 Nils W. Engel1*
Nils W. Engel1* Simon Schliffke1
Simon Schliffke1 Ulrich Schüller2,3,4Christian Frenzel1Carsten Bokemeyer1Christian Kubisch5
Ulrich Schüller2,3,4Christian Frenzel1Carsten Bokemeyer1Christian Kubisch5 Davor Lessel5*
Davor Lessel5*- 1Department of Oncology, Haematology and Bone Marrow Transplantation with Section Pneumology, Hubertus Wald Tumorzentrum, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 3Department of Pediatric Hematology and Oncology, University Medical Center, Hamburg-Eppendorf, Germany
- 4Research Institute Children's Cancer Center Hamburg, Hamburg, Germany
- 5Institute of Human Genetics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Unrecognized genome instability syndromes can potentially impede the rational treatment of cancer in rare patients. Identification of cancer patients with a hereditary condition is a compelling necessity for oncologists, giving varying hypersensitivities to various chemotherapeutic agents or radiation, depending on the underlying genetic cause. Omission of genetic testing in the setting of an overlooked hereditary syndrome may lead to unexpected and unbearable toxicity from oncological standard approaches. We present a case of a 33-year-old man with an early-onset stage IV intrahepatic cholangiocarcinoma, who experienced unusual bone marrow failure and neutropenic fever syndrome as a consequence of palliative chemotherapy containing cisplatin and gemcitabine, leading to a fatal outcome on day 25 of his first chemotherapeutic cycle. The constellation of bone marrow failure after exposure to the platinum-based agent cisplatin, the presence of an early-onset solid malignancy and the critical appraisal of further phenotypical features raised suspicion of a hereditary genome instability syndrome. Whole-exome sequencing from buccal swab DNA enabled the post mortem diagnosis of Fanconi anemia, most likely linked to the fatal outcome due to utilization of the DNA crosslinking agent cisplatin. The patient's phenotype was exceptional, as he never displayed significant hematologic abnormalities, which is the hallmark of Fanconi anemia. As such, this case stresses the importance to at least question the possibility of a hereditary basis in cases of relatively early-onset malignancy before defining an oncological treatment strategy.
Introduction
Genome instability syndromes are a group of inherited conditions caused by germline mutations in genes encoding DNA repair proteins involved in diverse DNA damage response (DDR) pathways, resulting in defects in genome maintenance, sensitivity toward various genotoxic agents, and early-onset cancer susceptibility (1, 2). Besides the increased incidence of various cancers, based on the underlying genetic cause, major symptoms of genome instability syndromes include developmental and craniofacial abnormalities, neurological deficiencies, immunodeficiency, cardiovascular diseases, metabolic abnormalities, and clinical signs of premature/accelerated aging. The phenotypic spectrum of these disorders and especially the hypersensitivity toward genotoxic agents, many of which are actually used in antineoplastic therapy, result from the mutation-induced and thereby gene-specific defects in different DDR pathways as each recognizes and/or repairs a highly specific class of DNA lesions (3). Known genome instability syndromes are, for example, caused by mutations in genes encoding DNA helicases like WRN in Werner Syndrome and BLM in Bloom syndrome, mutations in genes involved in nucleotide excision repair like XPA-HPV in Xeroderma pigmentosum and CSA and CSB in Cockayne syndrome, mutations in genes involved in DDR in response to the DNA double-strand breaks like ATM in Ataxia-telangiectasia and NBS1 in Nijmegen breakage syndrome, mutations in genes involved in protein-DNA crosslink repair like SPRTN in Ruijs-Aalfs syndrome, and mutations in genes involved in interstrand DNA crosslink repair like FANCA-FANCU in Fanconi anemia (1, 2).
Fanconi anemia (FA) is a multisystem disorder characterized by physical anomalies, hematologic abnormalities including progressive bone marrow failure and pancytopenia, early-onset leukemia and solid cancer susceptibility, as well as hypersensitivity toward DNA crosslinking agents such as mitomycin C, cisplatin and diepoxybutane (4, 5). The majority of patients have normal blood counts at birth, and develop hematological abnormalities including anemia, neutropenia, thrombocytopenia, and even pancytopenia within the first decade of life. Physical anomalies are present in around the half of affected individuals and can include skin hypopigmentation and café-au-lait spots, skeletal anomalies like short stature, malformations of the upper and lower limbs, malformed or absent kidneys, malformations of genitalia, urinary and reproductive system including (predominantly) male infertility, gastrointestinal abnormalities, ophthalmic abnormalities, heart defects, abnormalities of the central nervous system, microcephaly, malformed ears, and hearing loss. In addition, around 10% of patients develop mild to moderate intellectual disability.
Similar to the majority of the above-mentioned genome instability syndromes, FA is mostly inherited in an autosomal-recessive manner, Meaning that the parents of affected individuals are usually unaffected heterozygous carriers, bearing the mutation on only a single allele. So far, biallelic mutations in 19 genes, termed FANC(-)A to FANC(-)U, have been shown to cause FA. However, two exceptions to this inheritance pattern exist. Namely, mutations affecting the FANCB, located on the X-chromosome, are inherited in an X-linked manner, therefore the female carriers do not develop FA-associated clinical signs and symptoms. In addition, heterozygous de novo mutations in RAD51 have also been connected to FA, representing an autosomal-dominant mode of inheritance (4).
Here, we report a case of unusual presentation of FA with a fatal outcome.
Background
A 33-year-old man of Turkish descent with stage IV intrahepatic cholangiocarcinoma according to the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) staging criteria (6) with distant metastases (e.g., to lungs; Figure 1) was admitted to our emergency room with a clinical and laboratory constellation consistent with sepsis. The patient had a quick Sequential [Sepsis-related] Organ Failure Assessment (qSOFA) score (7) of three points (tachypnea, altered mentation, systolic blood pressure 64 mmHg), a measured body temperature of 38.4°C (tympanic), acute kidney injury stage 2 according to Kidney Disease: Improving Global Outcome (KDIGO) criteria (8) (serum creatinine 2.1 mg/dl), an elevated lactate level (lactate 5.9 mmol/l, pH 7.36) and markedly elevated markers of inflammation [C-reactive protein (CRP) 199 mg/dl, procalcitonin (PCT) 26 μg/l]. The patient's complete blood count (CBC) showed severe pancytopenia [hemoglobin (Hgb) 7.5 mg/dl, hematocrit (Hct) 20.9%, white blood cells (WBC) 0.2 × 109/l, platelets (PLT) 39 × 109/l], raising concern about a neutropenic fever syndrome. The patient's history revealed, that he was on day 11 of his first cycle of a standard chemotherapeutic regime of gemcitabine (500 mg/m2; dose reduction of 50% due to bilirubinemia prior to treatment initiation) and cisplatin (25 mg/m2) for advanced cholangiocarcinoma (9), which he had received in an outpatient setting on days 1 and 8. Thus, a neutropenic fever grade 4 according to Common Terminology Criteria for Adverse Events (CTCAE, version 5) (10) was considered as an unexpected complication to this particular palliative chemotherapy regimen. An empiric broad-spectrum anti-infective therapy according to our institutional guidelines comprising the intravenous administration of meropenem, metronidazole, and vancomycin was immediately initiated. Additionally, packed red blood cells and granulocyte-colony stimulating factor (G-CSF) from the day of admission were given and the patient was transferred to intensive care unit (ICU) for initial management and monitoring.
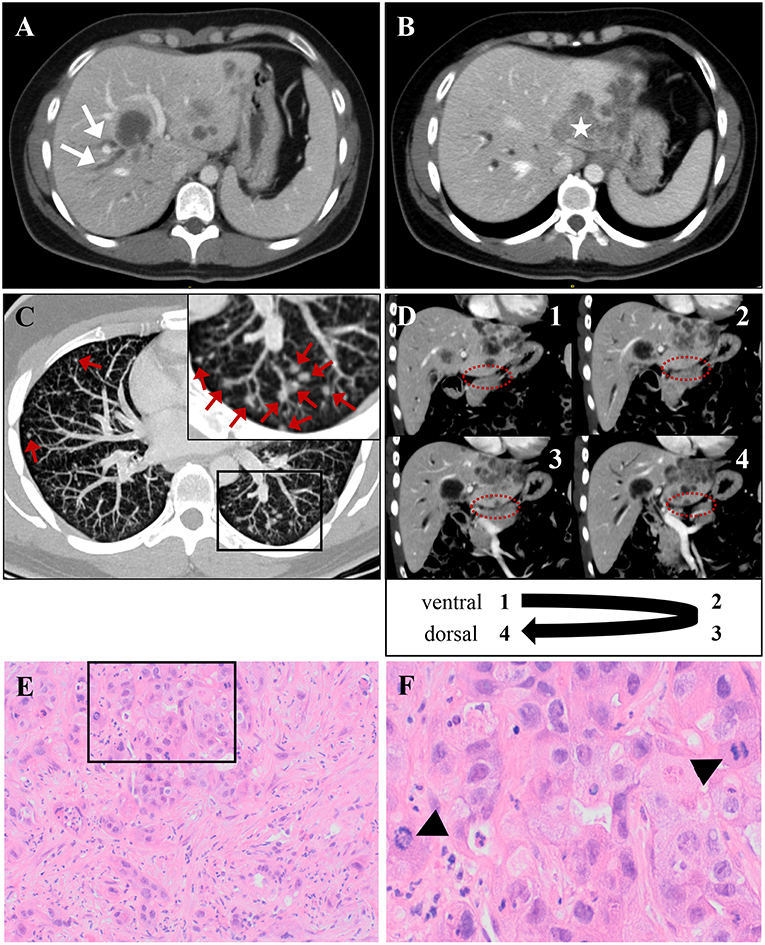
Figure 1. The patient's intrahepatic cholangiocarcinoma, which was classified as TNM: cT3, cN1, cM1, and stage IV tumor [according to AJCC/UICC staging criteria (6)]. (A,B) CT scan demonstrates multiple liver lesions, exemplary shown on two different axial layers. (A) Arrows indicate right-sided cholestasis, caused by a hypodense lesion in liver segments IV/VIII. (B) Asterisk marks a large, lobular-configured tumor mass in liver segment II, that dorsally crosses liver capsule. (C) A representative axial CT scan layer shows multiple bilateral lung noduli, interpreted as pulmonary metastases (indicated by red arrows). A framed image section is displayed as magnified inset in the top left corner of (C). A likely single spleen metastasis and enlargement of retroperitoneal lymph nodes are not shown. (D) The unaffected pancreas neighboring the infiltrated liver is depicted in four serial CT scan layers in coronal plane (consecutively numbered 1–4 from ventral to dorsal). The border between both organs (highlighted by dashed ovals) is not obviously violated by infiltrating tumor tissue, arguing against a primarily pancreatic origin of malignancy. (E,F) Histology from ultrasound-guided liver-biopsy. (E) H&E stain shows infiltrates of an adenocarcinoma with scattered duct formation and a desmoplastic stroma component. (F) High-power field from inset in (E) demonstrates frequent mitoses (indicated by arrow-heads). Immunohistochemistry (not shown) reveals positivity for CK7 and DPC4 and negativity for Hep Par 1, CK20, TTF1, CDX-2, and PSMA, consistent with a tumor of biliopancreatic origin.
Extensive diagnostic work-up for possible sites of infection included thorough physical examination, and repetitive blood and urine culture sampling. A computed tomography (CT) scan of chest and abdomen led to suspicion of neutropenic colitis. Stent placement therapy for a tumor-compression of the right hepatic duct (cf. Figure 1A) with moderate peripherical cholestasis was performed, but demonstrated no clear evidence for cholangitis. Bacteremia with vancomycin-resistant enterococcus species was detected over the course of hospital admission and addressed by appropriate adaption of antibiotic regimen (switch from vancomycin to linezolid). Moreover, empiric antifungal therapy using fluconazole was timely initiated. Transfusions of red blood cells and platelets were repeatedly performed. However, irrespective of therapeutic approach, the patient's clinical condition increasingly deteriorated with persisting signs of inflammation and no sustainable signs of bone marrow recovery until day 22 of chemotherapy cycle (Hgb 11.2 mg/dl, Hct 32.0%, WBC 0.2 × 109/l, PLT 16 × 109/l). Notwithstanding continuous G-CSF stimulation, not a single day with leukocytes >0.5 × 109/l was recorded during hospital stay. Given uncontrolled infection in the setting of refractory aplasia and the context of a palliative tumor setting, intensive care treatment was withheld, and the patient deceased on day 25 of his first cycle of chemotherapy and 14 days after hospital admission, making for an unusual grade five toxicity of a first cycle of palliative chemotherapy for this patient.
Phenotypic Features and Family History
Two days prior to death, the constellation of bone-marrow failure after exposure to a DNA crosslinking chemotherapeutic drug (cisplatin) and the presence of early-onset solid malignancy at the age of 33 years pointed to the possibility of an inherited DNA repair defect. A thorough evaluation of phenotypic features and past medical history identified several physical features suggestive of a hereditary syndrome. The patient displayed a short stature of 158 cm [1.8th percentile, −2.1 SD from average for Turkish men (11)] and had a solitary pelvic kidney representing a congenital kidney malformation diagnosed 7 years before. Additionally, his parents retrospectively reported a past surgery for a malformation of the left thumb at the age of 5 years and multiple surgeries for correction of bilateral ear canal malformations during childhood back in Turkey. A premature graying of the beard hair at the age of 18 years was also reported. However, the patient's CBC was never judged abnormal. It was completely normal 7 years before and only showed a moderate normocytic normochromic anemia prior to initiation of chemotherapy (Hgb 9.5 mg/dl, Hct 30.6%, WBC 9 × 109/l, PLT 464 × 109/l), which might also be explainable in the setting of advanced malignancy. The patient had a healthy 27-year-old brother of normal stature. The patient's parents and grandparents anamnestically displayed no phenotypic signs of hereditary syndromes. Besides, there was evidence in this patient's medical records for a long lasting unspecific chronic liver disease with undulating elevation of transaminases and cholestatic parameters for a period of ~5 years prior to diagnosis of cholangiocarcinoma, for which no clear cause had been established. A viral form of hepatitis, autoimmune biliary or liver diseases, hemochromatosis or secondary iron overload, gallstone disease, larger congenital malformations of the biliary tract as well as cirrhosis had been ruled out in previous hepatologic evaluations. The pathologist's interpretation of a liver biopsy, obtained 4.5 years prior to the diagnosis of cholangiocarcinoma, was toxic hepatitis, although a linked toxic agent could not be identified.
Genetic Testing
To unravel the putative genetic cause of patient's condition we performed whole-exome sequencing (WES) from DNA isolated from buccal swabs, as previously described (12). Buccal swab samples were collected 2 days prior to death (on day 12 after hospital admission). Bioinformatic filtering concentrated on rare variants within Mendelian cancer-risk DNA-repair genes, from the curated COSMIC germline cancer census gene set (v86; http://cancer.sanger.ac.uk/census). This analysis revealed two heterozygous variants in FANCA. Namely, a missense variant c.3391A>G, (p.Thr1131Ala), that had already been associated with FA (13–19), and a novel variant within the splice acceptor site of intron 7, c.710-3A>G. In silico prediction using NNSplice (20) indicated a disrupted splice acceptor site for this variant. Notably, a disrupting variant of the same splice acceptor (c.710-5T>C) had been previously described in a FA patient and was shown to induce an aberrant FANCA isoform by skipping of exon 8 (21). Segregation analysis with DNA samples of both healthy parents and his brother revealed the missense variant to be paternally inherited, whereas both the mother and his brother only bear the c.710-3A>G variant (Figure 2A), confirming the compound heterozygosity and an autosomal recessive inheritance. Further, RT-PCR analysis performed with RNA isolated from lymphocytes of the healthy brother as previously described (22) revealed that the c.710-3A>G variant causes skipping of exon 8 (Figure 2B), thus confirming its pathogenicity and allowing the post mortem diagnosis of FA in the index patient. Notably, primary dermal fibroblasts from a FA patient harboring the c.710-5T>C variant of FANCA (in compound heterozygosity with c.3558insG; FA-52 cells), that also causes skipping of exon 8 (21), demonstrated chromosomal instability after exposure to diepoxybutane (23). Furthermore, a previous study has shown that the c.3391A>G variant in FANCA significantly decreases FANCD2 monoubiquitination and the number of FANCD2 foci before and after mitomyocin C treatment (24), which is the critical event in the Fanconi/BRCA pathway for initiation of DNA repair (25). Taken together, the herein performed genetic analyses identified compound heterozygosity for two clearly pathogenic FANCA mutations thereby allowing the post-mortem clinical diagnosis of FA according to the published guidelines (4).
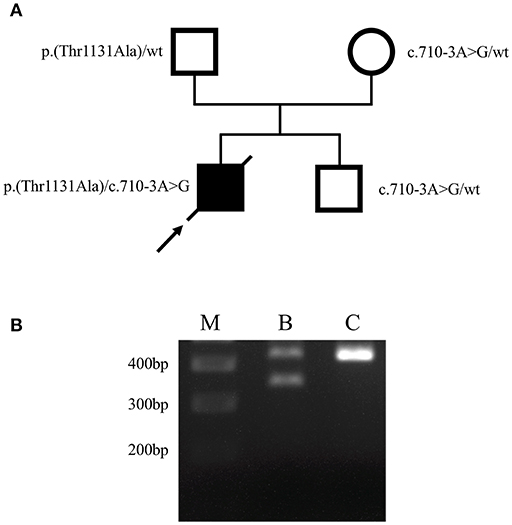
Figure 2. Identification of causative FANCA mutations. (A) Pedigree of the family. Filled and open symbols denote affected and healthy individuals, respectively; an arrow indicates the index patient, diagonal lines indicate deceased status. The mutation status is shown next to each symbol. (B) Agarose gel electrophoresis of FANCA RT-PCR products performed using lymphocyte-derived RNA of the patient's brother (B) who bears the heterozygous c.710-3A>G mutation, and a healthy individual (C), generated using primers F5′-AAGGCATTGTG AGCCTGCAAGA-3′ and R5′-ACAGGGCTGTGAGTGAGTATCTGA-3′. The 436-bp wild-type RT-PCR amplicon was amplified in both the patient's brother (B) and the control (C), using exon 8 flanking primers. A second, smaller PCR product of ~350 bp was only obtained in the patient's brother (B), corresponding to skipping of exon 8 (exon 8 contains 83 bp). The 400, 300, and 200 bp reference bands of the molecular marker (M) are indicated.
Discussion
Fanconi Anemia is typically considered a pediatric disorder [median age at diagnosis 4.8–7.5 years (26, 27)] and this patient's phenotypic presentation was exceptional in two aspects. First, he never presented with hematologic abnormalities until initiation of systemic tumor therapy, although the majority of FA patients develop pancytopenia as a pathognomonic hallmark within the first decade of life, and the cumulative incidence of bone marrow failure is reported to be 90% by the age of 40 (28). Second, even though the patient was diagnosed with a relatively early-onset solid malignancy [median age at diagnosis for intrahepatic cholangiocarcinoma 67 years (29)], raising concern of an inherited condition, this is to our knowledge the first reported case of cholangiocarcinoma associated with FA so far. Notwithstanding, FA patients are at high risk for the development of myelodysplastic syndrome, acute myeloid leukemia and various solid malignancies especially head and neck squamous cell carcinomas (HNSCCs) (26, 30). FA patients are also prone to the occurrence of liver tumors (comprising hepatocellular carcinoma, hepatic adenoma, and hepatoblastoma) (31–34) However, formation of liver tumors is mainly observed in FA patients receiving androgen treatment (32) for palliation of bone marrow failure. Notably, the herein described patient was never on androgen treatment nor ever had an indication to be put on. Biliary tract tumors in FA patients have not been reported in literature (35). However, this patient had displayed blood value alterations non-specifically indicative of chronic liver disease years before the onset of tumor formation, which is a known phenotype in FA (33). Notably, chronic liver disease of various etiologies may contribute to the risk of cholangiocarcinoma formation in the general population (36) and might have facilitated cholangiocarcinoma formation in this patient with FA background. Moreover, the patient‘s premature hair graying, although not a validated feature of FA, is a common clinical sign of many genome instability disorders (12, 37). Thus, the finding of premature graying of the beard hair in the herein presented patient actually further expands the phenotypic spectrum of FA.
The patient's rapid demise most likely resulted from his particular treatment, particularly utilizing the gemcitabine/cisplatin chemotherapeutic regime. Thus, a timely awareness of underlying FA would have resulted in a different therapeutic approach with a putatively better outcome. Hypersensitivity to DNA crosslinking agents, including cisplatin (38–41), is one of the major features of FA (42), even utilized in diagnostic procedures, and fatal toxicity in FA patients exposed to cisplatin has already been documented (43–45). The gemcitabine plus cisplatin combination therapy remains the mainstay for the treatment of advanced cholangiocarcinoma and allow for a median overall survival of ~12 months (9, 46, 47). By choosing the gemcitabine combination partner cisplatin, the actual survival of this patient was 24 days from start of therapy due to fatal myelotoxicity. Noteworthy, a speculative combination with either capecitabine or S-1 (which is available in Germany), that demonstrated comparable efficacy to cisplatin in sporadic cholangiocarcinoma (46, 47), although head-to-head comparison data of gemcitabine-combinations is missing, might have been better tolerated. Even a conservative approach with gemcitabine monotherapy allows for a median overall survival of ~7 months (9) in sporadic bile duct cancer. Gemcitabine, capecitabine and tegafur (antineoplastic component of S-1) all belong to the class of pyrimidine analogs which exert cytotoxicity by inhibiting DNA replication, a mechanism of action thought to be of better tolerability in FA patients compared to direct DNA damage by crosslinking agents (48, 49). Genotoxic agents of this group (e.g., fludarabine, cytarabine) were well-tolerated in stem cell transplantation conditioning regimes for pediatric FA patients treated for myeloid malignancies (48, 50).
This is not the first report of delayed diagnosis of FA, being diagnosed only after unforeseen toxicity of DNA crosslinking agents in a primarily presumed sporadic-onset malignancy (44, 51). Thus, FA should be ruled out in early-onset malignancies, especially, if a typically FA-associated malignancy is diagnosed. However, the onset of a typically non-FA associated tumor type, such as cholangiocarcinoma does not rule out atypical presentation of FA, as this case demonstrated. Thus, the possibility of an underlying hereditary syndrome in cases manifesting as early-onset malignancies should be considered prior to the start of treatment. Notably, excessive toxicity following antineoplastic therapy is not solely attributable to FA, but has been observed in a broad range of genome instability syndromes, including Nijmegen breakage syndrome (52), Xeroderma pigmentosum (53), Bloom syndrome (54), Werner syndrome (55), Ataxia-telangiectasia (56), and Ruijs–Aalfs syndrome (37). Although all of these conditions are exceedingly rare and many of these patients become diagnosed early in life due to the multisystem nature of these disorders, some might actually lack the exact diagnosis prior to onset of malignancy formation. A further diagnostic burden represents the fact that although all genome instability disorders can result in multi-organ symptoms, the primarily affected organs, clinical presentations, and courses are highly variable. Moreover, based on the underlying genetic cause and the affected genome maintenance system, resulting DNA lesions and hypersensitivity toward genotoxic agents highly differ between these syndromes (Table 1). Thus, the precise elucidation of the underlying genetic cause represents the prerequisite for the selection of optimal antineoplastic therapy in genome instability disorders. Modern genetic analyses, e.g., whole-genome sequencing (WGS) by next-generation sequencing (NGS) technologies, covering the complete human genome, can utilize successful differential diagnosis of a genetic disorder within 50-h (57). Therefore, it is theoretically possible to perform those analyses without accepting an unfavorable delay of treatment initiation in tumor patients. However, these tests can also identify variants of unknown significance (VUS), whose pathogenicity can sometimes not be deduced without time consuming follow-up analyses. A further burden, at least in Germany, is the fact that the health insurance covers only WES, which compared to WGS does not properly cover the whole genome and even results in the poorer coverage of coding regions (exons), not to mention that it takes considerably longer, up to 2 weeks. It is further worth noting that NGS-based testing of the tumor sample first, would have also likely identified two heterozygous FANCA mutations. Misinterpretation of this finding, which is not completely excluded without pairing to normal tissue, could have potentially led to misinterpretation with even recommending a cisplatin-based therapy in this patient as the onset of somatic Fanconi/BRCA pathway defects is a feature of some cisplatin-sensitive tumors (58, 59). For experienced investigators, such a fault is unlikely to make, though. Indeed, the vast majority of early-onset cancer patients, which are either non-syndromic patients or have a negative family history, receive genetic analyses of normal tissue only after a mutation in a gene known to be associated with germline cancer predisposition is first identified in the tumor sample (60). However, the targeted re-analysis of the normal sample takes additional time, either resulting in treatment delay or start of an inappropriate therapy. Therefore, we suggest that in cases of early-onset cancer a paired tumor and normal NGS-based analysis should be performed, which additionally reduces the number of identified VUS (60).
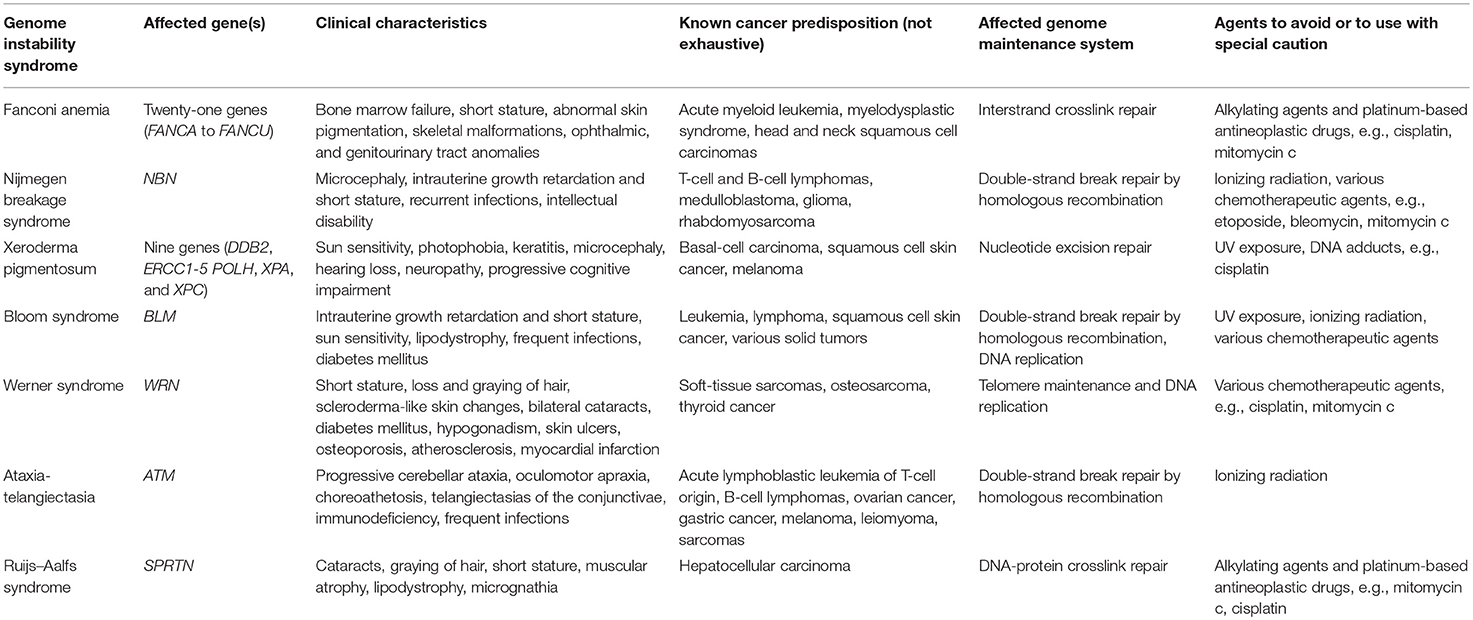
Table 1. Comparison of typical features of various genome instability syndromes.
Concluding Remarks
One major aspect of precision medicine in oncology is the identification of patients with germline cancer predisposing mutations, in order to optimize the initial therapy. The latter is further highlighted by the herein presented patient with normal hematologic values and a non-classically FA-associated early-onset cancer followed by fatal outcome due to therapy-induced myelotoxicity. In order to identify such patients, the clinical team should perform an in-depth investigation of patient's family history and previous medical records, followed by rapid and appropriate genetic analyses, in order to pinpoint or rule out a possibly underlying hereditary genome instability syndrome and to facilitate a rational treatment strategy in early-onset malignancies. However, it seems a vain attempt to derive a solid guideline for this rare clinical problem mostly grounded on experiences from sporadic case reports. Nevertheless, we propose a multidisciplinary management for any patient with an early-onset cancer (relatively with respect to patient's risk profile and epidemiologic data for the respective tumor entity). Those who additionally display at least subtle phenotypical features suggestive of a hereditary syndrome should be considered for paired tumor and normal NGS-based genomic testing.
Ethics Statement
Collection of biological samples and genetic analyses were conducted following written informed consent from studied individuals or his legal representatives. In addition, written informed consent for the publication of this case was obtained from the family of the deceased patient. The study was performed in accordance with the Declaration of Helsinki protocols.
Author Contributions
NE, SS, and DL designed the study. NE and DL prepared the manuscript. NE is responsible for clinical information gathering. NE and DL are responsible for sample collection and consent. US assisted with histology. CF, CB, CK, and DL supervised the study and data analysis.
Funding
This research was funded in part through the German Cancer Aid (Deutsche Krebshilfe) to DL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the family members for their participation.
References
1. Terabayashi T, Hanada K. Genome instability syndromes caused by impaired DNA repair and aberrant DNA damage responses. Cell Biol Toxicol. (2018) 34:337–50. doi: 10.1007/s10565-018-9429-x
2. Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JHJ, Robbins PD. Nuclear Genomic Instability and Aging. Annu Rev Biochem. (2018) 87:295–322. doi: 10.1146/annurev-biochem-062917-012239
3. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
4. Mehta PA, Tolar J. Fanconi Anemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews (R). Seattle, WA: University of Washington (2002).
5. D' Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. (2003) 3:23–34. doi: 10.1038/nrc970
6. The American Joint Committee on Cancer (AJCC). AJCC Cancer Staging Manual. 8th ed. Springer International Publishing (2017). Available online at: https://www.springer.com/us/book/9783319406176
7. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:762–74. doi: 10.1001/jama.2016.0288
8. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. (2012) 120:c179–84. doi: 10.1159/000339789
9. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. (2010) 362:1273–81. doi: 10.1056/NEJMoa0908721
10. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), Version 5.0. (2017). Available online at: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf accessed (April 16, 2019)
11. Iseri A, Arslan N. Obesity in adults in Turkey: age and regional effects. Eur J Public Health. (2009) 19:91–4. doi: 10.1093/eurpub/ckn107
12. Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, et al. Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Invest. (2017) 127:3598–608. doi: 10.1172/JCI92171
13. Levran O, Erlich T, Magdalena N, Gregory JJ, Batish SD, Verlander PC, et al. Sequence variation in the Fanconi anemia gene FAA. Proc Natl Acad Sci USA. (1997) 94:13051–6. doi: 10.1073/pnas.94.24.13051
14. Adachi D, Oda T, Yagasaki H, Nakasato K, Taniguchi T, D'andrea AD, et al. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum Mol Genet. (2002) 11:3125–34. doi: 10.1093/hmg/11.25.3125
15. Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. (2005) 37:931–3. doi: 10.1038/ng1624
16. Ameziane N, Errami A, Leveille F, Fontaine C, De Vries Y, Van Spaendonk RM, et al. Genetic subtyping of Fanconi anemia by comprehensive mutation screening. Hum Mutat. (2008) 29:159–66. doi: 10.1002/humu.20625
17. Moghrabi NN, Johnson MA, Yoshitomi MJ, Zhu X, Al-Dhalimy MJ, Olson SB, et al. Validation of Fanconi anemia complementation Group A assignment using molecular analysis. Genet Med. (2009) 11:183–92. doi: 10.1097/GIM.0b013e318193ba67
18. Pinto FO, Leblanc T, Chamousset D, Le Roux G, Brethon B, Cassinat B, et al. Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica. (2009) 94:487–95. doi: 10.3324/haematol.13592
19. Gille JJP, Floor K, Kerkhoven L, Ameziane N, Joenje H, De Winter JP. Diagnosis of Fanconi anemia: mutation analysis by multiplex ligation-dependent probe amplification and PCR-based sanger sequencing. Anemia. (2012) 2012:603253. doi: 10.1155/2012/603253
20. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. (1997) 4:311–23. doi: 10.1089/cmb.1997.4.311
21. Mattioli C, Pianigiani G, De Rocco D, Bianco AM, Cappelli E, Savoia A, et al. Unusual splice site mutations disrupt FANCA exon 8 definition. Biochim Biophys Acta. (2014) 1842:1052–8. doi: 10.1016/j.bbadis.2014.03.014
22. Girisha KM, Bidchol AM, Graul-Neumann L, Gupta A, Hehr U, Lessel D, et al. Phenotype and genotype in patients with Larsen syndrome: clinical homogeneity and allelic heterogeneity in seven patients. BMC Med Genet. (2016) 17:27. doi: 10.1186/s12881-016-0290-6
23. Rio P, Baños R, Lombardo A, Quintana-Bustamante O, Alvarez L, Garate Z, et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med. (2014) 6:835–48. doi: 10.15252/emmm.201303374
24. Wilkes DC, Sailer V, Xue H, Cheng H, Collins CC, Gleave M, et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harbor Mol Case Stud. (2017) 3:a001487. doi: 10.1101/mcs.a001487
25. Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. (2001) 7:249–62. doi: 10.1016/S1097-2765(01)00173-3
26. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. (2003) 101:822–6. doi: 10.1182/blood-2002-05-1498
27. Risitano AM, Marotta S, Calzone R, Grimaldi F, Zatterale A. Twenty years of the Italian Fanconi anemia Registry: where we stand and what remains to be learned. Haematologica. (2016) 101:319–27. doi: 10.3324/haematol.2015.133520
28. Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on the international fanconi anemia registry (IFAR). Blood. (2003) 101:1249–56. doi: 10.1182/blood-2002-07-2170
29. Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist. (2016) 21:594–99. doi: 10.1634/theoncologist.2015-0446
30. Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. (2003) 97:425–40. doi: 10.1002/cncr.11046
31. Touraine RL, Bertrand Y, Foray P, Gilly J, Philippe N. Hepatic tumours during androgen therapy in Fanconi anaemia. Eur. J. Pediatr. (1993) 152:691–3. doi: 10.1007/BF01955250
32. Velazquez I, Alter BP. Androgens and liver tumors: Fanconi's anemia and non-Fanconi's conditions. Am J Hematol. (2004) 77:257–67. doi: 10.1002/ajh.20183
33. Masserot-Lureau C, Adoui N, Degos F, De Bazelaire C, Soulier J, Chevret S, et al. Incidence of liver abnormalities in Fanconi anemia patients. Am J Hematol. (2012) 87:547–9. doi: 10.1002/ajh.23153
34. Paustian L, Chao MM, Hanenberg H, Schindler D, Neitzel H, Kratz CP, et al. Androgen therapy in Fanconi anemia: a retrospective analysis of 30 years in Germany. Pediatr Hematol Oncol. (2016) 33:5–12. doi: 10.3109/08880018.2015.1129567
35. Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. (2014) 27:214–21. doi: 10.1016/j.beha.2014.10.002
36. Welzel TM, Graubard BI, El-Serag HB, Shaib YH, Hsing AW, Davila JA, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clin Gastroenterol Hepatol. (2007) 5:1221–8. doi: 10.1016/j.cgh.2007.05.020
37. Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. (2014) 46:1239–44. doi: 10.1038/ng.3103
38. Poll EH, Arwert F, Joenje H, Eriksson AW. Cytogenetic toxicity of antitumor platinum compounds in Fanconi's anemia. Hum Genet. (1982) 61:228–30. doi: 10.1007/BF00296447
39. Poll EH, Arwert F, Joenje H, Wanamarta AH. Differential sensitivity of Fanconi anaemia lymphocytes to the clastogenic action of cis-diamminedichloroplatinum (II) and trans-diamminedichloroplatinum (II). Hum Genet. (1985) 71:206–10. doi: 10.1007/BF00284574
40. Plooy AC, Van Dijk M, Berends F, Lohman PH. Formation and repair of DNA interstrand cross-links in relation to cytotoxicity and unscheduled DNA synthesis induced in control and mutant human cells treated with cis-diamminedichloroplatinum(II). Cancer Res. (1985) 45:4178–84.
41. Ferrer M, Izeboud T, Ferreira CG, Span SW, Giaccone G, Kruyt FA. Cisplatin triggers apoptotic or nonapoptotic cell death in Fanconi anemia lymphoblasts in a concentration-dependent manner. Exp Cell Res. (2003) 286:381–95. doi: 10.1016/S0014-4827(03)00112-5
42. Sasaki MS, Tonomura A. A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. (1973) 33:1829–36.
43. Carvalho JP, Dias ML, Carvalho FM, Del Pilar Estevez Diz M, Petito JW. Squamous cell vulvar carcinoma associated with Fanconi's anemia: a case report. Int J Gynecol Cancer. (2002) 12:220–2. doi: 10.1046/j.1525-1438.2002.01090.x
44. Tan IB, Cutcutache I, Zang ZJ, Iqbal J, Yap SF, Hwang W, et al. Fanconi's anemia in adulthood: chemoradiation-induced bone marrow failure and a novel FANCA mutation identified by targeted deep sequencing. J Clin Oncol. (2011) 29:e591–4. doi: 10.1200/JCO.2011.35.1064
45. Spanier G, Pohl F, Giese T, Meier JK, Koelbl O, Reichert TE. Fatal course of tonsillar squamous cell carcinoma associated with Fanconi anaemia: a mini review. J Craniomaxillofac Surg. (2012) 40:510–5. doi: 10.1016/j.jcms.2011.08.013
46. Knox JJ, Hedley D, Oza A, Feld R, Siu LL, Chen E, et al. Combining gemcitabine and capecitabine in patients with advanced biliary cancer: a phase II trial. J Clin Oncol. (2005) 23:2332–8. doi: 10.1200/JCO.2005.51.008
47. Morizane C, Okusaka T, Mizusawa J, Katayama H, Ueno M, Ikeda M, et al. Randomized phase III study of gemcitabine plus S-1 combination therapy versus gencitabine plus cisplatin combination therpay in advanced biliary cancer: A japan oncology group study (JCOG1113, FUGA-BT) (abstract). J Clin Oncol. (2018) 36 (Suppl. 4):205. doi: 10.1200/JCO.2018.36.4-suppl.205
48. Mehta PA, Ileri T, Harris RE, Williams DA, Mo J, Smolarek T, et al. Chemotherapy for myeloid malignancy in children with Fanconi anemia. Pediatr Blood Cancer. (2007) 48:668–72. doi: 10.1002/pbc.20843
49. Yabe M, Yabe H, Hamanoue S, Inoue H, Matsumoto M, Koike T, et al. In vitro effect of fludarabine, cyclophosphamide, and cytosine arabinoside on chromosome breakage in Fanconi anemia patients: relevance to stem cell transplantation. Int J Hematol. (2007) 85:354–61. doi: 10.1532/IJH97.06191
50. Stepensky P, Shapira MY, Balashov D, Trakhtman P, Skorobogatova E, Rheingold L, et al. Bone marrow transplantation for Fanconi anemia using fludarabine-based conditioning. Biol Blood Marrow Transplant. (2011) 17:1282–8. doi: 10.1016/j.bbmt.2011.01.001
51. Huck K, Hanenberg H, Gudowius S, Fenk R, Kalb R, Neveling K, et al. Delayed diagnosis and complications of Fanconi anaemia at advanced age–a paradigm. Br J Haematol. (2006) 133:188–97. doi: 10.1111/j.1365-2141.2006.05998.x
52. Digweed M, Sperling K. Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair. (2004) 3:1207–17. doi: 10.1016/j.dnarep.2004.03.004
53. Sumiyoshi M, Soda H, Sadanaga N, Taniguchi H, Ikeda T, Maruta H, et al. Alert Regarding cisplatin-induced severe adverse events in cancer patients with Xeroderma Pigmentosum. Intern Med. (2017) 56:979–82. doi: 10.2169/internalmedicine.56.7866
54. Thomas ER, Shanley S, Walker L, Eeles R. Surveillance and treatment of malignancy in Bloom syndrome. Clin Oncol. (2008) 20:375–9. doi: 10.1016/j.clon.2008.01.007
55. Seiter K, Qureshi A, Liu D, Galvin-Parton P, Arshad M, Agoliati G, et al. Severe toxicity following induction chemotherapy for acute myelogenous leukemia in a patient with Werner's syndrome. Leuk Lymphoma. (2005) 46:1091–5. doi: 10.1080/10428190500102688
56. Yanofsky RA, Seshia SS, Dawson AJ, Stobart K, Greenberg CR, Booth FA, et al. Ataxia-telangiectasia: atypical presentation and toxicity of cancer treatment. Can J Neurol Sci. (2009) 36:462–7. doi: 10.1017/S0317167100007794
57. Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. (2012) 4:154ra135. doi: 10.1126/scitranslmed.3004041
58. Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. (2003) 9:568–74. doi: 10.1038/nm852
59. Verhagen CVM, Vossen DM, Borgmann K, Hageman F, Grenman R, Verwijs-Janssen M, et al. Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget. (2018) 9:18198–213. doi: 10.18632/oncotarget.24797
Keywords: fanconi anemia, FANCA, cholangiocarcinoma, genomic instability, myelotoxicity
Citation: Engel NW, Schliffke S, Schüller U, Frenzel C, Bokemeyer C, Kubisch C and Lessel D (2019) Fatal Myelotoxicity Following Palliative Chemotherapy With Cisplatin and Gemcitabine in a Patient With Stage IV Cholangiocarcinoma Linked to Post Mortem Diagnosis of Fanconi Anemia. Front. Oncol. 9:420. doi: 10.3389/fonc.2019.00420
Received: 04 January 2019; Accepted: 03 May 2019;
Published: 22 May 2019.
Edited by:
Christian Celia, Università degli Studi G. d'Annunzio Chieti e Pescara, ItalyReviewed by:
Jochen K. Lennerz, Harvard Medical School, United StatesGunjan Arora, National Institutes of Health (NIH), United States
Copyright © 2019 Engel, Schliffke, Schüller, Frenzel, Bokemeyer, Kubisch and Lessel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nils W. Engel, bi5lbmdlbEB1a2UuZGU=
Davor Lessel, ZC5sZXNzZWxAdWtlLmRl