 Andreia Peixoto1,2,3,4Marta Relvas-Santos1
Andreia Peixoto1,2,3,4Marta Relvas-Santos1 Rita Azevedo1,2
Rita Azevedo1,2 Lúcio Lara Santos1,5
Lúcio Lara Santos1,5 José Alexandre Ferreira1,2,6*
José Alexandre Ferreira1,2,6*- 1Experimental Pathology and Therapeutics Group, Portuguese Institute of Oncology, Porto, Portugal
- 2Institute of Biomedical Sciences Abel Salazar, University of Porto, Porto, Portugal
- 3Tumour and Microenvironment Interactions Group, INEB-Institute for Biomedical Engineering, Porto, Portugal
- 4Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto, Portugal
- 5Department of Surgical Oncology, Portuguese Institute of Oncology, Porto, Portugal
- 6Porto Comprehensive Cancer Center, Porto, Portugal
Decades of research have disclosed a plethora of alterations in protein glycosylation that decisively impact in all stages of disease and ultimately contribute to more aggressive cell phenotypes. The biosynthesis of cancer-associated glycans and its reflection in the glycoproteome is driven by microenvironmental cues and these events act synergistically toward disease evolution. Such intricate crosstalk provides the molecular foundations for the activation of relevant oncogenic pathways and leads to functional alterations driving invasion and disease dissemination. However, it also provides an important source of relevant glyco(neo)epitopes holding tremendous potential for clinical intervention. Therefore, we highlight the transversal nature of glycans throughout the currently accepted cancer hallmarks, with emphasis on the crosstalk between glycans and the tumor microenvironment stromal components. Focus is also set on the pressing need to include glycans and glycoconjugates in comprehensive panomics models envisaging molecular-based precision medicine capable of improving patient care. We foresee that this may provide the necessary rationale for more comprehensive studies and molecular-based intervention.
Introduction
Genetic and epigenetic alterations are considered primary causes of cancer development, with downstream phenotypic changes at the protein level being amongst the driving forces of cancer progression and dissemination. Specifically, post-translational modifications, as glycosylation, impact on protein trafficking, stability and folding, ultimately altering its biochemical, and biophysical properties (1, 2). Moreover, glycans dictate proteolysis patterns and directly mediate ligand-receptor interactions, oncogenic signaling transduction, immune recognition, migration and both cell-cell and cell-matrix adhesion (3–5). In addition, intracellular O-GlcNAc glycosylation (in Ser/Thr residues) of proteins plays a major role in cell physiology and signaling by direct competition with phosphorylation (6). As such, several studies have so far disclosed a plethora of glycans that confer selective advantage to tumor cells, while providing important surrogate biomarkers for specific biological milieus (7, 8). Moreover, while there are few evidences of mutations in genes involved in glycosylation pathways, it is well known that transcriptional and metabolic reprograming of cancer cells has tremendous impact on their glycome and glycoproteome, leading not only to the overexpression but also to the de novo expression of specific glycoepitopes (9, 10). Despite its sour side, cancer-specific alterations in protein glycosylation provide a unique opportunity for clinical intervention. The uniqueness of the created molecular features may be explored to selectively target tumor cells or may provide non-invasive biomarkers after secretion or shedding into body fluids from tumor sites (11, 12).
Building on these findings, the glycobiology field has been progressing toward a more functional understanding of glycosylation impact on cancer biology, disease progression, and dissemination. While specific details on the biosynthesis and diversity of cancer-associated glycans may be found in recent reviews (7, 8), the following sections attempts to highlight the transversal nature of glycans, glycoproteins, and glycan-binding proteins throughout currently accepted cancer hallmarks, with emphasis on the crosstalk between glycans and the stromal components of the tumor microenvironment (Figure 2). These comprehend: (i) sustained proliferative signaling; (ii) resistance to cell death; (iii) deregulated cellular energetics; (iv) evasion of growth suppressors; (v) genome instability and mutations; (vi) replicative immortality; (vii) induction of angiogenesis; (viii) activation of invasion and metastasis; (ix) tumor-promoting inflammation; and (x) immune escape (13). Moreover, we highlight the significance of the most promising protein glycosignatures in cancer arising from the cancer cells-microenvironment crosstalk, its relevance and main milestones facing clinical translation and personalized medicine, as well as the opportunities provided by high-throughput glycomics and glycoproteomics toward molecular-based precision oncology. We foresee that this may provide the necessary rationale for more comprehensive studies and molecular-based intervention.
Protein Glycosylation in Cancer
Glycosylation is the most common, structurally diverse and complex posttranslational modification of membrane-bound proteins, being a non-templated but highly regulated process that rapidly changes in response to physiological and pathological contexts. Glycans result from the highly coordinated action of nucleotide sugar transporters, glycosyltransferases (GTs) and glycosidases in the endoplasmic reticulum (ER) and Golgi apparatus (GA). Two main classes of glycans can be found in membrane and extracellular glycoproteins: (i) O-GalNAc glycans, initiated in the GA by the attachment of a GalNAc to the hydroxyl groups of serine (Ser) or threonine (Thr) residues, forming the simplest O-glycan Tn antigen (GalNAcα-Ser/Thr). The Tn antigen may be further elongated into different core structures that serve as scaffolds for more complex O-GalNAc glycans; (ii) N-glycans, whose biosynthesis starts in the ER with the addition of an oligosaccharide chain to an asparagine (Asn) residue in a peptide consensus sequence of Asn-X-Ser/Thr (X denotes any amino acid except proline). N-glycans experience further structural maturation in the GA to yield either partially unprocessed oligomannose antenna or, more frequently, complex or hybrid type structures, which frequently experience further elongation. Both O- and N-glycan chains are generally branched and/or elongated and may present sialic acids, Lewis blood group related antigens or ABO(H) blood group determinants as terminal structures (8). Further glycan diversity results from several modifications in individual sugars, including O-Acetylation of sialic acids and O-Sulfation of galactose and N-acetylglucosamine residues. Mature glycans may still experience structural remodeling at the cell-surface by extracellular glycosyltransferases and glycosidases freely circulating in the plasma or carried by platelets, further increasing the glycome's structural complexity and dynamic nature (14–16). In addition, other less abundant and far less studied classes of protein glycans can be found at the cell membrane, including O-Fucosylation, O-Mannosylation, O-glucosylation, and C-Mannosylation (17–19). This provides a wide array of potential posttranslational modifications that decisively contribute to define protein functional roles.
In addition to the structural modification of extracellular and cell membrane proteins, intracellular proteins can also be glycosylated with functional implications. Namely, intracellular glycosylation results from the reversible attachment of a N-acetylglucosamine moiety (β-linked GlcNAc) to Ser or Thr residues in cytoplasmic and nuclear proteins (20–22). The GlcNAc residue is generally not elongated or modified to generate complex structures (23). The dynamic cycling of O-GlcNAcylation is catalyzed by two ubiquitously expressed and highly conserved enzymes: uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase (O-GlcNAc transferase, OGT), which adds GlcNAc to the hydroxyl side chain of Ser and Thr, and N-acetyl-β-D-glucosaminidase (O-GlcNAcase, OGA), the enzyme that removes O-GlcNAc. This posttranslational modification has regulatory functions akin to phosphorylation, modulating protein conformation, stability, and reversible multimeric protein assembly (24). Moreover, it functions as a nutrient sensor, providing a biochemical switch to enable the cell adaptation to glucose level alterations and hormonal cues, while regulating a myriad of cellular processes like cellular adhesion, DNA transcription, translation, nuclear transport, and cytoskeletal assembly (25, 26). Interestingly; different isoforms of OGT and OGA vary in length and subcellular localization, suggesting that they target distinct subsets of the proteome (27).
It has been long known that advanced tumors present severe dysregulations in glycosylation pathways, with tumor-associated carbohydrates arising from incomplete or neo-synthesis processes (28). Of note, incomplete synthesis originating truncated structures is more common in early carcinogenesis (29, 30), while the de novo synthesis of neoantigens is more frequent in advanced stages of several cancers (31). The most reported alterations associated to cancer include the over- and/or de novo expression of short-chain O-GalNAc glycans (Tn, T, Sialyl-T, and Sialyl-Tn), Lewis blood group related antigens and their sialylated counterparts [sialyl-Lewis A (SLea) and X (SLex)], as well as complex branched N-glycans (32–34) (Figure 1). Many of these structural features are common to most advanced solid tumors and often associate with poor prognosis, suggesting common molecular mechanisms, which is yet to be proven. Nevertheless, distinct proteome signatures, glycosylation density, and glycosite distribution may ultimately dictate organ, cell-type and cancer-specific molecular signatures and clinically relevant glycoforms.
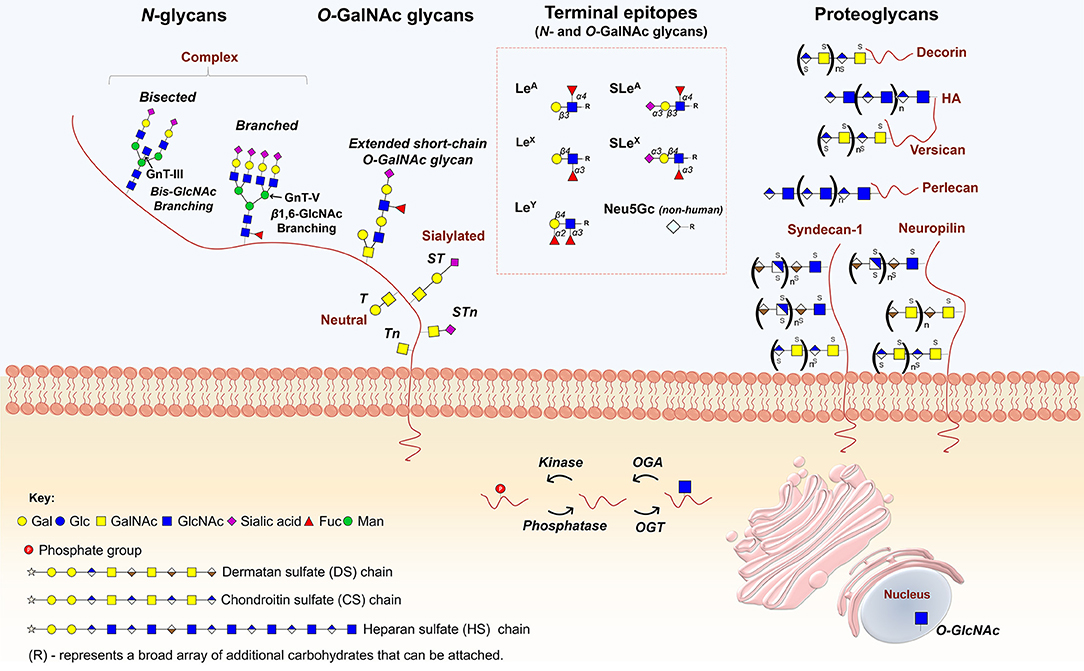
Figure 1. Main classes of glycans modulating cancer hallmarks. N-glycans, whose biosynthesis starts in the endoplasmic reticulum (ER) with the addition of an oligosaccharide chain to an asparagine (Asn) residue, experience further structural maturation in the golgi apparatus (GA) to yield complex bisected and branched structures. O-GalNAc glycans, initiated in the GA by the attachment of a GalNAc to the hydroxyl groups of serine (Ser) or threonine (Thr) residues, forming the simplest O-glycan Tn antigen (GalNAcα-Ser/Thr), may be further elongated into different core structures that serve as scaffolds for more complex O-GalNAc glycans. Both O- and N-glycan chains are generally branched and/or elongated and may present sialic acids, Lewis blood group related antigens and/or their sialylated counterparts as terminal structures. Proteoglycans constitute another class of functionally complex glycoconjugates found as transmembrane, basement membrane and extracellular matrix (ECM) components, exhibiting one or several high molecular weight glycosaminoglycan (GAG) chains covalently attached to a protein core. The figure highlights the structures of some of the most relevant glycans and glycoconjugates driving cancer hallmarks.
Another class of cell-surface glycoconjugates that populate the cell surface and extracellular matrix are proteoglycans, generally composed of one or several high molecular weight glycosaminoglycan (GAG) chains, and composed of sulphated disaccharide repeating units of chondroitin sulfate (CS), heparan sulfate (HS), or dermatan sulfate (DS) covalently attached to a protein core (Figure 1). These polymers can be found as transmembrane, basement membrane and extracellular matrix (ECM) components, presenting high affinities for various ECM constituents and cell adhesion molecules. As such, proteoglycans largely contribute to the acquisition of cancer hallmarks by playing a role in intercellular and ECM interactions, as well as in cellular signaling, especially as co-receptors for growth factors and tyrosine kinase receptors (35, 36).
Overall, the most widely occurring glycosylation modifications in cancer stem from alterations in glycan length, often toward shorter O-glycans and more branched N-glycans. This is accompanied by critical changes in glycans sialylation and fucosylation that impact on the nature of terminal epitopes at glycan chains. In addition, several changes in glycan chains have been reported for glycosaminoglycans (GAG). The structural nature of glycan alteration in cancer and underlying biosynthesis mechanisms have been comprehensively reviewed in recent years (7, 8, 37) and will not be covered in detail here. Aberrant glycosylation actively contributes to tumor progression by regulating tumor proliferation, invasion, metastasis, and angiogenesis (7, 38), being frequently cited as a hallmark of cancer (39). As such, we reinforce this notion by highlighting aberrant glycosylation as an integral part of all recognized cancer hallmark traits. Furthermore, we include the cabal contribution of stromal cells and microenvironmental features for tumor progression and aggressiveness.
Tumor Microenvironment and Glycosylation Crosstalk Toward the Hallmarks of Cancer
The glycocalyx, combining glycoproteins and sugar moieties located on the external side of the plasma membrane, drives the interplay between cancer cells and the tumor microenvironment (TME), a complex scaffold of extracellular matrix (ECM) and various cell types. Both glycans, glycoconjugates and the TME actively contribute to the acquisition of cancer hallmarks, adding another dimension of complexity to cancer progression by influencing cell adhesion and cell-cell recognition, as well as intracellular signaling and ECM interactions (8, 40, 41). Herein, we will highlight the glycosylation-mediated promotion of cancer hallmarks, including the role of stromal cells.
Sustained Proliferative Signaling
Malignant cells are characterized by uncontrolled proliferation, largely due to the loss of homeostasis in the production, release, and affinity for growth-promoting signals. That said, cancer cells may rely on autocrine proliferative signaling or stimulate stromal cells to supply them with mitotic factors to sustain proliferation. For instance, endothelial and infiltrating immune cells secrete growth-promoting factors that paracrinaly stimulate neoplastic cells proliferation independently from blood-borne factors (42, 43). Moreover, tumor and immune cells-promoted ECM remodeling uncages mitogenic agents while disabling growth suppressing adhesion complexes, thereby maintaining the proliferative potential of cancer cells (44). Furthermore, several ECM proteoglycans, mainly produced by cancer-associated fibroblast (CAF), regulate proliferative signaling in adjacent tumor cells (Figure 2A). For instance, CAF-derived proteoglycans syndecan-1 and versican promote proliferation of human breast cancer cells (45–47) and myeloma tumors (48), mainly by influencing EGF receptor signaling. Likewise, transmembrane syndecan-2 expression appears to be critical for colon carcinoma cell behavior by mediating increased adhesion and proliferation (49). Also, the ECM multifunctional heparan sulfate proteoglycan perlecan strongly augments the binding and mitogenic activity of basic fibroblast growth factor (bFGF), contributing to sustained tumor cell proliferation by FGF pathway activation (50). In line with this, fibroblast-derived hyaluronic acid (HA) paracrinally enhances the in vitro proliferation of melanoma cells, while proteins secreted by tumor cells further increase HA synthesis in CAFs in a phosphatidylinositol 3/mitogen-activated protein-kinase-dependent manner (51). On the other hand, the small leucine-rich proteoglycan decorin, expressed primarily by myofibroblast, autocrinally, and paracrinally reduces tumor growth and metastasis in murine xenograft models by downregulating EGFR and Met receptors (52), while inhibiting tumor growth factor β (TGF-β) signaling (53). Decorin also activates ERBB4, which blocks the phosphorylation of heterodimers containing either ERBB2 or ERBB3, thereby suppressing cell growth in mammary carcinoma cells (54). These findings suggest that CAF-derived proteoglycans mainly act as positive regulators of sustained proliferative signaling. In line with this, adipocyte-derived ECM collagen VI affects early mammary tumor progression in vivo via signaling through the NG2/chondroitin sulfate proteoglycan receptor expressed on tumor cells (55). Thereby, stromal adipocytes also constitute active players in driving tumor cell proliferation. Of note, the mechanisms through which proteoglycans enforce their action are not fully elucidated and the true implications of GAG chains are yet to be fully clarified. Given these insights, the reciprocal communication between neoplastic and stromal cells is essential to maintain mitogenic factors supply to sustain cellular proliferation.
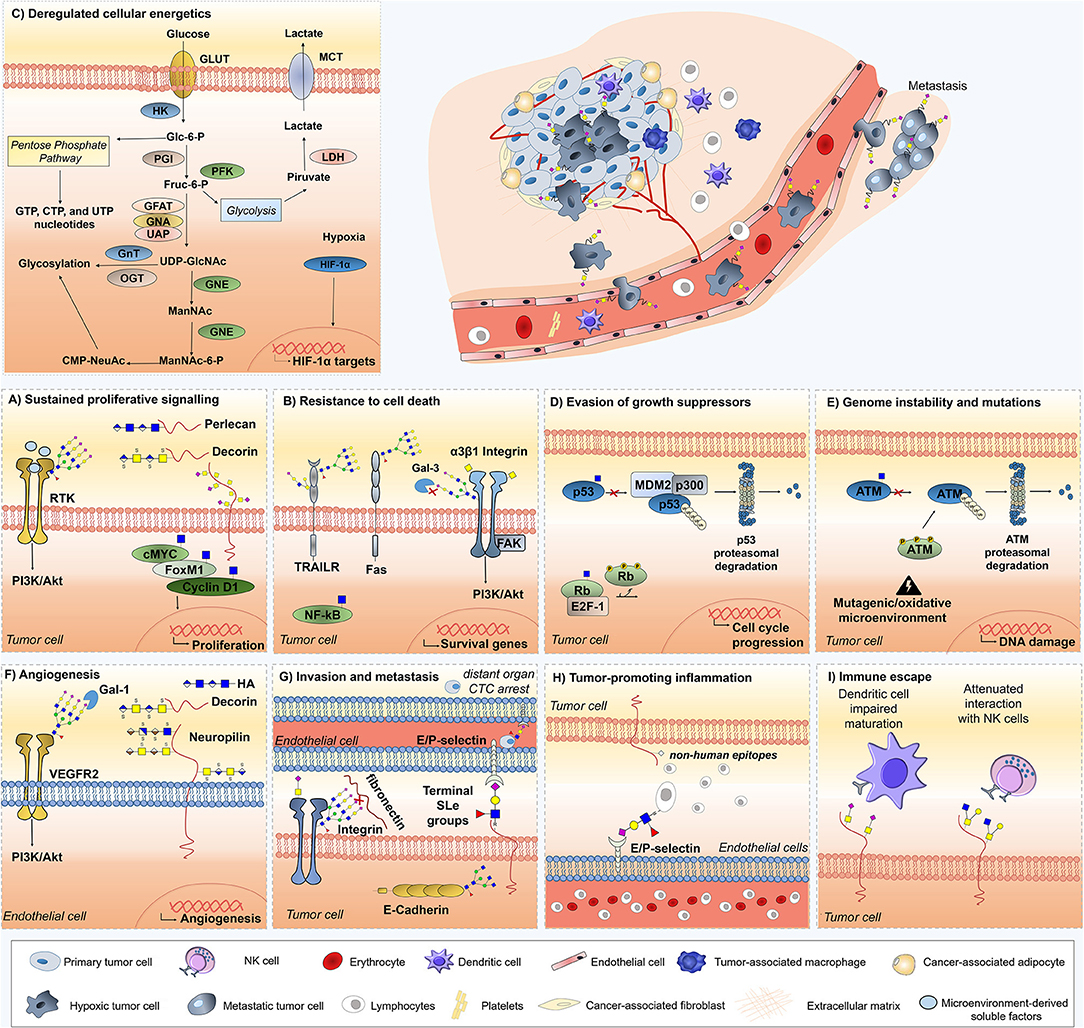
Figure 2. Role of glycans, glycoproteins, glycan-binding proteins, and proteoglycans across currently accepted cancer hallmarks. Glycans (sTn, sLeA/X, Neu5Gc,β1,6-branched N-glycans), glycoproteins (Fas, TRAIL-R, integrin α3β1, VEGFR2, ATM, p53, Rb), proteoglycans (decorin, neuropilin-1,-2, hyaluronic acid, versican, perlecan, hyaluronic acid), lectins (Gal-3 and Gal-1), and O-GlcNAcylated transcription factors (c-Myc, Fox M1, cyclin D1, NF-κB) are mechanistically implicated in cancer hallmarks acquisition and thus represented. Overall, the illustrations focus on particular molecular mechanisms driving hallmark acquisition; namely (A) sustaining proliferative signaling, (B) resistance to cell death, (C) deregulated cellular energetics; (D) evasion of growth suppression; (E) genome instability and mutation; (F) angiogenesis, (G) invasion and metastasis, (H) tumor-promoting inflammation, and (I) Immune scape. Stromal and immune cells providing the soluble factors driving cancer hallmarks are also highlighted, namely tumor-associated macrophages, dendritic cells, adipocytes, and fibroblasts.
Glycosylation adds a second level of proliferation regulation by mediating growth factor receptor activation and structural alterations (Figure 2A). Namely, the O-GlcNAc modification of transcription factors involved in cell cycle progression, such as factor forkhead protein M1 (FoxM1), cyclin D1, and c-MYC, stabilizes them and contributes to oncogenesis (56, 57) (Figure 2A). Moreover, numerous cell-surface tyrosine kinase receptors (RTK), including EGFR, FGFR, PDGF, c-MET, ERBB2/HER2, and IGFR are known to be regulated by cancer-associated glycans (58–60), glycosyltransferases (61), and proteoglycans (62–65). For instance, the degree of N-glycan branching of several RTKs contributes to its capability to induce or arrest cellular proliferation (66, 67). Showcasing this, studies with CHO cells demonstrated that the Asn418-linked N-glycan in ERBB3 plays an essential role in regulating receptor heterodimerization with ERBB2 (59), providing a pivotal checkpoint where N-glycans may regulate key cellular processes involved in cell proliferation and transformation.
Moreover, core 1 β1,3-galactosyltransferase (C1GALT1, responsible for Tn antigen biosynthesis) overexpression in hepatocellular carcinoma activates hepatocyte growth factor (HGF) signaling via modulation of MET kinase O-glycosylation and dimerization, thereby enhancing cell proliferation in vivo and in vitro (61). Contrastingly, overexpression of β1,4-N-acetylglucosaminyltransferase III (MGAT3), which adds β1,4 bisecting branches to N-glycans, appears to inhibit EGFR sensitivity to EGF in glioma cells (58), thereby reducing cellular response to the proliferative effects of EGF. In turn, β1,6-N-acetylglucosaminyltransferase V (MGAT5) knockout mice were shown less prone to mammary tumor growth and metastasis, while showing poor PI3K/AKT activation, emphasizing the importance of β1,6-GlcNAc-branched N-glycans in proliferative signaling pathways (68). Also, ABO glycosyltransferase mRNA downregulation in normal and malignant urothelium is associated with EGF stimulation, resulting in decreased cell proliferation (69). Together, these findings highlight the relevance of glycosyltransferases in tumor cell proliferative signaling.
In turn, the short-chain O-GalNAc STn antigen is mainly observed in non-proliferative tumor areas of highly proliferative bladder tumors (70), while being overexpressed in less proliferative hypoxic bladder cancer models (29), suggesting a yet unknown indirect regulation of proliferation by O-glycosylation in bladder cancer. In ovarian cancer cells, the knockout of core 1 synthase chaperone Cosmc, resulting in Tn and STn O-glycans expression, leads to a reduction in cellular proliferation compared to the parental cell lines (71). Moreover, the use of O-glycan inhibitors in colorectal cancer cell lines promptly blocks proliferation in a so far unexplored manner (72). Overall, short-chain O-glycans expression seem to reduce tumor cell growth. This process might actually confer selective advantage to tumor cells which are rendered less responsive to conventional chemotherapy that mostly targets highly proliferative clones (73).
In addition to alterations in core O- or N-glycans, changes in terminal glycan structures may likewise induce changes in cell proliferation. For instance, in aggressive non-small cell lung cancer cell lines, knockdown of α1,6-fucosyltransferase 8 (FUT8), catalyzing the addition of fucose in alpha 1-6 linkage to GlcNAc residues, significantly inhibits cell proliferation (74). Moreover, overexpression of sialyltransferases and α1,3-fucosyltransferases (FUT4 or FUT6) would suppress EGFR dimerization and phosphorylation upon EGF treatment, decreasing lung cancer cells proliferation (60). In line with this, enhanced α2–6 sialylation, secondary to overexpression of ganglioside-specific ST6GalNAcV, inhibits glioma growth in vivo (75, 76). Altogether, these findings demonstrate the pleiotropic and occasionally opposing effects of altered glycosylation in cell proliferation.
In summary, these examples demonstrate how the microenvironment and glycosylation can sustain proliferative signals. Overall, the crosstalk between neoplastic cells and the TME ensures the positive feedback look of growth factors supply and ECM remodeling, while glycosylation promotes the exposure and interaction of protein domains with RTKs as well as the constitutive activation of oncogenic pathways through kinases modification.
Resistance to Cell Death
The TME aids programmed cell death evasion by providing survival signals and offering a physical barrier against pro-apoptotic factors such as chemotherapy. First, endothelial cells establish vasculature to attenuate cell death that would otherwise result from hypoxia and lack of serum-derived nutrients (77). However, when neovascularization cannot keep up with nutrient demand, an hypoxic microenvironment is established where HIF-1α drives antiapoptotic changes (78). In addition, infiltrating macrophages circumvent apoptosis of cancer cells by shielding them from external apoptotic factors and chemotherapy (79). Similarly, CAFs are highly implicated in apoptotic signaling evasion by secreting paracrine survival factors and inducing ECM remodeling (80–82). Moreover, CAF-derived chondroitin sulfate proteoglycan serglycin (SRGN) induces lung cancer chemoresistance and anoikis-resistance, promoting malignant phenotypes through interaction with tumor cell receptor CD44 (83). In addition, ECM proteoglycans as the small leucine-rich lumican promote melanoma cells apoptosis, ultimately inhibiting metastasis to the lungs (84). Consistent with the changes in ECM composition and topography, expression of many ECM remodeling enzymes is often deregulated in human cancers as tumor cells acquire anchorage independence for survival (85). In this context, tumor cell-ECM interactions control malignant cells subversion of positional information and basement membrane dependence to evade apoptosis upon ECM detachment during cancer progression (86, 87). Furthermore, the ECM also aids tumor cells chemotherapy-induced apoptosis evasion (88–90). Likewise, cancer-associated adipocytes are an abundant source of pro-survival factors and extracellular matrix components, specially collagen VI which confers resistance to cisplatin-induced death in ovarian cancer cells (90, 91).
Glycosylation mostly influences the extrinsic apoptotic program, involving both TRAILR and Fas death receptors, as well as integrin and galectin-mediated signaling (Figure 2B). Several glycans, glycosyltransferases, and glycosidases play critical roles in programmed cell death (92) by hindering ligand–receptor interactions, which influences the formation of signaling complexes, and modulating ligand secretion from effector cells (92, 93). For instance, the tumor necrosis factor–related apoptosis-inducing ligand (Apo2L/TRAIL) promotes tumor cell apoptosis through the death receptors TRAIL-R1 and TRAIL-R2, whose O-glycosylation status determines its sensitivity to the ligand. Specifically, the O-glycosylation initiating enzyme GALNT14 showed a strong link to TRAIL sensitivity in pancreatic carcinoma, NSCLC and melanoma, whereas expression of GALNT3, along with the O-glycan processing enzymes FUT3 and FUT6, correlated with responsiveness in colorectal cancer cells, rendering helpful data for identifying cancer patients who are more likely to respond to TRAIL-based therapies (93). Consistent with these observations, a lower degree of fucosylation, which occurs by mutation of the GDP-mannose-4-6-dehydratase (GMDS) gene, increases resistance to TRAIL-induced apoptosis in colon cancer cells, followed by immune escape (94). Moreover, N-glycosylation also plays an important regulatory role in TRAIL-R1-mediated apoptosis, but not for TRAIL-R2, which is devoid of N-glycans. In this context, defective apoptotic signaling by N-glycan-deficient TRAIL receptors was associated with lower TRAIL receptor aggregation and reduced death-inducing signaling complex (DISC) formation, but not with reduced TRAIL-binding affinity (95).
In turn, the death receptor Fas (CD95/APO-1) has two N-glycosylation sites at N136 and N118 moderately affecting Fas-induced apoptosis. Specifically, the addition of sialic acids by ST6Gal-I in an α2-6 linkage to the N-glycans of Fas provides protection against Fas-mediated apoptosis in colon carcinoma cells. Namely, α2-6 sialylation of Fas prevents FasL-induced apoptosis by decreased activation of caspases 8 and 3, blockage of Fas–Fas-associated death domain (FADD) association with Fas cytoplasmic tails, and inhibition of Fas internalization (96). In line with this, high-grade tumors, which are known to express high levels of O-6 sialylation, significantly overexpress Fas, but are insensitive to Fas-ligand, thereby avoiding immune cell-mediated apoptosis (30, 97, 98). Moreover, N-deglycosylation of Fas leads to the slowing down of procaspase-8 activation at the DISC complex, with no impact on DISC formation or FADD recruitment (99). Overall, these findings demonstrate that, in contrast to the TRAIL-R O-linked glycan moiety, the Fas N-glycan structure contributes to a smaller extent to the initiation of the apoptotic signaling leading to cell death.
Glycosyltransferases, as N-acetylgalactosaminyltransferase 1 (GALNT1), also contribute to activate survival signals that supress apoptosis. Specifically, overexpression of N-acetylgalactosaminyltransferase 1 (GALNT1) contributes to aberrant glycosylation of integrin α3β1, changing the conformation of integrin heterodimers, and initiating signal transduction to induce focal adhesion kinase (FAK) activation in bladder cancer cells (100). Accordingly, both the knockdown of FAK and suppression of FAK phosphorylation were able to induce apoptosis in BC cells through caspase-3 recruitment and Src phosphorylation, respectively (101). The suppression of FAK phosphorylation also inhibited the PI3K/AKT signaling pathway, suggesting it acts downstream of FAK to regulate apoptosis (101). Interestingly, FAK is overexpressed in a variety of human tumors where it mediates survival signaling, and these findings might point an intervention strategy to regulate apoptotic stimuli through glycosyltransferases modulation.
In addition, several studies suggest that hyper-O-GlcNAcylation in cancer may play an anti-apoptotic role (Figure 2B). For instance, human pancreatic ductal adenocarcinoma cells are supported by oncogenic NF-κB transcriptional activity and both NF-κB p65 subunit and upstream kinases IKKα/IKKβ are O-GlcNAcylated. As such, reducing hyper-O-GlcNAcylation decreases NF-κB transcriptional activity and target gene expression, driving apoptosis (102). Furthermore, increasing O-GlcNAc in pancreatic cancer cells protects against suspension-induced apoptosis (102). Moreover, hyper-O-GlcNAcylation could contribute to cancer cell survival by mitigating ER stress through the inhibition of the folding enzyme chaperone CHOP (103).
Another important molecular mechanism relating protein glycosylation to apoptosis in cancer cells results from the crosstalk between lectins and death receptors. Classically, the effect of Galectin-3 (Gal-3) in the regulation of apoptosis depends on its subcellular localization. Accordingly, cytoplasmic Gal-3 is anti-apoptotic, whereas nuclear Gal-3 is pro-apoptotic (104). Upon extracellular secretion via a non-classical pathway (105), Gal-3 may bind to cell surface glycans, increasing cell signaling and cell-matrix interactions (106, 107). Interestingly, overexpression of STn results in decreased Gal-3 at the cell surface in colon cancer cells, promoting an accumulation of Gal-3 in the cytoplasm and reducing chemotherapy induced apoptosis (108). Moreover, it has been shown that O-6-sialylation of integrin β1 N-glycans, mediated by ST6Gal-I, completely blocked its recognition by Gal-3; conversely O-3-sialylation did not affect Gal-3 recognition in gastric cancer (108, 109). These observations suggest that Gal-3 binding to glycans is dependent on sialylation and that decoding the sialome of cancer cells may bring new insights on programmed cell death pathways.
Together, these findings demonstrate that both glycosidic and microenvironmental cues aid tumor cells to circumvent apoptosis. Interestingly, the tumor microenvironment mostly provides factors to evade intrinsic apoptotic signaling, while glycosylation mostly regulates the extrinsic signaling pathway initiated by binding of a death ligand to a death receptor on the cell surface.
Deregulated Cellular Energetics
The microenvironmental modulation of tumor cell energetics is crucial to drive metabolic adaptation and survival of neoplastic cells. As such, CAFs and endothelial cells are able to create collaborative metabolic domains by activating complementary metabolic pathways to buffer and recycle metabolites of tumor cells in order to maintain stromal and tumoral growth (110, 111). Adipocytes also engage in this metabolic crosstalk by providing fatty acids utilized by cancer cells to generate ATP via mitochondrial β-oxidation in metastatic ovarian cancer (112). Another pivotal microenvironmental feature driving energetic adaptation is hypoxia, resulting from uncontrolled proliferation and inefficient neovascularization. Hypoxic stress within a tumor leads to a shift from aerobic oxidative phosphorylation to anaerobic glycolysis, with high rates of glucose and glutamine uptake (the Warburg effect) (113). In this context, adaptation to hypoxia and cellular energetic reprograming occurs mostly in a HIF-1α-dependent manner, being frequently accompanied by cell dedifferentiation and acquisition of mesenchymal characteristics (29). Briefly, to compensate the reduction of intracellular ATP levels under hypoxic conditions, HIF-1α upregulates the expression of glucose transporters-1 and 3 (GLUT1, GLUT3), allowing the intracellular uptake and phosphorylation of glucose (114–116). Subsequently, Glc-6-P enters one of several possible biosynthetic pathways, namely glycolysis, hexosamine biosynthetic pathway (HBP), pentose phosphate pathway (PPP), or glycogen synthesis, all of which substantially regulated by HIF-1α (117–124) (Figure 2C). Simultaneously, HIF-1α decreases O2 consumption and reactive oxygen species (ROS) generation within the mitochondria (125–127) to circumvent oxidative stress.
By regulating the flux through the HBP and PPP pathways, HIF-1α dramatically affects glycosylation, either by altering precursor production or by governing enzymatic activity. Specifically, HIF-1α has significant impact on HBP by inhibiting the TCA cycle and suppressing the addition of acetyl groups, that would otherwise arise from that pathway, to glucosamine, leading to an overall reduction in the glycosylation precursor UDP-N-Acetylglucosamine (UDP-GlcNAc) production (128–130). Another branch of the HBP, the CMP-NeuAc nucleotide sugar biosynthesis pathway, is activated under hypoxia through the epimerization of UDP-GlcNAc by UDP-GlcNAc 2-epimerase (GNE), ultimately enabling cell surface sialylation in a HIF-1α-dependent manner (131) (Figure 2C).
Moreover, during acute hypoxia, the production of ATP, GTP, UTP, and CTP nucleotides through the PPP is decreased, compromising the addition of UDP to GlcNAc (132). Interestingly, while hypoxia causes downregulation of the rate limiting enzyme of the PPP Glucose-6-phosphate dehydrogenase (G6PD) in several cancers (133), glycosylation promotes G6PD activity and increases glucose flux through the PPP, providing precursors for nucleotide and lipid biosynthesis, and reducing equivalents for antioxidant defense. Particularly, G6PD is dynamically O-GlcNAcylated in response to hypoxia, and blocking G6PD glycosylation reduces cancer cell proliferation in vitro and in vivo (134), most likely through energetic unbalance. On the same note, blockage of hypoxia induced O-GlcNAcylation at serine 529 of phosphofructokinase 1 (PFK1) reduced cancer cell proliferation in vitro and impaired tumor formation in vivo (135). Of note, it has been reported that elevated O-GlcNAcylation in cancer cells stabilizes HIF-1α in an indirect manner, thereby reinforcing the Warburg effect (103) in what appears to be negative feedback loop toward homeostatic O-GlcNAcylation levels.
In addition to intracellular glucose metabolism modifications, decreased 1,2-fucosylation of cell-surface glycans, galectin overexpression, and glycosyltransferases as well as glycosidases modulation toward the expression of short-chain sialylated O-glycans are some consequences of the hypoxic tumor microenvironment. Additionally, increased expression of gangliosides carrying N-glycolyl sialic acids can also be significantly affected by hypoxia (29, 136). For all these reasons, it is possible to realize that hypoxia strongly alters glycobiologic events within tumors, resulting in increased O-GlcNAcylation and sialylation; thereby leading to more aggressive phenotypes (136–138).
Besides regulating glycolytic enzymes in the context of hypoxia, O-GlcNAcylation also governs transcription factors activity (ChREBP, carbohydrate-responsive element-binding protein, Sp, and c-MYC) toward increased aerobic glycolysis, anaplerotic resupply of TCA intermediates used in biosynthesis, nucleotide metabolism and lipogenesis (139–144). Together, these findings suggest that hyper-O-GlcNAcylation contributes to oncogenicity through metabolic reprograming and stabilization of oncogenic transcription factors.
Based on these insights, hypoxia is a major driving force of the energetic reprograming of cancer cells, largely affecting glycosylation in a HIF-1α-dependent manner. As such, both O-GlcNAc modifications and HIF-1α transcriptional activity emerge as key metabolic modulators, while stromal cells promote a metabolic symbiosis with tumor cells envisaging tumor survival and growth.
Evasion of Growth Suppressors
To prevail, cancer cells not only induce and maintain stimulatory growth signals but also develop the ability to evade the negative regulation of tumor suppressor genes (145). Even though tumor growth suppression is mostly regulated by intrinsic mechanisms involving p53 and retinoblastoma (RB) pathways, some stromal and microenvironmental components have been implicated in growth arrest evasion by inhibiting adhesion complexes and promoting clonal selection. Namely, proteolytic enzymes produced by stromal cells are able to disrupt cell-cell or cell-ECM adhesion complexes significantly contributing to uncontrolled cell proliferation and progressive distortion of normal tissue architecture (85, 146, 147). Moreover, tumor hypoxia selects clones expressing mutant p53, facilitating the clonal expansion of cells that have a dominant-negative effect on the wild-type cells, thus evading growth suppression (148).
Interestingly, the two canonical suppressors of cell proliferation, p53 and RB, are regulated by O-GlcNAcylation (149, 150) (Figure 2D). Particularly, it was demonstrated that p53 O-GlcNAcylation on Ser149 limits both ubiquitin-dependent proteasome degradation and the interaction with E3 ubiquitin-protein ligase MDM2 (149). Contrariwise, overexpression of O-GlcNAcase (OGA) results in increased MDM2 phosphorylation at Ser166, stimulating MDM2-p300 interactions and resulting in p53 degradation (151). In turn, RB activity is regulated by the dynamic crosstalk between O-GlcNAc modification and phosphorylation (150). Retinoblastoma binds E2F-1 transcription factor preventing co-activator complexes from binding E2F-1, thereby arresting cell cycle in the G1 phase. Particularly, RB is densely modified with O-GlcNAc in the G1 phase, which prevents its phosphorylation and sustains its activity. During mid- to late-G1, a shift toward increased phosphorylation leads to the release of E2F-1 from RB and E2F-1-dependent transcriptional activation of essential S-phase genes, allowing cell cycle progression (150).
In summary, cancer cells circumvent growth suppression by negatively regulating the two canonical suppressors of proliferation p53 and RB through glycosidic modifications, while stromal cells and hypoxia aid tumor cell growth by abrogating the suppressive role of adhesion complexes and selecting for more proliferative clones.
Genome Instability and Mutations
During uncontrolled cell division, random mutations, and chromosomal instability promote genomic alterations, which coupled with disruption of genome integrity checkpoints culminate in selective advantage of tumor cells (152). In this context, intratumoral hypoxia leads to increased mutation rates and altered DNA damage response, while HIF-1α interplays with oncoproteins such as c-MYC to drive malignant progression (153–155). In addition, recent evidence shows that oxidative stress in CAFs induces genomic instability in adjacent breast cancer cells via mutagenic evolution, potentially increasing their aggressive behavior (156). Together, these findings suggest that tumor progression is prompted by the orchestrated interaction of malignant cells and the TME, which promotes genetic instability toward more aggressive phenotypes.
It is known that the tumor suppressor p53 plays a central role in genomic stability maintenance (157). However, stabilization of previously mutated p53 by O-GlcNAcylation is not expected to lead to tumor suppression (149). Nevertheless, SILAC-based quantitative proteomics of O-GlcNAc transferase wild-type and Null cells has demonstrated the O-GlcNAcylation regulation of the ATM (ataxia-telangiectasia mutated)-mediated DNA damage response pathway through ATM and its downstream targets H2AX, and Chk2 (158) (Figure 2E). Other molecular studies have reinforced that ATM interacts with O-GlcNAc transferase, with its activation and recovery states being affected by O-GlcNAcylation (159).
Importantly, genetics is not the only factor contributing to genetic instability. Epigenetic modifications through DNA methylation, posttranslational modification of histone proteins, and interactions of non-coding RNAs with proteins or other nucleic acids also largely drive cancer progression (160, 161). Interestingly, histones H2A, H2B, and H4 are O-GlcNAcylated in vivo, making O-GlcNAc modifications a part of the histone code regulating gene transcription (162). Although no specific links between hyper-O-GlcNAcylation and cancer cell epigenetic contribution to transformation have been established, some clonal expansions may well be triggered by these non-mutational changes affecting the regulation of gene expression.
In summary, tumor microenvironmental features and stromal cells contribute to a mutagenic environment through the production of oxygen and nitrogen reactive species, while altering transcription and translation of several DNA damage response and repair genes. In turn, glycosylation modulates DNA damage response pathway components and possibly non-mutational changes affecting the regulation of gene expression.
Replicative Immortality
The maintenance of telomerase lengths by DNA polymerase telomerase is a key event contributing to the unlimited replicative potential of cancer cells (163). Recently, hotspot point mutations in the regulatory region of the telomerase reverse transcriptase (TERT) gene, encoding the core catalytic component of telomerase, was identified as a novel mechanism to activate telomerase in cancer (164, 165). Interestingly, there is currently no substantive evidence of microenvironmental contributions to telomere stabilization in cancer cells. However, there is evidence that hypoxia up-regulates telomerase activity in cancer cells via MAPK cascade signaling activation as a stress response against hypoxia-induced genotoxicity (166). Moreover, hypoxia induces c-MYC activation, which, in turn, transactivates TERT (167). So far, TERT has not been described as a glycoprotein; nevertheless, there could be an indirect link between glycosylation and telomerase activation through c-MYC O-GlcNAcylation regulation (57). As such, future studies should investigate whether O-GlcNAc-mediated stabilization of c-MYC can indirectly influence telomerase activation and contribute to replicative immortality.
In conclusion, both glycosylation and microenvironmental factors allow successive cell cycles mostly by circumventing cell death, while having little to do with avoiding senescence and regulating telomere length. However, tumor hypoxia might contribute to immortalization by indirectly influencing kinase cascades and transcriptions factors, while glycosylation modifications have a more modest impact in transcription factor regulation.
Angiogenesis
The formation of neovasculature through angiogenic processes is vital for cancer cell proliferation and tumor progression to metastasis (168). Historically, tumor angiogenesis was perceived as being primarily regulated by cancer cells expressing proangiogenic factors; however, now it becomes increasingly clear that the tumor microenvironment is a key factor inducing and sustaining chronic angiogenesis, including in a glycosylation-dependent manner. First, tumor hypoxia upregulates multiple pro-angiogenic pathways mediating key aspects of stromal, endothelial cell (EC) and vascular support cell biology to influence neovessel patterning, maturation, and function (169). Concomitantly, stromal innate immune cells and CAFs synthesize or release through ECM remodeling several angiogenic soluble factors driving the expansion of the pre-existing vascular supply (170–174). In line with this, stromal cells-derived proteoglycans and ECM molecules are also active angiogenesis regulators (Figure 2F). For instance, heparan sulfate (HS) proteoglycans inhibition hampers pro-angiogenic signaling and neovessel formation by effecting the bioactivity, diffusion, half-life and interaction of VEGF with its tyrosine kinase receptors (175, 176). In ovarian cancer, HS has also been shown to impact angiogenesis through EGF receptor signaling and by influencing the expression of angiogenic cytokines (177). Particularly, CAF-derived HS proteoglycan syndecan-1 expression stimulates breast tumor angiogenesis, being correlated with both vessel density and total vessel area (178). Furthermore, Neuropilin-1 (NRP-1) and Neuropilin-2 (NRP-2) transmembrane proteoglycans, as well as hyaluronic acid (HA) fragments resulting from the hydrolysis of carbohydrate chains in proteoglycans by HYAL hyaluronidase, also display pro-angiogenic properties in several cancer models (179–182). Contrastingly, stromal decorin angiogenic role seems to be context dependent. Namely, it blocks tumor cell-mediated angiogenesis by downregulating VEGFA production, as well as Met and downstream angiogenic networks in some tumor models (183, 184), while being required for efficient tube formation by EC and inflammation-induced angiogenesis in others (185). In turn, the basal lamina lumican, a class II small leucine-rich proteoglycan, inhibits melanoma angiogenesis by compromising the migratory capacity of EC and pseudotubes formation, supressing lung metastasis (84). Moreover, lumican affects angiogenesis by interfering with α2β1 integrin receptor activity and downregulating proteolytic activity associated with surface membranes of EC (186). In line with this, several studies highlight that lumican inhibits EC invasion, angiogenic sprouting, and vessel formation, while enhancing Fas mediated EC apoptosis (187–190). Collectively, these findings provide new insights into how ECM remodeling regulates angiogenesis activation and resolution, as well as identify proteoglycans as effectors modulating angiogenesis both in vitro and in vivo.
Glycans and glycan-binding proteins, as galectins, add another level of positive regulation of angiogenesis by modulating EC migration, branching, survival, and vascular permeability (191–193). For instance, a glycosylation-dependent pathway that preserves angiogenesis in response to VEGF blockade was identified, in which galectin-1 (Gal-1) binds β1-6GlcNAc branched N-glycans present on VEGFR2 in EC surface to activate a VEGF-like signaling (Figure 2F). Moreover, vessels within anti-VEGF-sensitive tumors exhibited high levels of α2-6-linked sialic acids, which prevented Gal-1 binding and VEGFR2 activation (192). Moreover, interruption of β1-6GlcNAc branching in EC or silencing of tumor-derived Gal-1 converted refractory tumors into anti-VEGF-sensitive (192). Importantly, this could allow pinpointing patients better served by anti-VEGF therapy and targeting glycosylation-dependent lectin-receptor interactions envisaging increased treatment efficacy in refractory patients (194, 195).
In addition, reduced O-GlcNAcylation in prostate cancer cells has been associated with decreased expression of several angiogenic factors, such as matrix metalloproteinases MMP-2 and MMP-9, and VEGF, resulting in inhibition of angiogenesis (196). Moreover, glycosydic cues as O-glucose, O-GlcNAc, and O-GalNAc glycans affect Notch signaling, thereby regulating angiogenesis (197). Also, α2,6-sialic acids are necessary for the cell-surface residency of platelet endothelial cell adhesion molecule (PECAM), a member of the immunoglobulin superfamily that plays multiple roles in EC adhesion, mechanical stress sensing, anti-apoptosis, and EC-mediated angiogenesis (198). Together these finding highlight the glycosylation modulation of tumor angiogenesis.
In summary, the tumor microenvironment ensures the supply of pro-angiogenic factors, while upregulating multiple pro-angiogenic pathways governing the maturation and survival of endothelial cells. In turn, glycans and glycoconjugates can be angiogenic per se or alter the affinity of angiogenic factor receptors for their ligands toward a pro-angiogenic phenotype of EC.
Invasion and Metastasis
Throughout the course of disease, cancer cells often acquire more motile phenotypes, as well as the capability to invade surrounding tissues and adjacent organs. Subsequently, cancer cells reach lymph and blood vessels, entering circulation and eventually metastasizing to distant locations. Interestingly, metastatic tumor cells may even travel from the primary site to the secondary location with stromal components, including activated fibroblasts, achieving a very favorable outcome in the colonization step of tumor progression (199). In this context, stroma, ECM, and microenvironmental cues often facilitate invasion and the establishment of metastatic colonies by tumor cells. For instance, tumor hypoxia aids migration and invasion of tumor cells by influencing angiogenesis, immune tolerance, epithelial-to-mesenchymal transition (EMT), and regulating adhesion molecules expression and glycosylation (200). At a distance, hypoxia contributes to the production of diffusible factors and exosomes involved in premetastatic niche formation, while regulating metabolic and survival pathways that allow cells to adapt to distant microenvironments (201). Within the tumor stroma, infiltrating immune cells and CAFs promote ECM remodeling while producing pro-invasive and EMT promoting factors (172, 202–204). Namely, the CAF-derived proteoglycans versican and serglycin promote tumor invasion and metastasis in breast, ovarian, and prostate cancer (47, 205, 206), as well as NSCLC cells EMT, migration, invasion and liver colonization, respectively (83). Similarly, the ECM hyaluronic acid (HA) and biglycan are directly involved in the metastatic potential of breast and prostate tumor cells (207, 208) as well as melanoma cells (209), respectively. Moreover, metastatic tumor cells must acquire the capability to autonomously synthesize, assemble, and process their own “portable” HA-rich microenvironments to survive in circulation, metastasize to ectopic sites, and escape therapeutic intervention. As such, strategies to disrupt the HA machinery of primary tumor and circulating tumor cells may enhance the effectiveness of current conventional and targeted therapies (210, 211). On the other hand, triple-negative orthotopic breast carcinoma systemic treatment with the proteoglycan decorin induced the tumor suppressor cell adhesion molecule 1 (Cadm1), favoring a less metastatic phenotype (212, 213). Altogether, these findings highlight stromal-derived proteoglycans as major players driving the metastatic potential of tumor cells. Concomitantly, in vitro studies suggested that stromal derived TGFβ-induced EMT alters glycogenes expression and consequently promotes N-glycan remodeling, including decreased bi-, tri- and tetra-antennary complex N-glycans and increased expression of hybrid-type N-glycans and fucosylation (214); thereby showing a correlation between microenvironmental soluble factors and glycosylation changes.
In line with glycoconjugate regulation of invasion and metastasis, glycans add another dimension of regulation to the acquisition of this cancer hallmark. Namely, it has been proposed that increased sialylation, accompanying malignant transformation, promotes cell detachment from the primary tumor through electrostatic repulsion of negative charges, physically disrupting cell adhesion (215, 216). In line with this, the STn antigen reduces cell adhesion in prostate cancer (217), while increasing migration and invasion in bladder (29), breast (218), and gastric (219, 220) carcinomas in a ST6GalNAc.I-dependent manner. Also, the increased and de novo expression of the STn antigen in bladder cancer cells is part of an array of molecular events underlying the establishment of mesenchymal traits (29). Moreover, STn was mainly found in densely O-glycosylated adhesion proteins such as integrins and cadherins (29, 30). It is likely that the transition from extended to shorter and heavily sialylated structures may impair these proteins normal function and induce molecular and spatial reorganization at the cell-cell and cell-matrix interfaces. In agreement with these observations, STn expressing cells are frequently simultaneously observed in invasion fronts, near blood vessels and corresponding lymph nodes, as well as in distant metastasis (70, 221). Moreover, it has been recently reported that most circulating tumor cells (CTC) in the blood of metastatic bladder cancer patients present a highly undifferentiated and more aggressive basal phenotype, while overexpressing the STn antigen (221). As such, STn expression seems to confer a competitive advantage to neoplastic bladder cells by enabling not only invasion but also the necessary mechanisms for successful cancer dissemination. Similarly, ST6Gal.I-mediated α2,6-sialylation of breast cancer cells mediates reduced cell-cell adhesion and enhanced invasion capacity (222). Overall, immature truncated O-glycophenotype of cancer cells directly induces oncogenic features, including enhanced migration and invasive capacity (223).
Reinforcing the key role played by sialic acids in cell-cell adhesion, sialylated α3β1 integrin, displaying numerous sialylated tetra-antennary complex type glycans, exhibited significantly lower fibronectin-binding capability than its unsialylated counterpart and showed migration ability through fibronectin in vitro (224). Apart from integrins, E-cadherin aberrant glycosylation highly affects its function and cellular localization, frequently culminating in epithelial cell invasion in gastric cancer (225, 226). Namely, N-acetylglucosaminyltransferase III (GnT-III, MGAT3) and N-acetylglucosaminyltransferase V (GnT-V, MGAT5) competitively modify E-cadherin N-glycans, adding bisecting GlcNAc structures and β1,6-GlcNAc branches, respectively. Wild-type E-cadherin positively regulates the metastasis suppressor MGAT3 gene, resulting in increased GnT-III expression and bisecting GlcNAc N-glycans addition to the plasma membrane-bound protein (225). Conversely, the addition of β1,6-GlcNAc branches by GnT-V, specially at Asn-554, drives E-cadherin translocation to the cytoplasm, alters cis-dimer formation and molecular assembly, and drives instability of the adherens junctions. Furthermore, preventing Asn-554 N-glycosylation, either by a mutation or by silencing GnT-V, resulted in a protective effect on E-cadherin, precluding its functional dysregulation and contributing to tumor suppression (226, 227). Another study demonstrated a novel pathway of GnT-V-mediated metastasis via the addition of β1,6-GlcNAc branches to matriptase, thereby stabilizing it and activating invasion effectors as urokinase-type plasminogen activator and hepatocyte growth factor (HGF) (228). Overall, these findings suggest that aberrant N-linked β1,6- GlcNAc branching occurring during oncogenesis can lessen cell-cell adhesion, contributing to increased cellular motility and invasiveness (Figure 2G). However, some glycosydic modifications can promote tumor cell adhesion and still favor tumor progression. For instance, tumor cells also overexpress SLea/x antigens, which are specific ligands for E- and P-selectins upregulated in activated endothelial cells. Selectins and SLea/x interactions are key regulators of the metastatic cascade by promoting the recruitment of malignant cells to vessels, rolling of tumor cells on the endothelial surface, and arrest of CTCs in distant locations (229–231) (Figure 2G). Besides the establishment of metastatic colonies, these ligands also mediate tumor growth, invasion, angiogenesis, and inflammation in numerous other tumor types (232–236). In addition, slightly altered forms of these antigens also have important biological features. Namely, the addition of a sulfate group at the sixth position of GlcNAc generates 6-sulfo-sLeX, which is considered the physiologic ligand for L-selectin (237) but also E-selectin in bladder cancer (238). Herein, it has a dual role by promoting tumor cell adhesion to vascular endothelial cells, while favoring lymphocyte recruitment to enhance anti-tumor immune responses (238). In agreement with these observations, Lex-positive cell lines from invasive bladder tumors with metastatic potential show high levels of alpha1,3-fucosyltransferase VI (FT-VI) and FT-VII, two enzymes involved in SLex synthesis, and display E-selectin dependent adhesion (232).
Glycosyltransferases may also play a key role in mediating cancer cell metastization. Namely, the sialyltransferase ST6GalNAcII was identified as a novel metastasis suppressor, while ST6GalNAcV and N-Acetylgalactosaminyltransferase GalNT9 identify metastatic potential in breast cancer (239–241).
In summary, cancer-associated glycosylation changes and stromal cells aid tumor cell invasion, distant organ colonization, and metastasis by supplying pro-metastatic factors, compromising vasculature integrity and the stromal barrier to tumor cell migration, promoting EMT and by tethering tumor cells to improve colonization at distant sites. Concomitantly, the highly regulated balance between loss of adhesive properties and the ability to anchor at metastatic sites defines the metastatic potential of tumor cells.
Tumor-Promoting Inflammation
Tumor-associated stromal cells have been found to secrete a variety of pro-inflammatory cytokines, chemokines and matrix-remodeling enzymes favoring the establishment of immune cell infiltrates (242, 243). Particularly, CAFs and mature adipocytes promote sustained inflammation by producing large amounts of pro-inflammatory IL-6, IL-1β, TNF-alpha, and CXCL1 to drive chemoattraction of monocytic immune cells (244), while favoring tumor growth and metastasis (245–250). Another pivotal microenvironmental factor driving cancer-associated inflammation is hypoxia, which is essential for granulocytes and monocytes/macrophages infiltration and activation in vivo in a HIF-1α-dependent manner (251).
Glycome alterations also decisively contribute to the establishment and maintenance of tumor-promoting inflammation. Namely, E-, P-, and L-Selectins interactions with SLea/x not only control the establishment of metastatic cancer cells colonies but also the recruitment of circulating lymphocytes into peripheral lymph nodes and inflamed tissues (238, 252, 253) (Figure 2H). Moreover, several inflammatory mediators are regulated by its glycosylation state. Namely, NF-κB is activated by O-GlcNAcylation at Ser350 of its c-Rel subunit (254), while the proinflammatory cytokine Cyclooxygenase-2 (COX-2) turnover depends on Asn570 glycosylation, negatively affecting the efficacy of certain COX-2 inhibitors (255, 256). Furthermore, recent studies have described that non-human N-glycolyl-neuraminic acid (Neu5Gc) can be incorporated into cell surface glycans instead of N-acetyl-neuraminic acid (Neu5Ac), leading to autoimmune systemic inflammation associated with cancer initiation and progression (257–259).
Importantly, in the same way glycans govern inflammation, the inflammatory tumor microenvironment is also able to induce changes in tumor cells glycosylation. For instance, pancreatic and gastric carcinomas are characterized by an abundant stroma containing several pro-inflammatory cytokines, as IL-1β and IL-6, which regulate the expression of biosynthetic glycosyltransferases to increase the expression sialylated antigens as SLea/x (260, 261). Furthermore, the extracellular matrix proteoglycan versican has been shown to promote bladder cancer-derived lung metastasis through enhanced tumor cell migration and creation of an inflammatory environment involving macrophages and pro-tumor CCL2/CCR2 signaling axis (262, 263), providing another the involvement of glycoconjugates in macrophage-mediated inflammation.
These findings highlight the relevance of tumor stromal cells, glycans, and glycoconjugates as mediators of tumor-promoting inflammation by providing pro-inflammatory factors and allowing the recruitment of circulating lymphocytes into tumor sites.
Immune Escape
Several stromal components of the tumor microenvironment aid tumor cell immune scape, either by recruiting immunosuppressive immune cells or by driving the acquisition of tolerogenic phenotypes. In this context, tumor-infiltrating immune cells frequently develop immunosuppressive activities, differentiating into regulatory T cells (Tregs), immature monocytes, and alternatively activated macrophages, mast cells, neutrophils, dendritic cells (DC), and T helper 2 (TH2)-CD4+ T cells, all of which producing a multitude of factors aiding tumor growth and survival (264). Specifically, endothelial cells lining the tumor vasculature can suppress T cell activity, target them for destruction, and block them from entering the tumor through the deregulation of adhesion molecules (265). Moreover, the CAF secretome can also shape T cell-dependent antitumor immune responses by negatively affecting DCs, myeloid-derived suppressor cells, TH17, and CD8+ T cells functions. Activated fibroblasts can also drive the switch of CD4+ T lymphocytes from a TH1 to a TH2 phenotype, while expressing some ligands of immune checkpoint receptors (266). CAF-derived proteoglycans, as decorin, further suppress immunomodulatory genes in triple-negative orthotopic breast carcinoma xenografts, including Siglec (Sialic acid binding Ig-like lectin), Lipg (IFNγ inducible GTPase), and Il1b (Interleukin 1β) (213). These findings suggest that targeting CAFs or their secretome may probably reduce immune effector cell dysfunctions as well as decrease the recruitment of immunosuppressive cells. Other ECM molecules, as HA, are known to determine the trafficking of tumor-associated macrophages (TAM) through tumor stromal areas. In line with this, HA deficiency in tumor stroma impairs not only macrophage trafficking but also tumor angiogenesis and lymphangiogenesis, ultimately compromising immune cells access to tumor sites and aiding immune scape (267). Furthermore, recent studies in myeloma tumors have demonstrated the immunomodulatory roles of the ECM proteoglycan versican proteolytic processing. In this context, the interplay between stromal cells and myeloid cells generates versikine, a novel bioactive damage-associated molecular pattern that may facilitate immune sensing of myeloma tumors and modulate the tolerogenic consequences of intact versican accumulation (268).
As described in previous sections, advanced stage tumors are frequently characterized by profound deregulations in glycosylation pathways, resulting in the presentation of aberrant structures at the cell surface. Importantly, these structures only render cancer cells mildly antigenic and rarely immunogenic (269). This may occur because most cancer-associated structures have an embryonic origin or are mildly expressed in healthy tissues, allowing them to be perceived as “self” by immune system effector cells (270). Furthermore, specialized B lymphocytes producing high-affinity antibodies against these structures might even be eliminated during development (271). However, glycans play a key role in the regulation of various aspects of immune response, ultimately enabling immune suppression by interacting with lectin receptors in immune cells. For instance, fucosylated blood group related Lewis antigens interact with C-type lectin DC-SIGN (dendritic cell-specific ICAM-3-grabbing non-integrin; also known as CD209) on macrophages and DC to upregulate the anti-inflammatory cytokines IL-10 and IL-27. This ultimately induces TH2, T follicular helper (TFH) or Treg cells, highlighting the immune suppressive nature of Lewis antigens (272, 273). Similarly to fucosylation, enhanced tumor sialylation often culminates in immune suppression and anti-inflammatory microenvironments. Accordingly, the presence of sialylated structures on melanoma cells impedes T cell mediated anti-tumor responses while promoting tumor-associated Treg cells and decreased NK cell activity (274) (Figure 2I). Moreover, sialoglycans interact with sialic acid-binding immunoglobulin-like lectins (SIGLECs) to induce an antigen-specific tolerogenic programming, enhancing Treg cells and reducing the generation and propagation of inflammatory T cells (275). For instance, macrophage associated Siglec-15 preferentially binds the STn antigen in myeloid tumor cells, resulting in increased TGF-β secretion into the tumor microenvironment and tumor progression (276). Moreover, in bladder cancer, STn expression has led to impaired DC maturation while significantly reducing the production of Th1-inducing cytokines IL-12 and TNF-α (277) (Figure 2I). Consistent with this tolerogenic profile, T cells primed by DCs pulsed with STn-expressing glycoproteins displayed a FoxP3(high) IFN-γ(low) phenotype and little capacity to trigger protective anti-tumor T cell responses (277). More importantly, blocking STn-MUC1 and CD44 glycoforms partially reverted DC maturation, suggesting that targeting STn-expressing glycoproteins may allow circumventing tumor-induced tolerogenic mechanisms. Similarly, sialylation of the T antigen in MUC1 on breast cancer cells creates the MUC1–ST antigen which engages Singlec-9 on tumor-associated macrophages to initiate inhibitory immune signaling through the activation of the MAPK/ERK pathway (278). In line with this, sialylated ligands of singlec-7 and−9 are expressed on cancer cells of different histological types and interactions between these lectin receptors and its ligands influence NK cell-dependent tumor immunosurveillance (279). Moreover, hypersialylation of tumor ligands for NKG2D receptors, expressed by NK cells, NK1.1+ T cells, γδ T cells, activated CD8+αβ T cells and macrophages, is thought to repulse their interaction via highly negative charge repulsions, hampering immune response (280, 281). Tumor-derived sialoglycans also inhibit CD8+ T cell cytotoxicity by interfering with lytic granule trafficking and exocytosis in response to TCR engagement (282). Thus, hypersialylation often observed on tumor cells may ultimately be amongst the mechanisms by which tumors evade immune system recognition (30, 70, 216, 283). Also, C2GnT-expressing bladder tumor cells express heavily core 2 O-glycosylated MUC1 which interacts with Gal-3 to attenuate the interaction of tumor cells with NK cells, allowing tumor cells to survive longer in host blood circulation and potentially metastasize (284). Given these insights, sialylated and fucosylated antigens contribute to create an immunosuppressive microenvironment toward tumor cell immune escape. Furthermore, the structure and function of well-known immune checkpoint molecules as PD-L1 can be stabilized by N-glycosylation, reducing its proteasomal degradation and consequently enhancing its immunosuppressive activity over T-cells (285). These findings highlight the disseminated role of glucans and glycoconjugates in tumor cell immune scape.
In summary, the tumor microenvironment increasingly becomes more immunosuppressive, resulting in tumor cell survival and metastasis. Concomitantly, tumor cells glycosylation promotes immune scape by being simple and “self”-like, by inducing tolerogenic immune cell phenotypes, and by effectively shielding tumor cells from effector immune cells, culminating in tumor progression.
Significance of Glycosignatures for Personalized Medicine
The previous sections have highlighted that changes in glycans and glycoconjugates drive several biological processes in tumor cells, culminating in the acquisition of cancer hallmarks and increasingly aggressive disease. Glycosylation changes reflect not only the genomic, transcriptomic, proteomic and metabolomic state of cells but also its external microenvironment, making glycosignatures highly context-specific and attractive targets for personalized medicine affecting tumor and stromal cells. At a systemic level, glycosignatures provide a global reflection on an individual's health/disease status and can function as predictive indicators for treatment success. In this context, several serological markers have emerged, with several FDA-approved cancer glycobiomarkers currently used in clinical practice recently revised by kirwan et al. (286). To circumvent relatively low specificity and sensitivity issues, more comprehensive approaches propose combinations of glycobiomarkers achieving remarkable sensitivity and specificity values (287). Another strategy to improve specificity consists in narrowing the cancer cell proteome to clinically relevant glycoforms. Showcasing this aspect, a recent targeted investigation of the bladder cancer glycoproteome highlighted that specific MUC16 glycoforms (CA125 antigen) could be used to define subsets of chemoresistant patients, whereas no associations could be found based solely on the presence of the protein (30). Moreover, the field of liquid biopsies is rapidly evolving from classical approaches, focusing on a single or few protein biomarkers, toward multiplex settings that will likely improve on these preliminary findings (Figure 3). The detection of minor amounts of circulating tumor nucleic acids, exosomes, circulating tumor cells (CTC) and stromal components, which decisively contribute to the pre-metastatic and metastatic niches, will pave the way for improving the management of advanced stage patients. In this context, deeper insights on their molecular nature may provide the necessary means for real-time disease monitoring and early intervention, guiding therapeutic decision and, more importantly, designing novel therapeutics (Figure 3). Accordingly, explorative studies have demonstrated that exosomes, responsible by pre-metastatic signaling, present distinct glycosylation patterns (288, 289). Furthermore, pioneer work using a recently developed microfluidics device has demonstrated that over 90% of bladder cancer CTC yield the STn antigen (221). More importantly, the STn antigen was not detected in blood cells from healthy individuals, reinforcing its cancer-associated nature. Downstream molecular analysis confirmed the basal nature of STn-positive CTC in molecular mimicry of the primary tumor and corresponding metastasis (221). Therefore, the STn may allow targeting bladder CTC, which has been a challenging enterprise given the scarce knowledge about their molecular nature.
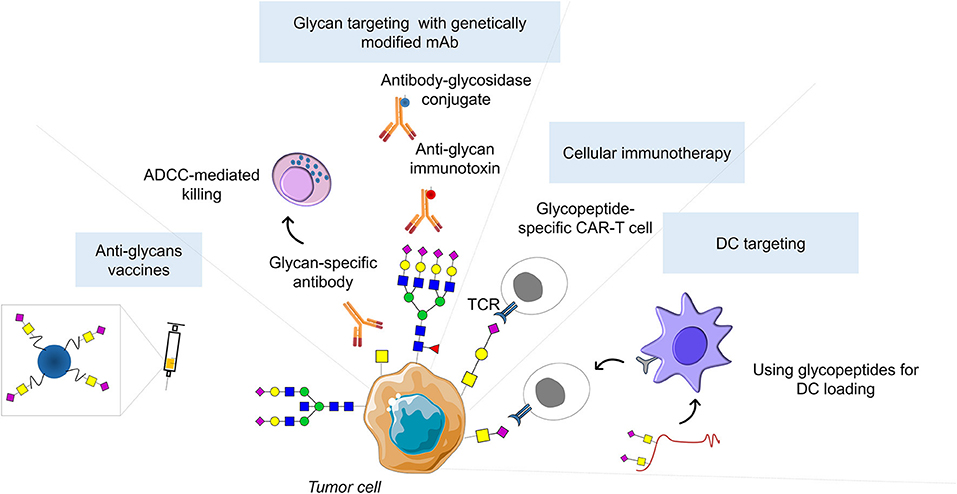
Figure 3. Glycan-based therapeutic strategies. Successful clinical implementation of glycan-based therapeutic strategies could include inhibitors of glycosyltransferases catalytic activity, as well as theragnostic antibodies against cancer glycoepitopes capable of cancer detection, antibody-dependent cellular cytotoxicity induction, and abrogation of immunotolerance generated by cancer-associated glycoconjugates. Moreover, glycan-based antibodies may be used to guide emerging nanotherapies or serve has basis for developing genetically modified T cells expressing chimeric antigen receptors (CART-T). In addition, glycopeptides can be used for in vivo targeting of dendritic cells (DCs) to induce tumor-specific CD4+ and CD8+ T cells. Finally, glycan antigens coupled to T-cell peptide epitopes or immunostimulant epitopes can form fully synthetic multicomponent glycoconjugate vaccines able to circumvent cancer immunotolerance.
Despite these promising advances, current diagnostic strategies are based on measuring protein marker concentrations, disregarding its glycosylation status, even though it might provide key information to improve diagnosis and stratify patients. This might be due to the lack of user-friendly tools allowing health care technicians to obtain this information in sufficient specificity and sensitivity within the standard capacities of a clinical laboratory. Moreover, the glyco-heterogeneity of protein markers, arising from multiple glycosylation sites and glycosylation patterns, might further hamper selectivity. As such, the profound knowledge of cancer-specific glycan signatures and glycosites, as well as its status within a healthy population represent the first crucial steps toward including glycosylation in the diagnostic process. From the bench side, current glycobiology rationale is mostly built on immunoaffinity-based studies addressing conventionally accepted glycan-biomarkers and involving small and often biased patient cohorts. Heterogeneous protocols, including different sample processing and detection methods, as well as the lack of endpoint standardization have also constituted major drawbacks. Moreover, most studies fail to provide complementary functional assays capable of pinpointing clinically relevant glycobiomarkers. These aspects are often further aggravated by the lack of untargeted approaches capable of broadening our understanding on the glycome and glycoproteome. Moreover, few efforts were undertaken to incorporate glycans in broad biomarker panels of different molecular natures, envisaging highly sensitive and specific detection methods. Facing these challenges, significant efforts are ongoing to standardize glycomics and glycoproteomics protocols and implement robust high-throughput mass spectrometry-based glycoanalytical platforms (290, 291). As such, it is now possible to extract significant structural information from minute amounts of clinical samples (nanomolar-fentomolar range), including from challenging starting materials such as formalin-fixed paraffin-embedded (FFPE) tissues available in many hospital archives (30, 292), which will enable large scale retrospective analysis of well characterized clinical samples. Moreover, advances in MALDI Imaging Mass Spectrometry has allowed obtaining structural information from glycans with significant spatial resolution (293). Important bioinformatics tools and databases are already available and novel improvements are emerging for supporting glycans and glycopeptide mass spectrometry data interpretation, which is a critical matter facing big datasets (294). Altogether, the technological set-up and structural knowledge envisaging the engagement in multicenter randomized glycan-based trials have been overcome; nevertheless, a more ambitious focus should be set on integrative panomics applications (295). This knowledge will foster the development of glycan-based therapeutic strategies and novel immunotherapeutics, including inhibitors of glycosyltransferases catalytic activity (296) and theragnostic antibodies against cancer-specific glycoepitopes. The later should be capable of inducing antibody-dependent cellular cytotoxicity and/or overcoming the immunotolerance generated by cancer-associated glycoconjugates and microenvironmental cues (297). Moreover, glycan-based antibodies may be used to guide emerging nanotherapies (298, 299) or serve has basis for developing genetically modified T cells expressing chimeric antigen receptors (CAR-T) (300), while allowing cancer detection and identification of patients better-served by these therapies. In addition, blocking tumor-associated glycan–lectin interactions could prevent the activation of inhibitory immune receptors toward more efficient immunotherapies. Regarding personalized immunotherapies, in recent years, the targeting of DCs has emerged as an interesting approach for the induction of antitumor immunity. Namely, glycopeptides targeting DC-SIGN in DCs are easily internalized and cross-presented to stimulate tumor-specific CD4+ and CD8+ T cell responses. Finally, anticancer multicomponent glycoconjugate vaccines, based on glycan antigens coupled to T-cell peptide epitopes or immunostimulant epitopes, have been demonstrated effective in circumventing cancer immunotolerance (301, 302), providing an appealing option for the much-awaited development of new glycan-based therapeutic agents.
In summary, analytical hurdles related with sample preparation, data acquisition and automated analysis that can also be handled by non-glycobiologists represent key steps to overcome to introduce glycomics and glycoproteomics as routine clinical parameters. To achieve this goal, the development of new and clinic-friendly techniques, as well as glycobiology-focused bioinformatics tools open new avenues to predict the tumor glyco-code. In addition, stratification and large-scale validation of potential diagnostic targets will also be indispensable to successfully translate promising research results into solid clinical tests. In a distant future, an inclusive approach combining the increasing amount of glycomics and glycoproteomics data with patient's genomics, transcriptomics, proteomics, and metabolomics will have a major impact on the unraveling of novel targets and strategies for early diagnosis, prognosis, patient stratification and improved cancer management.
Concluding Remarks
As thoroughly described in the previous sections, tumor stromal cells and ECM components have a preliminary regulatory role in the acquisition of hallmark capabilities, mostly by supplying the soluble factors that drive adaptation or shielding tumor cells from external stress. Glycosylation ads a second level of regulation by governing structural alterations in major receptors, by modifying soluble factors and/or by modulating intracellular kinase cascades (Figure 4). Showcasing this, proliferative signaling is sustained by stromal cells that supply mitogenic factors, while glycosylation promotes growth factor receptor activation and positively regulates intracellular kinases pathways. Besides sustained growth, tumor cells must circumvent programmed cell death to ensure cancer progression. Envisaging this, stromal cells and ECM remodeling provide diffusible paracrine survival factors and non-diffusible survival signals, while offering a physical barrier against pro-apoptotic factors such as chemotherapy. In line with this, glycosylation determines the sensitivity of death receptors to their ligands and drives the initiation of pro-survival cascades, while altering transcription factor activity. Concomitantly, sustained proliferation and programmed cell death evasion culminate in highly energy demanding tumors that establish symbiotic relationships with stromal cells that activate complementary metabolic pathways to buffer and recycle tumor-derived metabolites. Moreover, to sustain growth and survival in the face of hypoxia, HIF-1α strongly regulates glucose metabolism throughout the several biosynthesis pathways, culminating in altered glycosylation precursor expression as well as increased sialylation and O-GlcNAcylation toward more aggressive clones. Simultaneously, tumor cells evade growth suppression by abrogating the suppressive role of adhesion complexes with the ECM, mostly by the action of stromal-derived proteolytic enzymes. At the same time, the two canonical suppressors of proliferation p53 and RB are negatively regulated through O-GlcNAc modifications. All the above-mentioned events are largely driven by the genomic instability of cancer cells, culminating in advantageous random mutations. This variability thrives much as a consequence of the DNA damage promoted by the mutagenic/oxidative microenvironment indorsed by stromal cells. Also, hypoxia alters the transcription and translation of several DNA damage response and repair genes. In turn, glycosylation modulates DNA damage response pathway components, reinforcing the genomic instability of tumor cells. Interestingly, both the tumor microenvironment and glycosylation have little to do with the replicative immortality of tumor cells, their contribution is mainly based on the indirect regulation of the transcription factor c-MYC and kinase cascades. Importantly, to sustain proliferation and the energetic demands of ever-growing tumors, a pro-angiogenic environment must be established. As such, to ensure neovascularization, stromal cells supply pro-angiogenic factors and upregulate multiple angiogenic pathways culminating in the maturation and survival of endothelial cells. In turn, angiogenic glycans and glycoconjugates alter the affinity of angiogenic factor receptors for their ligands toward a pro-angiogenic phenotype of EC. Advanced stage tumors frequently progress to invasion and metastasis, which is facilitated by the compromised vascular and stromal barriers to tumor cell migration. Moreover, stromal cells can promote EMT in tumor cells and tether these cells to improve colonization at distant sites. Concomitantly, glycosylation changes in tumor cells physically disrupt cell adhesion by upregulating sialylated antigens and N-linked β1,6-GlcNAc branches, contributing to increased cellular motility and invasiveness. On the other hand, glycosylation can promote adhesion of tumor cells and still favor the establishment of metastatic colonies. Namely, tumor cells overexpressing SLea/x are able to roll on the endothelial surface and extravasate into circulation, while arresting its movement in distant locations by interacting with selectins expressed by endothelial cells. Some glycosyltransferases expression also defines the metastatic potential of tumor cells, acting as metastasis suppressors or enablers.
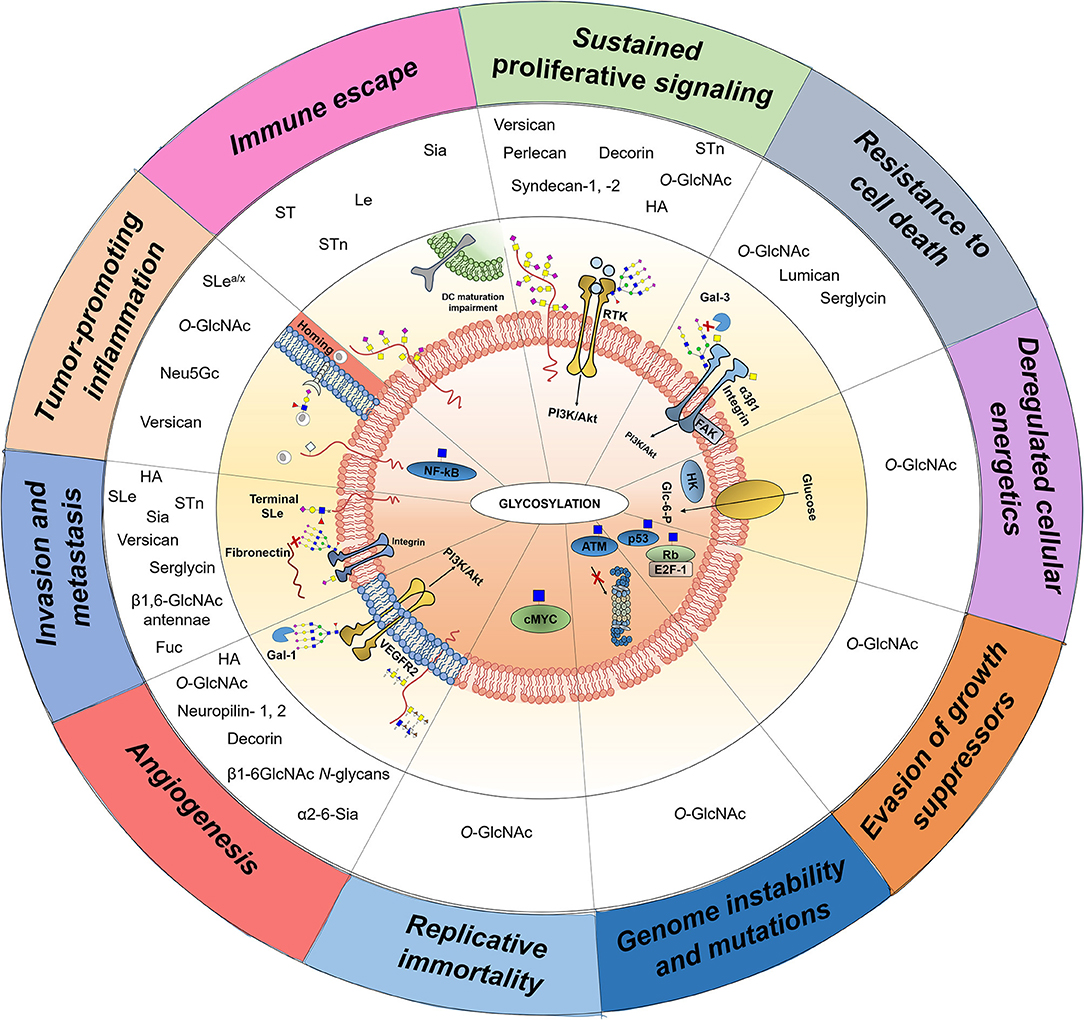
Figure 4. Transversal nature of glycans, glycoproteins, glycan-binding proteins, and proteoglycans throughout the 10 currently accepted cancer hallmarks. Aberrantly expressed glycans (O-GlcNAc, Le, SLe, Neu5Gc, Sialic acids (Sia), fucose residues (Fuc), ST, STn), glycoproteins (integrin α3β1, VEGFR2), and proteoglycans (Perlecan, Decorin, Neuropilin-1,-2, Syndecan-1,−2, Hyaluronic acid fragments (HA), Versican, Lumican, Serglycin) are mechanistically involved in cancer hallmarks acquisition. The illustration highlights the most common glycosylation modifications throughout the cancer hallmarks, transmitting the empiric notion that a great number of glycosylation aberrations mostly contribute to disease dissemination through increased angiogenesis and potentiation of invasion and metastasis. Moreover, the post-translational modification β-O-N-acetyl-d-glucosamine (O-GlcNAc) emerges as a key regulator of cellular activities through the modulation of signal transduction and protein stabilization. In conclusion, glycans and glycoconjugates are not bystanders to malignant transformation but major players, making then attractive targets to drive molecular-based clinical intervention.
In the meantime, tumor-associated stromal cells contribute to tumor-promoting inflammation by supplying several pro-inflammatory cytokines and chemokines, ultimately driving tumor growth, neovascularization, immune cell recruitment, and glycosyltransferases expression. Furthermore, glycosylation changes not only contribute to the recruitment of circulating lymphocytes into peripheral lymph nodes and inflamed tissues but also regulate the activity of several inflammatory mediators and the polarization of immune cells into immunosuppressor phenotypes. In line with this, the tumor microenvironment increasingly becomes populated with immunosuppressive immune cells. Concomitantly, tumor cells glycosylation, mostly characterized by hypersialylation, promotes immune scape by being simple and “self”-like, by inducing tolerogenic immune cell phenotypes, and by effectively shielding tumor cells from effector immune cells, culminating in tumor progression.
Based on these insights, glycosylation changes reflect not only the genomic, transcriptomic, proteomic, and metabolomic state of cells but also its external microenvironment, making glycosignatures highly context-specific and attractive targets for personalized medicine. Several evidences support the existence of a unique repertoire of glycans associated with disease progression and dissemination, decisively reflecting on virtually all cancer hallmarks (Figure 4). Changes in O-GlcNAcylation is the most common glycosylation modification throughout cancer hallmarks, providing a dynamic but highly regulated sensor driving protein stabilization and signal transduction. Sialic acids and, particularly sialylated short-chain O-glycans are also amongst the most common structures driving invasion and immune escape, clearly marking more aggressive tumor cell phenotypes. Moreover, the major bulk of glycosylation modifications accompanying malignant transformation seem to contribute to disease dissemination through increased angiogenesis and potentiation of invasion and metastasis. Notwithstanding, little is known about glycosylation contribution to key aspects of neoplastic transformation as the acquisition of genomic instability and replicative immortality, opening an avenue for novel research (Figure 4). The sweet side to this sour end resides on the possibility of exploring the extracellular nature of glycans for targeting tumor and stromal cells using more effective non-invasive tools. As such, we intend to reinforce the need to concentrate efforts to incorporate glycans in broad biomarker panels of different molecular natures, envisaging highly sensitive and specific detection methods for disease monitoring and early intervention. Moreover, by integrating microenvironmental information, glycosignatures will most likely provide the necessary key for designing highly specific cancer ligands envisaging theragnostic applications; thereby allowing guiding therapeutic decision and, more importantly, designing novel therapeutics. Notwithstanding, significant room lays beyond targeted approaches, specially facing the recent advances in glycomics and glycoproteomics. Therefore, it is now possible to engage on a comprehensive study of the glycome and glycoproteome envisaging the necessary glycobiology landscape for intervention. Of note, selectin and galectin antagonists, including glycomimetic compounds, antibodies, aptamers, and peptides are currently in FDA clinical trials and near-clinical trials for the treatment of blood-related cancers and solid tumors metastasis (303). Moreover, the high sensitivity and resolution of new generation mass-spectrometers will allow obtaining structural information almost to a single-cell level, enabling the analysis of exosomes, CTC, and stromal components, which will be crucial for addressing metastatic disease. Overall, we believe that the necessary context has been created to foster more in-depth studies on the glycobiology of tumors and its microenvironment envisaging molecular-based precision medicine and improved patient care.
Author Contributions
AP and JF wrote the manuscript. AP, MR-S, and RA produced the artwork. MR-S, RA, and LS revised it.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the Portuguese Foundation for Science and Technology (FCT) for the human resources grants: PhD grant SFRH/BD/111242/2015 (AP), and FCT auxiliary researcher grant CEECIND/03186/2017 (JF). FCT is co-financed by European Social Fund (ESF) under Human Potential Operation Programme (POPH) from National Strategic Reference Framework (NSRF). The authors also acknowledge the Portuguese Oncology Institute of Porto Research Centre (CI-IPOP-29-2014; CI-IPOP-58-2015), the PhD Program in Biomedical Sciences of ICBAS-University of Porto, and the Early stage cancer treatment, driven by context of molecular imaging (ESTIMA)framework (NORTE-01-0145-FEDER-000027). The authors were also supported by the CANCER project (NORTE-01-0145-FEDER-000029) co-funded through the NORTE-45-2015-02.
References
1. Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci USA. (2008) 105:8256–61. doi: 10.1073/pnas.0801340105
2. Vagin O, Kraut JA, Sachs G. Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. Am J Physiol Renal Physiol. (2009) 296:F459–69. doi: 10.1152/ajprenal.90340.2008
3. Ono M, Hakomori S. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J. (2004) 20:71–8. doi: 10.1023/B:GLYC.0000018019.22070.7d
4. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. (2006) 126:855–67. doi: 10.1016/j.cell.2006.08.019
5. Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. (2015) 10:473–510. doi: 10.1146/annurev-pathol-012414-040438
6. Leney AC, El Atmioui D, Wu W, Ovaa H, Heck AJR. Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc Natl Acad Sci USA. (2017) 114:E7255–61. doi: 10.1073/pnas.1620529114
7. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. (2015) 15:540–55. doi: 10.1038/nrc3982
8. Azevedo R, Peixoto A, Gaiteiro C, Fernandes E, Neves M, Lima L, et al. Over forty years of bladder cancer glycobiology: where do glycans stand facing precision oncology? Oncotarget. (2017) 2017:19433. doi: 10.18632/oncotarget.19433
9. Bennun SV, Yarema KJ, Betenbaugh MJ, Krambeck FJ. Integration of the transcriptome and glycome for identification of glycan cell signatures. PLoS Comput Biol. (2013) 9:e1002813. doi: 10.1371/journal.pcbi.1002813
10. Jozwiak P, Forma E, Brys M, Krzeslak A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front Endocrinol. (2014) 5:145. doi: 10.3389/fendo.2014.00145
11. Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. (2002) 1:845–67. doi: 10.1074/mcp.A300001-MCP200
12. Takeuchi M, Amano M, Kitamura H, Tsukamoto T, Masumori N. N- and O-glycome analysis of serum and urine from bladder cancer patients using a high-throughput glycoblotting method. J Glycomics Lipidomics. (2013) 3:108. doi: 10.4172/2153-0637.1000108
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
14. Weiser MM, Podolsky DK, Iselbacher KJ. Cancer-associated isoenzyme of serum galactosyltransferase. Proc Natl Acad Sci USA. (1976) 73:1319–22.
15. Ip C, Dao T. Alterations in serum glycosyltransferases and 5'-nucleotidase in breast cancer patients. Cancer Res. (1978) 38:723–8.
16. Lee-Sundlov MM, Ashline DJ, Hanneman AJ, Grozovsky R, Reinhold VN, Hoffmeister KM, et al. Circulating blood and platelets supply glycosyltransferases that enable extrinsic extracellular glycosylation. Glycobiology. (2017) 27:188–98. doi: 10.1093/glycob/cww108
17. Freeze HH, Haltiwanger RS. Other classes of ER/golgi-derived glycans. In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al. editors. Essentials of Glycobiology. New York, NY: Cold Spring Harbor (2009).
18. Praissman JL, Wells L. Mammalian O-mannosylation pathway: glycan structures, enzymes, and protein substrates. Biochemistry. (2014) 53:3066–78. doi: 10.1021/bi500153y
19. Vasudevan D, Haltiwanger RS. Novel roles for O-linked glycans in protein folding. Glycoconj J. (2014) 31:417–26. doi: 10.1007/s10719-014-9556-4
20. Vosseller K, Sakabe K, Wells L, Hart GW. Diverse regulation of protein function by O-GlcNAc: a nuclear and cytoplasmic carbohydrate post-translational modification. Curr Opin Chem Biol. (2002) 6:851–7. doi: 10.1016/S1367-5931(02)00384-8
21. Slawson C, Hart GW. Dynamic interplay between O-GlcNAc and O-phosphate: the sweet side of protein regulation. Curr Opin Struct Biol. (2003) 13:631–6. doi: 10.1016/j.sbi.2003.08.003
22. Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. (2010) 584:2526–38. doi: 10.1016/j.febslet.2010.04.044
23. Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics. (2014) 11:8. doi: 10.1186/1559-0275-11-8
24. Hart GW, Kreppel LK, Comer FI, Arnold CS, Snow DM, Ye Z, et al. O-GlcNAcylation of key nuclear and cytoskeletal proteins: reciprocity with O-phosphorylation and putative roles in protein multimerization. Glycobiology. (1996) 6:711–6.
25. Comer FI, Hart GW. O-Glycosylation of nuclear and cytosolic proteins. Dynamic interplay between O-GlcNAc and O-phosphate. J Biol Chem. (2000) 275:29179–82. doi: 10.1074/jbc.R000010200
26. Bektas M, Rubenstein DS. The role of intracellular protein O-glycosylation in cell adhesion and disease. J Biomed Res. (2011) 25:227–36. doi: 10.1016/S1674-8301(11)60031-6
27. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. (2017) 18:452–65. doi: 10.1038/nrm.2017.22
28. Hakomori S, Kannagi R. Glycosphingolipids as tumor-associated and differentiation markers. J Natl Cancer Inst. (1983) 71:231–51.
29. Peixoto A, Fernandes E, Gaiteiro C, Lima L, Azevedo R, Soares J, et al. Hypoxia enhances the malignant nature of bladder cancer cells and concomitantly antagonizes protein O-glycosylation extension. Oncotarget. (2016) 7:63138–57. doi: 10.18632/oncotarget.11257
30. Cotton S, Azevedo R, Gaiteiro C, Ferreira D, Lima L, Peixoto A, et al. Targeted O-glycoproteomics explored increased sialylation and identified MUC16 as a poor prognosis biomarker in advanced-stage bladder tumours. Mol Oncol. (2017) 11:895–912. doi: 10.1002/1878-0261.12035
31. Kannagi R, Yin J, Miyazaki K, Izawa M. Current relevance of incomplete synthesis and neo-synthesis for cancer-associated alteration of carbohydrate determinants–Hakomori's concepts revisited. Biochim Biophys Acta. (2008) 1780:525–31. doi: 10.1016/j.bbagen.2007.10.007
32. Limas C, Lange PH. Lewis antigens in normal and neoplastic urothelium. Am J Pathol. (1985) 121:176–83.
33. Orntoft TF, Nielsen MJ, Wolf H, Olsen S, Clausen H, Hakomori S, et al. Blood group ABO and Lewis antigen expression during neoplastic progression of human urothelium. Immunohistochemical study of type 1 chain structures. Cancer. (1987) 60:2641–8.
34. Fujii Y, Yoshida M, Chien LJ, Kihara K, Kageyama Y, Yasukochi Y, et al. Significance of carbohydrate antigen sialyl-Lewis X, sialyl-Lewis A, and possible unknown ligands to adhesion of human urothelial cancer cells to activated endothelium. Urol Int. (2000) 64:129–33. doi: 10.1159/000030512
35. He J, Baum LG. Galectin interactions with extracellular matrix and effects on cellular function. Methods Enzymol. (2006) 417:247–56. doi: 10.1016/S0076-6879(06)17017-2
36. Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. (2015) 42:11–55. doi: 10.1016/j.matbio.2015.02.003
37. Ferreira JA, Magalhaes A, Gomes J, Peixoto A, Gaiteiro C, Fernandes E, et al. Protein glycosylation in gastric and colorectal cancers: toward cancer detection and targeted therapeutics. Cancer Lett. (2017) 387:32–45. doi: 10.1016/j.canlet.2016.01.044
38. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. (2005) 5:526–42. doi: 10.1038/nrc1649
39. Munkley J, Elliott DJ. Hallmarks of glycosylation in cancer. Oncotarget. (2016) 7:35478–89. doi: 10.18632/oncotarget.8155
40. Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. (2008) 8:874–87. doi: 10.1038/nri2417
41. Varki A, Gagneux P. Biological functions of glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. editors. Essentials of Glycobiology. New York, NY: Cold Spring Harbor (2015). p. 77–88.
42. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. (2005) 7:211–7. doi: 10.1016/j.ccr.2005.02.013
43. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. (2010) 10:138–46. doi: 10.1038/nrc2791
44. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. (2011) 3:12. doi: 10.1101/cshperspect.a005058
45. Maeda T, Alexander CM, Friedl A. Induction of syndecan-1 expression in stromal fibroblasts promotes proliferation of human breast cancer cells. Cancer Res. (2004) 64:612–21. doi: 10.1158/0008-5472.CAN-03-2439
46. Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. (2007) 282:14906–15. doi: 10.1074/jbc.M611739200
47. Du WW, Yang BB, Shatseva TA, Yang BL, Deng Z, Shan SW, et al. Versican G3 promotes mouse mammary tumor cell growth, migration, and metastasis by influencing EGF receptor signaling. PLoS ONE. (2010) 5:e13828. doi: 10.1371/journal.pone.0013828
48. Yang Y, Yaccoby S, Liu W, Langford JK, Pumphrey CY, Theus A, et al. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. (2002) 100:610–7. doi: 10.1182/blood.V100.2.610
49. Park H, Kim Y, Lim Y, Han I, Oh ES. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J Biol Chem. (2002) 277:29730–6. doi: 10.1074/jbc.M202435200
50. Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell. (1994) 79:1005–13.
51. Willenberg A, Saalbach A, Simon JC, Anderegg U. Melanoma cells control HA synthesis in peritumoral fibroblasts via PDGF-AA and PDGF-CC: impact on melanoma cell proliferation. J Invest Dermatol. (2012) 132:385–93. doi: 10.1038/jid.2011.325
52. Csordas G, Santra M, Reed CC, Eichstetter I, McQuillan DJ, Gross D, et al. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J Biol Chem. (2000) 275:32879–87. doi: 10.1074/jbc.M005609200
53. Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. (1990) 346:281–4. doi: 10.1038/346281a0
54. Santra M, Eichstetter I, Iozzo RV. An anti-oncogenic role for decorin. Down-regulation of ErbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells. J Biol Chem. (2000) 275:35153–61. doi: 10.1074/jbc.M006821200
55. Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest. (2005) 115:1163–76. doi: 10.1172/JCI23424
56. Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. (2010) 29:2831–42. doi: 10.1038/onc.2010.41
57. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. (2013) 73:5277–87. doi: 10.1158/0008-5472.CAN-13-0549
58. Rebbaa A, Yamamoto H, Saito T, Meuillet E, Kim P, Kersey DS, et al. Gene transfection-mediated overexpression of beta1,4-N-acetylglucosamine bisecting oligosaccharides in glioma cell line U373 MG inhibits epidermal growth factor receptor function. J Biol Chem. (1997) 272:9275–9.
59. Yokoe S, Takahashi M, Asahi M, Lee SH, Li W, Osumi D, et al. The Asn418-linked N-glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res. (2007) 67:1935–42. doi: 10.1158/0008-5472.CAN-06-3023
60. Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci USA. (2011) 108:11332–7. doi: 10.1073/pnas.1107385108
61. Wu YM, Liu CH, Huang MJ, Lai HS, Lee PH, Hu RH, et al. C1GALT1 enhances proliferation of hepatocellular carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res. (2013) 73:5580–90. doi: 10.1158/0008-5472.CAN-13-0869
62. Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. (2011) 15:1013–31. doi: 10.1111/j.1582-4934.2010.01236.x
63. Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. (2011) 209:139–51. doi: 10.1530/JOE-10-0377
64. Niedworok C, Rock K, Kretschmer I, Freudenberger T, Nagy N, Szarvas T, et al. Inhibitory role of the small leucine-rich proteoglycan biglycan in bladder cancer. PLoS ONE. (2013) 8:e80084. doi: 10.1371/journal.pone.0080084
65. Neill T, Schaefer L, Iozzo RV. Decoding the matrix: instructive roles of proteoglycan receptors. Biochemistry. (2015) 54:4583–98. doi: 10.1021/acs.biochem.5b00653
66. Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. (2007) 129:123–34. doi: 10.1016/j.cell.2007.01.049
67. Stanley P. A method to the madness of N-glycan complexity? Cell. (2007) 129:27–9. doi: 10.1016/j.cell.2007.03.022
68. Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. (2000) 6:306–12. doi: 10.1038/73163
69. Orntoft TF, Meldgaard P, Pedersen B, Wolf H. The blood group ABO gene transcript is down-regulated in human bladder tumors and growth-stimulated urothelial cell lines. Cancer Res. (1996) 56:1031–6.
70. Ferreira JA, Videira PA, Lima L, Pereira S, Silva M, Carrascal M, et al. Overexpression of tumour-associated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol Oncol. (2013) 7:719–31. doi: 10.1016/j.molonc.2013.03.001
71. Coelho R, Marcos-Silva L, Mendes N, Pereira D, Brito C, Jacob F, et al. Mucins and truncated O-glycans unveil phenotypic discrepancies between serous ovarian cancer cell lines and primary tumours. Int J Mol Sci. (2018) 19:7. doi: 10.3390/ijms19072045
72. Patsos G, Robbe-Masselot C, Klein A, Hebbe-Viton V, Martin RS, Masselot D, et al. O-glycan regulation of apoptosis and proliferation in colorectal cancer cell lines. Biochem Soc Trans. (2007) 35(Pt 5):1372–4. doi: 10.1042/BST0351372
73. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. (2012) 23:1–6. doi: 10.1091/mbc.E10-04-0335
74. Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, et al. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci USA. (2013) 110:630–5. doi: 10.1073/pnas.1220425110
75. Yamamoto H, Oviedo A, Sweeley C, Saito T, Moskal JR. Alpha2,6-sialylation of cell-surface N-glycans inhibits glioma formation in vivo. Cancer Res. (2001) 61:6822–9.
76. Kroes RA, He H, Emmett MR, Nilsson CL, Leach FE III, Amster IJ, et al. Overexpression of ST6GalNAcV, a ganglioside-specific alpha2,6-sialyltransferase, inhibits glioma growth in vivo. Proc Natl Acad Sci USA. (2010) 107:12646–51. doi: 10.1073/pnas.0909862107
77. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. (2007) 26:489–502. doi: 10.1007/s10555-007-9094-7
78. Sendoel A, Hengartner MO. Apoptotic cell death under hypoxia. Physiology. (2014) 29:168–76. doi: 10.1152/physiol.00016.2013
79. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. (2011) 25:2465–79. doi: 10.1101/gad.180331.111
80. Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr Opin Genet Dev. (2009) 19:67–73. doi: 10.1016/j.gde.2009.01.003
81. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. (2016) 30:1002–19. doi: 10.1101/gad.279737.116
82. Itoh G, Chida S, Yanagihara K, Yashiro M, Aiba N, Tanaka M. Cancer-associated fibroblasts induce cancer cell apoptosis that regulates invasion mode of tumours. Oncogene. (2017) 36:4434–44. doi: 10.1038/onc.2017.49
83. Guo JY, Hsu HS, Tyan SW, Li FY, Shew JY, Lee WH, et al. Serglycin in tumor microenvironment promotes non-small cell lung cancer aggressiveness in a CD44-dependent manner. Oncogene. (2017) 36:2457–71. doi: 10.1038/onc.2016.404
84. Brezillon S, Zeltz C, Schneider L, Terryn C, Vuillermoz B, Ramont L, et al. Lumican inhibits B16F1 melanoma cell lung metastasis. J Physiol Pharmacol. (2009) 60(Suppl 4):15–22.
85. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. (2012) 196:395–406. doi: 10.1083/jcb.201102147
86. Gilmore AP, Metcalfe AD, Romer LH, Streuli CH. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J Cell Biol. (2000) 149:431–46. doi: 10.1083/jcb.149.2.431
87. Zahir N, Lakins JN, Russell A, Ming W, Chatterjee C, Rozenberg GI, et al. Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors. J Cell Biol. (2003) 163:1397–407. doi: 10.1083/jcb.200302023
88. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. (1999) 93:1658–67.
89. Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. (1999) 5:662–8. doi: 10.1038/9511
90. Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, et al. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int J Mol Sci. (2017) 18:7. doi: 10.3390/ijms18071586
91. Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: mature adipocytes, key actors in tumor progression and resistance. Oncotarget. (2017) 8:57622–41. doi: 10.18632/oncotarget.18038
92. Lichtenstein RG, Rabinovich GA. Glycobiology of cell death: when glycans and lectins govern cell fate. Cell Death Differ. (2013) 20:976–86. doi: 10.1038/cdd.2013.50
93. Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. (2007) 13:1070–7. doi: 10.1038/nm1627
94. Moriwaki K, Noda K, Furukawa Y, Ohshima K, Uchiyama A, Nakagawa T, et al. Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology. (2009) 137:188–198 e181–2. doi: 10.1053/j.gastro.2009.04.002
95. Dufour F, Rattier T, Shirley S, Picarda G, Constantinescu AA, Morle A, et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. (2017) 24:500–10. doi: 10.1038/cdd.2016.150
96. Swindall AF, Bellis SL. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem. (2011) 286:22982–90. doi: 10.1074/jbc.M110.211375
97. Lima L, Severino PF, Silva M, Miranda A, Tavares A, Pereira S, et al. Response of high-risk of recurrence/progression bladder tumours expressing sialyl-Tn and sialyl-6-T to BCG immunotherapy. Br J Cancer. (2013) 109:2106–14. doi: 10.1038/bjc.2013.571
98. Lima L, Ferreira JA, Tavares A, Oliveira D, Morais A, Videira PA, et al. FASL polymorphism is associated with response to bacillus Calmette-Guerin immunotherapy in bladder cancer. Urol Oncol. (2014) 32:44 e41–7. doi: 10.1016/j.urolonc.2013.05.009
99. Shatnyeva OM, Kubarenko AV, Weber CE, Pappa A, Schwartz-Albiez R, Weber AN, et al. Modulation of the CD95-induced apoptosis: the role of CD95 N-glycosylation. PLoS ONE. (2011) 6:e19927. doi: 10.1371/journal.pone.0019927
100. Li C, Yang Z, Du Y, Tang H, Chen J, Hu D, et al. BCMab1, a monoclonal antibody against aberrantly glycosylated integrin alpha3beta1, has potent antitumor activity of bladder cancer in vivo. Clin Cancer Res. (2014) 20:4001–13. doi: 10.1158/1078-0432.CCR-13-3397
101. Kong D, Chen F, Sima NI. Inhibition of focal adhesion kinase induces apoptosis in bladder cancer cells via Src and the phosphatidylinositol 3-kinase/Akt pathway. Exp Ther Med. (2015) 10:1725–31. doi: 10.3892/etm.2015.2745
102. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem. (2013) 288:15121–30. doi: 10.1074/jbc.M113.470047
103. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. (2014) 54:820–31. doi: 10.1016/j.molcel.2014.04.026
104. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. (2005) 5:29–41. doi: 10.1038/nrc1527
105. Hughes RC. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim Biophys Acta. (1999) 1473:172–85.
106. Ochieng J, Leite-Browning ML, Warfield P. Regulation of cellular adhesion to extracellular matrix proteins by galectin-3. Biochem Biophys Res Commun. (1998) 246:788–91. doi: 10.1006/bbrc.1998.8708
107. Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta. (2006) 1760:616–35. doi: 10.1016/j.bbagen.2005.12.020
108. Santos SN, Junqueira MS, Francisco G, Vilanova M, Magalhaes A, Dias Baruffi M, et al. O-glycan sialylation alters galectin-3 subcellular localization and decreases chemotherapy sensitivity in gastric cancer. Oncotarget. (2016) 7:83570–87. doi: 10.18632/oncotarget.13192
109. Zhuo Y, Chammas R, Bellis SL. Sialylation of beta1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J Biol Chem. (2008) 283:22177–85. doi: 10.1074/jbc.M8000015200
110. Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. (2006) 66:632–7. doi: 10.1158/0008-5472.CAN-05-3260
111. Rattigan YI, Patel BB, Ackerstaff E, Sukenick G, Koutcher JA, Glod JW, et al. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp Cell Res. (2012) 318:326–35. doi: 10.1016/j.yexcr.2011.11.014
112. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
114. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. (1997) 94:8104–9.
115. Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. (2001) 276:9519–25. doi: 10.1074/jbc.M010144200
116. Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. (2001) 276:43407–12. doi: 10.1074/jbc.M108181200
117. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. (1994) 269:23757–63.
118. Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J Biol Chem. (1995) 270:21021–7.
119. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. (1998) 12:149–62.
120. Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. (2003) 554:264–70.
121. Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. (2006) 281:9030–7. doi: 10.1074/jbc.M511397200
122. Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS ONE. (2010) 5:e9644. doi: 10.1371/journal.pone.0009644
123. Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouyssegur J, Mazure NM. Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Front Oncol. (2012) 2:18. doi: 10.3389/fonc.2012.00018
124. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. (2014) 2:3. doi: 10.1186/2049-3002-2-3
125. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. (2007) 129:111–22. doi: 10.1016/j.cell.2007.01.047
126. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. (2007) 11:407–20. doi: 10.1016/j.ccr.2007.04.001
127. Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. (2010) 29:4362–8. doi: 10.1038/onc.2010.193
128. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
129. Manzari B, Kudlow JE, Fardin P, Merello E, Ottaviano C, Puppo M, et al. Induction of macrophage glutamine: fructose-6-phosphate amidotransferase expression by hypoxia and by picolinic acid. Int J Immunopathol Pharmacol. (2007) 20, 47–58. doi: 10.1177/039463200702000106
130. Shirato K, Nakajima K, Korekane H, Takamatsu S, Gao C, Angata T, et al. Hypoxic regulation of glycosylation via the N-acetylglucosamine cycle. J Clin Biochem Nutr. (2011) 48:20–5. doi: 10.3164/jcbn.11-015FR
131. Keppler OT, Hinderlich S, Langner J, Schwartz-Albiez R, Reutter W, Pawlita M. UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science. (1999) 284:1372–6.
132. Hisanaga K, Onodera H, Kogure K. Changes in levels of purine and pyrimidine nucleotides during acute hypoxia and recovery in neonatal rat brain. J Neurochem. (1986) 47:1344–50.
133. Kathagen-Buhmann A, Schulte A, Weller J, Holz M, Herold-Mende C, Glass R, et al. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. (2016) 18:1219–29. doi: 10.1093/neuonc/now024
134. Rao X, Duan X, Mao W, Li X, Li Z, Li Q, et al. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat Commun. (2015) 6:8468. doi: 10.1038/ncomms9468
135. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA III, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. (2012) 337:975–80. doi: 10.1126/science.1222278
136. Silva-Filho AF, Sena WLB, Lima LRA, Carvalho LVN, Pereira MC, Santos LGS, et al. Glycobiology modifications in intratumoral hypoxia: the breathless side of glycans interaction. Cell Physiol Biochem. (2017) 41:1801–29. doi: 10.1159/000471912
137. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. (2008) 8:705–13. doi: 10.1038/nrc2468
138. Greville G, McCann A, Rudd PM, Saldova R. Epigenetic regulation of glycosylation and the impact on chemo-resistance in breast and ovarian cancer. Epigenetics. (2016) 11:845–57. doi: 10.1080/15592294.2016.1241932
139. Matsumura I, Tanaka H, Kanakura Y. E2F1 and c-Myc in cell growth and death. Cell Cycle. (2003) 2:333–8.
140. Kim JW, Dang CV. Cancer's molecular sweet tooth and the Warburg effect. Cancer Res. (2006) 66:8927–30. doi: 10.1158/0008-5472.CAN-06-1501
141. Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. (2008) 8:51–6. doi: 10.1038/nrc2274
142. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
143. Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, et al. The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem Biophys Res Commun. (2010) 402:784–9. doi: 10.1016/j.bbrc.2010.10.113
144. Penque BA, Hoggatt AM, Herring BP, Elmendorf JS. Hexosamine biosynthesis impairs insulin action via a cholesterolgenic response. Mol Endocrinol. (2013) 27:536–47. doi: 10.1210/me.2012-1213
145. Amin A, Karpowicz PA, Carey TE, Arbiser J, Nahta R, Chen ZG, et al. Evasion of anti-growth signaling: a key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol. (2015) 35(Suppl):S55–77. doi: 10.1016/j.semcancer.2015.02.005
147. Moh MC, Shen S. The roles of cell adhesion molecules in tumor suppression and cell migration: a new paradox. Cell Adh Migr. (2009) 3:334–6.
148. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. (1996) 379:88–91. doi: 10.1038/379088a0
149. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. (2006) 8:1074–83. doi: 10.1038/ncb1470
150. Wells L, Slawson C, Hart GW. The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids. (2011) 40:877–83. doi: 10.1007/s00726-010-0709-x
151. Soesanto YA, Luo B, Jones D, Taylor R, Gabrielsen JS, Parker G, et al. Regulation of Akt signaling by O-GlcNAc in euglycemia. Am J Physiol Endocrinol Metab. (2008) 295:E974–80. doi: 10.1152/ajpendo.90366.2008
152. Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev. (2004) 14:37–42. doi: 10.1016/j.gde.2003.12.008
153. Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, et al. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiother Oncol. (2005) 76:168–76. doi: 10.1016/j.radonc.2005.06.025
154. Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. (2012) 72:949–57. doi: 10.1158/0008-5472.CAN-11-2371
155. Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. (2013) 4:5. doi: 10.1186/2041-9414-4-5
156. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: a new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. (2010) 9:3256–76. doi: 10.4161/cc.9.16.12553
157. Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen. (2014) 5:165. doi: 10.4172/2157-2518.1000165
158. Zhong J, Martinez M, Sengupta S, Lee A, Wu X, Chaerkady R, et al. Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics. (2015) 15:591–607. doi: 10.1002/pmic.201400339
159. Miura Y, Sakurai Y, Endo T. O-GlcNAc modification affects the ATM-mediated DNA damage response. Biochim Biophys Acta. (2012) 1820:1678–85. doi: 10.1016/j.bbagen.2012.06.013
160. Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. (2012) 81:303–11. doi: 10.1111/j.1399-0004.2011.01809.x
161. Coyle KM, Boudreau JE, Marcato P. Genetic mutations and epigenetic modifications: driving cancer and informing precision medicine. Biomed Res Int. (2017) 2017:9620870. doi: 10.1155/2017/9620870
162. Sakabe K, Wang Z, Hart GW. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci USA. (2010) 107:19915–20. doi: 10.1073/pnas.1009023107
163. Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. (2016) 8:69. doi: 10.1186/s13073-016-0324-x
164. Rachakonda PS, Hosen I, de Verdier PJ, Fallah M, Heidenreich B, Ryk C, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci USA. (2013) 110:17426–31. doi: 10.1073/pnas.1310522110
165. Bell RJ, Rube HT, Xavier-Magalhaes A, Costa BM, Mancini A, Song JS, et al. Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res. (2016) 14:315–23. doi: 10.1158/1541-7786.MCR-16-0003
166. Seimiya H, Tanji M, Oh-hara T, Tomida A, Naasani I, Tsuruo T. Hypoxia up-regulates telomerase activity via mitogen-activated protein kinase signaling in human solid tumor cells. Biochem Biophys Res Commun. (1999) 260:365–70. doi: 10.1006/bbrc.1999.0910
167. Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, et al. Direct activation of TERT transcription by c-MYC. Nat Genet. (1999) 21:220–4. doi: 10.1038/6010
168. Fus LP, Gornicka B. Role of angiogenesis in urothelial bladder carcinoma. Cent Eur J Urol. (2016) 69:258–63. doi: 10.5173/ceju.2016.830
169. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. (2011) 2:1117–33. doi: 10.1177/1947601911423654
170. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. (2009) 15:21–34. doi: 10.1016/j.ccr.2008.12.004
171. Ribatti D, Crivellato E. Immune cells and angiogenesis. J Cell Mol Med. (2009) 13:2822–33. doi: 10.1111/j.1582-4934.2009.00810.x
172. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
173. Lederle W, Hartenstein B, Meides A, Kunzelmann H, Werb Z, Angel P, et al. MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis. (2010) 31:1175–84. doi: 10.1093/carcin/bgp248
174. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers. (2015) 7:2443–58. doi: 10.3390/cancers7040902
175. Stringer SE. The role of heparan sulphate proteoglycans in angiogenesis. Biochem Soc Trans. (2006) 34(Pt 3):451–3. doi: 10.1042/BST0340451
176. van Wijk XM, Thijssen VL, Lawrence R, van den Broek SA, Dona M, Naidu N, et al. Interfering with UDP-GlcNAc metabolism and heparan sulfate expression using a sugar analogue reduces angiogenesis. ACS Chem Biol. (2013) 8:2331–8. doi: 10.1021/cb4004332
177. Cole CL, Rushton G, Jayson GC, Avizienyte E. Ovarian cancer cell heparan sulfate 6-O-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (EGF)-like growth factor/EGF receptor signaling. J Biol Chem. (2014) 289:10488–501. doi: 10.1074/jbc.M113.534263
178. Maeda T, Desouky J, Friedl A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene. (2006) 25:1408–12. doi: 10.1038/sj.onc.1209168
179. Lokeshwar VB, Cerwinka WH, Isoyama T, Lokeshwar BL. HYAL1 hyaluronidase in prostate cancer: a tumor promoter and suppressor. Cancer Res. (2005) 65:7782–9. doi: 10.1158/0008-5472.CAN-05-1022
180. Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. (2007) 11:53–67. doi: 10.1016/j.ccr.2006.10.018
181. Caunt M, Mak J, Liang WC, Stawicki S, Pan Q, Tong RK, et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. (2008) 13:331–42. doi: 10.1016/j.ccr.2008.01.029
182. Golshani R, Lopez L, Estrella V, Kramer M, Iida N, Lokeshwar VB. Hyaluronic acid synthase-1 expression regulates bladder cancer growth, invasion, and angiogenesis through CD44. Cancer Res. (2008) 68:483–91. doi: 10.1158/0008-5472.CAN-07-2140
183. Grant DS, Yenisey C, Rose RW, Tootell M, Santra M, Iozzo RV. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene. (2002) 21:4765–77. doi: 10.1038/sj.onc.1205595
184. Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, et al. Decorin antagonizes the angiogenic network: concurrent inhibition of Met, hypoxia inducible factor 1alpha, vascular endothelial growth factor A, and induction of thrombospondin-1 and TIMP3. J Biol Chem. (2012) 287:5492–506. doi: 10.1074/jbc.M111.283499
185. El Behi M, Krumeich S, Lodillinsky C, Kamoun A, Tibaldi L, Sugano G, et al. An essential role for decorin in bladder cancer invasiveness. EMBO Mol Med. (2013) 5:1835–51. doi: 10.1002/emmm.201302655
186. Niewiarowska J, Brezillon S, Sacewicz-Hofman I, Bednarek R, Maquart FX, Malinowski M, et al. Lumican inhibits angiogenesis by interfering with alpha2beta1 receptor activity and downregulating MMP-14 expression. Thromb Res. (2011) 128:452–7. doi: 10.1016/j.thromres.2011.06.011
187. Hagedorn M, Javerzat S, Gilges D, Meyre A, de Lafarge B, Eichmann A, et al. Accessing key steps of human tumor progression in vivo by using an avian embryo model. Proc Natl Acad Sci USA. (2005) 102:1643–8. doi: 10.1073/pnas.0408622102
188. Albig AR, Roy TG, Becenti DJ, Schiemann WP. Transcriptome analysis of endothelial cell gene expression induced by growth on matrigel matrices: identification and characterization of MAGP-2 and lumican as novel regulators of angiogenesis. Angiogenesis. (2007) 10:197–216. doi: 10.1007/s10456-007-9075-z
189. Williams KE, Fulford LA, Albig AR. Lumican reduces tumor growth via induction of fas-mediated endothelial cell apoptosis. Cancer Microenviron. (2010) 4:115–26. doi: 10.1007/s12307-010-0056-1
190. Sharma B, Ramus MD, Kirkwood CT, Sperry EE, Chu PH, Kao WW, et al. Lumican exhibits anti-angiogenic activity in a context specific manner. Cancer Microenviron. (2013) 6:263–71. doi: 10.1007/s12307-013-0134-2
191. Thijssen VL, Rabinovich GA, Griffioen AW. Vascular galectins: regulators of tumor progression and targets for cancer therapy. Cytokine Growth Factor Rev. (2013) 24:547–58. doi: 10.1016/j.cytogfr.2013.07.003
192. Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell. (2014) 156:744–58. doi: 10.1016/j.cell.2014.01.043
193. Croci DO, Cerliani JP, Pinto NA, Morosi LG, Rabinovich GA. Regulatory role of glycans in the control of hypoxia-driven angiogenesis and sensitivity to anti-angiogenic treatment. Glycobiology. (2014) 24:1283–90. doi: 10.1093/glycob/cwu083
194. Mazzola CR, Chin J. Targeting the VEGF pathway in metastatic bladder cancer. Expert Opin Investig Drugs. (2015) 24:913–27. doi: 10.1517/13543784.2015.1041588
195. Narayanan S, Srinivas S. Incorporating VEGF-targeted therapy in advanced urothelial cancer. Ther Adv Med Oncol. (2017) 9:33–45. doi: 10.1177/1758834016667179
196. Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem. (2012) 287:11070–81. doi: 10.1074/jbc.M111.302547
197. Takeuchi H, Haltiwanger RS. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun. (2014) 453:235–42. doi: 10.1016/j.bbrc.2014.05.115
198. Kitazume S, Imamaki R, Ogawa K, Komi Y, Futakawa S, Kojima S, et al. Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J Biol Chem. (2010) 285:6515–21. doi: 10.1074/jbc.M109.073106
199. Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA. (2010) 107:21677–82. doi: 10.1073/pnas.1016234107
200. Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1alpha and HIF-2alpha differentially regulate metastatic success. Cancer Cell. (2012) 21:52–65. doi: 10.1016/j.ccr.2011.11.017
201. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. (2016) 352:175–80. doi: 10.1126/science.aaf4405
202. Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. (2006) 66:5242–50. doi: 10.1158/0008-5472.CAN-05-4463
203. Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol. (2010) 184:702–12. doi: 10.4049/jimmunol.0902360
204. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. (2011) 331:1559–64. doi: 10.1126/science.1203543
205. Ricciardelli C, Russell DL, Ween MP, Mayne K, Suwiwat S, Byers S, et al. Formation of hyaluronan- and versican-rich pericellular matrix by prostate cancer cells promotes cell motility. J Biol Chem. (2007) 282:10814–25. doi: 10.1074/jbc.M606991200
206. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. (2013) 73:5016–28. doi: 10.1158/0008-5472.CAN-13-0023
207. Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol. (2000) 156:529–36. doi: 10.1016/S0002-9440(10)64757-8
208. Lipponen P, Aaltomaa S, Tammi R, Tammi M, Agren U, Kosma VM. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur J Cancer. (2001) 37:849–56. doi: 10.1016/S0959-8049(00)00448-2
209. Andrlova H, Mastroianni J, Madl J, Kern JS, Melchinger W, Dierbach H, et al. Biglycan expression in the melanoma microenvironment promotes invasiveness via increased tissue stiffness inducing integrin-beta1 expression. Oncotarget. (2017) 8:42901–16. doi: 10.18632/oncotarget.17160
210. Turley EA, Wood DK, McCarthy JB. Carcinoma cell hyaluronan as a “portable” cancerized prometastatic microenvironment. Cancer Res. (2016) 76:2507–12. doi: 10.1158/0008-5472.CAN-15-3114
211. Shea DJ, Li YW, Stebe KJ, Konstantopoulos K. E-selectin-mediated rolling facilitates pancreatic cancer cell adhesion to hyaluronic acid. FASEB J. (2017) 31:5078–86. doi: 10.1096/fj.201700331R
212. Reed CC, Waterhouse A, Kirby S, Kay P, Owens RT, McQuillan DJ, et al. Decorin prevents metastatic spreading of breast cancer. Oncogene. (2005) 24:1104–10. doi: 10.1038/sj.onc.1208329
213. Buraschi S, Neill T, Owens RT, Iniguez LA, Purkins G, Vadigepalli R, et al. Decorin protein core affects the global gene expression profile of the tumor microenvironment in a triple-negative orthotopic breast carcinoma xenograft model. PLoS ONE. (2012) 7:e45559. doi: 10.1371/journal.pone.0045559
214. Guo J, Li X, Tan Z, Lu W, Yang G, Guan F. Alteration of N-glycans and expression of their related glycogenes in the epithelial-mesenchymal transition of HCV29 bladder epithelial cells. Molecules. (2014) 19:20073–90. doi: 10.3390/molecules191220073
215. Seidenfaden R, Krauter A, Schertzinger F, Gerardy-Schahn R, Hildebrandt H. Polysialic acid directs tumor cell growth by controlling heterophilic neural cell adhesion molecule interactions. Mol Cell Biol. (2003) 23:5908–18. doi: 10.1128/MCB.23.16.5908-5918.2003
216. Schultz MJ, Swindall AF, Bellis SL. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. (2012) 31:501–18. doi: 10.1007/s10555-012-9359-7
217. Munkley J, Oltean S, Vodak D, Wilson BT, Livermore KE, Zhou Y, et al. The androgen receptor controls expression of the cancer-associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget. (2015) 6:34358–74. doi: 10.18632/oncotarget.6024
218. Julien S, Adriaenssens E, Ottenberg K, Furlan A, Courtand G, Vercoutter-Edouart AS, et al. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology. (2006) 16:54–64. doi: 10.1093/glycob/cwj033
219. Pinho S, Marcos NT, Ferreira B, Carvalho AS, Oliveira MJ, Santos-Silva F, et al. Biological significance of cancer-associated sialyl-Tn antigen: modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett. (2007) 249:157–70. doi: 10.1016/j.canlet.2006.08.010
220. Ozaki H, Matsuzaki H, Ando H, Kaji H, Nakanishi H, Ikehara Y, et al. Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin Exp Metastasis. (2012) 29:229–38. doi: 10.1007/s10585-011-9445-1
221. Lima L, Neves M, Oliveira MI, Dieguez L, Freitas R, Azevedo R, et al. Sialyl-Tn identifies muscle-invasive bladder cancer basal and luminal subtypes facing decreased survival, being expressed by circulating tumor cells and metastases. Urol Oncol. (2017) 35:675.e1-675.e8. doi: 10.1016/j.urolonc.2017.08.012
222. Lin S, Kemmner W, Grigull S, Schlag PM. Cell surface alpha 2,6 sialylation affects adhesion of breast carcinoma cells. Exp Cell Res. (2002) 276:101–10. doi: 10.1006/excr.2002.5521
223. Radhakrishnan P, Dabelsteen S, Madsen FB, Francavilla C, Kopp KL, Steentoft C, et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci USA. (2014) 111:E4066–4075. doi: 10.1073/pnas.1406619111
224. Pochec E, Litynska A, Bubka M, Amoresano A, Casbarra A. Characterization of the oligosaccharide component of α3β1 integrin from human bladder carcinoma cell line T24 and its role in adhesion and migration. Eur J Cell Biol. (2006) 85:47–57. doi: 10.1016/j.ejcb.2005.08.010
225. Pinho SS, Reis CA, Paredes J, Magalhaes AM, Ferreira AC, Figueiredo J, et al. The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum Mol Genet. (2009) 18:2599–608. doi: 10.1093/hmg/ddp194
226. Carvalho S, Catarino TA, Dias AM, Kato M, Almeida A, Hessling B, et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene. (2016) 35:1619–31. doi: 10.1038/onc.2015.225
227. Guo HB, Lee I, Kamar M, Pierce M. N-acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J Biol Chem. (2003) 278:52412–24. doi: 10.1074/jbc.M308837200
228. Ihara S, Miyoshi E, Ko JH, Murata K, Nakahara S, Honke K, et al. Prometastatic effect of N-acetylglucosaminyltransferase V is due to modification and stabilization of active matriptase by adding beta 1-6 GlcNAc branching. J Biol Chem. (2002) 277:16960–7. doi: 10.1074/jbc.M200673200
229. Burdick MM, Henson KA, Delgadillo LF, Choi YE, Goetz DJ, Tees DF, et al. Expression of E-selectin ligands on circulating tumor cells: cross-regulation with cancer stem cell regulatory pathways? Front Oncol. (2012) 2:103. doi: 10.3389/fonc.2012.00103
230. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. (2012) 2:1091–9. doi: 10.1158/2159-8290.CD-12-0329
231. Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. (2013) 13:858–70. doi: 10.1038/nrc3628
232. Numahata K, Satoh M, Handa K, Saito S, Ohyama C, Ito A, et al. Sialosyl-Lex expression defines invasive and metastatic properties of bladder carcinoma. Cancer. (2002) 94:673–85. doi: 10.1002/cncr.10268
233. Hegele A, Mecklenburg V, Varga Z, Olbert P, Hofmann R, Barth P. CA19.9 and CEA in transitional cell carcinoma of the bladder: serological and immunohistochemical findings. Anticancer Res. (2010) 30:5195–200.
234. Borentain P, Carmona S, Mathieu S, Jouve E, El-Battari A, Gerolami R. Inhibition of E-selectin expression on the surface of endothelial cells inhibits hepatocellular carcinoma growth by preventing tumor angiogenesis. Cancer Chemother Pharmacol. (2016) 77:847–56. doi: 10.1007/s00280-016-3006-x
235. Cui HX, Wang H, Wang Y, Song J, Tian H, Xia C, et al. ST3Gal III modulates breast cancer cell adhesion and invasion by altering the expression of invasion-related molecules. Oncol Rep. (2016) 36:3317–24. doi: 10.3892/or.2016.5180
236. Liang JX, Gao W, Cai L. Fucosyltransferase VII promotes proliferation via the EGFR/AKT/mTOR pathway in A549 cells. Onco Targets Ther. (2017) 10:3971–8. doi: 10.2147/OTT.S140940
237. Kawashima H, Petryniak B, Hiraoka N, Mitoma J, Huckaby V, Nakayama J, et al. N-acetylglucosamine-6-O-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nat Immunol. (2005) 6:1096–104. doi: 10.1038/ni1259
238. Taga M, Hoshino H, Low S, Imamura Y, Ito H, Yokoyama O, et al. A potential role for 6-sulfo sialyl Lewis X in metastasis of bladder urothelial carcinoma. Urol Oncol. (2015) 33:496 e491–9. doi: 10.1016/j.urolonc.2015.05.026
239. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. (2009) 459:1005–9. doi: 10.1038/nature08021
240. Murugaesu N, Iravani M, van Weverwijk A, Ivetic A, Johnson DA, Antonopoulos A, et al. An in vivo functional screen identifies ST6GalNAc2 sialyltransferase as a breast cancer metastasis suppressor. Cancer Discov. (2014) 4:304–17. doi: 10.1158/2159-8290.CD-13-0287
241. Pangeni RP, Channathodiyil P, Huen DS, Eagles LW, Johal BK, Pasha D, et al. The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain. Clin Epigenet. (2015) 7:57. doi: 10.1186/s13148-015-0089-x
242. Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov. (2005) 4:477–88. doi: 10.1038/nrd1751
243. Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. (2010) 10:369–73. doi: 10.2174/156652410791316968
244. Katanov C, Lerrer S, Liubomirski Y, Leider-Trejo L, Meshel T, Bar J, et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: prominent roles for TNF-alpha and the NF-kappaB pathway. Stem Cell Res Ther. (2015) 6:87. doi: 10.1186/s13287-015-0080-7
245. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
246. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. (2010) 70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785
247. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. (2011) 71:2455–65. doi: 10.1158/0008-5472.CAN-10-3323
248. Fang WB, Mafuvadze B, Yao M, Zou A, Portsche M, Cheng N. TGF-beta negatively regulates CXCL1 chemokine expression in mammary fibroblasts through enhancement of Smad2/3 and suppression of HGF/c-Met signaling mechanisms. PLoS ONE. (2015) 10:e0135063. doi: 10.1371/journal.pone.0135063
249. Osuala KO, Sameni M, Shah S, Aggarwal N, Simonait ML, Franco OE, et al. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer. (2015) 15:584. doi: 10.1186/s12885-015-1576-3
250. Yeh CR, Hsu I, Song W, Chang H, Miyamoto H, Xiao GQ, et al. Fibroblast ERalpha promotes bladder cancer invasion via increasing the CCL1 and IL-6 signals in the tumor microenvironment. Am J Cancer Res. (2015) 5:1146–57.
251. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
252. Renkonen J, Tynninen O, Hayry P, Paavonen T, Renkonen R. Glycosylation might provide endothelial zip codes for organ-specific leukocyte traffic into inflammatory sites. Am J Pathol. (2002) 161:543–50. doi: 10.1016/S0002-9440(10)64210-1
253. Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets. (2007) 11:1473–91. doi: 10.1517/14728222.11.11.1473
254. Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci Signal. (2013) 6:290, ra75. doi: 10.1126/scisignal.2004097
255. Sevigny MB, Li CF, Alas M, Hughes-Fulford M. Glycosylation regulates turnover of cyclooxygenase-2. FEBS Lett. (2006) 580:6533–6. doi: 10.1016/j.febslet.2006.10.073
256. Sevigny MB, Graham K, Ponce E, Louie MC, Mitchell K. Glycosylation of human cyclooxygenase-2 (COX-2) decreases the efficacy of certain COX-2 inhibitors. Pharmacol Res. (2012) 65:445–50. doi: 10.1016/j.phrs.2012.01.001
257. Samraj A, Crittenden A, Banda K, Gregg CJ, Assar S, Diaz SL, et al. Diet-derived xeno-autoantigen sialic acid promotes inflammation - evidence for “xenosialitis.” FASEB J. (2013) 27(Suppl. 1):lb488.
258. Samraj AN, Laubli H, Varki N, Varki A. Involvement of a non-human sialic Acid in human cancer. Front Oncol. (2014) 4:33. doi: 10.3389/fonc.2014.00033
259. Samraj AN, Pearce OM, Laubli H, Crittenden AN, Bergfeld AK, Banda K, et al. A red meat-derived glycan promotes inflammation and cancer progression. Proc Natl Acad Sci USA. (2015) 112:542–7. doi: 10.1073/pnas.1417508112
260. Padro M, Mejias-Luque R, Cobler L, Garrido M, Perez-Garay M, Puig S, et al. Regulation of glycosyltransferases and Lewis antigens expression by IL-1beta and IL-6 in human gastric cancer cells. Glycoconj J. (2011) 28:99–110. doi: 10.1007/s10719-011-9327-4
261. Bassaganas S, Allende H, Cobler L, Ortiz MR, Llop E, de Bolos C, et al. Inflammatory cytokines regulate the expression of glycosyltransferases involved in the biosynthesis of tumor-associated sialylated glycans in pancreatic cancer cell lines. Cytokine. (2015) 75:197–206. doi: 10.1016/j.cyto.2015.04.006
262. Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest. (2012) 122:1503–18. doi: 10.1172/JCI61392
263. Said N, Theodorescu D. RhoGDI2 suppresses bladder cancer metastasis via reduction of inflammation in the tumor microenvironment. Oncoimmunology. (2012) 1:1175–7. doi: 10.4161/onci.20594
264. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev. (2011) 25:2559–72. doi: 10.1101/gad.169029.111
265. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
266. Ziani L, Chouaib S, Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. (2018) 9:414. doi: 10.3389/fimmu.2018.00414
267. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. (2010) 70:7073–83. doi: 10.1158/0008-5472.CAN-09-4687
268. Hope C, Foulcer S, Jagodinsky J, Chen SX, Jensen JL, Patel S, et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood. (2016) 128:680–5. doi: 10.1182/blood-2016-03-705780
269. Fukuda M. Possible roles of tumor-associated carbohydrate antigens. Cancer Res. (1996) 56:2237–44.
270. Speiser DE, Miranda R, Zakarian A, Bachmann MF, McKall-Faienza K, Odermatt B, et al. Self antigens expressed by solid tumors Do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J Exp Med. (1997) 186:645–53.
271. Berg JM, Tymoczko JL, Stryer L. Biochemistry, 5th edition, in Section 33.6, Immune Responses Against Self-Antigens Are Suppressed. New York, NY: W H Freeman (2002).
272. Garcia-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. (2014) 211:1465–83. doi: 10.1084/jem.20122192
273. Gringhuis SI, Kaptein TM, Wevers BA, van der Vlist M, Klaver EJ, van Die I, et al. Fucose-based PAMPs prime dendritic cells for follicular T helper cell polarization via DC-SIGN-dependent IL-27 production. Nat Commun. (2014) 5:5074. doi: 10.1038/ncomms6074
274. Perdicchio M, Cornelissen LA, Streng-Ouwehand I, Engels S, Verstege MI, Boon L, et al. Tumor sialylation impedes T cell mediated anti-tumor responses while promoting tumor associated-regulatory T cells. Oncotarget. (2016) 7:8771–82. doi: 10.18632/oncotarget.6822
275. Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci USA. (2016) 113:3329–34. doi: 10.1073/pnas.1507706113
276. Takamiya R, Ohtsubo K, Takamatsu S, Taniguchi N, Angata T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-beta secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology. (2013) 23:178–87. doi: 10.1093/glycob/cws139
277. Carrascal MA, Severino PF, Guadalupe Cabral M, Silva M, Ferreira JA, Calais F, et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol Oncol. (2014) 8:753–65. doi: 10.1016/j.molonc.2014.02.008
278. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. (2016) 17:1273–81. doi: 10.1038/ni.3552
279. Jandus C, Boligan KFChijioke O, Liu H, Dahlhaus M, Demoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. (2014) 124:1810–20. doi: 10.1172/J.C.I.65899
280. Cohen M, Elkabets M, Perlmutter M, Porgador A, Voronov E, Apte RN, et al. Sialylation of 3-methylcholanthrene-induced fibrosarcoma determines antitumor immune responses during immunoediting. J Immunol. (2010) 185:5869–78. doi: 10.4049/jimmunol.1001635
281. Lanier LL. NKG2D Receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
282. Lee HC, Wondimu A, Liu Y, Ma JS, Radoja S, Ladisch S. Ganglioside inhibition of CD8+ T cell cytotoxicity: interference with lytic granule trafficking and exocytosis. J Immunol. (2012) 189:3521–7. doi: 10.4049/jimmunol.1201256
283. Costa C, Pereira S, Lima L, Peixoto A, Fernandes E, Neves D, et al. Abnormal protein glycosylation and activated PI3K/Akt/mTOR pathway: role in bladder cancer prognosis and targeted therapeutics. PLoS ONE. (2015) 10:e0141253. doi: 10.1371/journal.pone.0141253
284. Suzuki Y, Sutoh M, Hatakeyama S, Mori K, Yamamoto H, Koie T, et al. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int J Oncol. (2012) 40:1831–8. doi: 10.3892/ijo.2012.1411
285. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. (2016) 7:12632. doi: 10.1038/ncomms12632
286. Kirwan A, Utratna M, O'Dwyer ME, Joshi L, Kilcoyne M. Glycosylation-based serum biomarkers for cancer diagnostics and prognostics. Biomed Res Int. (2015) 2015:490531. doi: 10.1155/2015/490531
287. Kailemia MJ, Park D, Lebrilla CB. Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem. (2017) 409:395–410. doi: 10.1007/s00216-016-9880-6
288. Escrevente C, Grammel N, Kandzia S, Zeiser J, Tranfield EM, Conradt HS, et al. Sialoglycoproteins and N-glycans from secreted exosomes of ovarian carcinoma cells. PLoS ONE. (2013) 8:e78631. doi: 10.1371/journal.pone.0078631
289. Saraswat M, Joenvaara S, Musante L, Peltoniemi H, Holthofer H, Renkonen R. N-linked (N-) glycoproteomics of urinary exosomes. Mol Cell Proteomics. (2015) 14:263–76. doi: 10.1074/mcp.M114.040345
290. York WS, Agravat S, Aoki-Kinoshita KF, McBride R, Campbell MP, Costello CE, et al. MIRAGE: the minimum information required for a glycomics experiment. Glycobiology. (2014) 24:402–6. doi: 10.1093/glycob/cwu018
291. Struwe WB, Agravat S, Aoki-Kinoshita KF, Campbell MP, Costello CE, Dell A, et al. The minimum information required for a glycomics experiment (MIRAGE) project: sample preparation guidelines for reliable reporting of glycomics datasets. Glycobiology. (2016) 26:907–10. doi: 10.1093/glycob/cww082
292. Hinneburg H, Schirmeister F, Korac P, Kolarich D. N- and O-glycomics from minor amounts of formalin-fixed, paraffin-embedded tissue samples. Methods Mol Biol. (2017) 1503:131–45. doi: 10.1007/978-1-4939-6493-2_11
293. Everest-Dass AV, Briggs MT, Kaur G, Oehler MK, Hoffmann P, Packer NH. N-glycan MALDI imaging mass spectrometry on formalin-fixed paraffin-embedded tissue enables the delineation of ovarian cancer tissues. Mol Cell Proteomics. (2016) 15:3003–16. doi: 10.1074/mcp.M116.059816
294. Baycin Hizal D, Wolozny D, Colao J, Jacobson E, Tian Y, Krag SS, et al. Glycoproteomic and glycomic databases. Clin Proteomics. (2014) 11:15. doi: 10.1186/1559-0275-11-15
295. Lima L, Oliveira D, Ferreira JA, Tavares A, Cruz R, Medeiros R, et al. The role of functional polymorphisms in immune response genes as biomarkers of bacille Calmette-Guerin (BCG) immunotherapy outcome in bladder cancer: establishment of a predictive profile in a Southern Europe population. BJU Int. (2015) 116:753–63. doi: 10.1111/bju.12844
296. Compain P, Martin OR. Carbohydrate mimetics-based glycosyltransferase inhibitors. Bioorg Med Chem. (2001) 9:3077–92. doi: 10.1016/S0968-0896(01)00176-6
297. Liu SD, Chalouni C, Young JC, Junttila TT, Sliwkowski MX, Lowe JB. Afucosylated antibodies increase activation of FcgammaRIIIa-dependent signaling components to intensify processes promoting ADCC. Cancer Immunol Res. (2015) 3:173–83. doi: 10.1158/2326-6066.CIR-14-0125
298. Azevedo R, Ferreira JA, Peixoto A, Neves M, Sousa N, Lima A, et al. Emerging antibody-based therapeutic strategies for bladder cancer: a systematic review. J Control Release. (2015) 214:40–61. doi: 10.1016/j.jconrel.2015.07.002
299. Fernandes E, Ferreira JA, Andreia P, Luis L, Barroso S, Sarmento B, et al. New trends in guided nanotherapies for digestive cancers: a systematic review. J Control Release. (2015) 209:288–307. doi: 10.1016/j.jconrel.2015.05.003
300. Posey ADJr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
301. Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, et al. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci USA. (2012) 109:261–6. doi: 10.1073/pnas.1115166109
302. Abdel-Aal AB, Lakshminarayanan V, Thompson P, Supekar N, Bradley JM, Wolfert MA, et al. Immune and anticancer responses elicited by fully synthetic aberrantly glycosylated MUC1 tripartite vaccines modified by a TLR2 or TLR9 agonist. Chembiochem. (2014) 15:1508–13. doi: 10.1002/cbic.201402077
Keywords: cancer, microenvironment, glycans, protein glycosylation, cancer hallmarks
Citation: Peixoto A, Relvas-Santos M, Azevedo R, Santos LL and Ferreira JA (2019) Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 9:380. doi: 10.3389/fonc.2019.00380
Received: 15 March 2019; Accepted: 23 April 2019;
Published: 14 May 2019.
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Feng Guan, Northwest University, ChinaMonica M. Burdick, Ohio University, United States
Copyright © 2019 Peixoto, Relvas-Santos, Azevedo, Santos and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Alexandre Ferreira, am9zZS5hLmZlcnJlaXJhQGlwb3BvcnRvLm1pbi1zYXVkZS5wdA==