MDH2, malate dehydrogenase 2; PHD, prolyl hydroxylase; Ub, ubiquitin.
In glioma, where HIF2α is now considered as a marker of CSC (44), acidic conditions were documented to increase both the expression of a panel of glioma stem cells (GSC)-associated genes, including POU5F1 (OCT4), OLIG2, and NANOG, independently of a restricted O2 availability (34), and the fraction of cells positive for the GSC markers CD133 and CD15 (33). Acidosis also promotes production of angiogenic factors such as VEGF and IL-8 in GSC that favor tumor growth through paracrine effects (34). Acidosis has been also correlated to stem cells through the role of mesenchymal stem cells (MSCs) within the tumor stroma. MSCs grown in acidic pH express a higher level of transforming growth factor-β (TGFβ) that induces an epithelial-to-mesenchymal transition (EMT)-like phenotype in melanoma cells (45). Acidosis-exposed MSCs also stimulate the invasive and clonogenic capacities of osteosarcoma (OS) cells via the secretion of a variety of factors, including colony-stimulating factor 2 (CSF2, also referred to as GM-CSF), CSF3 (also known as G-CSF), bone morphogenetic protein 2 (BMP2), and interleukins 6 and 8 (IL6 and IL8) (46). MSCs, under acidic pH conditions, can also promote a stem cell phenotype in OS, by enhancing the sphere formation capacity and chemoresistance, via the induction of octamer-binding protein 4 (OCT4) (46). Finally, some studies have shown that cancer cell exposure to acidic conditions was associated with changes in the epigenetic landscape, including histone acetylation levels (36, 47), as well as a reprogramming of the genome-wide transcriptome (48, 49). Further investigations are however needed to study in depth the influence of the acidic niche on CSC (epi)genetic pattern, in other cancer types, but also in preclinical in vivo models.
Tumor Acidosis and Multidrug Resistance Phenotype
As stated above, CSC are resistant to anti-cancer treatments and they support long-term cancer cell survival and tumor relapse in patients. Acidosis has been directly correlated with drug resistance since it can reduce the passive permeability of weak base chemotherapeutic agents (e.g., doxorubicin, paclitaxel, mitoxantrone) by increasing their protonation state (the so-called “ion trapping” phenomenon) (50) (Figure 4). Several studies have indeed shown that neutralization of tumor-derived acid with systemic buffers (e.g., sodium bicarbonate, imidazoles, and lysine) (51) or the reversal of the pH gradient with proton pump inhibitors (e.g., omeprazole, esomeprazole) (52–54) can restore the sensitivity of cancer cells to chemotherapeutic drugs, such as doxorubicin.
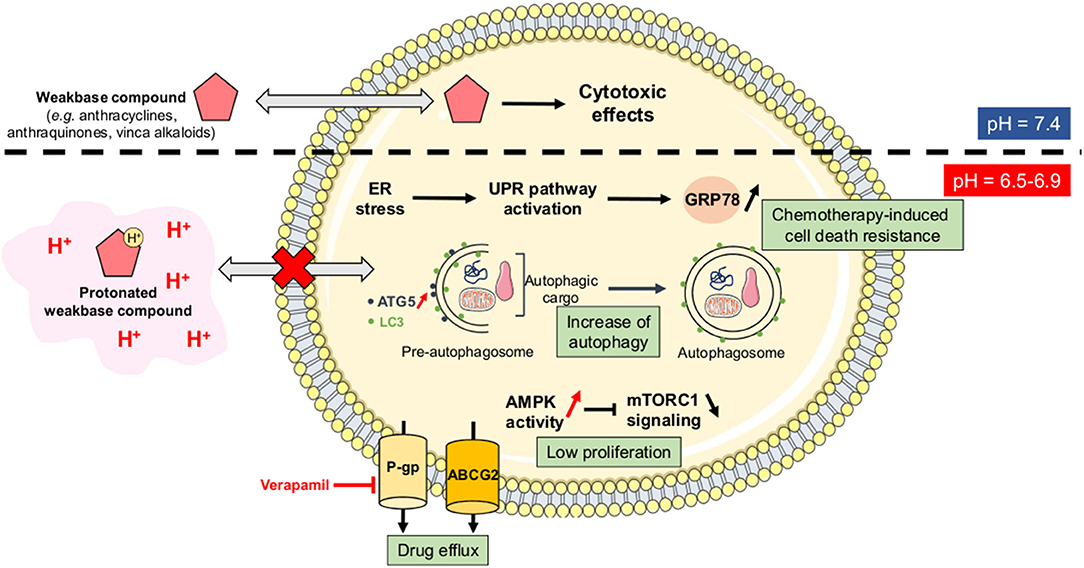
Figure 4. Ion trapping phenomenon and acidosis-induced multidrug resistance. Under acidic conditions, several weak base chemotherapeutic agents become positively charged species and therefore lose their ability to diffuse passively across cellular membranes (the so-called “ion trapping phenomenon”). Tumor acidosis also directly promotes a resistance-sustaining cell phenotype through several mechanisms including the upregulation of glucose-related protein 78 (GRP78) or Bip, an increased autophagic flux, a lower proliferation rate, and an increase of drug efflux capacities via the P-glycoprotein (Pgp) and the ATP-binding cassette protein ABCG2. AMPK, AMP-activated protein kinase; ATG5, autophagy-related protein 5; ER, endoplasmic reticulum; LC3, microtubule-associated protein light chain 3; mTORC1, mammalian target of rapamycin complex 1; UPR, unfolded protein response.
Besides this direct effect on the physico-chemical nature of anti-cancer drugs, acidosis can also promote a resistance-sustaining phenotype in cancer cells through different mechanisms. Indeed, while conventional treatments such as chemotherapies and/or radiation therapy are usually designed to eradicate highly proliferative cells, acidosis has been reported to reduce the proliferation status of cancer cells, that in some conditions even become relatively dormant (quiescent). Several studies have shown that cancer cells, exposed to acute acidic conditions, exhibit a low-proliferating phenotype as a consequence of a non-permissive intracellular acidification, an increased activity of the metabolic stress sensor AMP-activated protein kinase (AMPK) and a reduction of the multi-component mechanistic target of rapamycin complex 1 (mTORC1) signaling (Figure 4) (55–58). Another study reported that acidic conditions triggered a reduced proliferation state and high resistance to apoptosis in BRAFV600E mutant melanoma cells (59). Acidosis-mediated melanoma cell phenotype was also associated with an acquired resistance to vemurafenib, a BRAF inhibitor, that could be overcome by treatment with everolimus, an inhibitor of mTOR activity (59).
Acidosis can also increase drug efflux capacities, both in in vitro and in vivo cancer models, through the upregulation and activation of membrane transporters such as the ATP-binding cassette protein ABCG2 (60), and the P-glycoprotein (P-gp) (61–64). For the latter, acidosis-induced chemotherapy resistance is mainly mediated through p38 signaling and can be reversed by treatment with verapamil, a P-gp inhibitor (61, 63). Another mechanism reported to mediate acidosis-induced therapy resistance is the unfolded protein response (UPR) pathway. Indeed, acidic conditions can trigger endoplasmic reticulum stress, thereby resulting in UPR activation and overexpression of the glucose-regulated protein 78 (GRP78) chaperone that contributes to chemotherapy-induced cell death resistance (65–67) (Figure 4). Finally, autophagy has also been described as an adaptive survival mechanism for cancer cells under acidosis, in particular through the enhanced expression of autophagy-related protein 5 (ATG5) (68, 69). Although an increased autophagic flux has already been associated with chemotherapy resistance in a variety of cancers (70), Avnet et al. (52) have reported that acidosis-induced doxycycline resistance in OS cells is not supported by autophagy since ATG5 gene silencing cannot restore drug sensitivity. These observations suggest that acidosis-driven drug resistant phenotype might be tumor type-dependent and/or supported by a variety of mechanisms that are redundant and have therefore the ability to compensate for the inhibition of one of them.
Tumor Acidosis and Immune Escape
Besides their ability to resist conventional treatments, CSC needs also to evade immune surveillance to support cellular dormancy and long-term clinical relapse. Acidic pHe conditions have been reported to decrease T cell proliferation and their capacity to produce a variety of cytokines, including interleukin-2 (IL-2), interferon-γ (IFN-γ), granzyme B and perforin, in a dose-dependent manner (71). Tumor acidosis also impairs immune system functions by reducing dendritic cell maturation (72), monocyte-derived tumor necrosis factor (TNF) secretion (73), and natural killer (NK) activity (74). Indeed, tumor-derived H+ and/or lactate accumulation, in the extracellular compartment, supports the suppressive effect on T cell function by inhibiting the glycolytic pathway within T cells (71, 73). Moreover, inhibition of the transcription factor nuclear factor of activated T cells (NFAT) has been proposed to mediate the blockade of IFNγ production in T cells and NK cells, upon intracellular accumulation of H+ and lactate (75). The same authors also proposed a direct role of LDHA for lactate generation and the subsequent inhibition of tumor surveillance by T and NK cells (75). Mouse melanomas with reduced H+ and lactate generation (upon LDHA genetic knockdown) actually exhibit a lower growth rate than control tumors and show an increased infiltration of IFNγ-producing T and NK cells (75). Importantly, this effect was lost when LDHA-knockdown tumors were grown either in immunodeficient Rag2−/−γc−/− mice or in Ifng−/− mice. Another study has revealed that phosphoenolpyruvate (PEP), a glycolytic intermediate, can act as a metabolic checkpoint to sense glucose availability and modulate a Ca2+-NFAT signaling, such that a decrease of PEP intracellular concentration triggers a T cell anergy (76). A recent study also reported that extracellular acidification, within melanoma tumors, can be sensed by tumor-associated macrophages (TAMs), resulting in macrophage polarization and promotion of tumor growth (77). Mechanistically, a macrophage G-protein-coupled receptor (GCPR) can sense tumor acidification and leads to expression, by macrophages, of the inducible cyclic AMP early repressor (ICER), a transcriptional repressor that mediates the functional polarization into TAMs, which support tumor growth (77). Neutralization of tumor acidity with sodium bicarbonate (78), or with proton pump inhibitors (79) helps to improve the response to antitumor immunotherapy by restoring T cell cytolytic activity and cytokine secretion together with an increased tumor lymphocyte infiltration in mouse models but also in human cancer patients.
Tumor Acidosis and Metabolic Rewiring
Current controversy about the metabolic characteristics of CSC, described as either strictly glycolytic (80, 81) or instead dependent on mitochondrial metabolism (82, 83) may simply reflect their adaptability upon microenvironmental fluctuations. Here below, we will strictly focus on the current understanding of the influence of a low pH on cancer cell metabolism in an attempt to delineate the anticipated interplay between stemness and metabolism in the acidic TME niche.
Indeed, while cancer cells can use a variety of substrates to fulfill their need in energy and/or biosynthetic precursors (84, 85), we have recently documented, by using tumor cell lines chronically adapted to acidosis, a metabolic shift toward a preferential use of glutamine to the detriment of glucose utilization (32). HIF2α was found to drive glutamine metabolism by increasing expression of the glutamine transporter ASC-like Na+-dependent neutral amino acid transporter 2 (ASCT2) and glutaminase 1 (GLS1) (32). On the contrary, HIF1α activity and expression are reduced under chronic acidosis, thereby decreasing the expression of several target genes, including the glucose transporter GLUT1 and the monocarboxylate transporter 4 (MCT4) (32). Another study has also reported that several breast cancer cell lines, exposed to acute acidic conditions (24 h), show an increased glutaminolysis and redirection of glucose toward the oxidative branch of the pentose phosphate pathway (PPP), via a p53-dependent induction of glucose-6-phosphate dehydrogenase (G6PD), and glutaminase GLS2 expression (56). These metabolic changes certainly support an antioxidant response of acidosis-exposed cancer cells by increasing NADPH production and may have yet a broader impact considering how glycolysis inhibition may lead to various defects in protein glycosylation (86).
Besides changes in glutamine and glucose metabolism, tumor acidosis has also been related to profound alterations in lipid metabolism (Figure 5). Indeed, acidosis-induced reductive carboxylation of glutamine-derived α-ketoglutarate was reported as a source of acetyl-CoA from citrate to neo-synthesize fatty acids (36). Acetate was identified as another source of acetyl-CoA for fatty acid synthesis, under acidic conditions, in response to activation of sterol regulatory element-binding protein 2 (SREBP2) and subsequent upregulation of acyl-CoA synthetase short-chain family member 2 (ACSS2) (87). Importantly, fatty acid oxidation (FAO) is also stimulated in acidosis-exposed cancer cells (36, 56). This apparent juxtaposition of mitochondrial FA catabolism and cytosolic FA synthesis is rendered possible through a sirtuin-mediated histone deacetylation of the ACACB gene, encoding the mitochondrion-associated acetyl-CoA carboxylase 2 (ACC2) enzyme that normally prevents the degradation of neo-synthesized fatty acids in healthy tissues (Figure 5) (36).
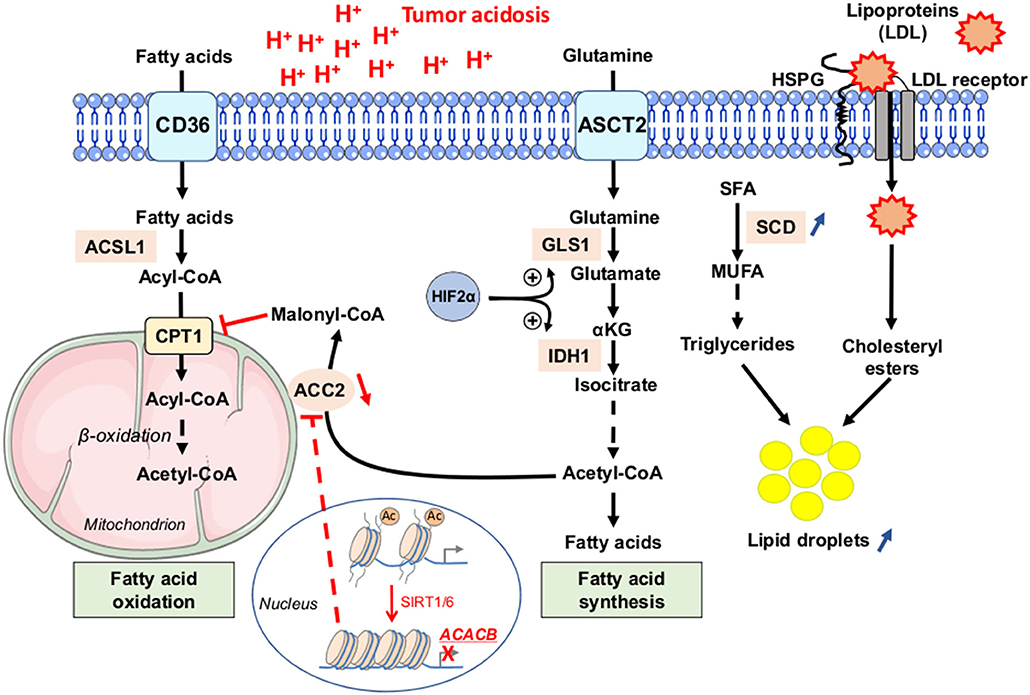
Figure 5. Fatty acid metabolism dysregulation as a phenotypic feature of cancer cells under acidic conditions. Tumor cells, exposed to low pH conditions, exhibit profound alterations in lipid metabolism, with a concomitance of fatty acid oxidation (FAO) in mitochondria and synthesis from glutamine (FAS) in the cytoplasm rendered possible through a sirtuin 1 and 6 (SIRT1/6)-mediated downregulation of the acetyl-CoA carboxylase 2 (ACC2). Saturated fatty acids (SFA) can be actively transformed by stearoyl-CoA desaturase enzyme (SCD) into mono-unsaturated FA (MUFA), and then into triglycerides. Acidosis-exposed tumor cells also increase lipoprotein uptake in a heparan sulfate proteoglycan (HSPG)-dependent manner. Increased accumulation of neutral lipids (i.e., triglycerides or LDL particle-derived cholesteryl esters) into lipid droplets is observed in cancer cells under acidic pH conditions. Ac, acetyl group; ACSL1, long-chain fatty acid CoA ligase 1; ASCT2, alanine serine cysteine-preferring transporter 2; CD36, cluster of differentiation 36; CPT1, carnitine palmitoyl transferase 1; GLS1, glutaminase 1; IDH1, isocitrate dehydrogenase 1; LDL, low-density lipoprotein.
Mild acidosis can also change mitochondrial morphology to preserve efficient ATP production regardless of O2 levels (88); these data are supportive of the concept of an acidic niche that shapes cancer cells toward an OXPHOS-dependent metabolic phenotype. Interestingly, compelling evidence indicates that cancer stem-like cells, including therapy-resistant tumor cells, mostly rely on mitochondrial respiration and OXPHOS for growth (82, 83, 89, 90). Moreover, a recent study reported the isolation and characterization of a new distinct subpopulation of proliferating CSC, called “energetic” CSC, showing a significantly increased oxidative metabolism and mitochondrial mass, as well as a strict reliance on OXPHOS when grown in 3D anchorage-independent conditions (91). All these studies position an elevated mitochondrial metabolism as an important phenotypic adaptation for cancer stem-like cells and expand on the anticancer potential of mitochondrial biogenesis inhibitors, such as doxycycline or tigecycline (92).
More precisely, the role of FA metabolism, together with the concept of an adipose tissue niche, has been reported to support tumor growth and resistance to chemotherapy (18, 93, 94). Indeed, some investigators have documented that cancer cells transiently exposed to low pH conditions may accumulate neutral lipids into lipid droplets (LD) (95, 96). More recently, Menard et al. (97) reported that acute exposure of cancer cells to acidosis increases the uptake of lipoproteins, in a heparan sulfate proteoglycan (HSPG)-dependent manner, which are then accumulated into LD. This LD-loaded phenotype is associated with enhanced spheroid-forming capacity in vitro and metastatic potential in vivo; pharmacological or genetic targeting of HSPG could fully reverse these effects (97). This acidosis-induced LD-loaded phenotype is reminiscent of the accumulation of neutral lipids observed in colorectal CSC populations (Figure 5) (98–100). High levels of LD were actually found as a distinctive mark of CSC in colorectal cancer, as revealed by label-free Raman spectroscopy, and they correlated with CSC markers such as CD133 and Wnt pathway activity (98). Finally, an elegant study revealed that increased lipid desaturation, via the activity of the stearoyl-CoA desaturase 1 (SCD1) enzyme, is essential to stem-like characteristics in ovarian cancer cells (94). Indeed, the authors have shown that ovarian CSC (ALDH+/CD133+) have a higher ratio of unsaturated to saturated fatty acids, and this ratio is essential for the cells to retain stemness. Further investigations are however needed to address whether acidic conditions in the TME also induce similar changes of the lipid profile in cancer (stem) cells.
Acidosis-Based Therapeutic Strategies to Tackle CSC Compartment
Therapeutic Strategies to Directly Manipulate/Exploit Extracellular pH
Utilization of systemic buffers, such as sodium bicarbonate, imidazoles and lysine, was proposed several years ago as an obvious strategy to directly neutralize the tumor-derived acid and hamper tumor cell aggressiveness (101–106). Importantly, all these studies actually showed that oral administration of such buffers reduces the metastatic dissemination of cancer cells in animal models without affecting primary tumor growth. Many groups have already documented that acidosis could facilitate migration/invasion of cancer cells in vitro as well as metastasis formation in vivo via the activation of proteases (101, 107, 108), the secretion of pro-angiogenic factors (109) or the promotion of an EMT-like phenotype (110, 111). Further investigations are however needed to address whether interfering with tumor acidification is directly correlated with a decrease in stem-like cell population. As stated above, several studies have shown that buffer therapy (51) can restore the sensitivity of cancer cells to chemotherapeutic drugs, such as doxorubicin. There is however no direct evidence for a straightforward modulation of acidosis-induced cancer cell phenotype (vs. changes of the physico-chemical properties of the drug) by systemic buffer administration.
Because of their relative small proportion into the tumor bulk and their phenotypic features strongly associated with the local microenvironment peculiarities, CSCs are indeed inherently difficult to isolate and to maintain in culture, making almost unfeasible a direct CSC-selective screening of small molecules. This obstacle has prompted the artificial induction of EMT to produce cells displaying CSC-like characteristics suitable for high-throughput phenotypic screening (112–114). Salinomycin, an ionophore antibiotic, was identified as a selective agent against experimentally-induced CSCs (113, 115). Interestingly, salinomycin-induced cytotoxic effects were enhanced under conditions of transient and chronic acidosis, with in particular an inhibition of autophagic flux in breast CSC-like cells (116).
Tumor acidosis can also be exploited in order to selectively deliver anti-cancer drugs (117). Over the years, a variety of pH-sensitive nano-systems, such as peptides, micelles, liposomes, nanoparticles and polymersomes, have been synthesized, as extensively reviewed elsewhere (118–120). Nevertheless, only few studies have reported the use of such nano-scale carriers, responding to an acidic pH, for the selective targeting of CSC. A pH-responsive prodrug (PEG-modified doxorubicin) has for instance been co-delivered with SN38, an active metabolite of irinotecan, to eradicate both breast CSC and non-CSC populations (121). Such stable nanomedicine with pH sensitivity enhances drug accumulation at the tumor site, thereby leading to a potent tumor growth inhibition, while reducing chemotherapy-induced adverse effects (121). Finally, pH low-insertion peptides (pHLIP) have recently emerged as new modalities for tumor-specific drug delivery, but also for tumor imaging (122). These water-soluble membrane peptides undergo pH-dependent folding that triggers insertion across the cell membrane (123, 124). A pHLIP can directly translocate cargo molecules (attached to its C-terminal tail) through cell membranes without binding to cell surface receptors or pore formation. Although systemic administration of pHLIP has been used for the translocation of a variety of molecules, including chemotherapeutic drugs such as paclitaxel and doxorubicin (125, 126), antimicrobial peptides (127), polar membrane-impermeable peptides (e.g., phalloidin and other toxins) (128, 129) and peptide nucleic acid antimiRs (130), none study has investigated a specific targeting of CSC-like tumor cells with pHLIP-conjugated anti-cancer drugs. pHLIP grafted with agents known to interfere with CSC phenotypic features could be particularly suited to selectively kill this small cell subpopulation.
Therapeutic Targets and Modalities to Exploit Acidosis-Induced Phenotypic Alterations
As stated above, tumor acidosis induces several CSC-like phenotypic features that could be directly targeted for a therapeutic purpose. Among them, acidosis-mediated metabolic rewiring has a huge potential to be genetically or pharmacologically targeted since many enzymes/transporters that sustain cancer cell growth under low pH conditions are known (see above). Indeed, systemic administration of chitosan-based nanoparticles loaded with siRNA targeting two key transporters of energy fuels for acidosis-adapted cancer cells, namely the lactate/acetate transporter MCT1 and the glutamine transporter ASCT2, could lead to significant in vivo antitumor effects (131). Moreover, in vitro and in vivo experiments revealed that acidosis accounts for a net increase in tumor sensitivity to BPTES, an inhibitor glutaminase GLS1 (32).
Dysregulated fatty acid metabolism is another critical determinant of acidosis-adapted cancer cell growth, with the simultaneous occurrence of FAO and FAS pathways. Inhibition of mitochondrial transport of acyl-CoA, via the blockade of carnitine palmitoyltransferase 1 (CPT1) activity with etomoxir, showed a selective growth inhibitory effect in acidosis-adapted cancer cells (36). This is in adequation with the important role of FAO to support tumor proliferation and survival in a wide panel of tumors, including triple-negative breast cancer, glioma, leukemia, and colon (132). It is noteworthy that some compounds of interest, able to interfere with FAO, are currently under clinical development or already in use (perhexiline, trimetazidine, ranolazine) for the treatment of cardiovascular diseases; the anticancer potential of these molecules could therefore be rapidly evaluated in clinical trials.
Finally, acidosis-induced (epi)genetic reprogramming is another feature that might be targeted to eradicate stem-like cancer cells. Dual inhibition of SIRT1/6, with EX-527 compound, could for instance trigger selective growth inhibition of acidosis-adapted cancer cells (32). This effect was indeed associated with the re-expression of ACC2 enzyme that prevents the concomitant occurrence of FA oxidation and synthesis in acidosis-adapted cancer cells (36). Another study documented that human osteosarcoma cells were more sensitive to the inhibitor of histone deacetylases MC 1742 under acidosis than under neutral pH (47). Of note, this compound was reported by others to suppress proliferation and induce apoptosis of CSC in the same cancer type (133). In view of the central role of HIF-2α signaling under acidosis (see above), the use of recently developed HIF2α-selective inhibitors, PT2399 and PT2385 (134, 135), also appears as a promising therapeutic approach to selectively kill cancer (stem) cells exposed to acidic conditions.
Concluding Remarks
Frequent occurrence of tumor relapse is a major limitation for the cure of many patients, and that despite major improvements in prevention, diagnosis, and treatments. It is now acknowledged that local microenvironmental conditions select stem-like cancer cell phenotypes that dictate therapy resistance and re-initiation of the disease at the primary site but also into distant organs after metastatic dissemination. Recent findings reviewed here point to acidosis as one of the major selection barriers in the TME forcing the outgrowth of adaptive fitter phenotypes, when therapy is applied. While hypoxia has been reported as a CSC-permissive niche for many years, effects of acidosis by itself on CSC-related features were investigated more recently, upon the compelling evidence that oxygen and pH gradients were not perfectly overlapping in tumors. The reliance of CSC on the acidic niche is mediated by several mechanisms, including gene reprogramming, metabolic rewiring, apoptosis evasion and immune surveillance escape. Because a low pHe is a common feature of most solid tumors (vs. healthy tissues), there is an obvious interest to identify new therapeutic modalities that aim to take advantage of acidosis to selectively deliver anti-cancer drugs into tumors and eradicate resistance-sustaining cell populations such as CSC.
Since environment-mediated phenotype of cancer (stem) cells evolves de facto with time and tumor development, relevant pre-clinical, experimentally tractable, models as well as innovative approaches are needed to explore the intimate relationship between TME (in particular acidosis), cancer cell phenotypic adaptations (e.g., metabolic preferences) and drug response. Indeed, despite the strong evidence supporting the CSC model in a variety of cancers, it is critical to acknowledge major limitations associated with the poor reliability of CSC identification based on cell-surface markers expression and the lack of direct evidence about their in vivo existence. Future challenges to tackle the contribution of CSC in tumor relapse and to evaluate their clinical significance during drug resistance, minimal residual disease and metastatic dissemination rely therefore on the capacity to better integrate and exploit the microenvironment-driven phenotypic changes (e.g., dormant, mesenchymal-like state), including specific metabolic alterations (e.g., dysregulated FA metabolism, OXPHOS dependence) in order to propose novel CSC-targeting therapeutic modalities.
Author Contributions
CV and CC contributed to conception and design of the figures and the manuscript. CC developed the study concept, obtained funding, and wrote the final version of the manuscript. All authors wrote sections, revised, read, and approved the submitted version.
Funding
This work was supported by grants from the Fonds de la Recherche Scientifique (F.R.S.-FNRS) and the Télévie. CV is a Télévie PhD fellow and CC is a F.R.S.-FNRS Postdoctoral Researcher.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the Servier website (https://smart.servier.com/) that provided medical illustrations used in some figures of the review.
References
1. Salgia R, Kulkarni P. The genetic/non-genetic duality of drug 'resistance' in cancer. Trends Cancer. (2018) 4:110–8. doi: 10.1016/j.trecan.2018.01.001
2. Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. (1999) 9:M57–60. doi: 10.1016/S0962-8924(99)01661-X
3. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. (2012) 12:487–93. doi: 10.1038/nrc3298
4. Enriquez-Navas PM, Wojtkowiak JW, Gatenby RA. Application of evolutionary principles to cancer therapy. Cancer Res. (2015) 75:4675–80. doi: 10.1158/0008-5472.CAN-15-1337
5. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
6. Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. (2013) 15:338–44. doi: 10.1038/ncb2717
7. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. (2012) 12:133–43. doi: 10.1038/nrc3184
8. Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. (2014) 514:54–8. doi: 10.1038/nature13556
9. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
10. Boesch M, Sopper S, Zeimet AG, Reimer D, Gastl G, Ludewig B, et al. Heterogeneity of cancer stem cells: rationale for targeting the stem cell niche. Biochim Biophys Acta. (2016) 1866:276–89. doi: 10.1016/j.bbcan.2016.10.003
11. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. (2009) 9:239–52. doi: 10.1038/nrc2618
12. Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. (2011) 71:634–9. doi: 10.1158/0008-5472.CAN-10-3220
13. Carnero A, Lleonart M. The hypoxic microenvironment: a determinant of cancer stem cell evolution. BioEssays. (2016) 38(Suppl. 1):S65–74. doi: 10.1002/bies.201670911
14. Corbet C. Stem cell metabolism in cancer and healthy tissues: pyruvate in the limelight. Front Pharmacol. (2017) 8:958. doi: 10.3389/fphar.2017.00958
15. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. (2007) 11:69–82. doi: 10.1016/j.ccr.2006.11.020
16. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. (2011) 121:3804–9. doi: 10.1172/JCI57099
17. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:e816. doi: 10.1016/j.cell.2018.01.009
18. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
19. Kallinowski F, Vaupel P. pH distributions in spontaneous and isotransplanted rat tumours. Br J Cancer. (1988) 58:314–21. doi: 10.1038/bjc.1988.210
20. Schornack PA, Gillies RJ. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia. (2003) 5:135–45. doi: 10.1016/S1476-5586(03)80005-2
21. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. (2017) 17:577–93. doi: 10.1038/nrc.2017.77
22. Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. (2015) 1847:171–81. doi: 10.1016/j.bbabio.2014.10.005
23. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. (1997) 3:177–82. doi: 10.1038/nm0297-177
24. Vaupel PW, Frinak S, Bicher HI. Heterogeneous oxygen partial pressure and pH distribution in C3H mouse mammary adenocarcinoma. Cancer Res. (1981) 41:2008–13.
25. Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res. (2002) 8:1284–91.
26. Newell K, Franchi A, Pouyssegur J, Tannock I. Studies with glycolysis-deficient cells suggest that production of lactic acid is not the only cause of tumor acidity. Proc Natl Acad Sci USA. (1993) 90:1127–31. doi: 10.1073/pnas.90.3.1127
27. Yamagata M, Hasuda K, Stamato T, Tannock IF. The contribution of lactic acid to acidification of tumours: studies of variant cells lacking lactate dehydrogenase. Br J Cancer. (1998) 77:1726–31. doi: 10.1038/bjc.1998.289
28. Hulikova A, Black N, Hsia LT, Wilding J, Bodmer WF, Swietach P. Stromal uptake and transmission of acid is a pathway for venting cancer cell-generated acid. Proc Natl Acad Sci USA. (2016) 113:E5344–53. doi: 10.1073/pnas.1610954113
29. De Los Angeles A, Ferrari F, Fujiwara Y, Mathieu R, Lee S, Lee S, et al. Failure to replicate the STAP cell phenomenon. Nature. (2015) 525:E6–9. doi: 10.1038/nature15513
30. Peppicelli S, Andreucci E, Ruzzolini J, Laurenzana A, Margheri F, Fibbi G, et al. The acidic microenvironment as a possible niche of dormant tumor cells. Cell Molecul Sci. (2017) 74:2761–71. doi: 10.1007/s00018-017-2496-y
31. Rovida E, Peppicelli S, Bono S, Bianchini F, Tusa I, Cheloni G, et al. The metabolically-modulated stem cell niche: a dynamic scenario regulating cancer cell phenotype and resistance to therapy. Cell Cycle. (2014) 13:3169–75. doi: 10.4161/15384101.2014.964107
32. Corbet C, Draoui N, Polet F, Pinto A, Drozak X, Riant O, et al. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. (2014) 74:5507–19. doi: 10.1158/0008-5472.CAN-14-0705
33. Filatova A, Seidel S, Bogurcu N, Graf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res. (2016) 76:5845–56. doi: 10.1158/0008-5472.CAN-15-2630
34. Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. (2011) 18:829–40. doi: 10.1038/cdd.2010.150
35. Mekhail K, Gunaratnam L, Bonicalzi ME, Lee S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat Cell Biol. (2004) 6:642–7. doi: 10.1038/ncb1144
36. Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. (2016) 24:311–23. doi: 10.1016/j.cmet.2016.07.003
37. Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. (2009) 324:1289–93. doi: 10.1126/science.1169956
38. Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol. (2017) 13:494–500. doi: 10.1038/nchembio.2307
39. Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH Is a metabolic switch for 2-hydroxyglutarate generation and signaling. J Biol Chem. (2016) 291:20188–97. doi: 10.1074/jbc.M116.738799
40. Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. (2015) 22:304–11. doi: 10.1016/j.cmet.2015.06.023
41. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. (2012) 483:474–8. doi: 10.1038/nature10860
42. Ye D, Guan KL, Xiong Y. Metabolism, activity, and targeting of D- and L-2-hydroxyglutarates. Trends Cancer. (2018) 4:151–65. doi: 10.1016/j.trecan.2017.12.005
43. Barth PG, Wanders RJ, Scholte HR, Abeling N, Jakobs C, Schutgens RB, et al. L-2-hydroxyglutaric aciduria and lactic acidosis. J Inherit Metabol Dis. (1998) 21:251–4. doi: 10.1023/A:1005316121584
44. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. (2009) 15:501–13. doi: 10.1016/j.ccr.2009.03.018
45. Peppicelli S, Bianchini F, Toti A, Laurenzana A, Fibbi G, Calorini L. Extracellular acidity strengthens mesenchymal stem cells to promote melanoma progression. Cell Cycle. (2015) 14:3088–100. doi: 10.1080/15384101.2015.1078032
46. Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, et al. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int J Cancer. (2017) 140:1331–45. doi: 10.1002/ijc.30540
47. Chano T, Avnet S, Kusuzaki K, Bonuccelli G, Sonveaux P, Rotili D, et al. Tumour-specific metabolic adaptation to acidosis is coupled to epigenetic stability in osteosarcoma cells. Am J Cancer Res. (2016) 6:859–75.
48. Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, et al. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. (2008) 4:e1000293. doi: 10.1371/journal.pgen.1000293
49. Tang X, Lucas JE, Chen JL, LaMonte G, Wu J, Wang MC, et al. Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res. (2012) 72:491–502. doi: 10.1158/0008-5472.CAN-11-2076
50. Mahoney BP, Raghunand N, Baggett B, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics. I Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem Pharmacol. (2003) 66:1207–18. doi: 10.1016/S0006-2952(03)00467-2
51. Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Molecul Pharmaceut. (2011) 8:2032–8. doi: 10.1021/mp200292c
52. Avnet S, Lemma S, Cortini M, Pellegrini P, Perut F, Zini N, et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget. (2016) 7:63408–23. doi: 10.18632/oncotarget.11503
53. Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. (2004) 96:1702–13. doi: 10.1093/jnci/djh305
54. Taylor S, Spugnini EP, Assaraf YG, Azzarito T, Rauch C, Fais S. Microenvironment acidity as a major determinant of tumor chemoresistance: proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat. (2015) 23:69–78. doi: 10.1016/j.drup.2015.08.004
55. Balgi AD, Diering GH, Donohue E, Lam KK, Fonseca BD, Zimmerman C, et al. Regulation of mTORC1 signaling by pH. PLoS ONE. (2011) 6:e21549. doi: 10.1371/journal.pone.0021549
56. Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metabol. (2013) 1:23. doi: 10.1186/2049-3002-1-23
57. Mendoza EE, Pocceschi MG, Kong X, Leeper DB, Caro J, Limesand KH, et al. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low pH. Transl Oncol. (2012) 5:208–16. doi: 10.1593/tlo.11319
58. Pouyssegur J, Franchi A, L'Allemain G, Paris S. Cytoplasmic pH, a key determinant of growth factor-induced DNA synthesis in quiescent fibroblasts. FEBS Lett. (1985) 190:115–9. doi: 10.1016/0014-5793(85)80439-7
59. Ruzzolini J, Peppicelli S, Andreucci E, Bianchini F, Margheri F, Laurenzana A, et al. Everolimus selectively targets vemurafenib resistant BRAF(V600E) melanoma cells adapted to low pH. Cancer Lett. (2017) 408:43–54. doi: 10.1016/j.canlet.2017.08.010
60. Cheng GM, To KK. Adverse cell culture conditions mimicking the tumor microenvironment upregulate ABCG2 to mediate multidrug resistance and a more malignant phenotype. ISRN Oncol. (2012) 2012:746025. doi: 10.5402/2012/746025
61. Sauvant C, Nowak M, Wirth C, Schneider B, Riemann A, Gekle M, et al. Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer. (2008) 123:2532–42. doi: 10.1002/ijc.23818
62. Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M. Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia. (2006) 8:143–52. doi: 10.1593/neo.05697
63. Thews O, Nowak M, Sauvant C, Gekle M. Hypoxia-induced extracellular acidosis increases p-glycoprotein activity and chemoresistance in tumors in vivo via p38 signaling pathway. Adv Exp Med Biol. (2011) 701:115–22. doi: 10.1007/978-1-4419-7756-4_16
64. Thews O, Riemann A, Nowak M, Gekle M. Impact of hypoxia-related tumor acidosis on cytotoxicity of different chemotherapeutic drugs in vitro and in vivo. Adv Exp Med Biol. (2014) 812:51–8. doi: 10.1007/978-1-4939-0620-8_7
65. Dong L, Krewson EA, Yang LV. Acidosis activates endoplasmic reticulum stress pathways through GPR4 in human vascular endothelial cells. Int J Molecul Sci. (2017) 18:E278. doi: 10.3390/ijms18020278
66. Sharma V, Kaur R, Bhatnagar A, Kaur J. Low-pH-induced apoptosis: role of endoplasmic reticulum stress-induced calcium permeability and mitochondria-dependent signaling. Cell Stress Chaperones. (2015) 20:431–40. doi: 10.1007/s12192-014-0568-6
67. Visioli F, Wang Y, Alam GN, Ning Y, Rados PV, Nor JE, et al. Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PLoS ONE. (2014) 9:e101053. doi: 10.1371/journal.pone.0101053
68. Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. (2012) 287:30664–76. doi: 10.1074/jbc.M112.339127
69. Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, et al. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. (2012) 72:3938–47. doi: 10.1158/0008-5472.CAN-11-3881
70. Das CK, Mandal M, Kogel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metast Rev. (2018) 37:749–66. doi: 10.1007/s10555-018-9727-z
71. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
72. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. (2006) 107:2013–21. doi: 10.1182/blood-2005-05-1795
73. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. (2010) 184:1200–9. doi: 10.4049/jimmunol.0902584
74. Liao YP, Schaue D, McBride WH. Modification of the tumor microenvironment to enhance immunity. Front Biosci. (2007) 12:3576–600. doi: 10.2741/2336
75. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
76. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
77. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. (2018) 19:1319–29. doi: 10.1038/s41590-018-0226-8
78. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. (2016) 76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743
79. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. (2012) 72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272
80. Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. (2014) 5:e1336. doi: 10.1038/cddis.2014.285
81. Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci USA. (2013) 110:8644–9. doi: 10.1073/pnas.1221478110
82. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. (2017) 23:1234–40. doi: 10.1038/nm.4399
83. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
84. Corbet C, Feron O. Metabolic and mind shifts: from glucose to glutamine and acetate addictions in cancer. Curr Opin Clin Nutr Metab Care. (2015) 18:346–53. doi: 10.1097/MCO.0000000000000178
85. Corbet C, Feron O. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta Rev Cancer. (2017) 1868:7–15. doi: 10.1016/j.bbcan.2017.01.002
86. Polet F, Martherus R, Corbet C, Pinto A, Feron O. Inhibition of glucose metabolism prevents glycosylation of the glutamine transporter ASCT2 and promotes compensatory LAT1 upregulation in leukemia cells. Oncotarget. (2016) 7:46371–83. doi: 10.18632/oncotarget.10131
87. Kondo A, Yamamoto S, Nakaki R, Shimamura T, Hamakubo T, Sakai J, et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. (2017) 18:2228–42. doi: 10.1016/j.celrep.2017.02.006
88. Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun. (2014) 5:3550. doi: 10.1038/ncomms4550
89. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
90. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. (2014) 514:628–32. doi: 10.1038/nature13611
91. Fiorillo M, Sotgia F, Lisanti MP. “Energetic” cancer stem cells (e-CSCs): a new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front Oncol. (2019) 8:677. doi: 10.3389/fonc.2018.00677
92. Scatena C, Roncella M, Di Paolo A, Aretini P, Menicagli M, Fanelli G, et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: a clinical pilot study. Front Oncol. (2018) 8:452. doi: 10.3389/fonc.2018.00452
93. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. (2018) 28:e105. doi: 10.1016/j.cmet.2018.05.005
94. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell. (2017) 20:e305. doi: 10.1016/j.stem.2016.11.004
95. Barba I, Cabanas ME, Arus C. The relationship between nuclear magnetic resonance-visible lipids, lipid droplets, and cell proliferation in cultured C6 cells. Cancer Res. (1999) 59:1861–8.
96. Delikatny EJ, Lander CM, Jeitner TM, Hancock R, Mountford CE. Modulation of MR-visible mobile lipid levels by cell culture conditions and correlations with chemotactic response. Int J Cancer. (1996) 65:238–45. doi: 10.1002/(SICI)1097-0215(19960117)65:2<238::AID-IJC18>3.0.CO;2-9
97. Menard JA, Christianson HC, Kucharzewska P, Bourseau-Guilmain E, Svensson KJ, Lindqvist E, et al. Metastasis stimulation by hypoxia and acidosis-induced extracellular lipid uptake is mediated by proteoglycan-dependent endocytosis. Cancer Res. (2016) 76:4828–40. doi: 10.1158/0008-5472.CAN-15-2831
98. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells. (2015) 33:35–44. doi: 10.1002/stem.1837
99. Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J, et al. An Overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. (2017) 2017:1656053. doi: 10.1155/2017/1656053
100. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging role of lipid metabolism alterations in cancer stem cells. J Exp Clin Cancer Res. (2018) 37:118. doi: 10.1186/s13046-018-0784-5
101. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
102. Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ, et al. Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol. (2012) 188:624–31. doi: 10.1016/j.juro.2012.03.113
103. Ibrahim-Hashim A, Wojtkowiak JW, de Lourdes Coelho Ribeiro M, Estrella V, Bailey KM, Cornnell HH, et al. Free base lysine increases survival and reduces metastasis in prostate cancer model. J Cancer Sci Ther. (2011) (Suppl. 1): JCST-S1-004. doi: 10.4172/1948-5956.S1-004
104. Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J, Lloyd M, et al. Reduction of metastasis using a non-volatile buffer. Clin Exp Metast. (2011) 28:841–9. doi: 10.1007/s10585-011-9415-7
105. Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. (2009) 69:2260–8. doi: 10.1158/0008-5472.CAN-07-5575
106. Silva AS, Yunes JA, Gillies RJ, Gatenby RA. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. (2009) 69:2677–84. doi: 10.1158/0008-5472.CAN-08-2394
107. Damaghi M, Tafreshi NK, Lloyd MC, Sprung R, Estrella V, Wojtkowiak JW, et al. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat Commun. (2015) 6:8752. doi: 10.1038/ncomms9752
108. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
109. Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. (2006) 66:6699–707. doi: 10.1158/0008-5472.CAN-06-0983
110. Peppicelli S, Bianchini F, Torre E, Calorini L. Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clin Exp Metast. (2014) 31:423–33. doi: 10.1007/s10585-014-9637-6
111. Suzuki A, Maeda T, Baba Y, Shimamura K, Kato Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int. (2014) 14:129. doi: 10.1186/s12935-014-0129-1
112. Germain AR, Carmody LC, Morgan B, Fernandez C, Forbeck E, Lewis TA, et al. Identification of a selective small molecule inhibitor of breast cancer stem cells. Bioorg Med Chem Lett. (2012) 22:3571–4. doi: 10.1016/j.bmcl.2012.01.035
113. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. (2009) 138:645–59. doi: 10.1016/j.cell.2009.06.034
114. Hartwell KA, Miller PG, Mukherjee S, Kahn AR, Stewart AL, Logan DJ, et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat Chem Biol. (2013) 9:840–8. doi: 10.1038/nchembio.1367
115. Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. (2017) 9:1025–33. doi: 10.1038/nchem.2778
116. Pellegrini P, Dyczynski M, Sbrana FV, Karlgren M, Buoncervello M, Hagg-Olofsson M, et al. Tumor acidosis enhances cytotoxic effects and autophagy inhibition by salinomycin on cancer cell lines and cancer stem cells. Oncotarget. (2016) 7:35703–23. doi: 10.18632/oncotarget.9601
117. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. (2010) 148:135–46. doi: 10.1016/j.jconrel.2010.08.027
118. He X, Li J, An S, Jiang C. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. (2013) 4:1499–510. doi: 10.4155/tde.13.120
119. Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. (2016) 85:152–67. doi: 10.1016/j.biomaterials.2016.01.061
120. Liu J, Huang Y, Kumar A, Tan A, Jin S, Mozhi A, et al. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol Adv. (2014) 32:693–710. doi: 10.1016/j.biotechadv.2013.11.009
121. Sun N, Zhao C, Cheng R, Liu Z, Li X, Lu A, et al. Cargo-free nanomedicine with pH sensitivity for codelivery of DOX conjugated prodrug with SN38 to synergistically eradicate breast cancer stem cells. Molecul Pharmaceut. (2018) 15:3343–55. doi: 10.1021/acs.molpharmaceut.8b00367
122. Wyatt LC, Lewis JS, Andreev OA, Reshetnyak YK, Engelman DM. Applications of pHLIP technology for cancer imaging and therapy: (Trends in Biotechnology 35, 653–664, 2017). Trends Biotechnol. (2018) 36:1300. doi: 10.1016/j.tibtech.2017.11.005
123. Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, et al. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc Natl Acad Sci USA. (2007) 104:7893–8. doi: 10.1073/pnas.0702439104
124. Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane. Proc Natl Acad Sci USA. (2008) 105:15340–5. doi: 10.1073/pnas.0804746105
125. Burns KE, Delehanty JB. Cellular delivery of doxorubicin mediated by disulfide reduction of a peptide-dendrimer bioconjugate. Int J Pharmaceut. (2018) 545:64–73. doi: 10.1016/j.ijpharm.2018.04.027
126. Onyango JO, Chung MS, Eng CH, Klees LM, Langenbacher R, Yao L, et al. Noncanonical amino acids to improve the pH response of pHLIP insertion at tumor acidity. Angew Chem. (2015) 54:3658–63. doi: 10.1002/anie.201409770
127. Burns KE, McCleerey TP, Thevenin D. pH-selective cytotoxicity of pHLIP-antimicrobial peptide conjugates. Sci Rep. (2016) 6:28465. doi: 10.1038/srep28465
128. An M, Wijesinghe D, Andreev OA, Reshetnyak YK, Engelman DM. pH-(low)-insertion-peptide (pHLIP) translocation of membrane impermeable phalloidin toxin inhibits cancer cell proliferation. Proc Natl Acad Sci USA. (2010) 107:20246–50. doi: 10.1073/pnas.1014403107
129. Burns KE, Robinson MK, Thevenin D. Inhibition of cancer cell proliferation and breast tumor targeting of pHLIP-monomethyl auristatin E conjugates. Molecul Pharmaceut. (2015) 12:1250–8. doi: 10.1021/mp500779k
130. Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. (2015) 518:107–10. doi: 10.1038/nature13905
131. Corbet C, Ragelle H, Pourcelle V, Vanvarenberg K, Marchand-Brynaert J, Preat V, et al. Delivery of siRNA targeting tumor metabolism using non-covalent PEGylated chitosan nanoparticles: identification of an optimal combination of ligand structure, linker and grafting method. J Control Release. (2016) 223:53–63. doi: 10.1016/j.jconrel.2015.12.020
132. Corbet C, Feron O. Emerging roles of lipid metabolism in cancer progression. Curr Opin Clin Nutr Metab Care. (2017) 20:254–60. doi: 10.1097/MCO.0000000000000381
133. Di Pompo G, Salerno M, Rotili D, Valente S, Zwergel C, Avnet S, et al. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J Med Chem. (2015) 58:4073–9. doi: 10.1021/acs.jmedchem.5b00126
134. Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. (2013) 9:271–6. doi: 10.1038/nchembio.1185
Keywords: cancer stem cell, acidosis, niche, microenvironment, drug resistance, metabolism, metastasis, dormancy
Citation: Vander Linden C and Corbet C (2019) Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 9:159. doi: 10.3389/fonc.2019.00159
Received: 27 December 2018; Accepted: 25 February 2019;
Published: 19 March 2019.
Edited by:
Annarosa Arcangeli, Università degli Studi di Firenze, ItalyReviewed by:
Michael P. Lisanti, University of Salford, United KingdomNor Eddine Sounni, University of Liège, Belgium
Lido Calorini, Università degli Studi di Firenze, Italy
Copyright © 2019 Vander Linden and Corbet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cyril Corbet, Y3lyaWwuY29yYmV0QHVjbG91dmFpbi5iZQ==