 Zhuo-Xian Rong1,2
Zhuo-Xian Rong1,2 Zhi Li1,2,3Jun-Ju He1,2Li-Yu Liu1,2Xin-Xin Ren1,2Jie Gao1,2Yun Mu1,2Yi-Di Guan1,2Yu-Mei Duan4Xiu-Ping Zhang5De-Xiang Zhang6Nan Li5*Yue-Zhen Deng1,2,3*
Zhi Li1,2,3Jun-Ju He1,2Li-Yu Liu1,2Xin-Xin Ren1,2Jie Gao1,2Yun Mu1,2Yi-Di Guan1,2Yu-Mei Duan4Xiu-Ping Zhang5De-Xiang Zhang6Nan Li5*Yue-Zhen Deng1,2,3* Lun-Quan Sun1,2,3*
Lun-Quan Sun1,2,3*- 1Center for Molecular Medicine, Xiangya Hospital, Central South University, Changsha, China
- 2Key Laboratory of Molecular Radiation Oncology, Changsha, China
- 3Hunan International Collaboration Base for Science and Technology, Changsha, China
- 4Department of Pathology, Xiangya Hospital, Central South University, Changsha, China
- 5Department of Hepatic Surgery VI (Ward I), Shanghai Eastern Hepatobiliary Surgery Hospital, Shanghai, China
- 6General Surgery Department, Zhongshan-Xuhui Hospital Affiliated to Fudan University, Shanghai, China
Intrahepatic cholangiocarcinoma (ICC) ranks as the second most malignant type of primary liver cancer with a high degree of incidence and a very poor prognosis. Fat mass and obesity-associated protein (FTO) functions as an eraser of the RNA m6A modification, but its roles in ICC tumorigenesis and development remain unknown. We showed here that the protein level of FTO was downregulated in clinical ICC samples and cell lines and that FTO expression was inversely correlated with the expression of CA19-9 and micro-vessel density (MVD). A Kaplan-Meier survival analysis showed that a low expression of FTO predicted poor prognosis in ICC. in vitro, decreased endogenous expression of FTO obviously reduced apoptosis of ICC cells. Moreover, FTO suppressed the anchorage-independent growth and mobility of ICC cells. Through mining the database, FTO was found to regulate the integrin signaling pathway, inflammation signaling pathway, epidermal growth factor receptor (EGFR) signaling pathway, angiogenesis, and the pyrimidine metabolism pathway. RNA decay assay showed that oncogene TEAD2 mRNA stability was impaired by FTO. In addition, the overexpression of FTO suppressed tumor growth in vivo. In conclusion, our study demonstrated the critical roles of FTO in ICC.
Introduction
Intrahepatic cholangiocarcinoma (ICC) is the second most malignant type of primary liver cancer, and its incidence is increasing (1, 2). Intrahepatic cholangiocarcinoma (ICC) originates from epithelial cells of the bile duct or hepatic ducts (3). Since some aspects of the clinical features are similar to those of hepatocellular carcinoma (HCC), ICC has insidious symptoms, a high degree of malignancy and very poor prognosis (4). ICC harbors many genetic aberrations, including mutations in isocitrate dehydrogenase1/2 (IDH1/2), epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), KRAS, and BRAF (5, 6), as well as amplifications of Cyclin D1 (CCND1). Strategies for ICC treatment include surgery, intervention, ablation treatment, radiotherapy, chemotherapy, immune targeting, and other comprehensive treatments (7–9). In principle, ICC is insensitive to radiotherapy and chemotherapy, and thus, the clinical outcomes are very poor.
N6-methyladenosine (m6A) represents a predominant RNA modification that, not surprisingly, regulates tumorigenesis, and tumor progression (10). The m6A RNA modification is a reversible process that is coordinated by methyltransferase (m6A “writers”), m6A reader proteins and demethylase (m6A “erasers”) (11). These members cover more than 13 enzymes. The m6A “writers” complex consists of METTL3, METTL14, WTAP, CBLL1, RBM15, ZC3H13, and VIRMA and is responsible for methylation of target RNA transcripts (12). Then, m6A readers, including YTHDF1-3, YTHDC1, IGF2BPs, and eIF3, discern these m6A modifications to direct RNA alternative splicing, translation, localization, and RNA stability, among other processes (13). However, as the m6A “erasers,” FTO and ALKBH5 remove m6A from the aforementioned target transcripts (14, 15). FTO, the first identified m6A demethylase, belongs to the AlkB family of Fe(II)/α-ketoglutarate-dependent dioxygenases (16). FTO mediates multiple RNA modifications, including m6A and m6Am in mRNA and snRNA as well as m1A in tRNA (17). Recent studies demonstrated that FTO plays an oncogenic role in cancers (18–20). For example, in one study, FTO significantly promoted leukemic cell proliferation, transformation, leukemogenesis, curtailed AML cell differentiation and apoptosis by targeting ASB2, and RARA (18). However, this process may be suppressed by R-2-hydroxyglutarate (R-2HG) and IDH mutations, which competitively inhibit FTO (21, 22). FTO also removes m6A from β-catenin to induce the chemo-radiotherapy resistance in cervical squamous cell carcinoma (CSCC) (20). In addition, the overexpression of FTO can promote breast cancer and gastric cancer progression (19, 23). These studies suggest that FTO participates in the regulation of various biological processes in cancer cells through modulation of the RNA transcripts which are critical to the respective pathways.
In the present study, we sought to explore the clinical relevance and biological functions of FTO in ICC.
Materials and Methods
Cell Lines and Reagents
HIBEPIC, HCCC-9810, RBE, TFK-1, and HuCC-T1 cells (ATCC, Manassas, VA, USA) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum. All cell lines were transfected with plasmids using DharmaFECT Duo Transfection Reagent (Thermo Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Vectors expressing short hairpin RNA (shRNA) sequences were provided by Sangon Biotechnology (Shanghai, China). Cisplatin was obtained from Qilu Pharm (Jinan, China) was obtained from Hansoh (Jiangsu, China).
Western Blotting (WB)
For WB, 5 × 106 cells were incubated with 100 μl cell lysis buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, and protease and phosphatase inhibitor cocktail (B14002&B15002, Biotool) at 4°C for 30 min. After centrifugation for 30 min, the supernatant was collected. All samples were mixed with 4 × SDS-PAGE loading buffer and analyzed by WB with the indicated antibody. The following primary antibodies were used in this study: Anti-FTO (1:4000, ab124892, Abcam), Anti-α-Tubulin (1:1000, sc-69969, Santa Cruz), and Anti-FLAG (1:5000, F1804, SIGMA).
Expression Plasmids and Clones
Human FTO was generated using the following primers: forward: 5′-ATGAAGCGCACCCCGACTGC-3′; reverse: 5′-CTAGGGTTTTGCTTCCAGAA-3′. siRNA sequence 1: 5′-TCACCAAGGAGACTGCTATTT-3′; siRNA sequence 2: 5′-CTAGGGTTTTGCTTCCAGAA-3′.
The vectors psPAX2 and pMD2.G (Addgene) were used for viral packaging. A small hairpin RNA (shRNA) expression plasmid pLVX-shRNA was used to construct lentivirus for the knockdown of FTO. The plasmids pLVX-IRES-Puro (Flag-SBP), pCDNA3.1+ and pRK5-FLAG were used for the overexpression of FTO.
Quantitative Reverse Transcriptase-PCR
RNA was extracted using TRIzol (Invitrogen). Then, a PrimeScript™ RT Reagent Kit with gDNA Eraser was used according to the manufacturer's protocol (TaKaRa). The primer sequences were as follows: FTO, forward primer: 5′-ACTTGGCTCCCTTATCTGACC-3′, reverse primer: 5′-TGTGCAGTGTGAGAAAGGCTT-3′. RT-PCR was performed using a CFX96 Real-Time PCR Detection System (Bio-Rad, Richmond, CA, USA). The 2−ΔΔCt method was used to calculate the expression of FTO. Three replicates were tested for each gene for each sample, and the mean value was calculated. The experiment was repeated three times.
Soft Agar Colony Forming Assay
Cells were seeded in 24-well culture plates (3 × 103 cells/well, 4 repetitions per group) with Roswell Park Memorial Institute 1640 (RPMI-1640) containing 20% FBS. Agar was autoclaved and placed in a 42°C water bath kettle. Lower layer: 0.7% agar was mixed with 2 × RPMI-1640 (containing 2 × antibiotic and 20% FBS) at the ratio of 1:1, and 200 μl was added to each well of the 24-well plate, which was allowed to solidify in a CO2 incubator for at least 30 min. Upper layer: The 1.2% agar was also mixed with the cell suspension at a ratio of 1:1, and 200 μl was added to each well of the 24-well plate, which was maintained in a CO2 incubator. The soft agar colony plate was incubated in a 5% CO2 incubator at 37°C for 10~14 days. Eight fields were randomly selected for colony counts (>50 μm diameter) under an inverted microscope (Leica DMI4000B, Germany) at 40 × magnification. The freeware ImageJ (National Institutes of Health, Bethesda, MD, USA) and Adobe Photoshop (Adobe Systems, San Jose, CA, USA) were used in colony diameter measurement. Thirteen colonies were randomly selected for diameter measurement. Colony volume was calculated using the formula: volume = (length × width2)/2.
Flow Cytometry
For the apoptosis analysis, 5 × 105 cells were digested with trypsin without EDTA and centrifuged (300 g, at 4°C for 5 min). Cells were washed twice in prechilled PBS. Then, 100 μl of 1 × Binding Buffer was added to resuspend the cells. Then, the cells were incubated with 5 μl Annexin V-FITC and 5 μl propidium iodide (PI) Staining Solution for 10 min at room temperature after gentle mixing. After adding 400 μl of 1 × Binding Buffer and mixing, the samples were examined for 1 h by flow cytometry. The apoptosis detection kit used in this experiment was purchased from BIomake (B32117, Houston, TX, USA).
Transwell Assay
The Transwell chamber (8 μm, Corning #3422, ME, USA) was placed into a 24-well plate. The transfected cells were diluted with 200 μl 0.1% FBS medium to generate a cell suspension with a density of 1 × 106 cells/mL and seeded in the upper chamber. Then, 600 μl RPMI-1640 containing 30% FBS was added to the basolateral chamber. Each group was tested in 3 replicates. After culture at 37°C with 5% CO2 for 72 h, the Transwell chambers were removed, the cells were rinsed with PBS twice, fixed and stained with 0.3% crystal violet for 15 min, and rinsed in ddH2O three times. Cells in the upper chamber were removed. The invasive cells were observed with an inverted microscope (DMI 3000, Leica, IL, USA) (×100), and 5 fields were randomly selected for cell counts.
Immunohistochemistry (IHC)
Paraffin-embedded sections were deparaffinized and rehydrated. Antigen retrieval was performed by boiling the sections in sodium citrate solution in a pressure-cooker for 3 min. Endogenous peroxidase was blocked by 3% H2O2 solution for 10 min at 37°C and washed with PBST. Then, the paraffin sections were incubated with the primary antibody at 4°C overnight. The next day, the paraffin sections were rewarmed at 37°C for 10 min. After two washes in PBS and treatment with an adjuvant for 20 min at 37°C, the sections were incubated with the second antibody for 30 min at 37°C. The immunohistochemical reaction was visualized with 3,3,0-diaminobenzidine (DAB) for 3 min. Sections were counterstained for 3 min in hematoxylin (zSGB-Bio, Beijing). Slides were scanned with the digital 3DHISTECH–Pannoramic MIDI (3DHISTECH Ltd, Budapest). The image analysis software ImageJ IHC Profiler was used for staining quantification (24).
Colony Formation Assay
ICC cells were seeded in 12-well plates at densities of 2 × 102 per well and. After 12 days culture, cells were treated with 0, 10 and 20 μM cisplatin. In 4 days later, cells were washed with cold phosphate buffered saline (PBS) and stained with 0.3% crystal violet (containing methanol). Colonies consisting of more than 50 cells were defined as surviving colonies. All viability measurements are normalized with the untreated group. Cell survival curves were fitted by GraphPad software.
RNA Decay Assay
FTO-overexpressing TFK1 cells, shFTO TFK1 cells, and corresponding control cells were cultured in 12-well plates. Then cells were treated with 8 μM actinomycin D (MedChemExpress, Monmouth Junction, NJ, USA) at 0, 2, and 4 h before RNA extraction.
Animal Assay
1.5 × 106 TFK1 cells with or without FTO overexpression were injected subcutaneously into the left and right flanks of 6-week-old female athymic nude mice (SJA, Changsha, China). After transplantation, tumor size was measured by caliper every other day. Tumor volume was calculated using the formula: volume = (length × width2)/2. The animals were sacrificed and the tumors were removed for WB and IHC assay. All animal work procedures were performed in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International and approved by the Animal Ethics Committee of Xiangya Hospital, Central South University.
Statistical Analyses
In our study, Data is presented as mean ± SD, and analyzed using the two-tailed Student t-test for two groups, ANOVA for multiple groups or Chi-square tests for Table 1. The statistical significance of Kaplan–Meier survival curves was assessed with a Log-rank (Mantel-Cox) Test (25). A value of p < 0.05 was considered as statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001. Student's t-tests, two-way ANOVA, curve fitting and box plots calculations were performed with GraphPad software (GraphPad, La Jolla, CA, USA). IBM SPSS Statistics 19 (IBM, Chicago, USA) is used for Chi-square test analysis (26).
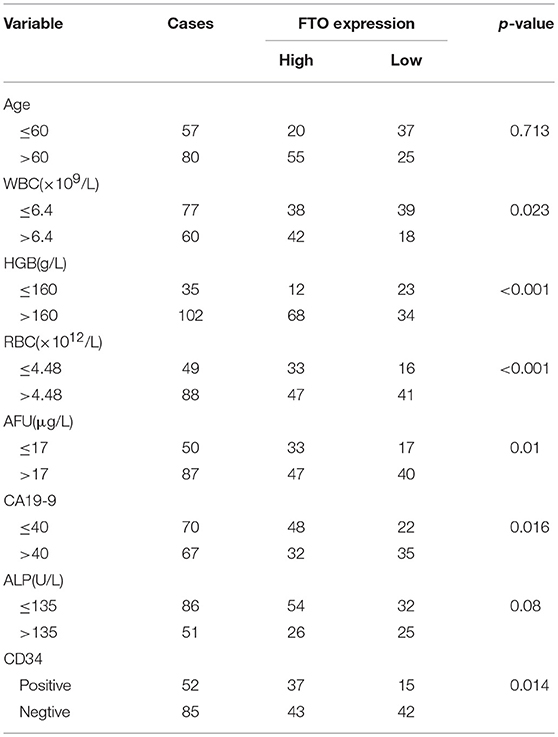
Table 1. Relationship between FTO expression in ICC and clinicopathologic features.
Results
FTO Protein Levels Are Decreased in ICC
To understand the regulation of m6A modification in ICC, we examined the expression of m6A-related enzymes in human paracancerous intrahepatic biliary epithelial cells (para-HIBECS) and ICCs by IHC (data not shown). After analyzing the tissue microarray containing 142 patient specimens, it was found that FTO was downregulated in ICC (Figures 1A–C). Although no correlation was observed between disease recurrence and FTO expression (p = 0.084, Figure 1D), the expression of FTO was positively correlated with tumor differentiation, and a lower protein level was observed in the poorly differentiated ICC clinical samples (p = 0.0029, Figure 1E).
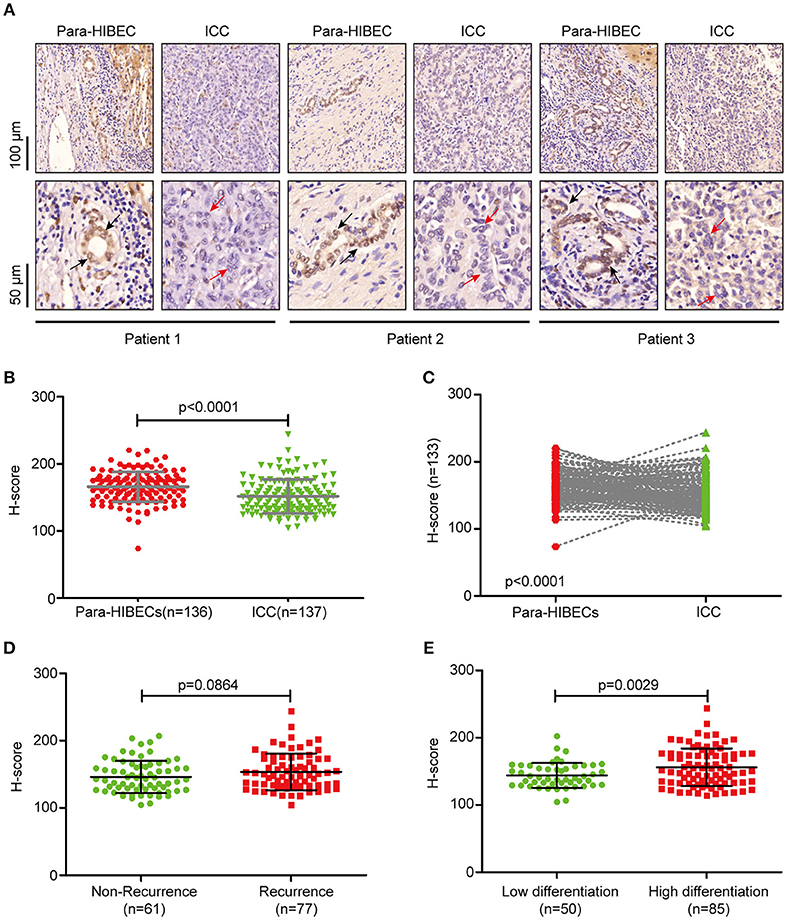
Figure 1. FTO was downregulated in intrahepatic cholangiocarcinoma. (A) Representative IHC staining of FTO in human ICC specimens. Black arrow indicates intrahepatic bile duct epithelial cells and red arrow indicates ICC cells. (B) Statistical analysis of FTO expression in ICC cases (n = 137) and human paracancerous intrahepatic biliary epithelial cells (para-HIBECS, n = 136). (C) Analysis of FTO expression between ICC cases and paired para-HIBECS (n = 133). (D) Comparison of FTO expression in the cases of recurrence (n = 77) with that of specimens from patients without recurrence (n = 6 1). (E) Differential expression of FTO between poorly-differentiated cases (n = 50) and highly differentiated cases (n = 85).
FTO Expression Is Correlated With the Clinicopathologic Features of ICC
In the further analysis, we assessed the association between FTO expression and the clinicopathologic features. No association was observed between FTO expression and clinicopathologic features such as age, gender, cirrhosis, primary tumor diameter, tumor number, necrosis, and p53 or E-cadherin expression. Carbohydrate antigen 19-9 (CA19-9), a mucin-type glycoprotein in the serum, is one of the tumor markers used for ICC diagnosis, classification, prognosis, and treatment guidance (27, 28). A higher CA19-9 level in the serum is associated with poor prognosis. Our data indicated that the expression of FTO was negatively correlated with CA19-9 concentration (p = 0.016, Table 1). CD34, another tumor marker, is involved in angiogenesis and is used as a quantitative indicator of micro-vessel density (MVD) (29). An association was found between low FTO expression and high CD34 expression in ICC (p = 0.014, Table 1). In addition, FTO expression was also significantly related to the number of WBCs (white blood cells, p = 0.023) and RBCs (red blood cells, p < 0.001), HGB levels (hemoglobin, p < 0.001), AFU levels (α-L-Fucosidase, p = 0.01), and ALP level (alkaline phosphatase, p = 0.008) in the serum.
Low Expression of FTO Predicts Poor Prognosis in ICC
Next, we analyzed the correlation between FTO expression and prognosis through the tissue microarray IHC staining described above. ICC patients with low expression of FTO showed both poorer overall survival (p = 0.0077) and poorer relapse-free survival (p = 0.0195) (Figures 2A,B). Moreover, in 88 patients with necrobiotic tumor, those with a lower expression of FTO also showed a poorer overall survival than those with a higher expression of FTO (p = 0.0273, Figure 2C). However, the overall survival of patients without necrosis did not differ between the lower FTO expression group and the higher FTO expression group (p = 0.1051, Figure 2D).
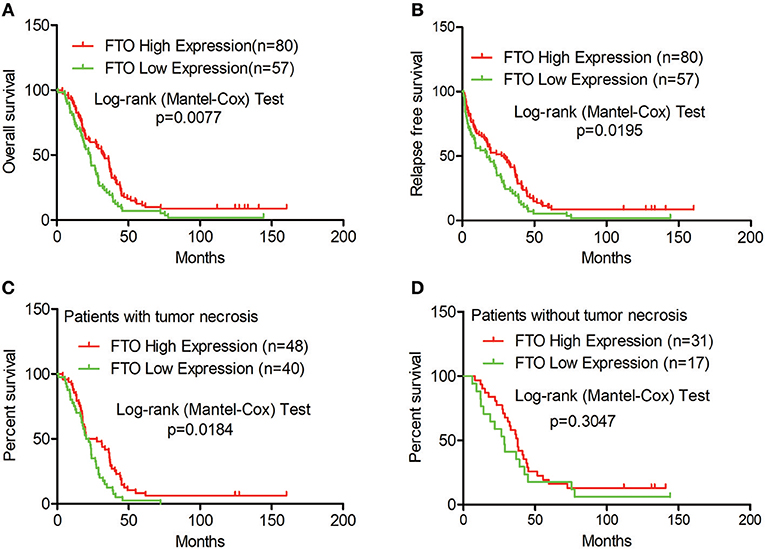
Figure 2. Low expression of FTO predicted a poor prognosis in ICC. (A–D) Kaplan-Meier survival analysis. Red line, FTO high expression; Green line, FTO low expression. (A) Overall survival analysis (n = 137), (B) Relapse-free survival analysis (n = 137), (C) Overall survival analysis in patients with tumor necrosis (n = 88) and (D) in tumor necrosis-free patients (n = 48).
FTO Promotes Cisplatin -Induced Apoptosis and Inhibits the Migration of ICC Cells in vitro
We next investigated the biological functions of FTO in ICC. We first analyzed FTO mutation status and copy number alterations (CNAs) in cholangiocarcinoma using the cBioPortal for Cancer Genomics (http://www.cbioportal.org). Neither mutations nor copy number alterations in FTO or other m6A modification enzymes were found in cholangiocarcinoma (Figure 3A). The mRNA level of FTO was lower in ICC cell lines (HCCC-9810, HuCC-T1, TFK1 and RBE) compared with normal human intrahepatic biliary epithelial cells (HIBEPIC, Figure 3B). TFK1 and HCCC-9810 cells were used for both gain- and loss-of-function assays (Figures 3C–E). Cisplatin, which is a standard chemotherapy regimen, has been extensively used for treating ICC and other cancers. To examine a potential function in chemosensitivity, we tested FTO knock-down cells in their response to cisplatin through assaying plating efficiency. Downregulating FTO insensitized the ICC cells to the cisplatin treatment (Figures 4A,B). Consistently, decreased endogenous expression of FTO obviously reduced apoptosis in ICC cell lines (Figures 4C,D), and increased expression of FTO synergized cisplatin-induced apoptosis in ICC cell lines (Figures 4E,F).
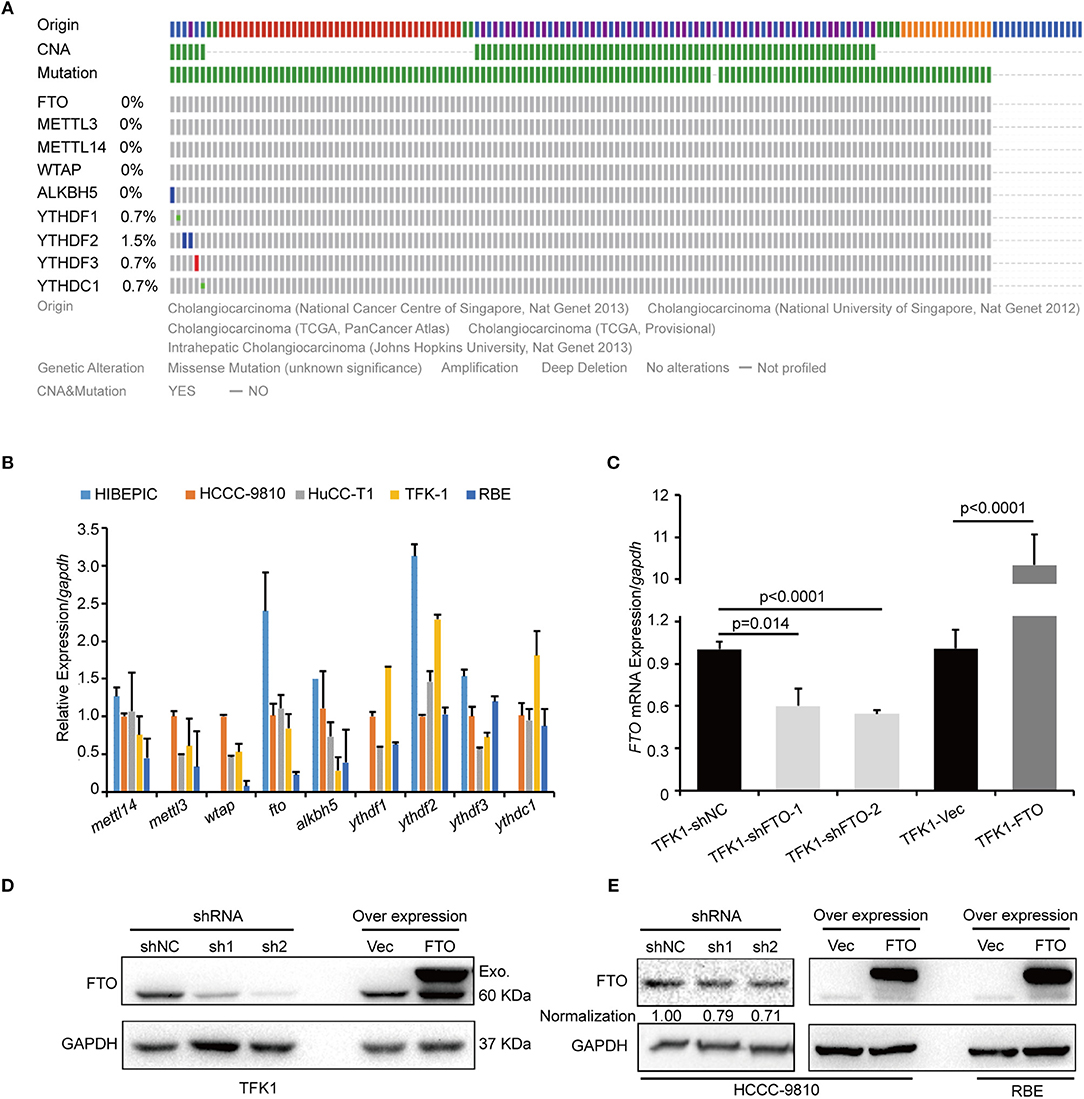
Figure 3. FTO mRNA level was low in ICC cells. (A) The genetic alterations of erasers, readers, and writers for the m6A modification in ICC were analyzed by cBioPortal. (B) The expression levels of erasers, readers and writers for m6A modification in ICC cell lines (n = 4) and the intrahepatic biliary epithelial cell line HIBEPIC were analyzed by q-PCR. (C) The mRNA levels of FTO in control, overexpression, and knockdown TFK1 cells were assessed by q-PCR. (D,E) The protein levels of FTO in control, overexpression and knockdown TFK1 and HCCC-9810 cells.
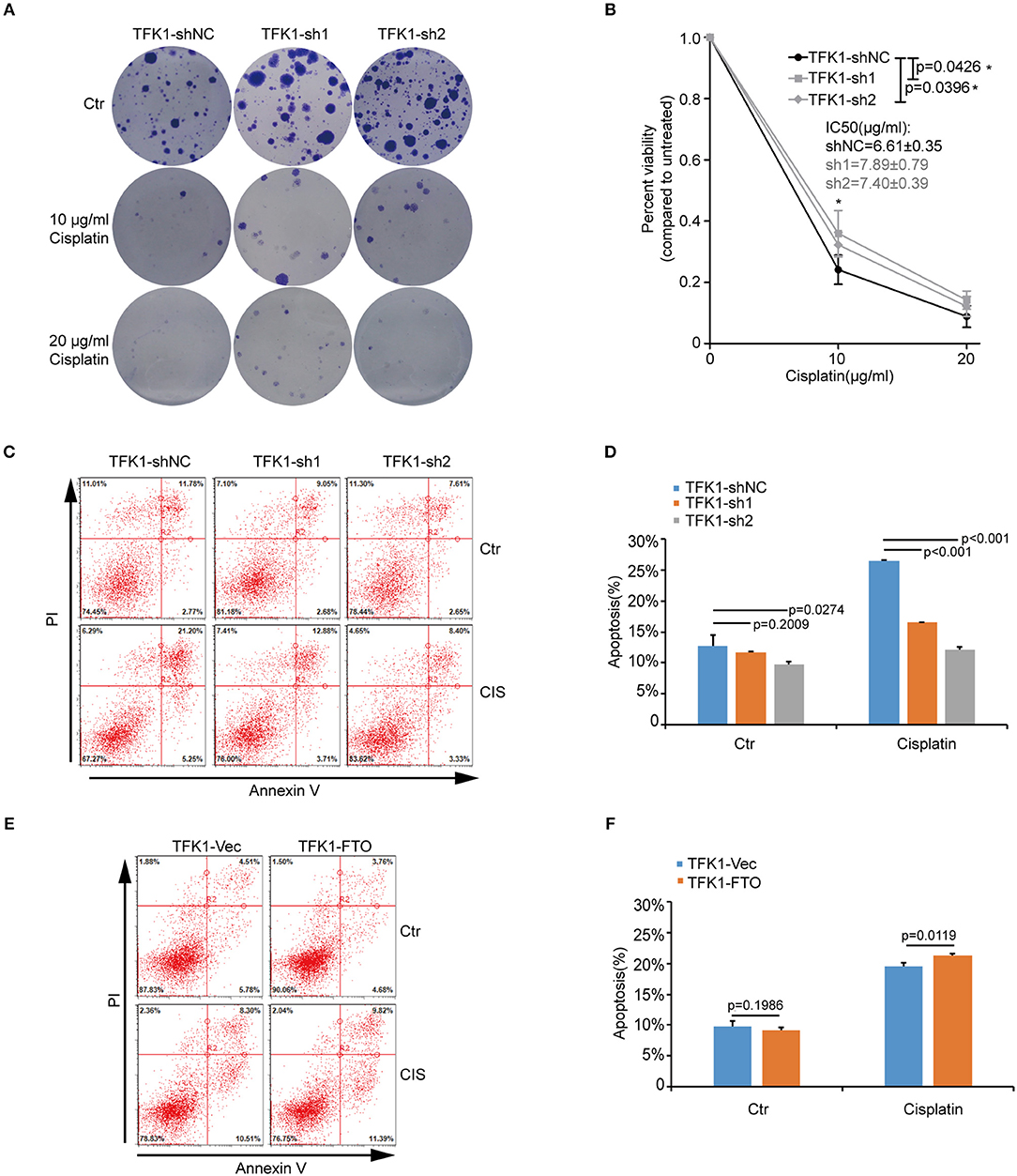
Figure 4. FTO promoted cisplatin-induced apoptosis of ICC cells. (A,B) Clonogenic survival assay of shFTO TFK1 cells and corresponding control cells treated with 0, 10, and 20 μM cisplatin. Data are presented as mean ± SD from n = 4. (C) The effects of FTO knockdown on the apoptosis of TFK1 cells treated with cisplatin (CIS, 30 ng/ml, 24 h) were examined using flow cytometric analysis. (D) The statistical analysis for (C). Data are presented as mean ± SD from n = 3. (E) The effects of FTO over-expression on the apoptosis of TFK1 cells treated with cisplatin (CIS, 30 ng/ml, 24 h) were examined using flow cytometric analysis. (F) The statistical analysis for (E), data are presented as mean ± SD from n = 3.
To examine the effects of FTO expression on the anchorage-independent growth and invasion of ICC cells, we performed soft agar colony assay and Transwell migration assay. Forced expression of FTO dramatically reduced colony numbers and colony size compared with the control (Figures 5A–C). Additionally, ectopic expression of FTO significantly hampered the invasiveness of TFK1 cells (Figures 5D,E), and knockdown of FTO promoted the invasiveness of HCCC-9810 and TFK1 cells (Figures 5F–I).
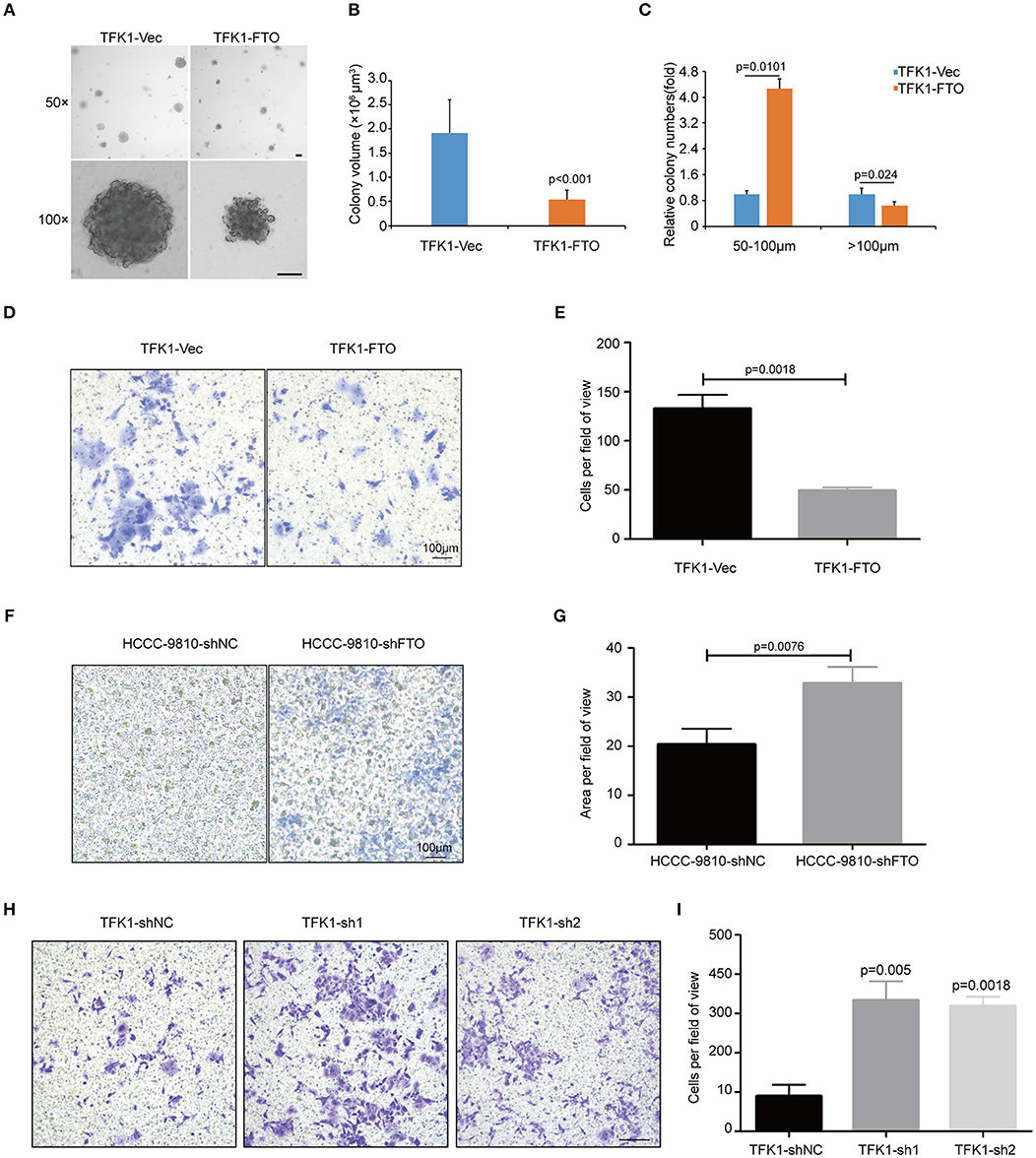
Figure 5. FTO inhibited the colony formation and migration of ICC cells in vitro. (A) The anchorage-independent viability of TFK1 cells was tested in a soft agar colony formation assay. (B) Statistical analysis of colony size. (C) Statistical analysis of colony numbers. (D,E) Transwell assays were performed to examine the effects of FTO overexpression on the invasiveness of FTK1 cells (scale: 100 μm). (F–I) Transwell assays were performed to examine the effects of FTO knockdown on the invasiveness of HCCC-9810 and TFK1 cells (scale: 100 μm).
FTO Regulates ICC Progression Through Multiple Key Oncogenes and Suppressors
To determine the effects of FTO expression on ICC growth in vivo, TFK1 cells (control cells and FTO overexpressing cells) were subcutaneously transplanted into nude mice. Consistently, overexpression of FTO suppressed the growth, volume and weight of the tumors (Figures 6A–C). In addition, the overexpression of FTO in tumors was further determined by western blot and immunohistochemistry (Figures 6D,E).
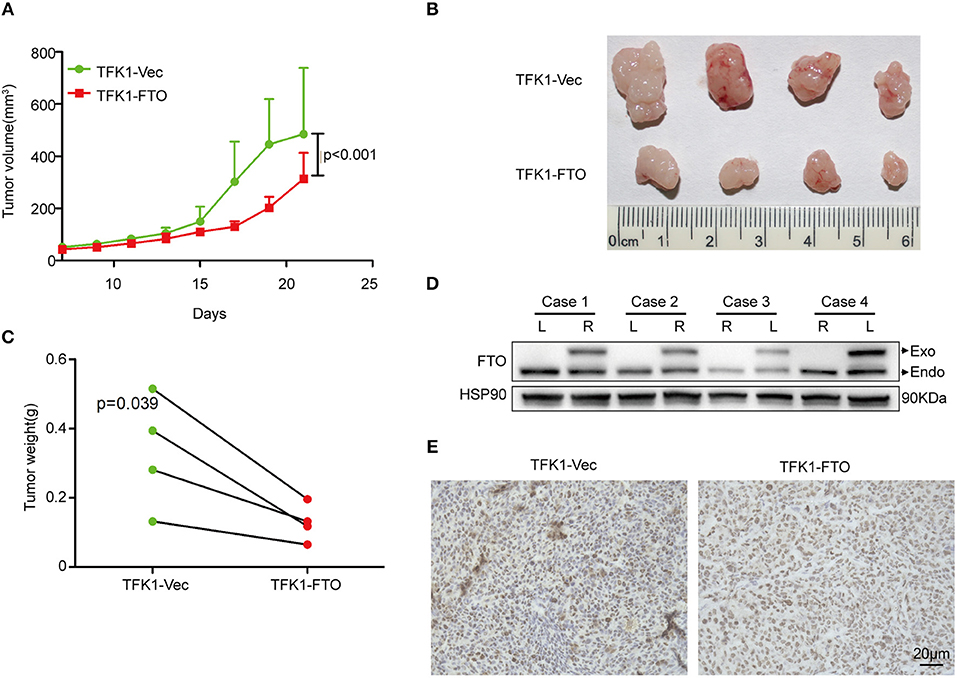
Figure 6. The overexpression of FTO suppressed tumor growth. (A) The growth curve of the tumor (n = 4). (B) FTO overexpression significantly suppressed tumor growth (n = 4). (C) Tumors weight (n = 4). (D,E) The FTO expression in tumors was determined by western blot (D) and immunohistochemistry (E).
To unravel the molecular mechanisms meditating the biological functions of FTO, we analyzed microarray data obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). In all, 1824 differentially expressed genes between FTO-depleted cells and scrambled cells were found by analyzing GSE33870 using GEO2R (p < 0.05, fold change>2, Figure 7A). The analysis of two ICC chips (GSE45001, 10 ICC samples and paired non-tumorous tissues; GSE32225, 149 ICC tissues and 6 non-tumorous tissues), (30, 31) revealed 3,246 differentially expressed genes in GSE45001 and 1580 differentially expressed genes in GSE32225 (p < 0.05, fold change>2, Figure 7A). Then, 35 differentially expressed genes were found to be overlapped from three databases (Figures 7A,B); these genes were determined to be involved in the integrin signaling pathway, inflammation mediated by chemokine and cytokine signaling pathways, the EGFR signaling pathway, angiogenesis and the pyrimidine metabolism pathway (Figure 7C). Further analysis of the expression status of 35 genes demonstrated that FTO knockdown induced the expression of COL8A1, TEAD2, and CMTM4, which were upregulated in ICC (Table 2) and that FTO knockdown inhibited the expression of HAO2, NR5A2, CCL19, TCF21, APOA2, NTRK2, DPT FGA, and SCML4, which were downregulated in ICC (Table 2). We further determined their roles in tumors. TEAD2, a transcriptional enhancer factor, contributes to EMT in breast cancer and pancreatic adenocarcinoma (PDAC) by directing its co-factor YAP1 to the nucleus (32, 33). CMTM4 has been identified as a regulator of PD-L1 protein stability in tumor cells (34). The tumor suppressor gene HAO2 inhibits hepatocellular carcinoma metastasis and predicts a favorable prognosis (35). Nuclear receptor NR5A2 is associated with PDAC and transcriptionally regulates inflammatory gene expression (36). Chemokine CCL19 inhibits colorectal cancer (CRC) angiogenesis in a CCR7-dependent manner (37). TCF21 also acts as a tumor suppressors in multiple tumors, such as renal tumors, ovarian cancer, colorectal cancer, and lung cancer, among others (38–40). SCML4 was reported to be downregulated in breast cancer stem cells (41). However, APOA2, NTRK2, and FGA are oncogenes in PDAC, glioblastoma, and gastric cancer (42–45). Thus, FTO may inhibit ICC progression by reducing oncogene expression (TEAD2 and CMTM4) and inducing tumor suppressor expression (HAO2, NR5A2, CCL19, TCF21, NTRK2, and SCML4). Consistent with our analysis, in TFK1 cells, downregulation of FTO increased the mRNA levels of TEAD2 (Figure 7D). Since FTO was demonstrated to regulate RNA stability, we tested the mRNA stability of TEAD2 through RNA decay assay. The result showed that TEAD2 mRNA stability was increased by inhibiting FTO in TFK1 cells (Figure 7E). Consistently, the mRNA stability of TEAD2 was impaired by over-expressing FTO in TFK1 cells (Figure 7F).
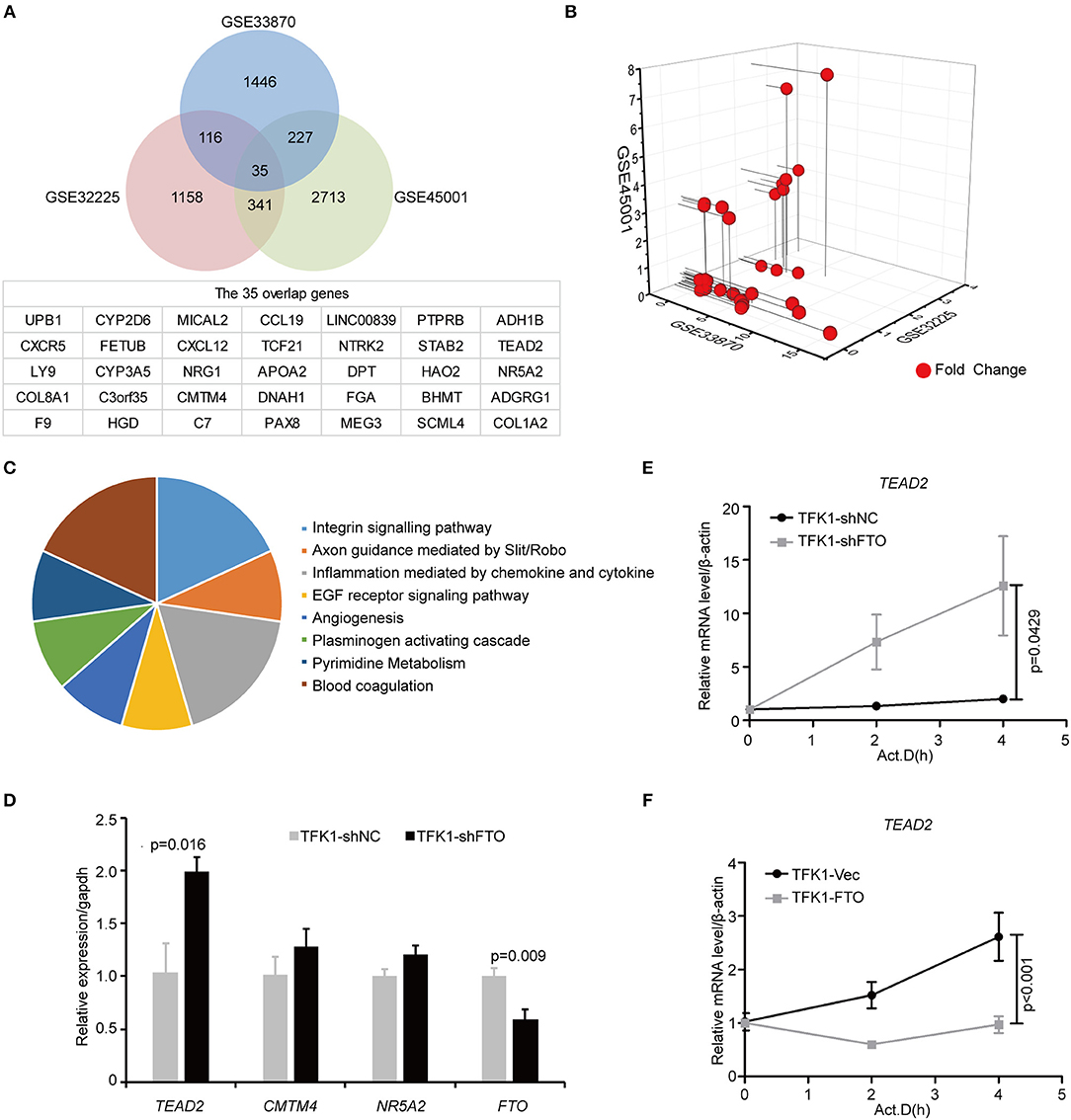
Figure 7. FTO thwarted ICC progression through regulation of multiple oncogenes and suppressors. (A) Mining the differentially expressed genes in GSE33870, GSE32225 and GSE45001 using GEO2R. (B) 3D scatter of 35 common differentially expressed genes in the three data sets. (C) GO pathway analysis of 35 common differentially expressed genes in the three data sets. (D) The mRNA expression of candidate genes was detected by RT-PCR in shFTO TFK1 cells and corresponding control cells. (E,F) The mRNA stability of candidate genes was examined in FTO-overexpressing TFK1 cells, shFTO TFK1 cells, and corresponding control cells by RNA decay assay.
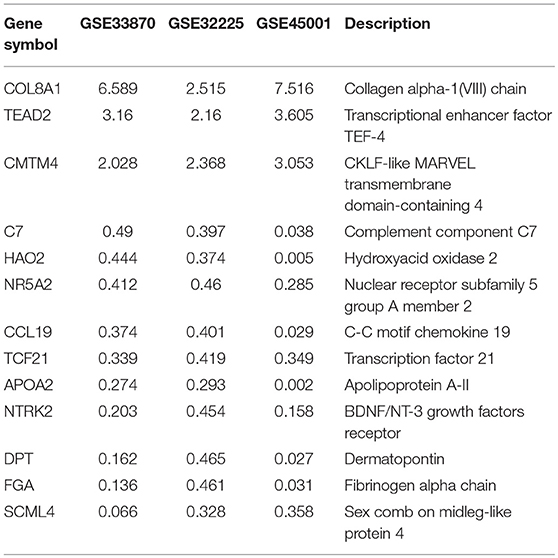
Table 2. Genes related with ICC development based on FTO regulation.
Discussion
The first m6A eraser, FTO, has long been considered an oncogene in tumors (18–20). However, in the present study, we showed that FTO functions as a tumor suppressor in ICC, which implies that FTO may be a context-dependent regulator in oncogenesis network. To explore the clinical relevance of the m6A modification in ICC, we screened the differentially expressed m6A readers, writers and erasers in para-HIBECS and ICC by IHC. FTO was found to be downregulated in ICC tissues and cell lines. In the clinical ICC samples, downregulated FTO was associated with the ICC biomarker CA19-9, which suggested that FTO negatively modulated the progression of ICC. Down-regulated FTO was also correlated with angiogenesis and MVD, which was demonstrated by CD34 expression. A Kaplan-Meier survival analysis showed that low FTO expression predicted poor prognosis in ICC. These clinical findings suggested that FTO played a suppressive role in ICC.
Previous study reported that FTO was highly expressed and functioned as an oncogene in certain subtypes of acute myeloid leukemia (AML) by targeting ASB2 and RARA through mRNA demethylation (18). However, their report also showed that FTO was downregulated in certain subtypes of AML, such as primary AML cases with MLL rearrangements/inv (16) or t(8, 21) (18). Shun Zhou et al. also showed that FTO promoted chemo-radiotherapy resistance in cutaneous squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation (20). These observations suggested that FTO might play dual roles in cancer. Furthermore, FTO and other m6A modification enzymes are also rarely mutated in cholangiocarcinoma, and their copy number changes are unknown. In the biological function analysis, forced expression of FTO markedly synergized cisplatin-induced apoptosis of ICC cells. Consistently, decreased endogenous expression of FTO reduced apoptosis of ICC cells. An anchorage-independent assay on soft agar demonstrated that FTO repressed the viability and growth of ICC cells. In addition, ectopic expression of FTO significantly hampered the mobility of ICC cells in vitro. Mechanistically, we turned to a GEO2R analysis using the public GEO microarray data. From the overlapped and differentially expressed genes from the three databases (GSE33870, 45001, 32225), we identified 35 differentially expressed genes. These genes broadly participate in multiple aspects of tumorigenesis, which suggests that FTO regulates the progression of ICC possibly through modulating the expression of these genes. Further research is needed to elucidate the mechanisms of FTO in the promotion of ICC. Recently, it has been reported that IDH mutations competitively inhibit RNA demethylation of FTO (21, 22). FTO is an α-ketoglutarate (α-KG)-dependent dioxygenase, which is competitively inhibited by R-2-hydroxyglutarate (R-2HG) and the structurally related metabolite D-2-hydorxyglutarate (D2-HG) (21, 22). In several studies, the IDH mutations promoted the accumulation of 2-HG in tumors, including ~20% of AMLs and ~15% of ICCs (5, 46). In ICC, the mutant IDH1/2 proteins convert alpha-ketoglutarate (αKG) to 2-hydroxyglutarate (2HG), which inhibits the activity of multiple αKG-dependent dioxygenases and results in alterations in cell differentiation and survival (47–49). These reports suggested that the mutated IDH1/2 promotes ICC by inhibiting the functions of FTO.
In summary, our study provides a novel insight into ICC development and suggests that down-regulation of FTO might establish a gene network that is in favor of ICC progression.
Data Availability
All datasets generated for this study are included in the manuscript.
Ethics Statement
All experimental protocols were approved by a Central South University institutional committee. Informed consent was obtained from all subjects. The study was reviewed and approved by the China national institutional animal care and use committee.
Author Contributions
Z-XR, NL, Y-ZD, and L-QS designed experiments. Z-XR, ZL, and J-JH carried out experiments. Z-XR, L-YL, X-XR, JG, YM, and Y-DG analyzed experimental results and sequencing data. Y-DG and Y-MD assisted Z-XR with IHC CHIP assay. X-PZ, D-XZ, and NL gathered information of all patients. Z-XR wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81572750, 81874200, 81602405, 81602405); Health and Family Planning Commission of Xuhui District, Shanghai-Jianfeng Clinical Medicine Grant (SHXH201703); Shanghai Education Committee of Shuguang Plan (No. 18SG32); The National Key Basic Research Program 973 project (No. 2015CB555400). The Strategy-Oriented Special Project of Central South University in China (No. ZLXD2017003).
References
1. Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol. (2014) 60:1268–89. doi: 10.1016/j.jhep.2014.01.021
2. Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. (2013) 145:1215–29. doi: 10.1053/j.gastro.2013.10.013
3. Chun YS, Javle M. Systemic and adjuvant therapies for intrahepatic cholangiocarcinoma. Cancer Control. 24:1073274817729241 (2017) doi: 10.1177/1073274817729241
4. Sirica AE, Gores GJ, Groopman JD, Selaru FM, Strazzabosco M, Wang XW, et al. Intrahepatic cholangiocarcinoma: continuing challenges and translational advances. Hepatology. (2018) 69:1803–15. doi: 10.1002/hep.30289
5. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res. (2016) 22:291–300. doi: 10.1158/1078-0432.CCR-14-3296
6. Yang H, Liu T, Wang J, Li TW, Fan W, Peng H, et al. Deregulated methionine adenosyltransferase alpha1, c-Myc, and Maf proteins together promote cholangiocarcinoma growth in mice and humans(double dagger). Hepatology. (2016) 64:439–55. doi: 10.1002/hep.28541
7. Zhang YM, Zhou ZT, Liu GM. Is recurrent pyogenic cholangitis an independent poor prognostic indicator for resectable intrahepatic cholangiocarcinoma? HPB. (2018) 20:1067–72. doi: 10.1016/j.hpb.2018.08.009
8. Gu TL, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS ONE. (2011) 6:e15640. doi: 10.1371/journal.pone.0015640
9. Wang X, Yan Y, Chen X, Zeng S, Qian L, Ren X, et al. The antitumor activities of marsdenia tenacissima. Front Oncol. (2018) 8:473. doi: 10.3389/fonc.2018.00473
10. Wang S, Chai P, Jia R, Jia R. Novel insights on m(6)A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. (2018) 17:101. doi: 10.1186/s12943-018-0847-4
11. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. (2015) 29:1343–55. doi: 10.1101/gad.262766.115
12. Roignant JY, Soller M. m(6)A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. (2017) 33:380–90. doi: 10.1016/j.tig.2017.04.003
13. Dezi V, Ivanov C, Haussmann IU, Soller M. Nucleotide modifications in messenger RNA and their role in development and disease. Biochem Soc Trans. (2016) 44:1385–93. doi: 10.1042/BST20160110
14. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
15. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m(6)Am in the 5' cap controls mRNA stability. Nature. (2017) 541:371–5. doi: 10.1038/nature21022
16. Deng X, Su R, Stanford S, Chen J. Critical enzymatic functions of FTO in obesity and cancer. Front Endocrinol. (2018) 9:396. doi: 10.3389/fendo.2018.00396
17. Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3'-end processing. Nucleic Acids Res. (2017) 45:11356–70. doi: 10.1093/nar/gkx778
18. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
19. Xu D, Shao W, Jiang Y, Wang X, Liu Y, Liu X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. (2017) 38:2285–92. doi: 10.3892/or.2017.5904
20. Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. (2018) 57:590–7. doi: 10.1002/mc.22782
21. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. (2018) 172:90–105 e23. doi: 10.1016/j.cell.2017.11.031
22. Elkashef SM, Lin AP, Myers J, Sill H, Jiang D, Dahia PLM, et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell. (2017) 31:619–20. doi: 10.1016/j.ccell.2017.04.001
23. Liu Y, Wang R, Zhang L, Li J, Lou K, Shi B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett. (2017) 13:4685–90. doi: 10.3892/ol.2017.6038
24. van den Bosch TP, Caliskan K, Kraaij MD, Constantinescu AA, Manintveld OC, Leenen PJ, et al. CD16+ monocytes and skewed macrophage polarization toward M2 type hallmark heart transplant acute cellular rejection. Front Immunol. (2017) 8:346. doi: 10.3389/fimmu.2017.00346
25. Zhu L, Huang F, Deng G, Nie W, Huang W, Xu H, et al. Knockdown of Sall4 inhibits intrahepatic cholangiocarcinoma cell migration and invasion in ICC-9810 cells. Onco Targets Ther. (2016) 9:5297–305. doi: 10.2147/OTT.S107214
26. Song Z, Qiu L, Hu Z, Liu J, Liu D, Hou D. Evaluation of the obesity genes FTO and MC4R for contribution to the risk of large artery atherosclerotic stroke in a chinese population. Obes Facts. (2016) 9:353–62. doi: 10.1159/000448588
27. Chen L, Yan HX, Yang W, Hu L, Yu LX, Liu Q, et al. The role of microRNA expression pattern in human intrahepatic cholangiocarcinoma. J Hepatol. (2009) 50:358–69. doi: 10.1016/j.jhep.2008.09.015
28. Li H, Wu JS, Wang XT, Lv P, Liu G, Tian BN, et al. Major hepatectomy is a safe modality for the treatment of intrahepatic cholangiocarcinoma in selected patients complicated with cirrhosis. J Gastrointest Surg. (2014) 18:194–9. doi: 10.1007/s11605-013-2363-6
29. Nanashima A, Abo T, Murakami G, Matsumoto A, Tou K, Takeshita H, et al. Intrahepatic cholangiocarcinoma, relationship between tumor imaging enhancement by measuring attenuation and clinicopathologic characteristics. Abdom Imaging. (2013) 38:785–92. doi: 10.1007/s00261-012-9974-3
30. Sulpice L, Desille M, Turlin B, Fautrel A, Boudjema K, Clement B, et al. Gene expression profiling of the tumor microenvironment in human intrahepatic cholangiocarcinoma. Genom Data. (2016) 7:229–32. doi: 10.1016/j.gdata.2016.01.012
31. Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. (2013) 144:829–40. doi: 10.1053/j.gastro.2013.01.001
32. Diepenbruck M, Waldmeier L, Ivanek R, Berninger P, Arnold P, van Nimwegen E, et al. Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial-mesenchymal transition. J Cell Sci. (2014) 127(Pt 7):1523–36. doi: 10.1242/jcs.139865
33. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. (2014) 158:185–97. doi: 10.1016/j.cell.2014.06.003
34. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. (2017) 549:106–10. doi: 10.1038/nature23669
35. Mattu S, Fornari F, Quagliata L, Perra A, Angioni MM, Petrelli A, et al. The metabolic gene HAO2 is downregulated in hepatocellular carcinoma and predicts metastasis and poor survival. J Hepatol. (2016) 64:891–8. doi: 10.1016/j.jhep.2015.11.029
36. Cobo I, Martinelli P, Flandez M, Bakiri L, Zhang M, Carrillo-de-Santa-Pau E, et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature. (2018) 554:533–7. doi: 10.1038/nature25751
37. Xu Z, Zhu C, Chen C, Zong Y, Feng H, Liu D, et al. CCL19 suppresses angiogenesis through promoting miR-206 and inhibiting Met/ERK/Elk-1/HIF-1alpha/VEGF-A pathway in colorectal cancer. Cell Death Dis. (2018) 9:974. doi: 10.1038/s41419-018-1010-2
38. Dai Y, Duan H, Duan C, Zhu H, Zhou R, Pei H, et al. TCF21 functions as a tumor suppressor in colorectal cancer through inactivation of PI3K/AKT signaling. Oncol Targets Ther. (2017) 10:1603–11. doi: 10.2147/OTT.S118151
39. Chen B, Zeng C, Ye Y, Wu D, Mu Z, Liu J, et al. Promoter methylation of TCF21 may repress autophagy in the progression of lung cancer. J Cell Commun Signal. (2018) 12:423–32. doi: 10.1007/s12079-017-0418-2
40. Wei J, Zhang L, Li J, Zhu S, Tai M, Mason CW, et al. MicroRNA-205 promotes cell invasion by repressing TCF21 in human ovarian cancer. J Ovarian Res. (2017) 10:33. doi: 10.1186/s13048-017-0328-1
41. da Silveira WA, Palma PVB, Sicchieri RD, Villacis RAR, Mandarano LRM, Oliveira TMG, et al. Transcription Factor Networks derived from Breast Cancer Stem Cells control the immune response in the Basal subtype. Sci Rep. (2017) 7:2851. doi: 10.1038/s41598-017-02761-6
42. Honda K, Katzke VA, Husing A, Okaya S, Shoji H, Onidani K, et al. CA19-9 and apolipoprotein-A2 isoforms as detection markers for pancreatic cancer: a prospective evaluation. Int J Cancer. (2018) 144:1877–87. doi: 10.1002/ijc.31900
43. Wang X, Prager BC, Wu Q, Kim LJY, Gimple RC, Shi Y, et al. Reciprocal signaling between glioblastoma stem cells and differentiated tumor cells promotes malignant progression. Cell Stem Cell. (2018) 22:514–28 e5. doi: 10.1016/j.stem.2018.03.011
44. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
45. Shi F, Wu H, Qu K, Sun Q, Li F, Shi C, et al. Identification of serum proteins AHSG, FGA and APOA-I as diagnostic biomarkers for gastric cancer. Clin Proteomics. (2018) 15:18. doi: 10.1186/s12014-018-9194-0
46. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. (2013) 3:730–41. doi: 10.1158/2159-8290.CD-13-0083
47. Saha SK, Gordan JD, Kleinstiver BP, Vu P, Najem MS, Yeo JC, et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. (2016) 6:727–39. doi: 10.1158/2159-8290.CD-15-1442
48. Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. (2014) 513:110–4. doi: 10.1038/nature13441
Keywords: FTO, ICC, RNA m6A modification, metastasis, TEAD2
Citation: Rong Z-X, Li Z, He J-J, Liu L-Y, Ren X-X, Gao J, Mu Y, Guan Y-D, Duan Y-M, Zhang X-P, Zhang D-X, Li N, Deng Y-Z and Sun L-Q (2019) Downregulation of Fat Mass and Obesity Associated (FTO) Promotes the Progression of Intrahepatic Cholangiocarcinoma. Front. Oncol. 9:369. doi: 10.3389/fonc.2019.00369
Received: 11 February 2019; Accepted: 23 April 2019;
Published: 09 May 2019.
Edited by:
Kuzhuvelil B. Harikumar, Rajiv Gandhi Centre for Biotechnology, IndiaReviewed by:
Paolo Armando Gagliardi, University of Bern, SwitzerlandBraden C. McFarland, University of Alabama at Birmingham, United States
Copyright © 2019 Rong, Li, He, Liu, Ren, Gao, Mu, Guan, Duan, Zhang, Zhang, Li, Deng and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Li, bGlwYXJpc2xpc2lAYWxpeXVuLmNvbQ==
Yue-Zhen Deng, eXVlemhlbmRlbmdAY3N1LmVkdS5jbg==
Lun-Quan Sun, bHVucXVhbnN1bkBjc3UuZWR1LmNu