 Shabnum Patel1
Shabnum Patel1 Rachel A. Burga1
Rachel A. Burga1 Allison B. Powell1
Allison B. Powell1 Elizabeth A. Chorvinsky2
Elizabeth A. Chorvinsky2 Nia Hoq1Sarah E. McCormack1Stacey N. Van Pelt1
Nia Hoq1Sarah E. McCormack1Stacey N. Van Pelt1 Patrick J. Hanley2
Patrick J. Hanley2 Conrad Russell Y. Cruz1,2*
Conrad Russell Y. Cruz1,2*- 1GW Cancer Center, The George Washington University, Washington, DC, United States
- 2Center for Cancer and Immunology Research, Children's National Health System, Washington, DC, United States
Background: Chimeric antigen receptor (CAR)-modified T cells have successfully harnessed T cell immunity against malignancies, but they are by no means the only cell therapies in development for cancer.
Main Text Summary: Systemic immunity is thought to play a key role in combatting neoplastic disease; in this vein, genetic modifications meant to explore other components of T cell immunity are being evaluated. In addition, other immune cells—from both the innate and adaptive compartments—are in various stages of clinical application. In this review, we focus on these non-CAR T cell immunotherapeutic approaches for malignancy. The first section describes engineering T cells to express non-CAR constructs, and the second section describes other gene-modified cells used to target malignancy.
Conclusions: CAR T cell therapies have demonstrated the clinical benefits of harnessing our body's own defenses to combat tumor cells. Similar research is being conducted on lesser known modifications and gene-modified immune cells, which we highlight in this review.
Introduction
Chimeric antigen receptors and engineered T cell receptors (based on previously identified high affinity T cell receptors) function by redirecting T cells to a predefined tumor-specific (or tumor-associated) target. Most of these modifications use retroviral or lentiviral vectors to integrate the construct, and most of the receptors feature a costimulatory signal—enhancing T cell activation following recognition of the target antigen. These modified T cells have collectively shown promising success rates, particularly against hematologic malignancies (1), with growing excitement for these novel treatments (2). Pioneering work at the NIH resulted in promising therapies for melanoma (3) and synovial sarcoma (4). Some of these therapies have been approved as licensed drugs.
CAR T cells targeting commonly overexpressed leukemia and lymphoma markers such as CD19 have shown promise in the prevention and treatment of malignancies such as Acute Lymphoblastic Leukemia (ALL), Chronic Lymphocytic Leukemia (CLL), Non-Hodgkin's lymphoma (NHL), Diffuse Large B cell lymphoma (DLBCL), and other B cell malignancies (5–8). These CD19-CAR Phase I and II trials have demonstrated safety and efficacy, with substantial partial and complete response rates (PR and CR, respectively). There are however, important concerns about toxicity—as resulting from on target off tumor effects, cytokine release syndromes, and neurotoxicity (9). Current CAR clinical trials are expanding to target other tumor-associated markers including GD2 (10), BCMA (11), CD20, CD30, CD33, CD7, HER2 (human epidermal growth factor receptor 2), and mesothelin (12–17). CAR T cells have been highlighted as Advance of the Year, by the American Society of Clinical Oncology in 2018 (18). A similar technology involves using high affinity T cell receptors (TCRs) and introducing these into cells (19). In the hopes of extending this success, other immune cell-based therapies are in current development.
The first group, non-CAR/non-TCR gene modified cell therapies for cancer, incorporates methods to overcome the barriers presented by cancer and the tumor microenvironment, as well as strategies for enhancing potency of T cell therapies. The second group focuses on immunotherapies generated from less frequently studied cell types including gamma-delta T cells, invariant natural killer T (iNKT) cells, natural killer (NK), and dendritic cells.
This review explores these lesser known cancer cell immunotherapy strategies, highlighting advances that have been made in recent preclinical and clinical efforts, and presents platforms for which they could demonstrate efficacy and may be critical for treating different cancer subtypes.
NON-CAR/TCR Modification of T Cells
The ease by which T cells can be genetically modified has led to other gene modifications that aim to further enhance activity of T cells [a strategy that some groups have labeled as “armored” CARs (20), initially dubbed as “TRUCKS” (21)], including modifications to introduce dominant negative receptors, chemokine receptors, cytokines, cytokine receptors, and checkpoint inhibitors Figure 1.
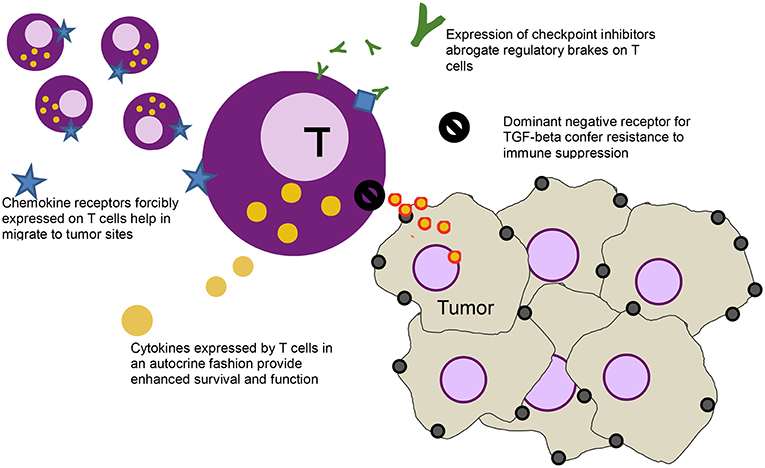
Figure 1. Schematic of a T cell (purple) and various modification - clockwise from left: forcibly expressed chemokine receptors (blue star) that help the cell migrate down the relevant chemokine gradient secreted by the tumor, secreted checkpoint inhibitors (green Ys) that bind to the checkpoint receptors on T cells (blue diamonds), abrogating their inhibitory function, a dominant negative receptor (black no sign) that helps block immune suppressive effects of cytokines like TGF-beta, and cytokines (yellow) that help stimulate the T cells in an autocrine function.
Dominant Negative Receptors
Translation of successful T cell therapies to solid tumors has been hampered by the immunosuppressive tumor microenvironment. Cancers secrete immunosuppressive cytokines which impair immune cell proliferation and function, and recruit regulatory T cells. These cytokines include TGFβ which inhibits the function of host immune cells (even those that successfully infiltrate the tumor), and induces epithelial-to-mesenchymal transition leading to cancer metastasis. Upregulation of TGFβ in the tumor microenvironment has been described in many aggressive malignancies including those of the brain, gastrointestinal tract, bone, breast, lung, and pancreas (22). TGFβ downregulates the secretion of critical Th1 cytokines, such as IFNγ, and impairs T cell and natural killer (NK) cell cytolytic activity and proliferation (23, 24).
A mutated form of the TGFβ receptor has previously been shown to exert a dominant-negative effect by abrogating the negative signaling cascade in cells that express this protein (25). This dominant negative receptor of the type II subunit (TGFβRII DNR) encompass the extracellular and transmembrane region of the endogenous cytokine receptor but exclude intracellular signaling domains, preventing downstream signaling when bound to ligand. Expression of this DNR has led to decrease in downstream signaling following TGFβ ligation—for example SMAD phosphorylation in the presence of TGFβ is abrogated by this receptor (26). T cells genetically engineered to express a TGFβRII dominant negative receptor (DNR) are resistant to the antiproliferative and anti-cytolytic effects of this cytokine (27). Genetically modified tumor antigen-associated T cells (in this case directed against Epstein-Barr virus antigens) expressing DNR show enhanced persistence and activity, resulting in superior antitumor activity (28). In this study, TGFβRII DNR restored proliferation of EBV-specific T cells in the presence of TGFβ, restored cytotoxicity against EBV-expressing lymphoblastoid cell lines, and demonstrated greater antitumor activity and migration in vivo (28). Other studies have also demonstrated the benefits of this DNR on the activity of T cells (see Table 1) (27, 29, 30, 32–34).

Table 1. Examples of preclinical research evaluating DNR-expressing T cells for the treatment of malignancies.
A dose escalation study (using TGFβRII DNR antigen-specific T cells directed against EBV) of patients with EBV-positive lymphoma showed that these T cells were resistant to the inhibitory cytokine, with increased signals from peripheral blood, corresponding to increased frequencies of T cells. Persistence extended to more than 4 years, and four of seven evaluable patients had clinical responses (28). Other clinical trials incorporating TGFβRII DNR expressing cells have targeted a number of cancers including nasopharyngeal carcinoma (using antigen-specific T cells directed against EBV), metastatic melanoma (using tumor infiltrating lymphocytes TILs), EBV-positive Hodgkin disease and non-Hodgkin lymphoma using antigen-specific T cells directed against EBV), and HER2+ breast cancer (using chimeric antigen receptors directed against HER2) (see Table 2).
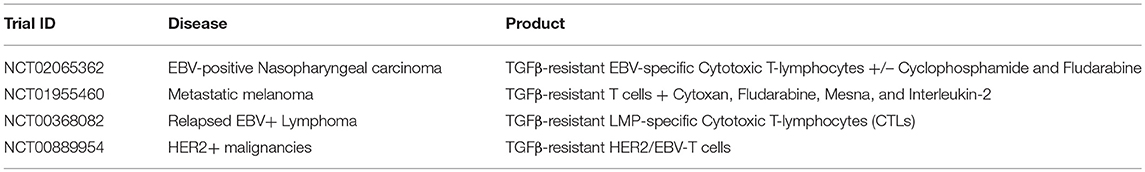
Table 2. Examples of clinical trials using various DNR-expressing T cells for the treatment of malignancies (35).
It is important to note that there may potentially be unintended consequences of conferring resistance to a regulatory cytokine: disruption of normal T cell homeostasis may result from expression of TGFβRII DNR. A study by Lucas et al. show that expression of the dominant negative receptor resulted in massive expansion of CD8 T cells in lymphoid organs (36). So far, no dysfunction has been observed in patients (28).
Cytokine Receptors
Besides TGFβ, other negative/regulatory cytokines in the tumor environment limit T cell persistence and activity—these include IL10, IL13, and IL4. Another approach to reversing the immunosuppressive effects of these cytokines are chimeric cytokine receptors (CcR) (37). CcR's use the extracellular binding domain of an immunosuppressive cytokine bound to the intracellular signaling domain of an immune-activating cytokine to reverse its signaling effects. The first use of a chimeric IL4 cytokine receptor was described by Wilkie et al. where a fusion of IL4 receptor alpha ectodomain was fused to the subunit used by IL-2 and IL-15; this resulted in expansion and enhanced killing of MUC1 CAR T cells (38). In another study combining the extracellular domain of IL-4 cytokine receptor and the intracellular signaling domain of IL-7 cytokine receptor, CcRs restored the anti-tumor cytotoxicity of autologous T cells against EBV-transformed B cell tumors in vivo (37). In this study, CcR expression induced phosphorylation of STAT5 (part of the native signaling cascade in IL7 signaling) after ligation with tumor-secreted IL4, and restored T cell proliferation in the presence of the cytokine (37). This chimeric cytokine receptor also showed efficacy in a pancreatic cancer model: T cells modified to express a chimeric antigen receptor targeting prostate stem cell antigen (PSCA), found in pancreatic tumors, maintained their antitumor activity in an IL4-rich tumor microenvironment when they are co-transduced with the IL4/IL7 CcR (39). Another example uses a tumor-derived cytokine, CSF-1, to stimulate T cells by modifying these cells to express CSF-1R. Acquired responsiveness to CSF-1 allowed for improved chemotaxis and proliferation (40).
A simpler construct involves overexpression of a native cytokine receptor to allow for improved persistence following exogenous administration of the cytokine. One of the major challenges in T cell therapies is enhancing persistence of the cells in vivo. Previously, IL2 was administered to maintain T cell proliferation and activity (41), but IL2 is also associated with adverse effects (42)—limiting its applicability. IL7, on the other hand, provides the same effects without the unwanted toxicities. T cells, however, lose expression of the IL7 receptor after prolonged culture. In one study, genetic modification of EBV-specific CTLs to forcibly express IL-7 receptor α chain (IL-7Rα) led to restoration of CTL responsiveness to IL-7, and their antitumor activity sustained in vivo and in vitro without the unwanted toxicities related to IL-2 administration (43). In another study, cytokine feedback loops were used to improve efficacy of T cells by modifying these cells to express IL-7 and IL-21 (44).
Cytokines
Select cytokines, like IL2, IL15, and IL12 perform stimulatory functions for T cells; in theory, autocrine secretion of these cytokines should help keep these cells persisting in vivo, even in the face of a hostile tumor environment (20).
In an example of this approach, CD19-CAR-specific T cells were modified to secrete IL15, and its anti-tumor efficacy evaluated using a xenogeneic model of lymphoma (45). In this study, IL-15 modified CD19 CAR T cells secreted IL15 following antigen stimulation, showed enhanced survival as a result of the transgenic cytokine, expanded better in vivo, and have better in vivo anti-tumor activity (45).
Other cell therapies incorporating cytokine secretion are listed in Table 3. One study, by Koneru et al. looked at MUC-16 specific T cells secreting IL12. Promising preclinical results (enhanced lysis of tumors and persistence in vivo) (49) led to its subsequent use in a phase I clinical trial for recurrent platinum-resistant ovarian cancer (50).

Table 3. Examples of preclinical research evaluating T cells expressing cytokines for the treatment of malignancies.
Chemokine Receptors
One relatively underappreciated requirement for improving T cell therapies is successful migration to the site of disease (51). In the setting of malignancies, a possible avenue for improvement relies on the fact that tumors secrete chemokines that can potentially be harnessed to lead T cells to the tumor site. Chemokine receptors corresponding to the chemokines that are secreted by tumor cells have been introduced into T cells, which maximized efficacy of these therapies by improving localization (51).
The first chemokine receptor-engineered T cells redirected cells using CXCR2: these allowed cells to migrate toward the Gro-alpha chemokine gradient, and induced interferon gamma secretion from transduced T cells (52).
Moon et al. transduced the chemokine receptor CCR2b into mesoCAR T cells to treat tumors that express CCL2 and mesothelin (53). These modified T cells improved tumor localization, a limitation of CAR-based approaches, and showed enhanced anti-tumor activity. Craddock et al showed that in neuroblastoma cell lines derived from six patients, modified activated T cells showed a 60% increase in the expression of CCR2b and co-expressed CCR2b and GD2-CAR showed a 10-fold improvement in migration to the tumor site compared to CCR2 negative activated T cells (54). Using the transgenic adenocarcinoma of mouse prostate (TRAMP) model, Garetto et al. (55) showed that expressing chemokine receptors on T cells tailored for chemokines that are strongly secreted in the tumor milieu can be used to improve targeting of T cells.
Additional examples are listed in Table 4.

Table 4. Examples of preclinical research evaluating T cells expressing chemokine receptors for the treatment of malignancies.
Checkpoint Inhibitors
In addition to T cell therapies, the introduction of checkpoint inhibitors has been responsible for the interest in immunotherapies. These molecules, typically antibodies directed against checkpoint receptors expressed on T cells, inhibit negative regulation of these cells—removing the “brakes” to their activity. Combinations of T cell therapies and checkpoint inhibitors are therefore particularly attractive. Administration of a PD-1 blocking antibody enhanced CAR T cell function against established tumors (58).
One way to coordinate spatiotemporal activity of these therapeutics is to have T cells directly secrete these inhibitors. One group engineered CD19 CAR T cells to secrete single chain variable fragments targeting PD1. T cells were shown to secrete functional anti-PD1 scFv (~600 ng/mL), capable of reversing PD1/PDL1 interactions and their negative effects on T cell function. This allowed for enhanced T cell expansion and effector function in vitro and in vivo (59). Another group also modified various CAR T cells to secrete PD1 blocking scFV and showed improved antitumor activity, as well as bystander tumor-specific T cell activity, in syngeneic and xenogeneic murine models of tumors expressing PDL1 (60). Other groups knocked down expression of PD-1 (61) or components of PD-1 signaling, to improve function of adoptively transferred cells (62).
Other Immune Cells
Although the specific, direct actions of gene-modified T cells are mostly responsible for the promising clinical results—indirect effects mediated through other immune cells also contributed to efficacy. In addition, there is an increasing body of evidence that suggests engagement of multiple arms of immunity are key toward longer lasting resolution of tumor.
The use of other immune cells as immunotherapies for cancer is therefore a necessary adjunct to the existing T cell therapies. Some of the more commonly studied cells include gamma-delta (γδ) T cells, invariant natural killer T (iNKT) cells, natural killer (NK), and dendritic cells. We limit this section to these endogenously occurring cells, though acknowledge that other cells that can be expanded ex vivo—e.g., cytokine induced killer cells (CIK)—may form a potentially efficacious immune therapeutic (Figure 2).
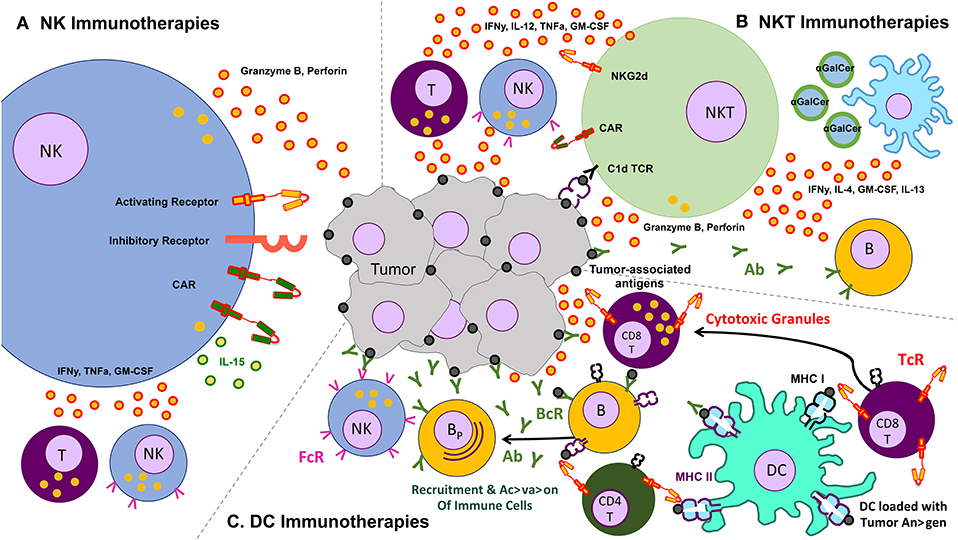
Figure 2. Schematic of various other cells and their effects on the tumor – clockwise from left: (A) natural killer (NK) cells, which lyse tumors without the need for identifying a known antigen, working through a balance of inhibitory and activating receptors (and also amenable to transduction with chimeric antigen receptors), as well as secrete cytokines that activate other components of the immune response, (B) natural killer T (NKT cells, which recognize lipid antigens and also help orchestrate the immune response, (C) dendritic cells (DC) which present antigen to T cells and help jump start immune responses like a vaccine.
Gamma-Delta T Cells
γδ T cells are a small subset of cells, whose functions make them attractive candidates for potential immunotherapies. γδ T cells have many innate like properties, and similar to other innate cells, such as NKs, γδ T cells express NK receptor NKG2D and show cytotoxicity to tumor cells (63). Two groups of γδ T cells are recognized, based on the TCR V delta usage: V delta 1 cells are located in mucosal tissue, and V delta 2 cells are located in the peripheral blood (64). V delta 2 cells are a source of proinflammatory cytokines once activated, including TNF- α and IFN-γ (64). The mechanisms by which γδ T cells recognize cancer are not fully understood. They can recognize tumor antigens via their TCRs and NK receptors, but it is unclear what specific antigens they respond to (65). γδ2 T cells typically recognize pyrophospate antigens produced by bacteria, while γδ1 T cells recognize MHC class I related molecules like MICA/MICB (64). In the cancer setting, it is thought that γδ T cells recognize stress induced self-like antigens, typically expressed by malignant cells and found to infiltrate tumors in some cases (66). These cells appear to mediate a graft vs. tumor response without eliciting GVHD (67).
In pre-clinical studies, γδ T cells have been expanded and have demonstrated cytotoxicity to a variety of tumor cell lines derived from lung carcinoma, liver cancer, and breast cancer, in an MHC-unrestricted manner (66). Deniger et al. demonstrated that they were able to see a 107-fold increase in γδ T cell numbers, despite a small starting population, suggesting it is possible to expand to clinically relevant numbers (68). Another study by Liu et al. show that γδ T cells have the ability to recognize and kill some forms of prostate cancer in vitro via innate mechanisms (69). In other preclinical studies, it was demonstrated that γδ T cells could be transduced to generate CAR-T cell products that maintained their natural tumor infiltration and killing abilities (70).
Some clinical trials using these cells are already underway In a Phase I study, autologous γδ T cells were infused in combination with IL-2 into 10 patients with metastatic renal cell carcinoma (mRCC) (71). This trial demonstrated safety, as infusions were tolerated with few serious adverse events related to the immunotherapy, with six patients showing stable disease. In another study, patients with hepatocellular carcinoma were given an injection of γδ T cells (NCT00562666).
Although γδ T cells have been well-tolerated in cancer patients, they are limited by difficulties in their isolation (65), and some questions surround their potential tumor-promoting effects (effects on angiogenesis and secretion of IL-17) (72, 73).
Natural Killer Cells
Natural killer cells were initially identified for their ability to target and kill tumor cells (74). They exhibit cytolytic function through the release of perforin and granzyme B as well as through FasL-TRAIL-mediated pathways, and NK cell activity is governed by a balance of signals from both activating and inhibitory receptors (75–78). NK cells are an possible option for adoptive immunotherapy because they do not require prior antigen exposure to elicit cytotoxicity. In addition, NK cells have limited persistence in vivo, a feature that appeals to clinicians and scientists alike. There is preclinical and clinical evidence that NK cells do not cause graft vs. host disease (GVHD) (79–83) or result in systemic toxicities associated with “cytokine storms” seen in T cell therapies (84–86). Similar to other new immunotherapies, an initial roadblock to the clinical use of NK cells was the inability to expand NK cells to clinically relevant numbers.
An additional challenge facing NK cells for adoptive therapy is the immunosuppressive tumor microenvironment, which directly nullifies the cytotoxicity of NK cells (87). Specifically, there is an abundance of immunosuppressive cell types such as myeloid-derived suppressor cells (MDSC) (88–90), tumor-associate macrophages (TAM) (91), and regulatory T cells (Treg) (26, 92–95), as well as cytokines such as transforming growth factor beta (TGFβ) and indoleamine 2,3 dioxygenase (IDO) (26, 96), that have been shown to interact with NK cells and cause phenotypic and functional dysfunction. Many groups have performed preclinical work in order to exploit the anti-tumor, cytotoxic, capabilities of NK cells, while addressing the challenges faced by adoptive cell therapy. For instance, Mentlik et al. focused on these combining NK cell therapy with monoclonal antibodies, boosting NK cell's ability to conduct ADCC (97); these combination therapies with antibodies or cytokines are the focus of other preclinical efforts (98).
Extensive effort has been put into generating and characterizing NK cells for adoptive cell therapy from both primary donor and immortalized NK line donor sources, with mixed results (Table 5). As with T cells, there is tremendous appeal for equipping cytotoxic cells with the ability to specifically recognize and kill a given tumor target—as such, there have been multiple attempts at generating CAR-NKs, that retain their cytotoxicity but are instead directed toward a specific antigen (122). CAR-NKs targeting B cell malignancies have demonstrated impressive in vivo cytolytic efficacy (123–126), and represent a promising transition of the technology to the clinic. Other modifications have been incorporated in NK cells—in one such study, cord blood NK cells engineered to express IL15 and a CD19 CAR showed marked increase in survival in a xenograft lymphoma model (127).
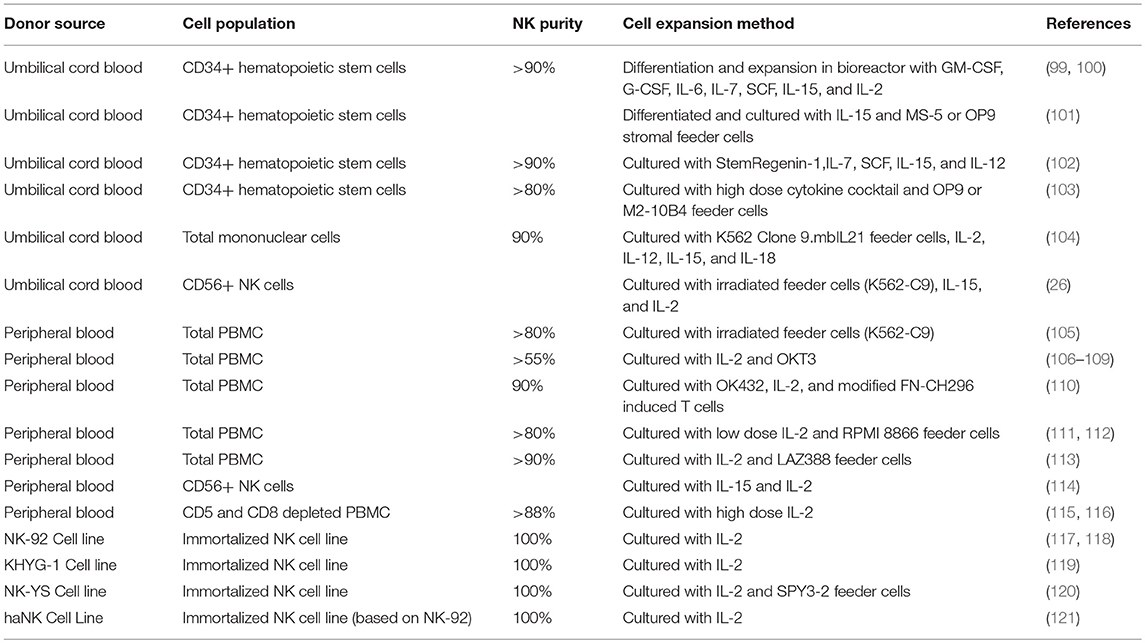
Table 5. NK cell production methods.
To date, three trials with genetically modified primary NK cells, and are currently active (NCT03056339, NCT00995137, NCT01974479). Existing clinical CAR-NK therapies borrow directly from the manufacturing schemes in the CAR-T cell field. One new approach involves substitution of the CD3ζ domain, which initiates TCR-based activation in T cells, with an intracellular domain that is specifically involved in NK cell activation. Indeed, NK-specific activation domains DNAX Activating Protein 10 (DAP10) and 12 (DAP12) have been introduced as the intracellular component in a CAR-NK in preclinical work, and promising results have demonstrated enhanced NK activation and function with this modification (128, 129).
In addition to the abovementioned CAR-NK clinical efforts, multiple clinical trials are underway using infusions of either autologous or allogenic NKs, with more promising results occurring in patients treated with allogenic NKs [reviewed in (130, 131)]. A study by Burns et al. using ex vivo activated NKs for treating patients with Hodgkin's and renal cell carcinoma was unable to demonstrate clinical efficacy (132), perhaps due to the autologous donor source. Furthering this claim were the results from multiple groups that demonstrated enhanced NK cell cytotoxicity occurring in patients if there was a killer immunoglobulin receptor-human leukocyte antigen (KIR-HLA) mismatch between donor and recipient cells (83, 93). One of the outstanding challenges for the use of adoptive NK cell therapy pertains to the cells' innate sensitivity to the freeze-thaw process. Indeed, preclinical reports have demonstrated impaired viability and cytotoxicity following cryopreservation (133, 134).
In addition to improving the manufacture end of NK cells therapies, developments are underway that aim to enhance the functionality and persistence of these therapies. For instance, focus for NK cell as well as other cell therapies has shifted toward modulating the suppressive tumor microenvironment concurrently with cell therapy in order to enhance efficacy (26). Moreover, a class of immunomodulatory drugs, such as thalidomide, have been found to modify the NK cells in the tumor environment by upregulating surface expression of TRAIL, which may increase NK-mediated apoptosis of target tumor cells (135–137). Miller et al. are developing bi-specific killer engagers (BiKEs) and tri-specific killer engagers (TriKEs) that can address many of the challenges facing NK cell therapy all in one construct (138–141). They have developed a platform by which NK cells are rendered specific for a given target antigen, while simultaneously increasing NK cell potency and persistence by incorporating CD16 single chain variable fragment (to increase ADCC-associated signaling) and an IL15 moiety (to increase NK activation and thus persistence). These findings that BiKE and TriKE-modified NK cells delivered potent anti-tumor responses in the setting of AML, ALL, and CLL, as well as the extensive number of ongoing clinical trials are only one example of how the field of immunotherapy is rapidly expanding to include a variety of non-T cell-based immunotherapies.
Natural Killer T Cells
NKTs represent an important link between the innate and adaptive immune system, as they can be activated by both antigen dependent and antigen-independent mechanisms. Divided into invariant (iNKT) or diverse (dNKT) subsets, they have a highly restricted TCR repertoire, only recognizing antigen in the context of the MHC class I-like CD1d molecule (142), and are uniquely classified by their ability to rapidly produce regulatory cytokines such as IFNγ, IL4, IL10, IL13, IL-17, GM-CSF, and TNFα in large quantities (143). These characteristics together contribute to the appeal of this cell subset as a form of immunotherapy. Although populations of iNKT cells isolated from cancer patients have been found to be decreased in quantity and defective (144–146), many groups have shown that this impaired phenotype is in fact reversible ex vivo (147–150). Additionally, preclinical studies have supported the promise of NKT therapy as a multimodal platform—the glycolipid alpha-galactosylceramide (αGalCer) can reactivate impaired NKTs ex vivo to result in restored cytokine production and anti-tumor responses (151–154). Further, inhibition of tumor progression has been demonstrated in models of colon carcinoma, lymphomas, sarcoma, melanoma, prostate cancer, and lung cancer, leading to resurgence of optimism in iNKT cells as agents of immunotherapy.
NKTs are of particular interest as a possible cell for CAR modification for two main reasons: first, because clinical data has indicated better outcomes occurring in patients with higher NKT cell tumor infiltrate (155, 156), and second because the CD1d restricted nature of NKT antigen recognition is able to limit the potential off-target toxicity and increase potential applicability in both the autologous and allogeneic setting (157). Because NKTs secrete a wide range of regulatory cytokines, they are able to both activate antigen presenting cells such as dendritic cells as well as cytotoxic cells such as CD8+ T cells and NK cells—further increasing their value as an agent of immunotherapy (Figure 2) (158–163). Heczey et al. generated CAR-modified NKT cells to target neuroblastoma (aGD2 CAR) and lymphoma (aCD19 CAR), with marked success. They found that their CAR NKT cells had highly potent and selective cytotoxic activity against tumor target antigen-expressing cells, and were able to efficiently proliferate and produce large amounts of cytokines in the tumor environment, thus mediating their efficacy (164). Rotolo et al. generated CAR CD19-modified NKT cells to better target CD19-expressing lymphomas that also express CD1d, the ligand for NKT (165).
Many attempts have been made to directly target and restore function to patients' endogenous NKT cells, and current trials are summarized in Table 6. This avenue has focused on the infusion of NKT cell activating or stimulating agents, largely αGalCer (166), or by combining these agents with APCs such as dendritic cells to enhance immune activation at the suppressed tumor site (167–173). Dendritic cells can be pulsed with glycolipid and reintroduced into patients, a strategy regularly used in vaccine development, which has been proven to induce activation and restore function to endogenous NKT cells in a range of cancer types (167, 174–176).
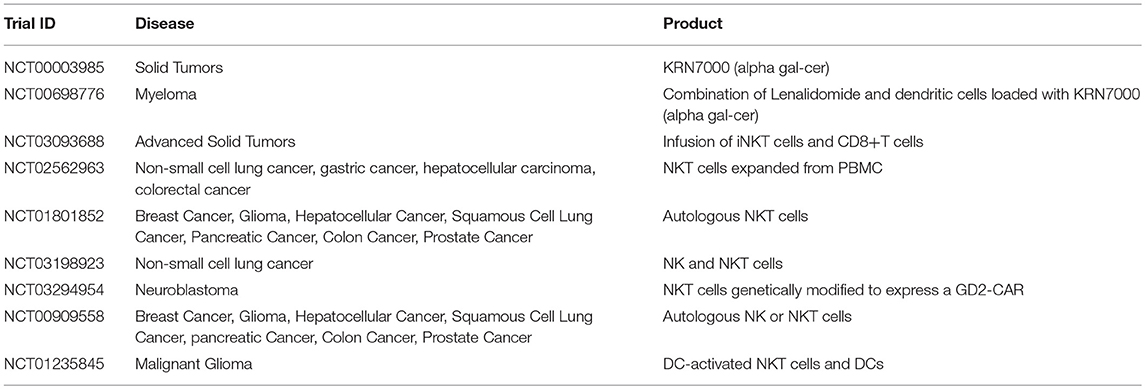
Table 6. Examples of clinical trials with iNKT cells (35).
The largest challenge facing the advancement of CAR and non-CAR NKT cell therapies is that of persistence; tumor progression negatively correlates with NKT cell functionality. Attempts to subvert this impairment in NKT function include efforts where autologous NKTs are expanded ex vivo with αGalCer prior to reinfusion, as previously described (177). Attempts have been made to classify the phenotype of NKT cells during tumor progression, and CD62L has been identified as a potential indicator of NKT cells most likely to demonstrate enhanced anti-tumor activity (178). Moreover, new approaches to drug or glycolipid delivery systems are currently in development, which aim to package agents causing activation of NKTs in enhanced nanoparticle-based constructs. Examples of this novel immunotherapy “associated agent,” such as αGalCer packaged into microspheres or liposomes, have demonstrated enhanced NKT functional responses as compared to the agent alone (179–182). These modifications to CAR and non-CAR NKTs speak to the tremendous promise of generating enhanced clinical NKT therapies.
Dendritic Cells
Dendritic cells (DCs), one of the professional antigen-presenting cells of the immune system, efficiently process antigens for presentation to T cells in order to activate the adaptive immune system (183). DCs naturally play a role in the control of immune responses and immune tolerance, both critical in anti-tumor immunity (183–185). Pre-clinical in vivo mouse models of cancer have demonstrated that DCs have the ability to home to tumor sites and capture tumor-associated antigens for processing. These DCs subsequently travel to nearby lymph nodes, where they present tumor antigens to T cells, generating tumor-specific T cells that can lead to clearance or tumor rejection (184, 185). Furthermore, DCs have the unique role of interacting with several subsets of the immune system, including both CD4 and CD8 T cell subsets in lymph nodes, resulting in downstream B cell activation into antibody-secreting cells, as well as activation of NKs and phagocytes. For example, in a murine model of melanoma, it was demonstrated that DCs interact with both cytotoxic T cells and NK cells to mediate tumor elimination (186, 187). However, NK depletion resulted in no tumor elimination, emphasizing the importance of DC-NK interactions in anti-tumor immunity (186, 187). This ability to interact with and regulate multiple immune cells make DCs an interesting candidate cell subset to be used in immunotherapy trials.
Due to their natural role in antigen processing and presentation, dendritic cells have been used in multiple Phase III clinical trials as an adjuvant or therapeutic vaccine for certain cancers including metastatic melanoma (NCT01875653), prostate cancer (below), renal cell carcinoma (NCT01582672), and glioblastoma multiforme (NCT00045968) (188, 189) (Table 7). The main objective of these studies was to deliver tumor antigens via DCs to stimulate and activate anti-tumor antigen-specific T cells, which subsequently eliminate cancerous cells and provide immunological memory to prevent tumor relapse. Furthermore, it has been demonstrated that the induction of anti-tumor T cell responses from DC-immunotherapies concurrently enhances natural killer immunity (187), underscoring the importance of DCs in regulating multiple immune cell subsets (Figure 2).
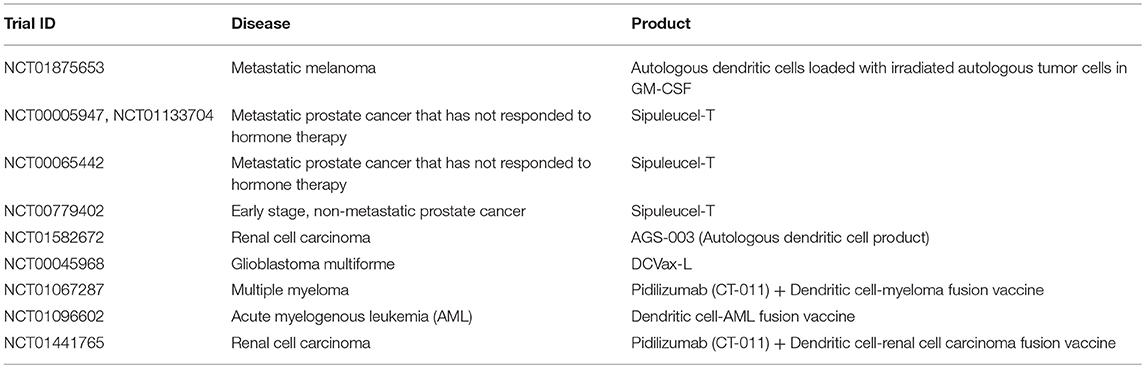
Table 7. Examples of clinical trials with dendritic cells (35).
One of the significant advantages with DC-based immunotherapies is the demonstration of safety across multiple clinical trials (188–191), with promising efficacy shown in certain cancer settings. For example, in a Phase 3 IMPACT study for prostate cancer, DC-based therapy, sipuleucel-T, demonstrated significantly better survival by 4 months, for patients with metastatic hormone-resistant prostate cancer compared to the placebo group (191). Multiple Phase 3 prostate cancer studies (NCT00005947, NCT00065442, NCT00779402, NCT01133704) with DC-immunotherapy sipuleucel-T have shown induction of antigen-specific immune responses correlate with better survival in patients (190–194) (Table 7). Because of observed improvements in survival, sipuleucel-T was FDA approved in 2010. It is interesting to note that this coincides with <5% patients achieving an objective response, or tumor reduction over time.
Current clinical strategies are looking to optimize DC immunotherapy through combinations with other agents, in an effort to improve tumor burden. For example, the immunosuppressive tumor environment may prevent DCs from effectively activating cytotoxic T cells and NK cells to eliminate the tumor. Consequently, immune checkpoint inhibitors such as pidilizumab, are currently being explored in combination with DC immunotherapies (NCT01067287, NCT01096602, NCT01441765) for multiple myeloma, acute myelogenous leukemia (AML), and renal cell carcinoma, in an effort to enhance activation of tumor-specific cytotoxic T cells by DCs (195, 196) (Table 7). Ultimately, DC immunotherapies have shown promise in certain cancer settings, and have the advantage of interacting with numerous immune cell subsets to mediate anti-tumor immunity. The efficacy of these DC immunotherapies may be improved upon through combination strategies with other agents and the targeting of immunosuppressive barriers to tumor eradication.
Conclusions
CAR T cell therapies have demonstrated the clinical benefits of harnessing our body's own defenses to combat tumor cells. Similar research is being conducted on lesser known modifications and gene-modified immune cells. Promising preclinical and clinical results point to a likely establishment of these therapeutics as another treatment modality against cancer. Because the field is a recent one, it is necessarily disjointed: different groups focus on their preferred immune effector and seldom compare efficacy with others, much less look at potential combinations. By presenting this review, the authors hope that researchers become more familiar with what is out there—and hope that more efforts at head-to-head comparisons between therapies and combination therapies (which is how the immune system is supposed to act) be explored.
Author Contributions
SP and CC conceptualized the review and made edits to the manuscript. SP, RB, EC, AP, SV, NH, SM, and CC wrote the body of the text. PH and CC made final edits to the manuscript.
Funding
Part of the work was supported by the Alex's Lemonade Stand Foundation A Award, given to CC.
Conflict of Interest Statement
PH and CC are co-founders of Mana Therapeutics, a biotech startup focusing on cell therapies. PH is a member of the board, and CC is a member of the scientific advisory board.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALL, Acute lymphocytic leukemia; AML, Acute myeloid leukemia; BCR, B cell Receptor; BMT, Bone Marrow Transplant; CAR, Chimeric Antigen Receptors; CLL, Chronic lymphocytic leukemia; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; DC, Dendritic Cell; DLBCL, Diffuse Large B Cell Lymphoma; DNR, Double Negative Receptor; ESC, Embryonic Stem Cell; FcR, Fc Receptor; HLA, Human Leukocyte Antigen; HSCT, Hematopoietic Stem Cell Transplant; iPSC, Induced Pluripotent Stem Cell; MHC, Major Histocompatability Complex; mRCC, metastatic renal cell carcinoma; NHL, Non-Hodgkin's Lymphoma; NK, Natural Killer Cell; NKT, Natural Killer T Cell; TALEN, Transcription Activator-Like Effector Nuclease; TCR, T cell Receptor; ZFN, Zinc Finger Nuclease.
References
1. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
2. Barrett DM, Grupp SA, June CH. Chimeric antigen receptor- and TCR-modified T cells enter main street and wall street. J Immunol. (2015) 195:755–61. doi: 10.4049/jimmunol.1500751
3. Phan GQ, Rosenberg SA. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control. (2013) 20:289–97. doi: 10.1177/107327481302000406
4. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. (2014) 257:56–71. doi: 10.1111/imr.12132
5. Abramson JS, Palomba L, Gordon LI, Lunning M, Arnason J, Forero-Torres A, et al. Transcend NHL 001: immunotherapy with the CD19-directed CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-Cell non-hodgkin lymphoma. Blood. (2016) 128:4192.
6. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
7. Neelapu SS. An interim analysis of the ZUMA-1 study of KTE-C19 in refractory, aggressive non-Hodgkin lymphoma. Clin Adv Hematol Oncol. (2017) 15:117–20.
8. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Primary analysis of juliet: a global, pivotal, phase 2 trial of CTL019 in adult patients with relapsed or refractory diffuse large B-cell lymphoma. Blood. (2017) 130:577.
9. Kalaitsidou M, Kueberuwa G, Schutt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy. (2015) 7:487–97. doi: 10.2217/imt.14.123
10. Rossig C, Kailayangiri S, Jamitzky S, Altvater B Carbohydrate targets for CAR T cells in solid childhood cancers. Front Oncol. (2018) 8:513. doi: 10.3389/fonc.2018.00513
11. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
12. Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE. (2013) 8:e82742. doi: 10.1371/journal.pone.0082742
13. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. (2017) 130:285–96. doi: 10.1182/blood-2017-01-761320
14. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. (2013) 121:1165–74. doi: 10.1182/blood-2012-06-438002
15. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. (2017) 127:3462–71. doi: 10.1172/JCI94306
16. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. (2012) 119:3940–50. doi: 10.1182/blood-2011-10-387969
17. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
18. American Society of Clinical Oncology. AR T-Cell Immunotherapy Named Advance of the Year in Annual ASCO Report (2018).
19. Bonini C, Mondino A. Adoptive T-cell therapy for cancer: the era of engineered T cells. Eur J Immunol. (2015) 45:2457–69. doi: 10.1002/eji.201545552
20. Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. (2014) 20:127–33. doi: 10.1097/PPO.0000000000000034
21. Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. (2012) 61:1269–77. doi: 10.1007/s00262-012-1202-z
22. Gold LI. The role for transforming growth factor-beta (TGF-beta) in human cancer. Crit Rev Oncogen. (1999) 10:303–60.
23. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. (2011) 17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557
24. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. (1999) 100:216–32. doi: 10.1016/S0165-5728(99)00203-9
25. Weis-Garcia F, Massague J. Complementation between kinase-defective and activation-defective TGF-beta receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. (1996) 15:276–89. doi: 10.1002/j.1460-2075.1996.tb00358.x
26. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. (2017) 19:408–18. doi: 10.1016/j.jcyt.2016.12.005
27. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. (2008) 31:500–5. doi: 10.1097/CJI.0b013e318177092b
28. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
29. Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. (2013) 20:575–80. doi: 10.1038/gt.2012.75
30. Bendle GM, Linnemann C, Bies L, Song JY, Schumacher TN. Blockade of TGF-beta signaling greatly enhances the efficacy of TCR gene therapy of cancer. J Immunol. (2013) 191:3232–9. doi: 10.4049/jimmunol.1301270
31. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
32. Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. (2005) 201:737–46. doi: 10.1084/jem.20040685
33. Qin W, Tian F, Wang F, Song B, Wang H, Zhang Q, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive cytolytic T cells for treatment of established mouse Renca tumors. Urology. (2008) 72:943–7. doi: 10.1016/j.urology.2008.04.017
34. Quatromoni JG, Wang Y, Vo DD, Morris LF, Jazirehi AR, McBride W, et al. T cell receptor (TCR)-transgenic CD8 lymphocytes rendered insensitive to transforming growth factor beta (TGFbeta) signaling mediate superior tumor regression in an animal model of adoptive cell therapy. J Transl Med. (2012) 10:127. doi: 10.1186/1479-5876-10-127
35. ClinicalTrials.gov. Bethesda, MD: National Library of Medicine (US). Available online at: http://clinicaltrials.gov/
36. Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. (2000) 191:1187–96. doi: 10.1084/jem.191.7.1187
37. Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. (2014) 22:1211–20. doi: 10.1038/mt.2014.47
38. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. (2010) 285:25538–44. doi: 10.1074/jbc.M110.127951
39. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T Cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. (2017) 25:249–58. doi: 10.1016/j.ymthe.2016.10.016
40. Lo AS, Taylor JR, Farzaneh F, Kemeny DM, Dibb NJ, Maher J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol Immunol. (2008) 45:1276–87. doi: 10.1016/j.molimm.2007.09.010
41. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. (2016) 22:3734–45. doi: 10.1158/1078-0432.CCR-15-1879
42. Butler LD, Mohler KM, Layman NK, Cain RL, Riedl PE, Puckett LD, et al. Interleukin-2 induced systemic toxicity: induction of mediators and immunopharmacologic intervention. Immunopharmacol Immunotoxicol. (1989) 11:445–87. doi: 10.3109/08923978909005379
43. Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. (2009) 17:880–8. doi: 10.1038/mt.2009.34
44. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. (2010) 115:3508–19. doi: 10.1182/blood-2009-09-241398
45. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
46. Treisman J, Hwu P, Minamoto S, Shafer GE, Cowherd R, Morgan RA, et al. Interleukin-2-transduced lymphocytes grow in an autocrine fashion and remain responsive to antigen. Blood. (1995) 85:139–45.
47. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. (2011) 19:751–9. doi: 10.1038/mt.2010.313
48. Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol. (2005) 175:7226–34. doi: 10.4049/jimmunol.175.11.7226
49. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. (2015) 4:e994446. doi: 10.4161/2162402X.2014.994446
50. Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. (2015) 13:102. doi: 10.1186/s12967-015-0460-x
51. Di Stasi A, De Angelis B, Savoldo B. Gene therapy to improve migration of T cells to the tumor site. Methods Mol Biol. (2010) 651:103–18. doi: 10.1007/978-1-60761-786-0_7
52. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. (2002) 13:1971–80. doi: 10.1089/10430340260355374
53. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
54. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
55. Garetto S, Sardi C, Martini E, Roselli G, Morone D, Angioni R, et al. Tailored chemokine receptor modification improves homing of adoptive therapy T cells in a spontaneous tumor model. Oncotarget. (2016) 7:43010–26. doi: 10.18632/oncotarget.9280
56. Siddiqui I, Erreni M, van Brakel M, Debets R, Allavena P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J Immunother Cancer. (2016) 4:21. doi: 10.1186/s40425-016-0125-1
57. Asai H, Fujiwara H, An J, Ochi T, Miyazaki Y, Nagai K, et al. Co-introduced functional CCR2 potentiates in vivo anti-lung cancer functionality mediated by T cells double gene-modified to express WT1-specific T-cell receptor. PLoS ONE. (2013) 8:e56820. doi: 10.1371/journal.pone.0056820
58. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
59. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. (2017) 23:6982–92. doi: 10.1158/1078-0432.CCR-17-0867
60. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–856. doi: 10.1038/nbt.4195
61. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2018) 78:4692. doi: 10.1007/s00262-018-2281-2
62. Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer research (2018) 78:4692–4703. doi: 10.1158/0008-5472.CAN-18-0030
63. Wu YL, Ding YP, Tanaka Y, Shen LW, Wei CH, Minato N, et al. Gammadelta T cells and their potential for immunotherapy. Int J Biol Sci. (2014) 10:119–35. doi: 10.7150/ijbs.7823
64. Kabelitz D, Marischen L, Oberg HH, Holtmeier W, Wesch D. Epithelial defence by gamma delta T cells. Int Arch Allergy Immunol. (2005) 137:73–81. doi: 10.1159/000085107
65. Silva-Santos B, Serre K, Norell H. Gammadelta T cells in cancer. Nat Rev Immunol. (2015) 15:683–91. doi: 10.1038/nri3904
66. Lamb LS Jr, Lopez RD. Gammadelta T cells: a new frontier for immunotherapy? Biol Blood Marrow Transplant. (2005) 11:161–8. doi: 10.1016/j.bbmt.2004.11.015
67. Minculescu L, Sengelov H. The role of gamma delta T cells in haematopoietic stem cell transplantation. Scand J Immunol. (2015) 81:459–68. doi: 10.1111/sji.12289
68. Deniger DC, Maiti SN, Mi T, Switzer KC, Ramachandran V, Hurton LV, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin Cancer Res. (2014) 20:5708–19. doi: 10.1158/1078-0432.CCR-13-3451
69. Liu Z, Guo BL, Gehrs BC, Nan L, Lopez RD. Ex vivo expanded human Vgamma9Vdelta2+ gammadelta-T cells mediate innate antitumor activity against human prostate cancer cells in vitro. J Urol. (2005) 173:1552–6. doi: 10.1097/01.ju.0000154355.45816.0b
70. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. (2018) 26:354–365. doi: 10.1016/j.ymthe.2017.12.001
71. Bennouna J, Bompas E, Neidhardt EM, Rolland F, Philip I, Galea C, et al. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. (2008) 57:1599–609. doi: 10.1007/s00262-008-0491-8
72. Lawand M, Dechanet-Merville J, Dieu-Nosjean MC. Key features of gamma-delta T-cell subsets in human diseases and their immunotherapeutic implications. Front Immunol. (2017) 8:761. doi: 10.3389/fimmu.2017.00761
73. Zhao Y, Niu C, Cui J. Gamma-delta (gammadelta) T cells: friend or foe in cancer development? J Transl Med. (2018) 16:3. doi: 10.1186/s12967-017-1378-2
74. Hellstrom I, Hellstrom KE, Pierce GE, Yang JP. Cellular and humoral immunity to different types of human neoplasms. Nature. (1968) 220:1352–4. doi: 10.1038/2201352a0
75. Burga RA, Nguyen T, Zulovich J, Madonna S, Ylisastigui L, Fernandes R, et al. Improving efficacy of cancer immunotherapy by genetic modification of natural killer cells. Cytotherapy. (2016) 18:1410–21. doi: 10.1016/j.jcyt.2016.05.018
76. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. (1986) 319:675–8. doi: 10.1038/319675a0
77. Lanier LL, Ruitenberg JJ, Phillips JH. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J Immunol. (1988) 141:3478–85.
78. Parham P. Taking license with natural killer cell maturation and repertoire development. Immunol Rev. (2006) 214:155–60. doi: 10.1111/j.1600-065X.2006.00462.x
79. Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. (2010) 115:4293–301. doi: 10.1182/blood-2009-05-222190
80. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. (2010) 28:955–9. doi: 10.1200/JCO.2009.24.4590
81. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
82. Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. Semin Immunol. (2014) 26:173–9. doi: 10.1016/j.smim.2014.02.004
83. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
84. Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr. (2014) 26:43–9. doi: 10.1097/MOP.0000000000000043
85. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. (2016) 3:16011. doi: 10.1038/mto.2016.11
86. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
87. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, et al. Critical Role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. (2015) 6:482. doi: 10.3389/fimmu.2015.00482
88. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
89. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425; author reply 426. doi: 10.1158/0008-5472.CAN-06-3037
90. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. (2014) 20:4096–106. doi: 10.1158/1078-0432.CCR-14-0635
91. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE. (2011) 6:e19495. doi: 10.1371/journal.pone.0019495
92. Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cancer immunology, immunotherapy : CII (2010) 59:1739–44. doi: 10.1007/s00262-010-0896-z
93. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
94. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
95. Gasteiger G, Hemmers S, Firth MA, Le Floc'h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. The J Exp Med. (2013) 210:1167–78. doi: 10.1084/jem.20122462
96. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. (2012) 72:1407–15. doi: 10.1158/0008-5472.CAN-11-2544
97. Mentlik James A, Cohen AD, Campbell KS. Combination immune therapies to enhance anti-tumor responses by NK cells. Front Immunol. (2013) 4:481. doi: 10.3389/fimmu.2013.00481
98. Lopes de Menezes DE, Denis-Mize K, Tang Y, Ye H, Kunich JC, Garrett EN, et al. Recombinant interleukin-2 significantly augments activity of rituximab in human tumor xenograft models of B-cell non-Hodgkin lymphoma. J Immunother. (2007) 30:64–74. doi: 10.1097/01.cji.0000211315.21116.07
99. Cany J, van der Waart AB, Spanholtz J, Tordoir M, Jansen JH, van der Voort R, et al. Combined IL-15 and IL-12 drives the generation of CD34(+)-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology. (2015) 4:e1017701. doi: 10.1080/2162402X.2015.1017701
100. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS ONE. (2011) 6:e20740. doi: 10.1371/journal.pone.0020740
101. Sathe P, Pang SHM, Delconte R, Elwood N, Huntington ND Identification of novel human NK cell progenitor subsets. Int J Mol Sci. (2017). 18:2716. doi: 10.3390/ijms18122716
102. Hoogstad-van Evert JS, Cany J, van den Brand D, Oudenampsen M, Brock R, Torensma R, et al. Umbilical cord blood CD34(+) progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rg(null) mice. Oncoimmunology. (2017) 6:e1320630. doi: 10.1080/2162402X.2017.1320630
103. Herrera L, Salcedo JM, Santos S, Vesga MA, Borrego F, Eguizabal C OP9 feeder cells are superior to M2-10B4 Cells for the generation of mature and functional natural killer cells from umbilical cord hematopoietic progenitors. Front Immunol. (2017) 8:755. doi: 10.3389/fimmu.2017.00755
104. Nham T, Poznanski SM, Fan IY, Vahedi F, Shenouda MM, Lee AJ, et al. Ex vivo-expanded natural killer cells derived from long-term cryopreserved cord blood are cytotoxic against primary breast cancer cells. J Immunother. (2018) 41:64–72. doi: 10.1097/CJI.0000000000000192
105. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. (2009) 69:4010–7. doi: 10.1158/0008-5472.CAN-08-3712
106. Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy. (2010) 12:1044–55. doi: 10.3109/14653249.2010.504770
107. Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. (2008) 111:3155–62. doi: 10.1182/blood-2007-09-110312
108. Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, et al. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol. (2001) 62:1092–8. doi: 10.1016/S0198-8859(01)00313-5
109. Guven H, Gilljam M, Chambers BJ, Ljunggren HG, Christensson B, Kimby E, et al. Expansion of natural killer (NK) and natural killer-like T (NKT)-cell populations derived from patients with B-chronic lymphocytic leukemia (B-CLL): a potential source for cellular immunotherapy. Leukemia. (2003) 17:1973–80. doi: 10.1038/sj.leu.2403083
110. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. (2015) 13:277. doi: 10.1186/s12967-015-0632-8
111. Torelli GF, Guarini A, Maggio R, Alfieri C, Vitale A, Foa R. Expansion of natural killer cells with lytic activity against autologous blasts from adult and pediatric acute lymphoid leukemia patients in complete hematologic remission. Haematologica. (2005) 90:785–92.
112. Torelli GF, Guarini A, Palmieri G, Breccia M, Vitale A, Santoni A, et al. Expansion of cytotoxic effectors with lytic activity against autologous blasts from acute myeloid leukaemia patients in complete haematological remission. Br J Haematol. (2002) 116:299–307. doi: 10.1046/j.1365-2141.2002.03277.x
113. Hercend T, Farace F, Baume D, Charpentier F, Droz JP, Triebel F, et al. Immunotherapy with lymphokine-activated natural killer cells and recombinant interleukin-2: a feasibility trial in metastatic renal cell carcinoma. J Biol Response Mod. (1990) 9:546–55.
114. Klingemann HG, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy. (2004) 6:15–22. doi: 10.1080/14653240310004548
115. Pierson BA, Europa AF, Hu WS, Miller JS. Production of human natural killer cells for adoptive immunotherapy using a computer-controlled stirred-tank bioreactor. J Hematother. (1996) 5:475–83. doi: 10.1089/scd.1.1996.5.475
116. Miller JS, Klingsporn S, Lund J, Perry EH, Verfaillie C, McGlave P. Large scale ex vivo expansion and activation of human natural killer cells for autologous therapy. Bone Marrow Transplant. (1994) 14:555–62.
117. Tam YK, Martinson JA, Doligosa K, Klingemann HG. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy. (2003) 5:259–72. doi: 10.1080/14653240310001523
118. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. (2008) 10:625–32. doi: 10.1080/14653240802301872
119. Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. (2000) 14:922–30. doi: 10.1038/sj.leu.2401769
120. Tsuchiyama J, Yoshino T, Mori M, Kondoh E, Oka T, Akagi T, et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood. (1998) 92:1374–83.
121. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM II. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
122. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. (2017) 25:1769–81. doi: 10.1016/j.ymthe.2017.06.012
123. Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. (2012) 14:830–40. doi: 10.3109/14653249.2012.671519
124. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell non-hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. (2015) 3:333–44. doi: 10.1158/2326-6066.CIR-14-0114
125. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. (2005) 106:376–83. doi: 10.1182/blood-2004-12-4797
126. Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. (2010) 17:147–54. doi: 10.1038/cgt.2009.61
127. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
128. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
129. Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. (2015) 194:3201–12. doi: 10.4049/jimmunol.1400330
130. Lim O, Jung MY, Hwang YK, Shin EC. Present and Future of allogeneic natural killer cell therapy. Front Immunol. (2015) 6:286. doi: 10.3389/fimmu.2015.00286
131. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: recent innovations and future developments. Front Immunol. (2017) 8:631. doi: 10.3389/fimmu.2017.00631
132. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant. (2003) 32:177–86. doi: 10.1038/sj.bmt.1704086
133. Klingemann H. Challenges of cancer therapy with natural killer cells. Cytotherapy. (2015) 17:245–9. doi: 10.1016/j.jcyt.2014.09.007
134. Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. (2012) 14:1131–43. doi: 10.3109/14653249.2012.700767
135. Jungkunz-Stier I, Zekl M, Stuhmer T, Einsele H, Seggewiss-Bernhardt R. Modulation of natural killer cell effector functions through lenalidomide/dasatinib and their combined effects against multiple myeloma cells. Leuk Lymphoma. (2014) 55:168–76. doi: 10.3109/10428194.2013.794270
136. Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene. (2013) 32:4191–202. doi: 10.1038/onc.2012.599
137. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. (2008) 14:4650–7. doi: 10.1158/1078-0432.CCR-07-4405
138. Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. (2014) 123:3016–26. doi: 10.1182/blood-2013-10-533398
139. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. (2012) 11:2674–84. doi: 10.1158/1535-7163.MCT-12-0692
140. Vallera DA, Zhang B, Gleason MK, Oh S, Weiner LM, Kaufman DS, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharmaceut. (2013) 28:274–82. doi: 10.1089/cbr.2012.1329
141. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. (2013) 19:3844–55. doi: 10.1158/1078-0432.CCR-13-0505
142. Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. (2004) 114:1379–88. doi: 10.1172/JCI200423594
143. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
144. Molling JW, Langius JA, Langendijk JA, Leemans CR, Bontkes HJ, van der Vliet HJ, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol. (2007) 25:862–8. doi: 10.1200/JCO.2006.08.5787
145. Motohashi S, Okamoto Y, Yoshino I, Nakayama T. Anti-tumor immune responses induced by iNKT cell-based immunotherapy for lung cancer and head and neck cancer. Clin Immunol. (2011) 140:167–76. doi: 10.1016/j.clim.2011.01.009
146. Schneiders FL, Scheper RJ, von Blomberg BM, Woltman AM, Janssen HL, van den Eertwegh AJ, et al. Clinical experience with alpha-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol. (2011) 140:130–41. doi: 10.1016/j.clim.2010.11.010
147. Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med. (2003) 197:1667–76. doi: 10.1084/jem.20021650
148. Fujii S, Shimizu K, Klimek V, Geller MD, Nimer SD, Dhodapkar MV. Severe and selective deficiency of interferon-gamma-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br J Haematol. (2003) 122:617–22. doi: 10.1046/j.1365-2141.2003.04465.x
149. Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, et al. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J Immunol. (2001) 167:4046–50. doi: 10.4049/jimmunol.167.7.4046
150. Yanagisawa K, Seino K, Ishikawa Y, Nozue M, Todoroki T, Fukao K. Impaired proliferative response of V alpha 24 NKT cells from cancer patients against alpha-galactosylceramide. J Immunol. (2002) 168:6494–9. doi: 10.4049/jimmunol.168.12.6494
151. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. (1997) 278:1626–9. doi: 10.1126/science.278.5343.1626
152. Nakagawa R, Motoki K, Ueno H, Iijima R, Nakamura H, Kobayashi E, et al. Treatment of hepatic metastasis of the colon26 adenocarcinoma with an alpha-galactosylceramide, KRN7000. Cancer Res. (1998) 58:1202–7.
153. Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, et al.The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol. (2000) 12:987–94. doi: 10.1093/intimm/12.7.987
154. Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, et al. Defective NKT cell activation by CD1d+ TRAMP prostate tumor cells is corrected by interleukin-12 with alpha-galactosylceramide. PLoS ONE. (2010) 5:e11311. doi: 10.1371/journal.pone.0011311
155. Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med. (2004) 199:1213–21. doi: 10.1084/jem.20031462
156. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
157. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. (1998) 188:1521–8. doi: 10.1084/jem.188.8.1521
158. Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. (1999) 163:4647–50.
159. Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci USA. (2008) 105:11287–92. doi: 10.1073/pnas.0801631105
160. Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med. (2005) 202:1279–88. doi: 10.1084/jem.20050953
161. Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev. (2007) 220:183–98. doi: 10.1111/j.1600-065X.2007.00561.x
162. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. (2003) 171:5140–7. doi: 10.4049/jimmunol.171.10.5140
163. Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, et al. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood. (2002) 99:1259–66. doi: 10.1182/blood.V99.4.1259
164. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. (2014) 124:2824–33. doi: 10.1182/blood-2013-11-541235
165. Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. (2018) 34:596–610 e11. doi: 10.1016/j.ccell.2018.08.017
166. Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. (2002) 8:3702–9.
167. Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. (2005) 201:1503–17. doi: 10.1084/jem.20042592
168. Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2005) 11:1910–7. doi: 10.1158/1078-0432.CCR-04-1453
169. Motohashi S, Nagato K, Kunii N, Yamamoto H, Yamasaki K, Okita K, et al. A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol. (2009) 182:2492–501. doi: 10.4049/jimmunol.0800126
170. Nicol AJ, Tazbirkova A, Nieda M. Comparison of clinical and immunological effects of intravenous and intradermal administration of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells. Clin Cancer Res. (2011) 17:5140–51. doi: 10.1158/1078-0432.CCR-10-3105
171. Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, et al. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. (2004) 103:383–9. doi: 10.1182/blood-2003-04-1155
172. Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, et al. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood. (2013) 121:423–30. doi: 10.1182/blood-2012-06-435503
173. Shin T, Nakayama T, Akutsu Y, Motohashi S, Shibata Y, Harada M, et al. Inhibition of tumor metastasis by adoptive transfer of IL-12-activated Valpha14 NKT cells. Int J Cancer. (2001) 91:523–8. doi: 10.1002/1097-0215(20010215)91:4<523::AID-IJC1087>3.0.CO;2-L
174. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. (2002) 3:867–74. doi: 10.1038/ni827
175. Toura I, Kawano T, Akutsu Y, Nakayama T, Ochiai T, Taniguchi M. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with alpha-galactosylceramide. J Immunol. (1999) 163:2387–91.
176. van der Vliet HJ, Molling JW, Nishi N, Masterson AJ, Kolgen W, Porcelli SA, et al. Polarization of Valpha24+ Vbeta11+ natural killer T cells of healthy volunteers and cancer patients using alpha-galactosylceramide-loaded and environmentally instructed dendritic cells. Cancer Res. (2003) 63:4101–6.
177. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical trial. Clin Cancer Res. (2017) 23:3510–9. doi: 10.1158/1078-0432.CCR-16-0600
178. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest. (2016) 126:2341–55. doi: 10.1172/JCI83476
179. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. (2010) 11:303–12. doi: 10.1038/ni.1853
180. Fujii S, Goto A, Shimizu K. Antigen mRNA-transfected, allogeneic fibroblasts loaded with NKT-cell ligand confer antitumor immunity. Blood. (2009) 113:4262–72. doi: 10.1182/blood-2008-08-176446
181. Macho Fernandez E, Chang J, Fontaine J, Bialecki E, Rodriguez F, Werkmeister E, et al. Activation of invariant Natural Killer T lymphocytes in response to the alpha-galactosylceramide analogue KRN7000 encapsulated in PLGA-based nanoparticles and microparticles. Int J Pharmaceut. (2012) 423:45–54. doi: 10.1016/j.ijpharm.2011.04.068
182. Nakamura T, Yamazaki D, Yamauchi J, Harashima H. The nanoparticulation by octaarginine-modified liposome improves alpha-galactosylceramide-mediated antitumor therapy via systemic administration. J Controlled Release. (2013) 171:216–24. doi: 10.1016/j.jconrel.2013.07.004
183. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
184. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. doi: 10.1084/jem.20101158
185. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
186. Boudreau JE, Bridle BW, Stephenson KB, Jenkins KM, Brunelliere J, Bramson JL, et al. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol Ther. (2009) 17:1465–72. doi: 10.1038/mt.2009.95
187. Lion E, Smits EL, Berneman ZN, Van Tendeloo VF. NK cells: key to success of DC-based cancer vaccines? Oncologist. (2012) 17:1256–70. doi: 10.1634/theoncologist.2011-0122
188. Draube A, Klein-Gonzalez N, Mattheus S, Brillant C, Hellmich M, Engert A, et al. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS ONE. (2011) 6:e18801. doi: 10.1371/journal.pone.0018801
189. Leonhartsberger N, Ramoner R, Falkensammer C, Rahm A, Gander H, Holtl L, et al. Quality of life during dendritic cell vaccination against metastatic renal cell carcinoma. Cancer Immunol Immunother. (2012) 61:1407–13. doi: 10.1007/s00262-012-1207-7
190. Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. (2009) 115:3670–9. doi: 10.1002/cncr.24429
191. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
192. Schellhammer PF, Chodak G, Whitmore JB, Sims R, Frohlich MW, Kantoff PW. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology. (2013) 81:1297–302. doi: 10.1016/j.urology.2013.01.061
193. Sheikh NA, Petrylak D, Kantoff PW, Dela Rosa C, Stewart FP, Kuan LY, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother. (2013) 62:137–47. doi: 10.1007/s00262-012-1317-2
194. Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. (2006) 24:3089–94. doi: 10.1200/JCO.2005.04.5252
195. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
Keywords: cell therapy, gene modified cells, immunotherapy, gamma delta T cells, NK cells, NKT cells, dendritic cells
Citation: Patel S, Burga RA, Powell AB, Chorvinsky EA, Hoq N, McCormack SE, Van Pelt SN, Hanley PJ and Cruz CRY (2019) Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies Against Cancer. Front. Oncol. 9:196. doi: 10.3389/fonc.2019.00196
Received: 02 January 2019; Accepted: 07 March 2019;
Published: 10 April 2019.
Edited by:
Matteo Bellone, San Raffaele Hospital (IRCCS), ItalyReviewed by:
John-Maher, King's College London, United KingdomDario Sangiolo, University of Turin, Italy
Copyright © 2019 Patel, Burga, Powell, Chorvinsky, Hoq, McCormack, Van Pelt, Hanley and Cruz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Conrad Russell Y. Cruz, Y3JjcnV6QGVtYWlsLmd3dS5lZHU=