 Giuseppe A. Palumbo1*
Giuseppe A. Palumbo1* Stefania Stella2,3
Stefania Stella2,3 Maria Stella Pennisi2,3
Maria Stella Pennisi2,3 Cristina Pirosa4
Cristina Pirosa4 Elisa Fermo5Sonia Fabris5
Elisa Fermo5Sonia Fabris5 Daniele Cattaneo5
Daniele Cattaneo5 Alessandra Iurlo6
Alessandra Iurlo6- 1Department of Scienze Mediche, Chirurgiche e Tecnologie Avanzate “G.F. Ingrassia,” University of Catania, Catania, Italy
- 2Center of Experimental Oncology and Hematology, A.O.U. Policlinico-Vittorio Emanuele, Catania, Italy
- 3Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 4Postgraduate School of Hematology, University of Catania, Catania, Italy
- 5Hematology Division, Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
- 6Hematology Division, Myeloproliferative Syndromes Unit, Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
The hallmark of BCR-ABL1-negative myeloproliferative neoplasms (MPNs) is the presence of a driver mutation in JAK2, CALR, or MPL gene. These genetic alterations represent a key feature, useful for diagnostic, prognostic and therapeutical approaches. Molecular biology tests are now widely available with different specificity and sensitivity. Recently, the allele burden quantification of driver mutations has become a useful tool, both for prognostication and efficacy evaluation of therapies. Moreover, other sub-clonal mutations have been reported in MPN patients, which are associated with poorer prognosis. ASXL1 mutation appears to be the worst amongst them. Both driver and sub-clonal mutations are now taken into consideration in new prognostic scoring systems and may be better investigated using next generation sequence (NGS) technology. In this review we summarize the value of NGS and its contribution in providing a comprehensive picture of mutational landscape to guide treatment decisions. Finally, discussing the role that NGS has in defining the potential risk of disease development, we forecast NGS as the standard molecular biology technique for evaluating these patients.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal disorders of the hematopoietic stem cell, mainly characterized by proliferative bone marrow with varying degrees of reticulin/collagen fibrosis, extramedullary hematopoiesis, abnormal peripheral blood count, and constitutional symptoms that are secondary to abnormally expressed inflammatory cytokines (1). Among the so-called “BCR-ABL1-negative MPNs” polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) are included. However, unlike chronic myeloid leukemia (which is always characterized by the BCR–ABL1 fusion gene), they have not yet been associated with any specific genetic abnormalities.
The discovery in 2005 of the JAK2V617F point mutation (2–5) and the subsequent identification of other specific abnormalities, such as JAK2 exon 12 (6, 7), MPL exon 10 (8–10) and CALR exon 9 (11, 12), gave an improvement in understanding their genetic basis. All of them are now included in the molecular diagnostic and prognostic algorithms for MPNs, leading to several revisions of the diagnostic criteria for these diseases. In addition, they are used as markers of disease burden and as a measure of assessing response to various therapeutic interventions that can target the mutant clone.
The MPN driver mutations are often mutually exclusive and, after the detection of the common JAK2V617F mutation, generally no further testing is performed. Nevertheless, in recent years, several reports have suggested that driver mutations indeed do rarely coexist (13–16), but additional studies are needed to clarify the clinical implications of double-mutated cases.
Importantly, additional sub-clonal driver and non-driver mutations in genes such as ASXL1, SRSF2, EZH2, IDH1, and IDH2, among others, have been identified as being associated with disease progression (17, 18). A wide choice of techniques is currently available for the detection of MPN mutations, and a continuous evolution of molecular diagnostic applications and platforms is now ongoing.
JAK2
JAK2 is a non-receptor tyrosine kinase, which, upon ligand binding to specific cytokine receptors, is phosphorylated and activated, leading to regulation of gene expression involved in cell proliferation and survival. The JAK2V617F mutation is a G to T somatic mutation at nucleotide 1849 in exon 14, resulting in the substitution of valine to phenylalanine at codon 617, which triggers constitutive activation of downstream signaling and uncontrolled cell growth (Figure 1A).
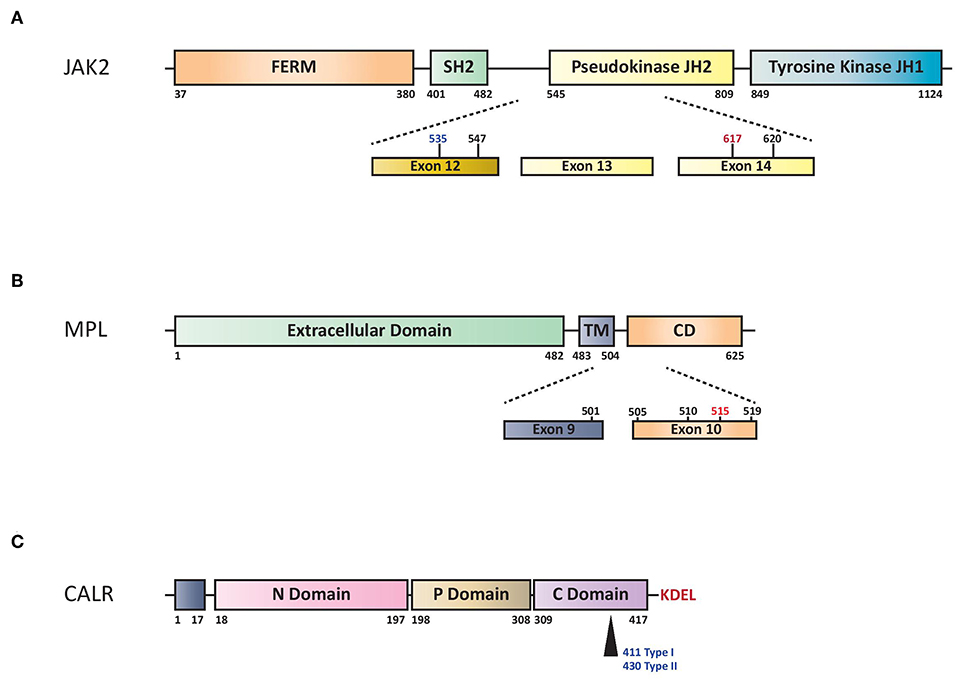
Figure 1. (A) Schematic representation of structural domains of the JAK2. FERM indicates 4.1 Ezrin, Radixin, Moesin domain; JH1 and JH2 signal (JAK Homology 1 and 2) domains refer to tyrosine kinase and pseudo-kinase domains, respectively. SH2 indicates Src Homology 2 domain. Numbers represent amino acid positions within the JAK2 protein: red indicates V617F mutation, in black other SNP mutations and in blue the region for insertions/deletions. (B) Structural domains and mutations of the MPL. Extracellular domain indicates NH2 amino terminal region, TM transmembrane domain and CD cytoplasmic domain. Dashed lines highlights exons reported mutations of MPL. W515L or W515K are indicated in red while the others hot spots variant mutations in black. Numbers represent amino acid positions. (C) Structural domains of CALR protein. CALR includes NH2 domain (N-Domain) and Proline-rich domain (P-Domain) with chaperone lectin-like function. COOH domain (C Domain) has aminoacids responsible for Ca++-buffering. Arrow indicates Type I (deletion) or Type II (insertion) alterations that determine loss of KDEL aminoacids and generation of a new tail with low calcium buffering.
V617F mutation in exon 14 of JAK2 gene is present in the majority of patients with PV (more than 95%) and in 60% of those with ET or PMF (2–5). Rare insertions and deletions in exon 12 have been described in 2 to 3% of patients with PV (6, 7) (Figure 1A). The most widely used method for JAK2V617F detection is based on allele-specific PCR (2). Quantitative PCR methods (qPCR) are preferred over qualitative ones because of greater reproducibility and sensitivity and because of the need of quantifying the mutated clone in MPN patients.
Actually, V617F allelic burden at diagnosis provides important prognostic information, being found to be associated with phenotypic presentation and severity of MPNs (19–21), the risk of thrombotic events (20, 22) and progression to secondary myelofibrosis (MF) (23, 24). In particular, in PMF patients JAK2V617F mutation is associated with clinical characteristics which include older age, higher hemoglobin level, leukocytosis, and lower platelet count (20; (25) and a low JAK2V617F allele burden may represent a favorable prognostic factor (26). With regards to PV, a higher JAK2V617F mutant allele burden has been associated with more frequent thrombotic complications (20), pruritus and fibrotic transformation (27). Moreover, V617F allelic burden measured during the follow-up is currently used in the course of treatment with alpha-interferon and JAK1/2 inhibitors (28–30), as well as for minimal residual disease (MRD) evaluation after allogeneic stem cells transplantation (31). In fact, although the ideal therapy should be able to eradicate the malignant MPN clone, this aim has not been reached with the current available treatments in contrast to the striking efficacy of tyrosine kinase inhibitor monotherapy in chronic myeloid leukemia (32).
Several quantitative methods have been developed in the years, most of them based on real-time allele-specific PCR, with sensitivity ranging from 0.05 to 1%, and specificity of 100% (33–36). Digital PCR (dPCR) has also been proposed with the aim to achieve an absolute quantification of the target gene without the need for a standard curve, with comparable or higher performances compared to qPCR (37–39). Recently, Next Generation Sequencing (NGS) has been shown to allow the detection of the V617F mutation with comparable performances, but weaker sensitivity to qPCR, with the advantage of detection of new potentially pathogenic JAK2 variants (40).
Regarding JAK2 exon 12 variants detection, different approaches can be adopted; Sanger Sequencing (SS) is the most frequently used because of the rarity and heterogeneity of these mutations (41, 42). Nested High-Resolution Melting (HRM) curve analysis has been proposed as highly sensitive screening method eventually followed by SS for the precise characterization of the mutation (36, 43).
MPL
The myeloproliferative leukemia virus oncogene (MPL) is located on chromosome 1p34 and encodes for the thrombopoietin (TPO) receptor, thus assuming a crucial role in the regulation of megakaryocyte growth and survival. In 2006, a somatic activating mutation in exon 10 of this gene, MPLW515L, was described in JAK2V617F-negative ET/PMF (44, 45). This mutation is characterized by a G to T transition at nucleotide 1544, resulting in a tryptophan to leucine substitution at codon 515 of the transmembrane region of MPL, inducing constitutive activation of the TPO receptor in a cytokine-independent fashion (Figure 1B).
Activating mutations in MPL are reported in ~5–10% of all PMF patients and 1–4% of those with ET (44–46). All of them cluster in exon 10 and in the majority of cases affect a tryptophan in position 515 (W515L, W515K, W515A, and W515R).
The methods applied for the detection of MPL mutations can be grouped according to different strategies: targeted identification of specific mutations or sequencing of the entire exon 10. Targeted analysis include allele-specific PCR, allele-specific qPCR and Amplification Refractory Mutation System (ARMS)-PCR; among them qPCR displays the highest sensitivity (0.1–0.5%) (8, 9, 47–49). A multiplexed allele-specific PCR assay for the four most frequent MPL exon 10 mutations (W515L, W515K, W515A, and S505N) has been developed, with 100% specificity and 2.5% sensitivity (50). Analysis of the entire exon 10 allows the detection of all the known and potentially new mutations and can be achieved by SS, pyrosequencing or HRM curve. The sensitivity of these approaches is lower, reaching 2–5% for the latter method (10, 51).
Calreticulin
Calreticulin (CALR) mutations were reported for the first time at the end of 2013 (11, 12). These mutations represent the second most common genetic abnormality in MPNs after JAK2V617F, even though they are absent in PV patients. On the other hand, CALR mutations partially addressed the molecular gap in JAK2/MPL-unmutated ET and PMF patients, accounting for 20–25% of the overall somatic mutations. The remaining cases (i.e., negative for JAK2, CALR, and MPL) are termed “triple-negative,” representing the 5–10% of all BCR-ABL1-negative MPN patients.
CALR is a multi-functional Ca2+ binding protein with chaperone activity mainly localized in the endoplasmic reticulum. The localization and retention of CALR are defined by the N-terminal signal sequence and the C-terminal ER-retention sequence KDEL (Figure 1C). It is involved in numerous intracellular (cytoplasm and nucleus), cell surface, and extracellular functions such as protein quality control, calcium metabolism, immune response, phagocytosis, cell adhesion and others (52). CALR mutations were phenotypic drivers in the pathogenesis of MPNs (53). Recently, studies concerning the role of CALR mutated proteins demonstrated that they are able to bind to the MPL receptor inducing JAK-STAT activation and the positive aminoacid charge of the mutant C terminus is required to mediate this interaction (54–57).
More than 50 different CALR mutations have been described so far, with type 1 (L367fs*46) resulted from 52-bp deletion and type 2 (K385fs*47) from 5-bp TTGTC insertion accounting for ~80% of all the cases. More type 1 (53%) than type 2 (32%) abnormalities are found in MPN patients (11), whereas the remaining cases are classified as either type 1-like, type 2-like, or “other type,” based on their structural similarities to the classical mutations. Their distribution is 57% type 1(-like) and 39% type 2(-like) in ET and 83% type 1(-like) and 15% type 2(-like) in PMF (58). All these abnormalities frequently consist of insertions or deletions involving exon 9 of the gene, generating a frameshift to a unique alternative reading frame; it results in a novel C-terminus peptide sequence enriched for positively charged residues. Furthermore, the mutated protein lacks the KDEL signal, leading to a partial dislocation of CALR from the endoplasmic reticulum (11).
In PMF patients the favorable prognostic impact is limited to CALR type 1/type 1-like mutations, whereas type 2/type 2-like are associated with a worse prognosis, similar to that of JAK2-positive patients (59). On the contrary, in ET CALR type 1 and type 1-like mutations are associated with an higher risk of MF transformation (58).
As previously published for JAK2 mutations, association between CALR mutation allele burden and disease phenotype has been reported (60, 61). In particular, an association between CALR allele burden, leukocyte and platelet counts, hemoglobin and lactate dehydrogenase levels was described. Furthermore, the median CALR allele burden remained steady over time; interestingly, differently from JAK2-positive cases, acute myeloid leukemias (AML) evolving from CALR-mutant MPNs commonly maintained their mutational profile (61). Additional studies are needed to better clarify the CALR mutant allele burden clinical implications.
Given their high frequency in MPNs, screening for CALR mutations is recommended in all JAK2V617F-negative patients with suspected ET or PMF (36, 62, 63). Several methods have been proposed to detect CALR mutations, including SS, fragment length analysis, real-time qPCR, HRM, ddPCR, pyrosequencing and NGS (64–69). Due to the heterogeneity of CALR insertions and deletions detected, direct SS is considered to be the primary method of testing. Indeed, SS of exon 9, providing specific sequence change information, allowed to identify the exact type of CALR mutation and detailed procedures, described in previous publications, were the most used conditions in diagnostic routine screening laboratories.
Role of Next-Generation Sequencing in MPNs
Nowadays, NGS has played an important role in understanding the genetic alterations of different human cancers. There are several number of available NGS platforms using different sequencing technologies, which perform sequencing of millions of small fragments of DNA in parallel (70). Nevertheless, different sequencing chemistry and methods for signal detection, the obtained results are comparable. Bioinformatics analyses are used to piece together these fragments by mapping the individual reads to the human reference genome.
This method provides several advantages compared to different sequencing methods. First of all, NGS is a high-throughput method as it detects concomitant mutations in the same run. Then, the analysis requires low input of DNA/RNA sample as compared to traditional sequencing methods (e.g., SS or Pyrosequencing). Moreover, NGS discriminates genomic aberrations, which are screened simultaneously, such as single/multiple nucleotide variants (SNVs), small and large insertions and deletions (ins/dels) and copy-number variations (CNV) with high sensitivity and accuracy, so reducing data analysis and clinical reporting time (Table 1) (70).
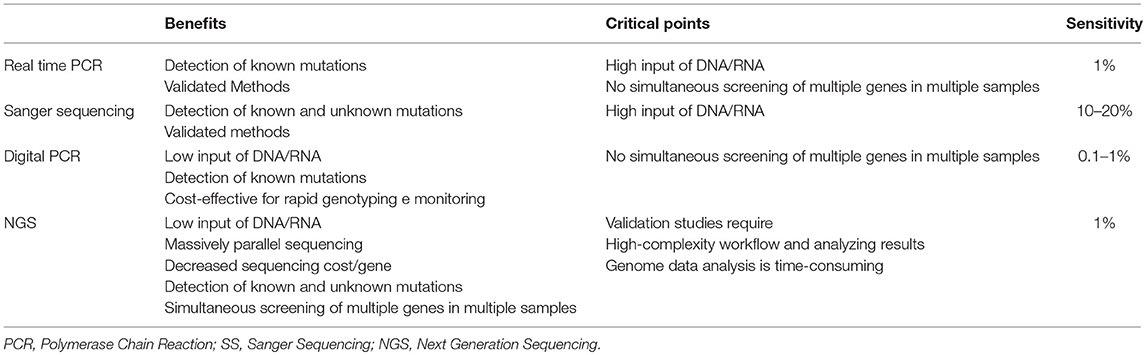
Table 1. Comparison of Real Time PCR, Digital PCR, SS, and NGS technologies in clinical molecular diagnostics.
In the last few years, NGS have been applied to many hematological disorders such as for establishing T-cell clonality, recurrent cytogenetic translocations and prognosis of Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL) (70). In addition, NGS approaches can be used in diagnostic samples to understand the possible evolution of MRD in clonal IGH and TCR rearrangements in lymphoproliferative disorders (71). A comparison study between the most common technologies used to lymphoproliferative disorders, underlined that NGS method was able to identify mutant or clonal DNA in few tumor circulating cells. For this reason, NGS may be used to improve MRD in post-therapy monitoring (72).
In MPNs, it was demonstrated that JAK2 V617F allele burden identified patients with different clinical course: in PV and in ET, variant allele frequency of JAK2 V617F, was associated with a higher ratio of fibrotic transformation. In patients in PMF high variant allele frequency was correlated to recurrent thrombosis event, while low variant allele frequency was associated to short leukemia-free survival and overall survival, probably due to acquisition of additional driver mutations. In fact, new NGS panels are used as predict methods to describe alterations of several genes involved in transcriptional regulation and cell signaling pathways (Table 2). These information can be used to predict the risk leukemic transformation, to guide target treatment and to speculate a personalized clinical trial (32, 73, 84). Using the high sensitivity and specificity of NGS technology, several groups analyzed and deeply studied the genetic and molecular profile of MPN subjects, with the aim of assessing not only non-driver mutations, but also infrequent variants of driver mutations (Table 2). In MPN patients negative for JAK2, CALR, and MPL (triple-negative, TN), mutations in LNK, TET2, DNMT3A, IDH1/2, CBL, and ASXL1 genes but also atypical mutation in MPL (S204P) have been identified (83, 85). In this contest, Chang et al. identified 30 missense mutations in 12 of 16 triple-negative MPN patients. In particular, in this study NGS platform was able to identify low allelic burden and atypical mutations of JAK2 (JAK2V626F and JAK2F556V) and MPL (MPLS204P and MPLY591N) (86).
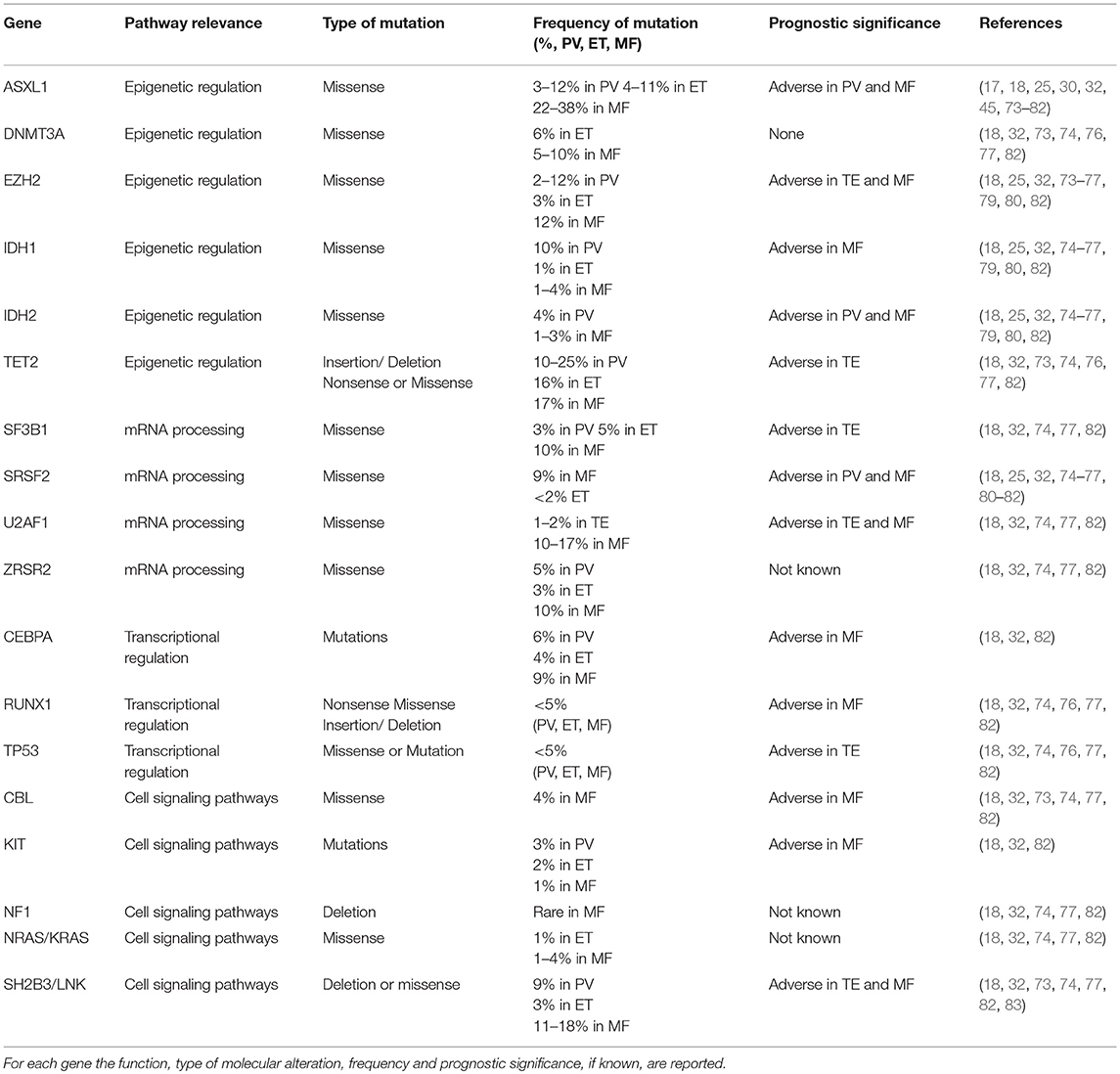
Table 2. Mutated genes in BCR-ABL1-negative myeloproliferative neoplasms (MPNs).
Furthermore, Cabagnols et al. noticed that an atypical mutation of MPL (MPLS204P) was associated with other alterations in ASXL1 and SRSF2 genes. The subjects with more than one alteration were classified as myelodysplastic syndrome (MDS) with thrombocytosis rather than ET. The MPLS204P is a weak gain-of-function mutation that induces constitutive STAT activation and more prolonged ligand-induced STAT phosphorylation than wild-type MPL, while MPLY591N is associated with MPLW515A and in a mouse model induced a more aggressive MPN behavior than that associated with a single MPLY591N mutant (87).
Additionally, applying NGS methodology on a group of 197 MPN patients, Lundberg et al. observed several alterations concerning genes involved in DNA methylation (TET2, DNMT3A, IDH1) and chromatin structure (EZH2, ASXL1) regulation. Moreover, they analyzed the impact of somatic mutations on clinical outcomes and found that the concomitant somatic mutations in TP53 or TET2 were correlated to both reduction of overall survival (OS) and increased risk of leukemic evolution (88). Also Agarwal et al., using a customized 26-gene NGS panel in a series of 171 MPN patients, highlighted that alterations in ASXL1, EZH2, and IDH1/2 were associated with an increasing risk of disease progression and a shorter OS in both ET and PMF patients. In this work the authors found that among JAK2-mutated patients, 88% of subjects presented a single JAK2 mutation while the remaining 12% showed additional mutations in TET2, ASXL1, and SF3B1 genes.
Finally, very recently Grinfeld et al. (89) sequencing coding exons from 69 myeloid cancer genes in a large and representative series of 2035 MPN patients, identified different genetic subgroups with distinct clinical phenotypes, including blood counts, risk of leukemic transformation, and event-free survival. Integration of 63 clinical and genomic variables allowed the authors to develop a prognostic model (https://cancer.sanger.ac.uk/mpn-multistage/) capable of generating personally tailored predictions of clinical outcomes in MPN patients, even within individual categories of existing prognostic schemas. In particular, they identified a first subgroup with TP53 disruption or aneuploidy characterized by a poor outcomes and high risk of transformation to AML; a second subgroup with chromatin or spliceosome mutations that showed an increased risk for transformation to MF and shorter event-free survival. Patients who are not included in these two subgroups were defined according to their dominant phenotypic driver mutation and were the following: patients with CALR, MPL, heterozygous JAK2, and homozygous JAK2 or NFE2 mutations. The remaining two subgroups included instead patients with no detectable driver mutations and those with additional driver mutations not identified in the other six subgroups. Thus, the model provides considerable discriminatory power that accurately generalizes to other real-world cohorts.
Focusing on CALR-mutated cases, 56/62 (90%) of subjects showed single mutation in CALR gene while 10% had additional mutations in TET2 and ASXL1 (CALR type 1 mutations were more represented in this cohort of patients than type 2 mutations). On the contrary, no additional mutations were noticed in MPL-mutated patients (90). In addition, Song et al. analyzed 135 MPN patients by NGS and found that JAK2, ASXL1, and TET2 were frequently mutated in PMF, PV, and ET; interestingly, the comparison between mutational and cytogenetic profiles identified a possibility role in triaging and guiding different treatments (91).
Different studies have demonstrated the utility of NGS technologies to analyze the genetic profiles in AML patients. This technique is applied not only to study primary but also secondary AML, a heterogeneous group of disorders arising from MDS or MDS/MPN. In fact, secondary AMLs are characterized by several cytogenetic abnormalities and genetic alterations. In particular, Hussaini et al. (92), examining 187 subjects with a diagnosis of AML, highlighted the frequency of related-gene mutations, where ASXL1 was the highest mutated gene, followed by TET2, RUNX1, DNMT3A, TP53, IDH2, NRAS, FLT3, and NPM1. Moreover, NGS analysis identified co-mutated genes (ASXL1 with RUNX1 or TET2 or NRAS) and was able to discriminate somatic mutations associated with MDS/AML.
The prognostic impact of certain co-occurring mutations has been associated with MPNs disease progression as well as with the development of secondary AML (74, 93). Accordingly, the value of NGS at present lays also in the risk stratification of leukemic evolutions, in particular in critical and difficult cases, thus guiding treatment decision. In this respect, in PMF patients ASXL1 seems to be the worst sub-clonal mutation (25, 45). Indeed, the presence of this mutation or any other among SRSF2 and IDH1/2 gives a High Molecular Risk (HMR) to the patient and is included in the Mutation-Enhanced International Prognostic Score System (MIPSS70) and MIPSS70-plus, where the number of HMR mutations is also weighted together with driver mutation status and clinical data. The presence of two or more HMR mutations has been associated with highly adverse prognosis and rapid leukemic transformation (75, 76). The association of HMR and worse prognosis has also been confirmed in a retrospective series of elderly MF patients (77). Similarly, in the Genetically Inspired Prognostic Scoring System (GIPSS), only the molecular features are taken into consideration and IDH1/2 are substituted by U2AF1 mutations (78). In the same way, in ET and PV patients IDH2, U2AF1, EZH2, TP53, SH2B3, and SF3B1 mutations were all reported to give an adverse prognostic value (93).
Finally, somatic mutations with frequency ranging from 10 to 20% have been described in elderly subjects without any clinical evidence of myeloid diseases. In 2016, Bartels et al. used a customized NGS panel with 23 genes mutated in both MDS and MPNs to profile 192 formalin-fixed and paraffin-embedded (FFPE) patient samples. In this study, the authors found overall 269 pathogenic mutations in 125 of 185 analyzed patients and several of these exhibited more one-gene variants. Although they used FFPE bone marrow samples, the study demonstrated that NGS improved diagnostic accuracy and contributed to understanding the development, progression and therapy of myeloid diseases (94). NGS technology provide additional information to define the potential risk of development a myeloid malignancy, understand the clinical course, select the appropriate target therapy and predict potential drug-resistance mechanisms.
Conclusions
The discovery of JAK2V617F mutation in BCR-ABL1-negative MPNs by four different international cooperative groups in 2005 (2–5) led to significant insights on the pathogenesis of these disorders. In fact, this mutation results in a gain-of-function with activation of cytokine and growth factor receptors, and thus of the downstream JAK-STAT pathway (79, 95–98). The JAK2 point mutation in exon 12, present in a small percentage of patients with PV, is able to induce the MPN phenotype through the same pathogenic mechanism (6, 7).
In 2006 the MPLW515L/K was reported in ET and PMF patients (44, 45) and demonstrated to be able to aberrantly activate JAK-STAT pathway through a gain-of function similar to that of JAK2, thus leading to megakaryocytic proliferation (8–10).
More recently, in 2013 two different groups demonstrated a spectrum of mutations in CALR gene that cause frameshifts of one base pair in the last coding exon with a generation of a protein with new C terminus and a tail of 36 aminoacids (11, 12). Although it was soon clear that JAK-STAT signaling pathway was consistently activated in CALR mutated cells (53, 99), only recently it has been demonstrated that the mutant CALR protein is able to bind to the MPL receptor and activate it independently of the TPO presence itself (54–57). Consistently with the fact that all these mutations cause JAK-STAT pathway activation, ruxolitinib, the first JAK1/2 inhibitor, is able to exert clinical results in MF patients independently of the type of mutation (80).
As BCR-ABL1-negative MPNs are initiated by a driver mutation in one of JAK2, CALR or MPL genes in more than 95% of cases, there is a key relevance in diagnosis, prognosis and therapy (12, 61, 81). Due to the fact that these mutations are mutually exclusive, a sequential search for diagnosing purposes starting from JAK2 is suggested (36, 62–64). Furthermore, each of these mutations has a different prognostic impact. In ET, JAK2 is taken into consideration in the IPSET prognostic scores (100, 101). In JAK2-positive PMF, allele burden is relevant, with a worse prognosis for patients with <50%, while type 1/type 1-like CALR mutations has a favorable impact (22, 26). In the new MYSEC-PM prognostic score developed for secondary MF, CALR mutations absence is associated with a worse outcome (102).
Since NGS studies have been performed in BCR-ABL1-negative MPNs, especially in PMF, other mutations than the driver ones have been reported with few particularly relevant for prognosis and prediction of response. Sub-clonal mutations in genes of the spliceosome machinery, regulators of chromatin structure and histone modification and epigenetic regulators of DNA methylation are now taken into consideration in the new prognostic scores together with driver mutation status and clinical data in MIPSS70 and MIPSS70-plus (75) or in the GIPSS, where only the molecular features are evaluated (45).
NGS analysis during the disease natural history is of key importance to underline the eventual clonal evolution, thus uncovering an aggressive course and possibly suggesting a change in the therapeutic strategy (103, 104). Thus, in a near future, due to the progressive lowering of costs and availability in more reference laboratories, NGS will become the standard to evaluate BCR-ABL1-negative MPN patients.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
2. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. (2005) 365:1054–61. doi: 10.1016/S0140-6736(05)71142-9
3. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
4. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. (2005) 7:387–97. doi: 10.1016/j.ccr.2005.03.023
5. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. (2005) 434:1144–8. doi: 10.1038/nature03546
6. Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. (2007) 356:459–68. doi: 10.1056/NEJMoa065202
7. Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. (2008) 111:1686–9. doi: 10.1182/blood-2007-07-101576
8. Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. (2008) 112:141–9. doi: 10.1182/blood-2008-01-131664
9. Pancrazzi A, Guglielmelli P, Ponziani V, Bergamaschi G, Bosi A, Barosi G, et al. A sensitive detection method for MPLW515L or MPLW515K mutation in chronic myeloproliferative disorders with locked nucleic acid-modified probes and real-time polymerase chain reaction. J Mol Diagnost JMD. (2008) 10:435–41. doi: 10.2353/jmoldx.2008.080015
10. Schnittger S, Bacher U, Haferlach C, Beelen D, Bojko P, Burkle D, et al. Characterization of 35 new cases with four different MPLW515 mutations and essential thrombocytosis or primary myelofibrosis. Haematologica. (2009) 94:141–4. doi: 10.3324/haematol.13224
11. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. (2013) 369:2379–90. doi: 10.1056/NEJMoa1311347
12. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
13. Mansier O, Migeon M, Etienne G, Bidet A, Lippert E. JAK2V617F and CALR double mutations are more frequently encountered in patients with low JAK2V617F allelic burdens. Leukemia Lymphoma. (2016) 57:1949–51. doi: 10.3109/10428194.2015.1116122
14. Usseglio F, Beaufils N, Calleja A, Raynaud S, Gabert J. Detection of CALR and MPL mutations in low allelic burden JAK2 V617F essential thrombocythemia. J Mol Diagnost JMD. (2017) 19:92–8. doi: 10.1016/j.jmoldx.2016.08.006
15. Nussenzveig RH, Pham HT, Perkins SL, Prchal JT, Agarwal AM, Salama ME. Increased frequency of co-existing JAK2 exon-12 or MPL exon-10 mutations in patients with low JAK2(V617F) allelic burden. Leukemia Lymphoma. (2016) 57:1429–35. doi: 10.3109/10428194.2015.1091932
16. Haunstrup LM, Ebbesen LH, Hansen M, Severinsen MT, Aggerholm A. Skewed ratio between type 1 and type 2 calreticulin mutations in essential thrombocytosis patients with concomitant Janus kinase 2 V617F mutation. Exp Hematol. (2018) 68:62–5. doi: 10.1016/j.exphem.2018.09.007
17. Tefferi A, Guglielmelli P, Lasho TL, Rotunno G, Finke C, Mannarelli C, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. (2014) 28:1494–500. doi: 10.1038/leu.2014.57
18. Tefferi A, Lasho TL, Finke CM, Elala Y, Hanson CA, Ketterling RP, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. (2016) 1:105–11. doi: 10.1182/bloodadvances.2016000208
19. Larsen TS, Pallisgaard N, Moller MB, Hasselbalch HC. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis–impact on disease phenotype. Eur J Haematol. (2007) 79:508–15. doi: 10.1111/j.1600-0609.2007.00960.x
20. Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. (2008) 22:1299–307. doi: 10.1038/leu.2008.113
21. Perricone M, Palandri F, Ottaviani E, Angelini M, Bagli L, Bellesia E, et al. Assessment of the interlaboratory variability and robustness of JAK2V617F mutation assays: a study involving a consortium of 19 Italian laboratories. Oncotarget. (2017) 8:32608–17. doi: 10.18632/oncotarget.15940
22. Guglielmelli P, Barosi G, Specchia G, Rambaldi A, Lo Coco F, Antonioli E, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. (2009) 114:1477–83. doi: 10.1182/blood-2009-04-216044
23. Koren-Michowitz M, Landman J, Cohen Y, Rahimi-Levene N, Salomon O, Michael M, et al. JAK2V617F allele burden is associated with transformation to myelofibrosis. Leukemia Lymphoma. (2012) 53:2210–3. doi: 10.3109/10428194.2012.682308
24. Latagliata R, Polverelli N, Tieghi A, Palumbo GA, Breccia M, Sabattini E, et al. Comparison of JAK2(V617F) -positive essential thrombocythaemia and early primary myelofibrosis: the impact of mutation burden and histology. Hematol Oncol. (2018) 36:269–75. doi: 10.1002/hon.2430
25. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. (2013) 27:1861–9. doi: 10.1038/leu.2013.119
26. Rozovski U, Verstovsek S, Manshouri T, Dembitz V, Bozinovic K, Newberry K, et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematologica. (2017) 102:79–84. doi: 10.3324/haematol.2016.149765
27. Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. (2010) 24:1574–9. doi: 10.1038/leu.2010.148
28. Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. (2008) 112:3065–72. doi: 10.1182/blood-2008-03-143537
29. Quintas-Cardama A, Abdel-Wahab O, Manshouri T, Kilpivaara O, Cortes J, Roupie AL, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha-2a. Blood. (2013) 122:893–901. doi: 10.1182/blood-2012-07-442012
30. Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, Naim A, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. (2017) 96:1113–20. doi: 10.1007/s00277-017-2994-x
31. Kroger N, Badbaran A, Holler E, Hahn J, Kobbe G, Bornhauser M, et al. Monitoring of the JAK2-V617F mutation by highly sensitive quantitative real-time PCR after allogeneic stem cell transplantation in patients with myelofibrosis. Blood. (2007) 109:1316–21. doi: 10.1182/blood-2006-08-039909
32. Greenfield G, McPherson S, Mills K, McMullin MF. The ruxolitinib effect: understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J Transl Med. (2018) 16:360. doi: 10.1186/s12967-018-1729-7
33. Lippert E, Girodon F, Hammond E, Jelinek J, Reading NS, Fehse B, et al. Concordance of assays designed for the quantification of JAK2V617F: a multicenter study. Haematologica. (2009) 94:38–45. doi: 10.3324/haematol.13486
34. Merker JD, Jones CD, Oh ST, Schrijver I, Gotlib J, Zehnder JL. Design and evaluation of a real-time PCR assay for quantification of JAK2 V617F and wild-type JAK2 transcript levels in the clinical laboratory. J Mol Diagnostics JMD. (2010) 12:58–64. doi: 10.2353/jmoldx.2010.090068
35. Jovanovic JV, Ivey A, Vannucchi AM, Lippert E, Oppliger Leibundgut E, Cassinat B, et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia. (2013) 27:2032–9. doi: 10.1038/leu.2013.219
36. Guglielmelli P, Pietra D, Pane F, Pancrazzi A, Cazzola M, Vannucchi AM, et al. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders-A consensus project of the Italian Society of Hematology. Leukemia Res. (2017) 58:63–72. doi: 10.1016/j.leukres.2017.04.006
37. Waterhouse M, Follo M, Pfeifer D, von Bubnoff N, Duyster J, Bertz H, et al. Sensitive and accurate quantification of JAK2 V617F mutation in chronic myeloproliferative neoplasms by droplet digital PCR. Ann Hematol. (2016) 95:739–44. doi: 10.1007/s00277-016-2623-0
38. Fontanelli G, Barate C, Ciabatti E, Guerrini F, Grassi S, Del Re M, et al. Real-Time PCR and Droplet Digital PCR: two techniques for detection of the JAK2(V617F) mutation in Philadelphia-negative chronic myeloproliferative neoplasms. Int J Lab Hematol. (2015) 37:766–73. doi: 10.1111/ijlh.12404
39. Link-Lenczowska D, Pallisgaard N, Cordua S, Zawada M, Czekalska S, Krochmalczyk D, et al. A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann Hematol. (2018) 97:2299–308. doi: 10.1007/s00277-018-3451-1
40. Maslah N, Verger E, Schlageter MH, Miclea JM, Kiladjian JJ, Giraudier S, et al. Next-generation sequencing for JAK2 mutation testing: advantages and pitfalls. Ann Hematol. (2018). doi: 10.1007/s00277-018-3499-y
41. Passamonti F, Elena C, Schnittger S, Skoda RC, Green AR, Girodon F, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. (2011) 117:2813–6. doi: 10.1182/blood-2010-11-316810
42. Furtado LV, Weigelin HC, Elenitoba-Johnson KS, Betz BL. A multiplexed fragment analysis-based assay for detection of JAK2 exon 12 mutations. J Mol Diagnost JMD. (2013) 15:592–9. doi: 10.1016/j.jmoldx.2013.04.006
43. Carillo S, Henry L, Lippert E, Girodon F, Guiraud I, Richard C, et al. Nested high-resolution melting curve analysis a highly sensitive, reliable, and simple method for detection of JAK2 exon 12 mutations–clinical relevance in the monitoring of polycythemia. J Mol Diagnost JMD. (2011) 13:263–70. doi: 10.1016/j.jmoldx.2010.12.002
44. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. (2006) 3:e270. doi: 10.1371/journal.pmed.0030270
45. Tefferi A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. (2018) 93:1551–60. doi: 10.1002/ajh.25230
46. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. (2006) 108:3472–6. doi: 10.1182/blood-2006-04-018879
47. Pietra D, Brisci A, Rumi E, Boggi S, Elena C, Pietrelli A, et al. Deep sequencing reveals double mutations in cis of MPL exon 10 in myeloproliferative neoplasms. Haematologica. (2011) 96:607–11. doi: 10.3324/haematol.2010.034793
48. Ghaderi M, Stromberg O, Porwit A. Rapid real-time PCR assay for detection of MPL W515L mutation in patients with chronic myeloproliferative disorders. Int J Lab Hematol. (2010) 32(1 Pt 2):122–6. doi: 10.1111/j.1751-553X.2008.01118.x
49. Zhuge J, Zhang W, Zhang W, Xu M, Hoffman R. Sensitive detection of MPLW515L/K mutations by amplification refractory mutation system (ARMS)-PCR. Clin Chim Acta Int J Clin Chem. (2010) 411(1-2):122–3. doi: 10.1016/j.cca.2009.10.012
50. Furtado LV, Weigelin HC, Elenitoba-Johnson KS, Betz BL. Detection of MPL mutations by a novel allele-specific PCR-based strategy. J Mol Diagnost JMD. (2013) 15:810–8. doi: 10.1016/j.jmoldx.2013.07.006
51. Boyd EM, Bench AJ, Goday-Fernandez A, Anand S, Vaghela KJ, Beer P, et al. Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br J Haematol. (2010) 149:250–7. doi: 10.1111/j.1365-2141.2010.08083.x
52. Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J Offic Publ Federation Am Soc Exp Biol. (2010) 24:665–83. doi: 10.1096/fj.09-145482
53. Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. (2016) 127:1317–24. doi: 10.1182/blood-2015-11-679571
54. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. (2016) 127:1325–35. doi: 10.1182/blood-2015-11-681932
55. Elf S, Abdelfattah NS, Chen E, Perales-Paton J, Rosen EA, Ko A, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Disc. (2016) 6:368–81. doi: 10.1158/2159-8290.CD-15-1434
56. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. (2016) 127:1307–16. doi: 10.1182/blood-2015-09-671172
57. Elf S, Abdelfattah NS, Baral AJ, Beeson D, Rivera JF, Ko A, et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood. (2018) 131:782–6. doi: 10.1182/blood-2017-08-800896
58. Pietra D, Rumi E, Ferretti VV, Di Buduo CA, Milanesi C, Cavalloni C, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. (2016) 30:431–8. doi: 10.1038/leu.2015.277
59. Tefferi A, Lasho TL, Tischer A, Wassie EA, Finke CM, Belachew AA, et al. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood. (2014) 124:2465–6. doi: 10.1182/blood-2014-07-588426
60. Oh Y, Song IC, Kim J, Kwon GC, Koo SH, Kim SY. Pyrosequencing-based quantitative measurement of CALR mutation allele burdens and their clinical implications in patients with myeloproliferative neoplasms. Clin Chim Acta Int J Clin Chem. (2018) 483:183–91. doi: 10.1016/j.cca.2018.05.001
61. Szuber N, Tefferi A. Driver mutations in primary myelofibrosis and their implications. Curr Opin Hematol. (2018) 25:129–35. doi: 10.1097/MOH.0000000000000406
62. Mesa R, Jamieson C, Bhatia R, Deininger MW, Gerds AT, Gojo I, et al. Myeloproliferative Neoplasms, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Comprehensive Cancer Netw JNCCN. (2016) 14:1572–611
63. Busque L, Porwit A, Day R, Olney HJ, Leber B, Ethier V, et al. Laboratory investigation of Myeloproliferative Neoplasms (MPNs): recommendations of the canadian Mpn group. Am J Clin Pathol. (2016) 146:408–22. doi: 10.1093/ajcp/aqw131
64. Tefferi A, Pardanani A. Genetics: CALR mutations and a new diagnostic algorithm for MPN. Nat Rev Clin Oncol. (2014) 11:125–6. doi: 10.1038/nrclinonc.2014.16
65. Maier CL, Fisher KE, Jones HH, Hill CE, Mann KP, Zhang L. Development and validation of CALR mutation testing for clinical diagnosis. Am J Clin Pathol. (2015) 144:738–45. doi: 10.1309/AJCPXPA83MVCTSOQ
66. Mehrotra M, Luthra R, Singh RR, Barkoh BA, Galbincea J, Mehta P, et al. Clinical validation of a multipurpose assay for detection and genotyping of CALR mutations in myeloproliferative neoplasms. Am J Clin Pathol. (2015) 144:746–55. doi: 10.1309/AJCP5LA2LDDNQNNC
67. Murugesan G, Guenther-Johnson J, Mularo F, Cook JR, Daly TM. Validation of a molecular diagnostic assay for CALR exon 9 indels in myeloproliferative neoplasms: identification of coexisting JAK2 and CALR mutations and a novel 9 bp deletion in CALR. Int J Lab Hematol. (2016) 38:284–97. doi: 10.1111/ijlh.12484
68. Jones AV, Ward D, Lyon M, Leung W, Callaway A, Chase A, et al. Evaluation of methods to detect CALR mutations in myeloproliferative neoplasms. Leukemia Res. (2015) 39:82–7. doi: 10.1016/j.leukres.2014.11.019
69. Mansier O, Migeon M, Saint-Lezer A, James C, Verger E, Robin M, et al. Quantification of the mutant CALR allelic burden by digital PCR: application to minimal residual disease evaluation after bone marrow transplantation. J Mol Diagnost JMD. (2016) 18:68–74. doi: 10.1016/j.jmoldx.2015.07.007
70. Serrati S, De Summa S, Pilato B, Petriella D, Lacalamita R, Tommasi S, et al. Next-generation sequencing: advances and applications in cancer diagnosis. OncoTargets Ther. (2016) 9:7355–65. doi: 10.2147/OTT.S99807
71. Ladetto M, Bruggemann M, Monitillo L, Ferrero S, Pepin F, Drandi D, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. (2014) 28:1299–307. doi: 10.1038/leu.2013.375
72. Gazzola A, Mannu C, Rossi M, Laginestra MA, Sapienza MR, Fuligni F, et al. The evolution of clonality testing in the diagnosis and monitoring of hematological malignancies. Therapeutic Adv Hematol. (2014) 5:35–47. doi: 10.1177/2040620713519729
73. Saeidi K. Myeloproliferative neoplasms: current molecular biology and genetics. Crit Rev Oncol Hematol. (2016) 98:375–89. doi: 10.1016/j.critrevonc.2015.11.004
74. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. (2017) 129:667–79. doi: 10.1182/blood-2016-10-695940
75. Guglielmelli P, Lasho TL, Rotunno G, Mudireddy M, Mannarelli C, Nicolosi M, et al. MIPSS70: Mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J Clin Oncol Offic J Am Soc Clin Oncol. (2018) 36:310–8. doi: 10.1200/JCO.2017.76.4886
76. Patel KP, Newberry KJ, Luthra R, Jabbour E, Pierce S, Cortes J, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. (2015) 126:790–7. doi: 10.1182/blood-2015-03-633404
77. Palandri F, Catani L, Bonifacio M, Benevolo G, Heidel F, Palumbo GA, et al. Ruxolitinib in elderly patients with myelofibrosis: impact of age and genotype. A multicentre study on 291 elderly patients. Br J Haematol. (2018) 183:35–46. doi: 10.1111/bjh.15497
78. Tefferi A, Lasho TL, Finke C, Gangat N, Hanson CA, Ketterling RP, et al. Prognostic significance of ASXL1 mutation types and allele burden in myelofibrosis. Leukemia. (2018) 32:837–9. doi: 10.1038/leu.2017.318
79. Mascarenhas J, Roper N, Chaurasia P, Hoffman R. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenet. (2011) 2:197–212. doi: 10.1007/s13148-011-0050-6
80. Guglielmelli P, Biamonte F, Rotunno G, Artusi V, Artuso L, Bernardis I, et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood. (2014) 123:2157–60. doi: 10.1182/blood-2013-11-536557
81. Passamonti F, Mora B, Maffioli M. New molecular genetics in the diagnosis and treatment of myeloproliferative neoplasms. Curr Opin Hematol. (2016) 23:137–43. doi: 10.1097/MOH.0000000000000218
82. Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. (2016) 1:21–30. doi: 10.1182/bloodadvances.2016000216
83. Pardanani A, Lasho T, Finke C, Oh ST, Gotlib J, Tefferi A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia. (2010) 24:1713–8. doi: 10.1038/leu.2010.163
84. Sallman DA, Padron E. Integrating mutation variant allele frequency into clinical practice in myeloid malignancies. Hematol Oncol Stem Cell Ther. (2016) 9:89–95. doi: 10.1016/j.hemonc.2016.04.003
85. Tefferi A, Pardanani A. Myeloproliferative Neoplasms: a contemporary review. JAMA Oncol. (2015) 1:97–105. doi: 10.1001/jamaoncol.2015.89
86. Chang YC, Lin HC, Chiang YH, Chen CG, Huang L, Wang WT, et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med Oncol. (2017) 34:83. doi: 10.1007/s12032-017-0944-z
87. Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, Ianotto JC, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. (2016) 127:333–42. doi: 10.1182/blood-2015-07-661983
88. Gardner JA, Peterson JD, Turner SA, Soares BL, Lancor CR, Dos Santos LL, et al. Detection of CALR mutation in clonal and nonclonal hematologic diseases using fragment analysis and next-generation sequencing. Am J Clin Pathol. (2016) 146:448–55. doi: 10.1093/ajcp/aqw129
89. Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. (2018) 379:1416–30. doi: 10.1056/NEJMoa1716614
90. Agarwal R, Blombery P, McBean M, Jones K, Fellowes A, Doig K, et al. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: data from targeted next-generation sequencing in the diagnostic laboratory. Ann Hematol. (2017) 96:725–32. doi: 10.1007/s00277-017-2937-6
91. Song J, Hussaini M, Zhang H, Shao H, Qin D, Zhang X, et al. Comparison of the mutational profiles of primary myelofibrosis, polycythemia vera, and essential thrombocytosis. Am J Clin Pathol. (2017) 147:444–52. doi: 10.1093/ajcp/aqw222
92. Hussaini MO, Mirza AS, Komrokji R, Lancet J, Padron E, Song J. Genetic landscape of acute myeloid leukemia interrogated by next-generation sequencing: a large cancer center experience. Cancer Genom Proteom. (2018) 15:121–6. doi: 10.21873/cgp.20070
93. Bacher U, Shumilov E, Flach J, Porret N, Joncourt R, Wiedemann G, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. (2018) 8:113. doi: 10.1038/s41408-018-0148-6
94. Bartels S, Schipper E, Hasemeier B, Kreipe H, Lehmann U. Routine clinical mutation profiling using next generation sequencing and a customized gene panel improves diagnostic precision in myeloid neoplasms. Oncotarget. (2016) 7:30084–93. doi: 10.18632/oncotarget.8310
95. Cross NC. Genetic and epigenetic complexity in myeloproliferative neoplasms. Hematol Am Soc Hematol Educ Progr. (2011) 2011:208–14. doi: 10.1182/asheducation-2011.1.208
96. Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. (2008) 19:385–93. doi: 10.1016/j.semcdb.2008.07.002
97. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol Offic J Am Soc Clin Oncol. (2011) 29:1356–63. doi: 10.1200/JCO.2010.32.9490
98. Vannucchi AM. From palliation to targeted therapy in myelofibrosis. N Engl J Med. (2010) 363:1180–2. doi: 10.1056/NEJMe1005856
99. Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. (2014) 123:e123–33. doi: 10.1182/blood-2014-02-554634
100. Barbui T, Thiele J, Carobbio A, Passamonti F, Rumi E, Randi ML, et al. Disease characteristics and clinical outcome in young adults with essential thrombocythemia versus early/prefibrotic primary myelofibrosis. Blood. (2012) 120:569–71. doi: 10.1182/blood-2012-01-407981
101. Haider M, Gangat N, Lasho T, Abou Hussein AK, Elala YC, Hanson C, et al. Validation of the revised International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET-thrombosis) in 585 Mayo Clinic patients. Am J Hematol. (2016) 91:390–4. doi: 10.1002/ajh.24293
102. Passamonti F, Giorgino T, Mora B, Guglielmelli P, Rumi E, Maffioli M, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. (2017) 31:2726–31. doi: 10.1038/leu.2017.169
103. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. (2014) 123:2220–8. doi: 10.1182/blood-2013-11-537167
Keywords: BCR-ABL1-negative myeloproliferative neoplasms (MPNs), myelofibrosis (MF), JAK2 mutations, calreticulin (CALR), MPL (W515K/L), ASXL1 mutation, High molecular risk (HMR) mutations, next generation sequencing (NGS)
Citation: Palumbo GA, Stella S, Pennisi MS, Pirosa C, Fermo E, Fabris S, Cattaneo D and Iurlo A (2019) The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 9:321. doi: 10.3389/fonc.2019.00321
Received: 14 January 2019; Accepted: 09 April 2019;
Published: 26 April 2019.
Edited by:
Onder Alpdogan, Thomas Jefferson University, United StatesReviewed by:
Jerald Gong, Thomas Jefferson University, United StatesMary Frances McMullin, Queen's University Belfast, United Kingdom
Copyright © 2019 Palumbo, Stella, Pennisi, Pirosa, Fermo, Fabris, Cattaneo and Iurlo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe A. Palumbo, cGFsdW1iby5nYUBnbWFpbC5jb20=